

Abstract
Second-generation chimeric antigen receptors (CARs) are powerful tools to redirect antigen-specific T cells independently of HLA-restriction. Recent clinical studies evaluating CD19-targeted T cells in patients with B-cell malignancies demonstrate the potency of CAR-engineered T cells. With results from 28 subjects enrolled by five centers conducting studies in patients with chronic lymphocytic leukemia (CLL) or lymphoma, some insights into the parameters that determine T-cell function and clinical outcome of CAR-based approaches are emerging. These parameters involve CAR design, T-cell production methods, conditioning chemotherapy as well as patient selection. Here, we discuss the potential relevance of these findings and in particular the interplay between the adoptive transfer of T cells and pre-transfer patient conditioning.
Keywords: CD19, adoptive T-cell therapy, chimeric antigen receptors, gene therapy
Introduction
Chimeric antigen receptors (CARs) are emerging as powerful tools for reprogramming T-cell specificity and function.1-3 CARs are hybrid receptors comprising a ligand for a cell-surface molecule, most often consisting of a single-chain variable fragment (scFv) derived from a monoclonal antibody or an antigen-binding fragment (Fab) fused to signaling domains assembled to redirect T-cell function.4 Unlike transduced T cell receptors (TCRs), CARs endow T cells with a new specificity that is independent of HLA restriction and do so without competing with the endogenous TCR for the rate-limiting CD3 complex. First-generation CARs mediated limited T-cell activation, enabling cytotoxicity but only short-term T-cell expansion. Second-generation CARs, which combine activating and co-stimulatory signaling domains, enable improved cytokine secretion, T-cell expansion upon repeated antigen exposure and T-cell persistence.1,5 CARs have been generated against a large number of cell surface molecules,4 including CD19, HER2, GD2, prostate-specific membrane antigen (PSMA) and mesothelin, and many of them are presently under evaluation in over 30 phase I clinical trials (www.clinicaltrials.gov). To date, the most promising clinical outcomes of this technology have been reported in patients treated with autologous CAR-modified T cells targeting CD19.6-10 CD19 is an attractive target for CAR-based therapy as it is expressed by most B-cell leukemias and lymphomas but not in tissues other than normal B lineage cells.11,12 In pre-clinical settings, CD19+ malignancies were the first cancers to be eliminated by CAR-engineered human T cells administered intravenously to systemic tumor-bearing mice.13 Successful B-cell tumor eradication was eventually obtained with different CD19-directed CARs,14-17 paving the way for multiple clinical studies and making the targeting of CD19 a paradigm for evaluating CAR technology.18 Here, we review and compare recently published results from clinical trials involving patients treated with CD19-targeted, CAR-modified T cells. These results identify at least some of the requirements for effective CAR therapy that should inform the design of future clinical studies.
Clinical Outcomes in the First Six Clinical Trials Targeting CD19 with CARs
The results of 6 clinical trials targeting CD19+ malignancies utilizing CAR-targeted autologous T cells have been recently reported.6-10,19,20 A total of 28 patients were treated, including 22 with chronic lymphocytic leukemia (CLL, Table 1). Jensen, et al.19 reported of two patients with relapsed follicular lymphoma who were treated with multiple infusions of CD19-targeted clonal T cells. Both patients developed progressive disease (PD) within 6 mo after the last T-cell infusion.19 Savoldo, et al.20 reported results from six patients with indolent or aggressive lymphomas, of whom two had stable disease (SD), the longest duration being 10 mo, while the other four developed PD. Kochenderfer, et al.6 reported the first promising clinical outcome with CD19-targeted T-cell therapy in a patient with relapsed follicular lymphoma who achieved a partial response (PR) as well as B-cell aplasia, a surrogate marker for CAR-modified T-cell functionality in vivo. Three studies from the Abramson Family Cancer Research Institute at the University of Pennsylvania (UPenn), Memorial Sloan-Kettering Cancer (MSKCC), and the National Cancer Institute (NCI) published in late 2011 used approaches that were overall similar but differed in some aspects of CAR design, T-cell manufacturing, patient selection and patient conditioning, setting the stage for insightful comparisons. The NCI group reported on the retreatment of their first patient6 and an additional cohort of seven patients (four with CLL, three with follicular lymphoma and one with marginal zone lymphoma).10 One patient showed a complete response (CR) and another SD. The remaining evaluable patients achieved PRs. Four of the eight patients receiving CAR-modified T cells exhibited B cell aplasias. Brentjens, et al.9 reported the results from two trials involving 8 patients with CLL and one patient with B-cell acute lymphoblastic leukemia (B-ALL). In the CLL cohort, two patients manifested SD and one patient demonstrated a substantial reduction in lymph node mass. None of the CLL patients developed B-cell aplasia, in contrast to the patient with relapsed B-ALL, who was in remission at the time of therapy and promptly developed this surrogate marker of T-cell functionality. June and colleagues7,8 treated three CLL patients: two achieved a CR and the third demonstrated a PR following T-cell therapy. One of the patients developed a sustained B-cell aplasia. Collectively, patients tolerated the infusion of autologous CD19-targeted T cells well, with common toxicities including fever, hypotension, lymphopenia and delayed tumor lysis syndrome.6-10,19,20 No deaths that could be directly attributed to the infusion of CD19-targeted T cells have been reported.
Table 1. Comparison of tumor burdens and outcome after infusion of anti-CD19 T cells into chronic lymphocytic leukemia (CLL) patients.
| Patient* | CAR+ T-cell dose (per kg) | CD4+/CD8+ ratio | Tumor burden** | E:T ratio*** | Outcome | Max VCN (per μg DNA) |
Peak CAR detection (day) |
|---|---|---|---|---|---|---|---|
| MSKCC01 |
31 × 106 |
94/5 |
4.2 × 1012 |
6.0 × 10−4 |
PD |
43 |
14 |
| MSKCC02 |
15 × 106 |
96/5 |
**** |
**** |
PD |
0 |
NE |
| MSKCC03 |
15 × 106 |
93/8 |
2.9 × 1012 |
3.7 × 10−4 |
PD |
0 |
NE |
| MSKCC05 |
5.2 × 106 |
87/12 |
2.0 × 1012 |
2.0 × 10−4 |
LN reduction |
257 |
6 |
| MSKCC06 |
4.6 × 106 |
79/21 |
2.9 × 1012 |
1.4 × 10−4 |
PD |
14 |
1 |
| MSKCC07 |
8.1 × 106 |
58/27 |
6.6 × 1011 |
1.1 × 10−3 |
SD |
6143 |
8 |
| MSKCC08 |
11 × 106 |
92/8 |
1.2 × 1012 |
1.1 × 10−3 |
SD |
1143 |
1 |
| UPENN01 |
16 × 106 |
NR |
1.7 × 1012 |
6.5 × 10−4 |
CR |
200000 |
15 |
| UPENN02 |
10 × 106 |
NR |
3.5 × 1012 |
1.7 × 10−4 |
PR |
1000 |
110 |
| UPENN03 |
0.2 × 106 |
NR |
8.8 × 1011 |
1.6 × 10−5 |
CR |
10000 |
23 |
| NCI03 |
11 × 106 |
35/53 |
NR |
|
CR |
NR |
7 |
| NCI05 |
3 × 106 |
87/12 |
NR |
|
SD |
NR |
7 |
| NCI06 |
17 × 106 |
37/57 |
NR |
|
PR |
NR |
7 |
| NCI07 | 28 × 106 | 58/41 | NR | PR | NR | 9 |
CLL, UPENN, and NCI refer to patients treated at MSKCC, UPenn, and NCI, respectively. Two CLL patients have been excluded from this table: one due to a history of Epstein-Barr virus (EBV)+ non-Hodgkin’s lymphoma,20 and one owing to early death.9 **Tumor burden for bone marrow and blood is calculated as described by Kalos, et al.7 ***The E:T ratio is calculated as the number of infused anti-CD19 T cells divided by the tumor burden. ****Bone marrow aspirate and biopsy did not include cellularity so tumor burden could not be calculated. Abbreviations: CR, complete remission; NE, not evaluable; NR, not reported; PD, progressive disease; PR, partial remission; SD, stable disease
While these clinical trials all follow a common immunotherapeutic approach (Fig. 1, inner circle), they differ with regard to several parameters (Fig. 1, outer boxes), including CAR design, T-cell manufacturing, conditioning chemotherapy, tumor burden, tumor chemo-sensitivity and T-cell dosage. A careful analysis of disease outcome in these trials provides valuable insights for refining CAR-based cancer immunotherapy.
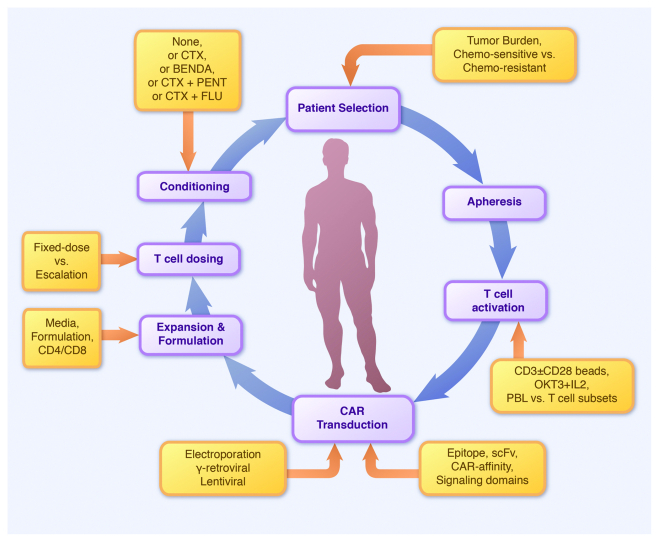
Figure 1. The mechanics of chimeric antigen receptor (CAR)-based trials. Inner circle (purple): key steps in patient preparation and T-cell manufacture. Outer circle (orange): key differences between studies targeting CD19+ malignancies with CARs. BENDA, bendamustine; CTX, cyclophosphamide; FLU, fludarabine. PENT, pentostatin.
CAR Design
CARs have considerably evolved over the past decade.4 First generation CARs, comprising an activation domain as the sole signaling component,21,22 effectively redirected cytotoxicity but showed major limitations in sustaining T-cell function.23,24 The introduction of dual-signaling receptors, combining activation and co-stimulatory signaling domains,5 paved the way for generating more potent and persisting immune responses. In an elegant side-by-side comparison, Savoldo, et al.20 demonstrated the greater persistence of T cells expressing a CD28/CD3ζ-based CAR as compared with concomitantly administered CD3ζ CAR-transduced T cells, validating earlier comparisons of first and second generation CARs in mouse models.14
Second generation CARs comprise the signaling domain of co-stimulatory receptors such as CD28, 4–1BB, OX-40, DAP10 and others,14,15,25-28 but have not been extensively compared with each other. In one study treating the CD19+ pre-B ALL cell line NALM-6 in SCID/beige mice,14 the CD28/CD3ζ construct outperformed a panel of other second generation CARs in terms of therapeutic efficacy. Milone, et al.,15 who also utilized a B-ALL model in NSG mice, found that CD28/CD3ζ- and 4–1BB/CD3ζ−based CARs are similar in terms of therapeutic efficacy, but that the 4–1BB/CD3ζ CAR-transduced T cells exhibit greater accumulation over time, possibly due to antigen-independent T-cell proliferation and persistence. CAR comparisons in xenogenic mouse models are important to study the biology of CARs and guide therapeutic choices, but they are complex to interpret. First, the xenogenic nature of these models does not recapitulate all the cell interactions that affect T-cell function, trafficking and persistence. Second, all CARs of a given kind, e.g., CD28/CD3ζ CARs, are not equal (for example, some require more interleukin-2 stimulation than others),5,29 These considerations command caution in the comparison of CARs of different types (optimized CARs representative of their category should be used for valid comparisons). In this respect, third-generation CARs combining CD28 and 4–1BB co-stimulatory signals in addition to CD3ζ-mediated activation are even more complex.25,28,30-32 Third, the role of other components of the CAR should not be underestimated. Indeed, the nature of the scFv or Fab, the topology of the targeted epitope and its distance relative to the cell surface, as well as the affinity of CARs, represent additional variables that profoundly influence CAR function.
The CD19-targeting CARs tested in CLL patients in the aforementioned clinical trials are shown in Figure 2. The constructs used at the MSKCC and NCI were based on the same CD28/CD3ζ structure,5 whereas the construct employed at UPenn utilizes a 4–1BB/CD3ζ motif.33 The NCI and UPenn groups selected the same scFv, which is different from that used at MSKCC. The three constructs thus differ in antigen recognition and/or signaling properties, but the degree to which these differences contribute to different outcomes needs to be analyzed in the context of other important parameters, as discussed below.
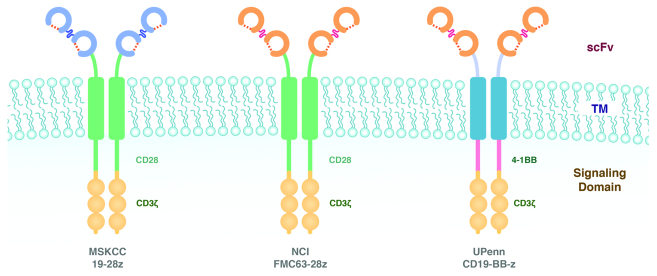
Figure 2. Schematic diagram of chimeric antigen receptor (CARs) used to treat chronic lymphocytic leukemia (CLL) patients at MSKCC, NCI and UPenn. (A) 19–28ζ (MSKCC). (B) FMC63–28ζ (NCI). (C) 19-BB-ζ (UPenn). Groups at MSKCC and NCI utilized the CD28/ζ design described by Maher, et al.5 The UPenn group used the 4–1BBζ design described by Imai, et al.33 The MSKCC group used the SJ single-chain variable fragment (scFv)13 while researchers at NCI and UPenn used the FMC63 scFv.44 TM, transmembrane.
T-Cell Manufacture
There are important differences in the T-cell production processes employed at different centers (Table 2). T-cell doses were generally obtained within 10 d to 3 weeks of ex vivo culture,7,9,20 although some approaches required a longer culture time.19 All centers use anti-CD3 antibody stimulation for T-cell activation in combination with either anti-CD28 antibody co-stimulation7,9 or co-culture with peripheral blood mononuclear cells (PBMCs).6,10,19 Although one run at UPenn failed to reach the required cell dose, the limited amount of infused T cells (14 × 106) was sufficient to achieve a ≥ 10 mo-long CR.8 The characterization of the T-cell subsets performed at 3 of the centers prior to infusion shows various levels of CD45RA, CD62L, CCR7 and CD28 expression, underscoring the variable composition in effector and central memory T cells of the administered product.6,9,10,20 Overall, all non-clonal infusion products encompassed CD4+ and CD8+ T cells, albeit with a relatively high CD4/CD8 ratio (Table 1), particularly in the MSKCC study.
Table 2. Comparison of T-cell production and phenotype at infusion.
| Center | T-cell activation | Gene delivery and expression methods | EOP T-cell phenotype | Range days in culture | Ref. |
|---|---|---|---|---|---|
| UPenn |
Anti-CD3/Anti-CD28 stimulation |
Lentiviral vector (EF-1α promoter) |
NA |
10–14 |
7, 8 |
| NCI |
Anti-CD3 (OKT3) + autologous PBMCs |
MSCV-Gammaretroviral vector |
CD45RA+ (5–26%), CD62L+(4–35%) CCR7+(5–37%) |
24 |
6, 10 |
| MSKCC |
Anti-CD3/Anti-CD28 stimulation |
SFG-Gammaretroviral vector |
CD62L+ (9–78%) CCR7+ (1–36%) CD28+ (43–94%) CD25+CD4+ FOXP3+ (0.6–2.4%) |
11–19 |
9 |
| Baylor |
Anti-CD3 (OKT3) |
SFG-Gammaretroviral vector |
CD45RA+ (0–15%) CD62L+ (15–90%) CCR7+ (0%) CD28+ (15–90%) |
6–18 |
20 |
| City of Hope | Anti-CD3 (OKT3) + PBMCs/lymphoblastoid cell lines | Plasmid electroporation and hygromycin B selection | NA | ≥ 55 | 19 |
Abbreviations: EOP, end of production; NA, not available; PBMC, peripheral blood mononuclear cell
The CD19-specific CARs were introduced in T cells by lentiviral or gamma-retroviral vector gene transfer or by electroporation. The efficiency of gene transfer is higher upon transduction with gamma-retroviral vectors than with lentiviral vectors, ranging from 4–71% and 4.7–23%, respectively.6-10,20 However, the lower transduction efficiencies do not appear to attenuate CAR-modified T-cell function, as one patient treated at UPenn developed a CR after infusion with a T-cell product exhibiting a low transduction efficiency (4.7%) and one of our patients at MSKCC had a significant decrease in lymphadenopathy after infusion with a T-cell product with one of the lowest transduction efficiencies observed in our center (32%).7-9 The wide range of transduction efficiency observed suggests that there is a large variability from patient to patient. We address this variability in our trial by normalizing the T-cell dose to CAR+ T cells, so that patients may receive different total T-cell doses but they are all infused with the same amount of CAR+ T cells.
Gamma-retroviral and lentiviral gene-transfer systems can produce active CAR-modified T cells despite highly variable gene-transfer efficiencies, thereby obviating the need for drug selection to create a T-cell product that is uniformly CAR+. Plasmid DNA electroporation followed by drug selection has been forsaken as a method for T-cell production as it may undermine the biological quality of the final cell product.19,34 It remains to be precisely determined how the distinct modes for gene transfer affect CAR expression by T cells over time, upon infusion into patients.
The long-term impact of different gene transfer modalities still remains difficult to apprehend. Scholler, et al.35 have recently demonstrated the presence of T cells harboring a gamma-retroviral encoded CD4/CD3ζ fusion receptor up to 7 y after infusion, the expression of which could be detected upon ex vivo T-cell activation. Burns, et al.36 also reported that TCR transgenes are still expressed in patients with melanoma 2 to 10 mo post-infusion. The transduced vector was likewise detected for up to 9 y in patients treated with donor-derived Epstein-Barr virus-specific cytotoxic T lymphocytes (EBV-CTLs).3 Additional insights into the in vivo expression levels of CARs over time are needed to discern the impact of viral and non-viral vectors. Controlled studies evaluating the manufacturing process are required to determine the extent to which T-cell production conditions determine the clinical outcome of CAR-based immunotherapeutic strategies.
Tumor Burden, T-Cell Persistence and Clinical Response
In our initial studies at MSKCC,9 we noted an inverse correlation between a detectable persistence of CAR-modified T cells and disease burden at the time of T-cell infusion. Moving this analysis forward, we reviewed three patients treated at UPenn7,8 and note a similar inverse correlation between disease burden and the degree of clinical benefit (Table 1). While there is no uniform standard to measure tumor burden in patients with B-cell malignancies, June and colleagues7 estimated CLL tumor burden by taking into account circulating tumor cell counts, the amount of tumor cells in the bone marrow and peripheral tumor masses (i.e., lymph nodes infiltrated by tumor cells). Calculations made the additional assumption that 1 × 1012 CLL tumor cells held an equivalent weight of 1 Kg. Utilizing this tumor burden calculation, the authors concluded that patient tumor loads in the bone marrow and blood ranged from 8.8 × 1011 to 3.5 × 1012 CLL tumor cells (Table 1). The degree of treatment response in this small sample size was inversely proportional to tumor burden, with the patient bearing the lowest tumor mass achieving the best clinical response (Table 1). Utilizing the same algorithm, we calculated tumor burden in our patients (Table 1).9 Acknowledging the caveat that tumor burdens are not uniformly measured at the same time point prior to CAR-modified T-cell infusion in the UPenn and MSKCC studies, we nonetheless found a similar inverse correlation between treatment response and initial tumor burden. Nevertheless, these studies indicate that large tumor burdens are not totally insensitive, and can even be eradicated by CAR-based therapy. Hence, while overall responses are greater when tumor burdens are smaller, tumor burden is not the sole predictor of response and should not be used to exclude patients from trials. Limited clinical data from the Baylor and NCI studies did not allow us to conduct similar retrospective analyses on these cohorts.
Pre-infusion Chemotherapy: Tumor Reduction, Tumor Conditioning or Lymphodepletion?
Most preclinical in vivo studies utilizing human CD19-targeted T cells that have been reported so far were conducted in immunocompromised mice bearing xenotransplanted human CD19+ tumors.13,15,17 Cell interactions that are closer to the physiological setting can be investigated in immunocompetent mice bearing syngenic CD19+ tumor cells treated with syngeneic CD19-targeted T cells.37-39 These studies demonstrated that pre-cell infusion conditioning chemotherapy is required to enable meaningful antitumor responses by CAR-modified T cells. The results of these preclinical studies are consistent with those from clinical studies performed at the NCI in melanoma patients treated with autologous tumor infiltrating lymphocytes (TILs) expanded ex vivo.40 In the Baylor study,20 patients were treated with CD19-targeted T cells in the absence of conditioning chemotherapy. In spite of interesting biological observations relative to T-cell persistence, the clinical outcomes of this study were poor. Of note, the design of the MSKCC study9 allowed for a direct comparison of CAR modified T-cell infusions given with and without conditioning chemotherapy. In particular, three patients were treated without prior conditioning chemotherapy, while a second cohort of patients was given cyclophosphamide 1.5 gm/m2 before cell transfer. In this setting, conditioning chemotherapy enhanced both T-cell persistence and disease outcome.9
However, it is essential to note that all patients treated at MSKCC had previously received the conditioning agent (cyclophosphamide) in one or more cycles of unsuccessful conventional chemotherapy. Therefore, the conditioning regimen probably mediated a lymphodepleting effect but had marginal activity against cyclophosphamide-refractory tumor cells.9
In contrast to conditioning based on a chemotherapeutic agent to which the underlying tumor is refractory, patients treated at UPenn received a conditioning chemotherapeutic regimen containing agents with high antitumor efficacy.7,8,41 In fact, the eligibility criteria for the UPenn trial require that patients manifest either reduction or SD in response to the most recent cycle of chemotherapy.7 As a consequence, all patients treated at UPenn received effective second-line chemotherapy agents prior to CD19-targeted T cells.41,42 One patient treated with bendamustine had previously been treated only with the antibody alemtuzumab, and therefore could arguably be deemed chemotherapy-naïve. Another patient7,8 was conditioned with cyclophosphamide in combination with pentostatin, a highly effective second-line agent for relapsed CLL patients41 that the patient had never received earlier. The patients treated at UPenn had advanced CLL tumors with TP53 deletions,7,8 but the prognostic significance of this parameter is different depending on whether this deletion was present at diagnosis or relapse.43
The first patient from the NCI who received CD19-targeted T cells was a heavily pretreated patient with follicular lymphoma.6,10 Also in this setting, conditioning chemotherapy included a robust regimen combining high dose cyclophosphamide (2.5 mg/m2 × 2 d) and fludarabine (25 mg/m2 × 5 d), a drug that is efficient against low-grade B-cell malignancies to which the patient had never been exposed. In recently reported clinical data involving seven additional patients,10 prior chemotherapy regimens were not specified, limiting the analysis of tumor sensitivity to the conditioning therapy and subsequent clinical responses.
Based on these collective results, one can conclude that the nature of pre-infusion conditioning chemotherapy plays a critical role in the efficacy of targeted T-cell therapy. In contrast to the clinical outcomes observed at MSKCC in patients whose tumors were refractory to the conditioning regimen, the remarkable results obtained at NCI and UPenn6-8,10 were seen in the setting of a conditioning chemotherapy that probably resulted in lymphodepletion, in direct antitumor effects and possibly in other tumor modifications.
Does T-Cell Dose Matter?
Phase I clinical trials testing conventional drugs often adopt a dose escalation scheme to identify the maximal tolerated dose (MTD). However, in contrast to chemotherapeutic agents, infused modified T cells may undergo significant expansion under optimal conditions, as well as rapid disappearance under suboptimal conditions. Furthermore, the nature of the T cells that constitute the final T-cell product is likely to vary to considerable extents from one individual to another. One may therefore question whether a dose escalation paradigm in early studies with CAR-modified T cells is appropriate. The MSKCC studies9 followed this paradigm, while the UPenn study did not.8 Given the outcomes of CD19-targeted CAR modified T cells from various centers, we are now able to reflect upon this question in a more evidence-based manner. Cumulated data suggest that there is no correlation between T-cell dose and clinical response (Table 1). We noted optimal T-cell persistence and antitumor efficacy at the planned -1 T-cell dose (1 × 107 19–28z T cells/kg) as compared with patients treated with the initial dose level 1 (3 × 107 19–28z T cells/kg).9 In the UPenn study, similarly dramatic clinical responses were noted in a patient infused with a standard dose of T cells as well as in another patient treated with an amount of T cells that was 2 logs lower.7,8 Further, in the NCI studies,6,10 two patients receiving a 10-fold higher T-cell dose than the patient who achieved a CR failed to exhibit as good an outcome. In summary, adoptive therapy with CD19-targeted T cells appears to be less dependent on T-cell dose than on other factors discussed above.
Perspectives
The introduction of second-generation CARs in the clinic is showing the first signs of success. The concept of T-cell potency, achieved through a combination of T-cell targeting and engineered co-stimulatory support, is supported by remarkable tumor regressions induced in patients with bulky disease. Yet, despite being born out of extensive preclinical molecular and animal modeling, how CARs work remains an enigma. Our early interim analysis of results obtained in 28 patients treated with CD19-targeted T cells at 5 centers places the spotlight not only on the CAR themselves but also on pre-infusion conditioning and individual patient characteristics. The impact of tumor burden and tumor chemosensitivity needs to be better defined. The importance of T-cell manufacturing, gene-transfer modality and T-cell subset composition of the infusion product are likewise important to evaluate. While mouse models can address these questions, at least in part, definitive answers are more likely to come from additional, well-designed clinical trials. The success of this research effort will benefit from inter-institutional collaborations to enable multi-center comparisons, accelerate patient enrollment and ensure homogenous patient selection. Such concerted efforts will eventually lead to the optimal clinical exploitation of the CAR technology.
Acknowledgments
Our work was supported by the NCI (K08 CA148821), ASH-AMFDP, ACGT, TFF, the Goodwyn Commonwealth Fund, the Sanders Fund, the Major Fund and the Mallah Fund.
Glossary
Abbreviations:
- B-ALL
B-cell acute lymphoblastic leukemia
- BENDA
bendamustine
- CAR
chimeric antigen receptor
- CLL
chronic lymphocytic leukemia
- CTX
cyclophosphamide
- EOP
end of production
- FLU
fludarabine
- IL-2
interleukin-2
- MSKCC
Memorial Sloan-Kettering Cancer Center
- NCI
National Cancer Institute
- PENT
pentostatin
- scFv
single-chain variable fragment
- TCR
T -cell receptor
- UPenn
University of Pennsylvania
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/22524
References
- 1.Sadelain M, Rivière I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3:35–45. doi: 10.1038/nrc971. [DOI] [PubMed] [Google Scholar]
- 2.Ho WY, Blattman JN, Dossett ML, Yee C, Greenberg PD. Adoptive immunotherapy: engineering T cell responses as biologic weapons for tumor mass destruction. Cancer Cell. 2003;3:431–7. doi: 10.1016/S1535-6108(03)00113-2. [DOI] [PubMed] [Google Scholar]
- 3.Brenner MK, Heslop HE. Adoptive T cell therapy of cancer. Curr Opin Immunol. 2010;22:251–7. doi: 10.1016/j.coi.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sadelain M, Brentjens R, Rivière I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. 2009;21:215–23. doi: 10.1016/j.coi.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maher J, Brentjens RJ, Gunset G, Rivière I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat Biotechnol. 2002;20:70–5. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 6.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–28. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2011;119:2709–20. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li YS, Wasserman R, Hayakawa K, Hardy RR. Identification of the earliest B lineage stage in mouse bone marrow. Immunity. 1996;5:527–35. doi: 10.1016/S1074-7613(00)80268-X. [DOI] [PubMed] [Google Scholar]
- 12.Li YS, Hayakawa K, Hardy RR. The regulated expression of B lineage associated genes during B cell differentiation in bone marrow and fetal liver. J Exp Med. 1993;178:951–60. doi: 10.1084/jem.178.3.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brentjens RJ, Latouche JB, Santos E, Marti F, Gong MC, Lyddane C, et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med. 2003;9:279–86. doi: 10.1038/nm827. [DOI] [PubMed] [Google Scholar]
- 14.Brentjens RJ, Santos E, Nikhamin Y, Yeh R, Matsushita M, La Perle K, et al. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res. 2007;13:5426–35. doi: 10.1158/1078-0432.CCR-07-0674. [DOI] [PubMed] [Google Scholar]
- 15.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17:1453–64. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006;66:10995–1004. doi: 10.1158/0008-5472.CAN-06-0160. [DOI] [PubMed] [Google Scholar]
- 17.Cooper LJ, Topp MS, Serrano LM, Gonzalez S, Chang WC, Naranjo A, et al. T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood. 2003;101:1637–44. doi: 10.1182/blood-2002-07-1989. [DOI] [PubMed] [Google Scholar]
- 18.Kohn DB, Dotti G, Brentjens R, Savoldo B, Jensen M, Cooper LJ, et al. CARs on track in the clinic. Mol Ther. 2011;19:432–8. doi: 10.1038/mt.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16:1245–56. doi: 10.1016/j.bbmt.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–6. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86:10024–8. doi: 10.1073/pnas.86.24.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Romeo C, Seed B. Cellular immunity to HIV activated by CD4 fused to T cell or Fc receptor polypeptides. Cell. 1991;64:1037–46. doi: 10.1016/0092-8674(91)90327-U. [DOI] [PubMed] [Google Scholar]
- 23.Brocker T, Karjalainen K. Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. J Exp Med. 1995;181:1653–9. doi: 10.1084/jem.181.5.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gong MC, Latouche JB, Krause A, Heston WD, Bander NH, Sadelain M. Cancer patient T cells genetically targeted to prostate-specific membrane antigen specifically lyse prostate cancer cells and release cytokines in response to prostate-specific membrane antigen. Neoplasia. 1999;1:123–7. doi: 10.1038/sj.neo.7900018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. 2010;18:413–20. doi: 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009;106:3360–5. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song DG, Ye Q, Poussin M, Harms GM, Figini M, Powell DJ., Jr. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood. 2012;119:696–706. doi: 10.1182/blood-2011-03-344275. [DOI] [PubMed] [Google Scholar]
- 28.Tammana S, Huang X, Wong M, Milone MC, Ma L, Levine BL, et al. 4-1BB and CD28 signaling plays a synergistic role in redirecting umbilical cord blood T cells against B-cell malignancies. Hum Gene Ther. 2010;21:75–86. doi: 10.1089/hum.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pulè MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. 2005;12:933–41. doi: 10.1016/j.ymthe.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 30.Stephan MT, Ponomarev V, Brentjens RJ, Chang AH, Dobrenkov KV, Heller G, et al. T cell-encoded CD80 and 4-1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat Med. 2007;13:1440–9. doi: 10.1038/nm1676. [DOI] [PubMed] [Google Scholar]
- 31.Wang J, Jensen M, Lin Y, Sui X, Chen E, Lindgren CG, et al. Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum Gene Ther. 2007;18:712–25. doi: 10.1089/hum.2007.028. [DOI] [PubMed] [Google Scholar]
- 32.Zhao Y, Wang QJ, Yang S, Kochenderfer JN, Zheng Z, Zhong X, et al. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J Immunol. 2009;183:5563–74. doi: 10.4049/jimmunol.0900447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18:676–84. doi: 10.1038/sj.leu.2403302. [DOI] [PubMed] [Google Scholar]
- 34.Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. 2012;119:3940–50. doi: 10.1182/blood-2011-10-387969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med. 2012;4:132ra53. doi: 10.1126/scitranslmed.3003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burns WR, Zheng Z, Rosenberg SA, Morgan RA. Lack of specific gamma-retroviral vector long terminal repeat promoter silencing in patients receiving genetically engineered lymphocytes and activation upon lymphocyte restimulation. Blood. 2009;114:2888–99. doi: 10.1182/blood-2009-01-199216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119:4133–41. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kochenderfer JN, Yu Z, Frasheri D, Restifo NP, Rosenberg SA. Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood. 2010;116:3875–86. doi: 10.1182/blood-2010-01-265041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheadle EJ, Hawkins RE, Batha H, O’Neill AL, Dovedi SJ, Gilham DE. Natural expression of the CD19 antigen impacts the long-term engraftment but not antitumor activity of CD19-specific engineered T cells. J Immunol. 2010;184:1885–96. doi: 10.4049/jimmunol.0901440. [DOI] [PubMed] [Google Scholar]
- 40.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26:5233–9. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lamanna N, Kalaycio M, Maslak P, Jurcic JG, Heaney M, Brentjens R, et al. Pentostatin, cyclophosphamide, and rituximab is an active, well-tolerated regimen for patients with previously treated chronic lymphocytic leukemia. J Clin Oncol. 2006;24:1575–81. doi: 10.1200/JCO.2005.04.3836. [DOI] [PubMed] [Google Scholar]
- 42.Iannitto E, Morabito F, Mancuso S, Gentile M, Montanini A, Augello A, et al. Bendamustine with or without rituximab in the treatment of relapsed chronic lymphocytic leukaemia: an Italian retrospective study. Br J Haematol. 2011;153:351–7. doi: 10.1111/j.1365-2141.2011.08597.x. [DOI] [PubMed] [Google Scholar]
- 43.Delgado J, Espinet B, Oliveira AC, Abrisqueta P, de la Serna J, Collado R, et al. on behalf of the Grupo Español de Leucemia Linfatica Cronica (GELLC) y Grupo Español de Citogenetica Hematologica (GECGH) Chronic lymphocytic leukaemia with 17p deletion: a retrospective analysis of prognostic factors and therapy results. Br J Haematol. 2012 doi: 10.1111/j.1365-2141.2011.09000.x. In Press. [DOI] [PubMed] [Google Scholar]
- 44.Zola H, MacArdle PJ, Bradford T, Weedon H, Yasui H, Kurosawa Y. Preparation and characterization of a chimeric CD19 monoclonal antibody. Immunol Cell Biol. 1991;69:411–22. doi: 10.1038/icb.1991.58. [DOI] [PubMed] [Google Scholar]