

Abstract
Autophagy is an evolutionarily ancient pathway for survival during different forms of cellular stress, including infection with viruses and other intracellular pathogens. Autophagy may protect against viral infection through degradation of viral components (xenophagy), by promoting the survival or death of infected cells, through delivery of Toll-like receptor (TLR) ligands to endosomes to activate innate immunity, or by feeding antigens to MHC class II compartments to activate adaptive immunity. Given this integral role of autophagy in innate and adaptive antiviral immunity, selective pressure likely promoted the emergence of escape mechanisms by pathogenic viruses. This review will briefly summarize the current understanding of autophagy as an antiviral pathway, and then discuss strategies that viruses may utilize to evade this host defense mechanism.
Keywords: autophagy, virus, xenophagy, immunity, viral evasion
Introduction
The mammalian immune system is composed of cellular, humoral, and innate effector arms, with each arm representing a target for evasion by pathogens.1 The cellular lysosomal degradation pathway of autophagy is gaining recognition as a central component of the host innate and adaptive immune response to intracellular pathogens.2,3 Moreover, a growing list of pathogens, including bacteria, parasites and viruses, have been shown to be targeted for autophagic degradation. Therefore, it is not surprising that intracellular pathogens have devised numerous mechanisms to outsmart host autophagy. Some intracelluar bacteria (e.g., Coxiella burnetti, Legionella pneumophila) and viruses (e.g., poliovirus) co-opt the autophagy machinery to utilize autophagy protein-dependent dynamic membrane rearrangements to their own replicative advantage.4 More commonly, successful intracellular pathogens modulate the signaling pathways that regulate autophagy or block the membrane trafficking events required for autophagy-mediated pathogen delivery to endosomal or lysosomal compartments. After providing background on the role of autophagy in antiviral immunity, this review will describe specific strategies that viruses utilize to counteract host autophagy and will identify unanswered questions about the battle between host autophagy and viral infection.
Autophagy Combats Viral Infection
The earliest published link between viral infection and autophagy suggested that herpes simplex virus type 1 (HSV-1) and cytomegalovirus (CMV) virions were present in autophagosomes.5 The discovery of autophagy (ATG) genes facilitated further investigation of the role of autophagy in viral infection. Studies involving over-expression or knock-down of ATG genes or upstream autophagy-regulatory signaling molecules have demonstrated an essential role for autophagy in innate antiviral defense.6,7 Notably, the first characterization of a mammalian orthologue (Beclin 1) of a yeast Atg protein (Atg6) suggested a neuroprotective role of autophagy in a mouse model of Sindbis virus encephalitis.8 Additionally, plant ATG genes (e.g., BECLIN 1, ATG3 and ATG7) prevent the spread of cell death during the hypersensitive response and reduce viral replication during tobacco mosaic virus infection.9 Innate antiviral signaling also regulates autophagy; in response to HSV-1 infection, activation of the type I interferon (IFN)-inducible antiviral molecule, protein kinase R (PKR) is required for autophagy induction.10,11 In contrast to this protective role against viral infection, the autophagic machinery may also support the replication of certain viruses, a subject which has been reviewed recently elsewhere12 and in this issue.
Autophagy as a Multi-Pronged Defense Against Viral Infection
While the precise mechanism(s) whereby autophagy functions to protect against viral infection remain undefined, a number of studies indicate that autophagic sequestration and delivery of cytosolic contents (including viral components) to the endo/lysosomal system may serve a pleiotropic role in innate antiviral defense (Fig. 1). During stress conditions, autophagy provides building blocks for cellular metabolism through its catabolic actions, but also functions to remove superfluous or damaged organelles and aggregate-prone proteins (especially cellular structures that are too large to be degraded by the ubiquitin-proteasome system). The term xenophagy was coined to describe the targeting of microorganisms (bacteria, viruses, fungi and parasites) by the autophagy pathway for degradation.13 Conceptually, targeting virions for degradation might benefit the host both by restricting viral replication and also by “reclaiming” host molecules parasitized by viruses. Experimental evidence for xenophagic degradation of virions exists for HSV-1 (Fig. 2) and the neurotropic alphavirus, Sindbis virus. Virion sequestration within double-membraned autophagosomes and degradation within autolysosomes is observed ultrastructurally and autophagy-dependent viral protein degradation is observed biochemically.6,7,11
Figure 1.

Functions of autophagy in antiviral immunity and potential targets of viral evasion. The sequestration of virions and viral replication intermediates in autophagosomes may result in: (1) delivery of viral nucleic acids to TLR-7-containing endosomes, and subsequent activation of Type I IFN production; (2) loading of endogenously synthesized viral peptides into MHC class II compartments for antigen presentation to CD4+ T cells; and (3) xenophagic delivery to degradative lysosomes resulting in virion disposal and modulation of cell death. Identified viral mechanisms for evading these anti-viral effects include: (a) inhibition of PKR-mediated autophagy induction through reversal of eIF2α phosphorylation (HSV-1 ICP34.5); and (b) direct antagonism of the host autophagy protein Beclin 1 (HSV-1 ICP34.5, viral Bcl-2 family members). Potential mechanisms (not yet demonstrated, labeled by question marks) include: (c) antagonism of IFN production or PKR activity (by proteins other than HSV-1 ICP34.5); (d) prevention of the maturation of autolysosomes or fusion events of autophagosomes with TLR-containing and MHC class II compartments; or (e) selective disruption of virion sequestration within autophagosomes.
Figure 2.
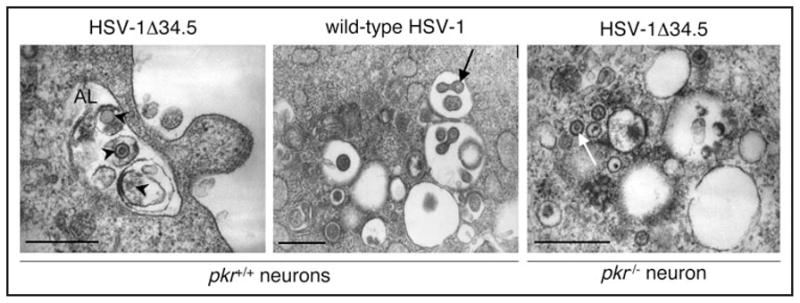
PKR signaling is required for xenophagic degradation of HSV-1 virions and is antagonized by HSV-1 ICP34.5. In wild-type neurons infected with HSV-1 deleted of the autophagy inhibitory gene, ICP34.5, numerous HSV-1 virion-containing autophagosomes and autolyososmes are present. Shown here (left panel) is an example of an autolysosome (AL) that contains HSV-1 virions (arrowheads) in different stages of degradation. In wild-type neurons infected with HSV-1 that contains the autophagy- inhibitory gene (middle panel) or in autophagy-deficient pkr−/− neurons infected with HSV-1 lacking ICP34.5 (right panel), few autophagosomes are seen. The cytoplasm contains numerous single-membraned viral vesicles (white arrow) that are intermediates in viral egress or free intracytoplasmic virions (black arrow). Scale bars, 0.5 microns. (Adapted with permission from Tallóczy, et al 2006).
In addition to functioning as a degradative pathway, autophagic sequestration and bridging of cytosolic and endosomal compartments may function to deliver viral ligands for innate and adaptive immune activation (Fig. 1). With respect to adaptive immunity, autophagy may target long-lived proteasome-resistant viral antigens for MHC class II presentation to CD4+ T cells, including certain Epstein-Barr virus nuclear antigens.3 In addition, antigen presenting cells may constitutively deliver cytosolic antigens to MHC class II-positive compartments through autophagy.14 With respect to innate immunity, a recent study suggests that autophagy delivers cytosolic Sendai virus and vesicular stomatis virus replication intermediates to the endosome for TLR-7 activation (and subsequent type I IFN production) in plasmacytoid dendritic cells.15 Thus, autophagy may serve as an integral component of antiviral immunity through direct xenophagic elimination of viral components, as well as through activation of innate and adaptive immune responses.
Autophagy is also integrally linked to cellular survival, functioning either as a survival pathway or as an alternative means of cell death, depending on the context.16 The fate of virally-infected cells may also differ between tissues in complex multicellular organisms. For example, the importance of survival of terminally differentiated neurons may take precedence over the need to eliminate infected cells, while continually renewing epithelial cells may be disposed of with little consequence. In the former case, the survival function of autophagy may prevent pathological tissue destruction by virus infection; while in the latter situation, the death function of autophagy may facilitate the clearance of infected cells. Indeed, a protective role for autophagy in promoting the survival of infected neurons is supported by the observation that enforced neuronal expression of Beclin 1 decreases neuronal apoptosis and increases survival in mice infected intracerebrally with Sindbis virus.8 In the plant hypersensitive response, ATG genes do not modify the death of virus-infected cells (which serves to clear the virus) but prevent the unwanted death of uninfected bystander cells, thereby promoting organismal survival during infection.9
Antiviral Signaling Molecules Direct Autophagy-Mediated Defense
As noted above, genes encoding components of autophagy machinery play an important role in protection against viral infection. The regulation of autophagy by antiviral immune signaling pathways further underscores a potential role of autophagy in antiviral immunity. The first indication that innate immune pathways regulate autophagy came from studies on PKR during HSV-1 infection. In yeast, mammalian PKR activity is functionally redundant with the GCN2 kinase, both of which phosphorylate elongation and initiation factor 2α (eIF2α), to activate not only general translational shutdown, but also starvation-induced autophagy.10 Moreover, PKR induces autophagy in response to HSV-1 infection in mammalian cells, and this response requires the Ser-51 phosphorylation site of eIF2α.10 Additionally, xenophagic sequestration and degradation of virions in cells infected with an HSV-1 strain deleted of the infected cell protein 34.5 (ICP34.5) neurovirulence factor (discussed below) require PKR-mediated phosphorylation of eIF2α.11 Therefore, at least one known IFN-inducible stress-response and viral recognition pathway - PKR/eIF2α signaling - upregulates autophagy.
Other studies indirectly raise the possibility that additional components of viral recognition pathways may stimulate autophagy. For example, TLR4 activation in response to bacterial lipopolysac-charide was recently reported to induce autophagy through the downstream signaling adaptor TRIF.17 Viral components activate diverse innate immune sensors, including the RNA helicases RIG-I and MDA-5, and members of the TLR family (TLRs 2, 3, 4, 7, 8 and 9).18 While the exact mechanism of autophagy activation by TRIF signaling is unclear, it is intriguing to speculate that TLR3 and 4 signaling through TRIF may also function to activate autophagy in response to viral ligands. In addition, TRIF activation leads to IRF-3 nuclear translocation and type I IFN production, which may further contribute to an anti-viral cellular state through autophagy induction, for example through PKR activation.
There is also evidence for negative regulation of autophagy by antiviral molecules and for negative regulation of antiviral signaling by Atg proteins. The nuclear factor (NF)-kappa B (NFκB) transcription factor family plays a central role in promoting the expression of genes involved in inflammatory, immune, and apoptotic processes during viral infection.19 Recent studies suggest that NFκB functions as a negative regulator of autophagy.20 Yet, autophagy may selectively target the upstream IκB kinase (IKK)21 and NFκB inducing kinase (NIK)22 for degradation, resulting in negative regulation of the canonical and non-canonical NFκB signaling pathway, respectively. Thus, NFκB signaling and the autophagy pathway may negatively regulate each other, which may ensure that viral antagonism of one pathway (i.e., NFκB blockade) leads to increased activation and antiviral effects of the other (i.e., enhanced autophagic activity), providing cells with a safety-net against viral inhibition of the host response. Somewhat counterintuitive, given the presumed role of autophagy in antiviral immunity, is the recent evidence that the Atg5-Atg12 complex (which functions in autophagosome expansion) negatively regulates RIG-I signaling through the mitochondrial signaling adaptor, IFNβ promoter stimulator (IPS-1).23 It is not yet clear, however, whether this represents a negative feedback mechanism, a true immunosuppressive role of autophagy during viral infection, or an autophagy-independent function of the Atg5-Atg12 complex.
Viruses Turn the Tables
Successful pathogens employ numerous measures for evading host immune responses, including autophagy. Viruses are known to antagonize autophagy by inhibiting upstream regulatory pathways and/or directly targeting the host autophagy machinery. An example of a virulence protein that contains both of these activities is HSV-1 ICP34.5.
HSV-1, a double-stranded DNA α-herpesvirus, infects the majority of the human population, and is the leading cause of sporadic viral encephalitis in adults.24 ICP34.5 is an important HSV-1 neurovirulence factor,25 and strains deleted of the ICP34.5 gene are severely neuroattenuated in mice and humans.26 ICP34.5 recruits the cellular protein phosphatase 1α to antagonize PKR-mediated phosphorylation of eIF2α and reverses host cell translational shutoff.27 However, mutant HSV-1 strains that retain the ability to reverse translational shutoff remain avirulent, suggesting that ICP34.5 has additional functions that confer neurovirulence.28,29 Recent studies provide strong evidence that autophagy evasion is one such function.
ICP34.5 evades autophagy by at least two distinct mechanisms. Tallóczy et al found that ICP34.5 antagonizes PKR-mediated autophagy induction during HSV-1 infection.10 By comparing HSV-1 replication in wild-type and PKR-deficient cells infected with wild-type HSV-1 or a mutant lacking ICP34.5, they determined that ICP34.5-mediated evasion of PKR-induced autophagy prevents HSV-1 degradation.11 While ICP34.5 antagonizes PKR signaling through dephosphorylation of eIF2α, it also antagonizes autophagy by interacting with the mammalian autophagy protein, Beclin 1.30 A region of ICP34.5 distinct from the GADD34 domain (that is responsible for mediating eIF2α dephosphorylation) is required for its interaction with Beclin 1 and for its autophagy-inhibitory activity, and a mutant virus lacking this region is severely neuroattenuated in a mouse model of HSV-1 encephalitis.30 Furthermore, the neurovirulence of the Beclin 1 binding-deficient mutant virus is restored in PKR knockout mice, demonstrating in vivo that ICP34.5 antagonism of Beclin 1-mediated autophagy lies downstream of PKR signaling.30
Taken together, these studies indicate that autophagy may play a role in restricting viral replication through autophagic degradation of virions and that viral evasion of autophagy may be an important factor in viral pathogenesis. The ability of a single HSV-1 neurovirulence protein to target both an autophagy-activating pathway and the autophagy execution machinery strongly suggests a requirement for autophagy inhibition to ensure successful infection. Further, the reduced neurovirulence of an HSV-1 mutant strain deficient in ICP34.5 binding to Beclin 1 (but wild-type with respect to reversal of host cell shutoff ) suggests a critical role for autophagy evasion per se (independently of ICP34.5-mediated blockade of PKR-dependent host cell shutoff ) in viral virulence.30 The precise mechanisms by which ICP34.5 antagonizes the autophagy function of Beclin 1 and mediates neurovirulence remain undefined. Nonetheless, based on the findings in mice infected with a mutant virus lacking the Beclin 1-binding domain of ICP34.5, it is reasonable to hypothesize that disruption of the HSV-1 ICP34.5/Beclin 1 interaction may be a novel useful therapeutic approach for treating serious HSV-1 infections.
Studies with other herpesvirus proteins suggest that evasion of autophagy may be a common strategy employed not only by α-herpesviruses but also by γ-herpesviruses. A member of the γ-herpesvirus virus family, Kaposi’s sarcoma-associated virus (KSHV) is associated with a number of neoplasms in immunosuppressed patients.31 Similar to its cellular counterpart, viral Bcl-2 (v-Bcl-2) from KSHV negatively regulates autophagy through its interaction with Beclin 1.32 Over-expression of KSHV vBcl-2 suppresses starvation-induced autophagy, whereas the inhibitory effects of v-Bcl-2 are lost in cells expressing Bcl-2 binding-defective forms of Beclin 1.32 Murine γ-herpesvirus-68 (γHV-68) shares a similar genome organization with human γ-herpesviruses and contains homologous genes, including a Bcl-2 homolog.33 v-Bcl-2 from the γHV-68 also binds to and inhibits Beclin 1-mediated autophagy, and studies of this interaction have revealed an additional positive cellular regulator of Beclin 1 activity, UVRAG.34 The role of autophagy evasion in γ-herpesvirus disease pathogenesis is unknown, although v-Bcl-2 binding to Beclin 1 may promote the survival of infected cells and/or support viral latency. Since Beclin 1-mediated autophagy also plays an important role in tumor suppression,35–38 it is tempting to speculate that viral inhibition of Beclin 1 function may contribute to the oncogenic potential of viruses such as KSHV and γHV68.
Intriguingly, recent studies suggest that viral Bcl-2 proteins may have evolved to inhibit Beclin 1-dependent autophagy more effectively than their cellular counterparts do. Viral Bcl-2 proteins lack the non-structured phosphorylation loop of cellular Bcl-2 that serves to regulate its release from Beclin 1 in response to physiological stimuli that activate autophagy. As a result, the binding of cellular Bcl-2 to Beclin 1 (and autophagy-inhibitory activity) is disrupted by phosphorylation during starvation whereas the binding (and autophagy-inhibitory activity) of viral Bcl-2 is not.39 Thus, by evading physiological regulatory mechanisms, viral Bcl-2 may function as a “super-repressor” of autophagy.
Interestingly, the α-herpesvirus-encoded gene product ICP34.5 and γ-herpesvirus-encoded v-Bcl-2’s likely interact with different regions of Beclin 1 (the C- and N-terminus of Beclin 1, respectively).30,32,34 These differences in binding site preference may reflect differential regulation of Beclin 1 activity but both proteins potently inhibit autophagy. Thus, the evolution of structurally distinct herpesvirus protein interactions with a common cellular autophagy target likely underscores the central importance of autophagy evasion during viral infection. Moreover, the targeting of Beclin 1, an essential upstream executioner in the autophagy pathway may suggest that these viral proteins disrupt multiple downstream anti-viral effector functions of autophagy, including not only xenophagy (as already demonstrated for HSV-1 ICP34.5) but also activation of innate and adaptive immunity (Fig. 1). An important question is whether proteins encoded by viruses outside of the herpesvirus family also target Beclin 1 (or other components of the host autophagy machinery). In view of a preliminary report that an HIV pathogenic protein also binds to Beclin 1, it seems that the targeting of Beclin 1 may be a common strategy employed by diverse viruses.40
Another important question is whether viral evasion of PKR/eIF2α signaling is a more universal strategy utilized by viruses to evade host autophagy. Prior to the identification of an autophagy-stimulatory role of this antiviral pathway, it was shown that numerous viral gene products possess diverse strategies to block PKR-mediated host cell shutoff.41 These include interfering with dsRNA-mediated activation of PKR (e.g., vaccinia virus E3, influenza virus NS1, HSV-1 US11, reovirus σ3, rotavirus NSP3), interfering with kinase dimerization (e.g., hepatitis C virus NS5A, vaccinia virus E3), blocking the kinase catalytic site and PKR-substrate interactions (e.g., vaccinia virus K3L, HIV tat), regulating eIF2α phosphorylation levels (e.g., HSV-1 ICP34.5), and affecting components downstream of eFI2α phosphorylation (e.g., SV40 T Ag). The prediction is that, like HSV-1 ICP34.5, all other viral gene products that antagonize PKR/eIF2α signaling will also antagonize host autophagy. If so, this could have important implications for understanding the role of viral evasion of autophagy in the pathogenesis of diseases caused by viruses encoding these gene products.
Cellular pathways governing cell growth and proliferation such as the Class I PI3-K/Akt/mTOR signaling pathway also negatively regulate autophagy, and this pathway is commonly activated by viruses, especially those associated with cancer. For example, oncogenic retroviruses encode active subunits of PI3-K and AKT, and a number gene products from other viruses (e.g., hepatitis B virus, hepatitis C virus, Epstein Barr virus, KSHV) activate the Class I PI3-K/Akt/mTOR pathway.6 Therefore, as with v-Bcl-2 interactions with Beclin 1 discussed above, it is reasonable to speculate that viral inhibition of autophagy through activation of these pathways may contribute to viral oncogenesis.
Viruses May Fight Autophagy on Multiple Fronts
Thus far, it has been demonstrated that viruses block autophagy by targeting the IFN-inducible PKR signaling pathway and the autophagy execution protein, Beclin 1. In the case of HSV-1 ICP34.5, such antagonism is known to block xenophagic degradation of herpes simplex virions and contribute to viral neurovirulence. A critical question is whether viral modulation of other aspects of innate and adaptive immunity also alters host autophagy and its contribution to antiviral immunity. Similarly, it remains to be determined whether viruses target discrete steps in the autophagy pathway to orchestrate a cellular state conducive to viral replication.
Figure 1 outlines the autophagy pathway, indicating points where viruses may have evolved mechanisms to modulate the pathway. As discussed above, it has already been shown that ICP34.5 blocks PKR-dependent autophagy, and it is likely that other known inhibitors of PKR signaling also block autophagy. Interestingly, autophagy-dependent TLR7 activation leads to type I IFN production, which may further enhance the autophagic response by inducing upstream regulators such as PKR (Fig. 1). Therefore, the activation and effector functions of autophagy may act synergistically in response to viral infection, and the ability to shutdown these pathways early during infection would likely benefit viral replication (Fig. 1c). Indeed, viruses have evolved an assortment of means to inhibit not only PKR signaling, but also IFN production. As the functions of the hundreds of other interferon-stimulated genes (ISGs) come to light, it will be exciting to determine whether they —like PKR—also intersect with the autophagy pathway. Further studies are required to dissect the contribution of autophagy as a downstream antiviral pathway mediated by type I IFN production, and to determine to what extent other viral IFN-antagonists (besides HSV-1 ICP34.5) exert their effects through interference with the autophagy pathway. A related question is whether the consequences of viral modulation of NFκB signaling19 are mediated partly through regulatory effects on autophagy.
If viruses target upstream autophagy regulatory pathways or autophagy execution genes, the prediction is that they will inhibit all antiviral effector functions of autophagy (unless downstream compensatory mechanisms occur). To date, it has only been shown that ICP34.5 blocks xenophagic degradation of HSV-1 virions, though it seems likely that ICP34.5—and other potential antagonists of autophagy regulators and execution proteins—may also block TLR7-dependent interferon production in dendritic cells, class II MHC antigen presentation in antigen-presenting cells, and any yet-to-be identified functions of autophagy in immunity. Thus, viral blockade of autophagy initiation may not only serve to protect the virus from being “eaten” by autophagolysosomal digestion, but may also orchestrate a multi-pronged attack on diverse aspects of host antiviral immunity.
An interesting question is whether viruses also possess mechanisms to block the fusion of autophagosomes with endo/lysosomal compartments (Fig. 1d), a process that is necessary for successful pathogen degradation, innate immune signaling, and class II MHC antigen presentation. Several human diseases are now known to be associated with defects in autophagosomal movement and/or autophagome maturation (e.g., spinal bulbar muscular atrophy, neuronal lipofuscinosis, Danon’s myopathy, Parkinson’s disease).42 It is possible that viral antagonism of autophagosomal movement or fusion with endosomal and lysosomal compartments in infected cells prevents autophagy from executing its immunoregulatory effects. Such a blockade might also contribute to viral-induced cellular pathology, as genetic defects in these processes tend to result in cellular degeneration, presumably due to a “traffic jam” in an important protein/organelle quality control and disposal pathway.
An additional possibility is that viruses may possess mechanisms to selectively block their own recognition and sequestration (independently of effects on the formation of autophagosomes) (Fig. 1e). The molecular determinants of autophagosomal recognition of viruses are unknown, but there is increasing evidence that exquisite molecular specificity may occur to permit autophagosomes to selectively target damaged cellular organelles, proteins involved in NFκB signaling and oxidative stress, and intracellular bacteria.43–46 It seems logical that selectivity may also exist for autophagic targeting of virions or viral components, and if so, that viruses may have evolved mechanisms to block this. If bulk degradation of cytoplasm can occur unimpeded in virally-infected cells without accompanying degradation of virions, this might further benefit the virus by providing a source of metabolites for viral replication.
Conclusion
Millions of years of coevolution between viruses and their hosts gave rise to a multifaceted host defense response, with the development of viral evasion strategies that mirror this complexity. Clearly, we are just in the infancy stages of understanding the role of autophagy in antiviral immunity and the strategies that viruses utilize to disarm host autophagy. Nonetheless, there are already hints that viral evasion of autophagy may be extremely important for viruses; a single viral neurovirulence protein (e.g., HSV-1 ICP34.5) possesses two different strategies for targeting autophagy and its inhibition of the Beclin 1 autophagy protein is essential for fatal encephalitis. Similarly, a single autophagy protein (Beclin 1) is targeted by virulence factors encoded by diverse viruses, including herpes simplex virus, oncogenic γ-herpesviruses, and HIV. Additional virulence factors that target autophagy at different steps in the pathway (and thereby function in viral evasion) will likely be identified in the near future. It will be interesting to see whether select points in the autophagy pathway may be ‘fine tuned’ by viruses to their own advantage, so that “virus-friendly” outcomes of autophagy (e.g., cell survival, metabolite generation) remain active, while antiviral functions (e.g., innate immune activation, antigen presentation, and virion degradation) are suppressed. It will also be important to figure out how to “turn the tables”—in the other direction—so that viruses are no longer ahead in the battle with host autophagy. The precise identification of the molecular strategies that viral proteins use to disarm host autophagy will likely provide such an opportunity.
Acknowledgments
The work from the authors’ laboratory was supported by NIH grants ROI AI051367 (B.L.) and T32 AI007520 (A.O.) and an Ellison Medical Foundation Senior Scholars Award in Infectious Diseases (A.O., B.L.). We thank Renee Talley for administrative assistance.
References
- 1.Finlay BB, McFadden G. Anti-immunology: Evasion of the host immune system by bacterial and viral pathogens. Cell. 2006;124:767–82. doi: 10.1016/j.cell.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 2.Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767–77. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmid D, Munz C. Innate and adaptive immunity through autophagy. Immunity. 2007;27:11–21. doi: 10.1016/j.immuni.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kirkegaard K, Taylor MP, Jackson WT. Cellular autophagy: Surrender, avoidance and subversion by microorganisms. Nat Rev Microbiol. 2004;2:301–14. doi: 10.1038/nrmicro865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith JD, de Harven E. Herpes simplex virus and human cytomegalovirus replication in WI-38 cells. III. Cytochemical localization of lysosomal enzymes in infected cells. J Virol. 1978;26:102–9. doi: 10.1128/jvi.26.1.102-109.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seay M, Dinesh-Kumar S, Levine B. In: Modulation of Host Gene Expression and Innate Immunity by Viruses. Palese P, editor. Springer, Dordrecht; The Netherlands: 2005. pp. 245–79. [Google Scholar]
- 7.Levine B. Autophagy in antiviral host defense. In: Deretic V, editor. Autophagy in Immunity and Infection. Weinheim, Germany: Wiley-Vch; 2006. pp. 227–41. [Google Scholar]
- 8.Liang XH, et al. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72:8586–96. doi: 10.1128/jvi.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y, et al. Autophagy regulates programmed cell death during the plant innate immune response. Cell. 2005;121:567–77. doi: 10.1016/j.cell.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 10.Talloczy Z, et al. Regulation of starvation- and virus-induced autophagy by the eIF2α kinase signaling pathway. Proc Natl Acad Sci USA. 2002;99:190–5. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Talloczy Z, Virgin HWt, Levine B. PKR-Dependent autophagic degradation of herpes simplex virus type 1. Autophagy. 2006;2:24–9. doi: 10.4161/auto.2176. [DOI] [PubMed] [Google Scholar]
- 12.Espert L, Codogno P, Biard-Piechaczyk M. Involvement of autophagy in viral infections: Antiviral function and subversion by viruses. J Mol Med. 2007;85:811–23. doi: 10.1007/s00109-007-0173-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levine B. Eating oneself and uninvited guests: Autophagy-related pathways in cellular defense. Cell. 2005;120:159–62. doi: 10.1016/j.cell.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 14.Schmid D, Pypaert M, Munz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 2007;26:79–92. doi: 10.1016/j.immuni.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315:1398–401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- 16.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–52. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 17.Xu Y, et al. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135–44. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saito T, Gale M., Jr Principles of intracellular viral recognition. Curr Opin Immunol. 2007;19:17–23. doi: 10.1016/j.coi.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 19.Hiscott J, Nguyen TL, Arguello M, Nakhaei P, Paz S. Manipulation of the nuclear factor-κB pathway and the innate immune response by viruses. Oncogene. 2006;25:6844–67. doi: 10.1038/sj.onc.1209941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Djavaheri-Mergny M, et al. NF-κB activation represses tumor necrosis factor-alpha-induced autophagy. J Biol Chem. 2006;281:30373–82. doi: 10.1074/jbc.M602097200. [DOI] [PubMed] [Google Scholar]
- 21.Qing G, Yan P, Xiao G. Hsp90 inhibition results in autophagy-mediated proteasome-independent degradation of I κB kinase (IKK) Cell Res. 2006;16:895–901. doi: 10.1038/sj.cr.7310109. [DOI] [PubMed] [Google Scholar]
- 22.Qing G, Yan P, Qu Z, Liu H, Xiao G. Hsp90 regulates processing of NF-κB 2 p100 involving protection of NF-κB -inducing kinase (NIK) from autophagy-mediated degradation. Cell Res. 2007;17:520–30. doi: 10.1038/cr.2007.47. [DOI] [PubMed] [Google Scholar]
- 23.Jounai N, et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci USA. 2007;104:14050–5. doi: 10.1073/pnas.0704014104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whitley RJ, Roizman B. Herpes simplex virus infections. The Lancet. 2001;357:1513–1518. doi: 10.1016/S0140-6736(00)04638-9. [DOI] [PubMed] [Google Scholar]
- 25.Chou J, Kern ER, Whitley RJ, Roizman B. Mapping of herpes simplex virus-1 neurovirulence to γ134.5, a gene nonessential for growth in culture. Science. 1990;250:1262–6. doi: 10.1126/science.2173860. [DOI] [PubMed] [Google Scholar]
- 26.Harrow S, et al. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: Safety data and long-term survival. Gene Ther. 2004;11:1648–58. doi: 10.1038/sj.gt.3302289. [DOI] [PubMed] [Google Scholar]
- 27.He B, Gross M, Roizman B. The γ134. 5 protein of herpes simplex virus 1 complexes with protein phosphatase 1α to dephosphorylate the α subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci USA. 1997;94:843–8. doi: 10.1073/pnas.94.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He B, Chou J, Liebermann DA, Hoffman B, Roizman B. The carboxyl terminus of the murine MyD116 gene substitutes for the corresponding domain of the gene of γ134.5 herpes simplex virus to preclude the premature shutoff of total protein synthesis in infected human cells. J Virol. 1996;70:84–90. doi: 10.1128/jvi.70.1.84-90.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Markovitz NS, Baunoch D, Roizman B. The range and distribution of murine central nervous system cells infected with the γ134. 5- mutant of herpes simplex virus 1. J Virol. 1997;71:5560–9. doi: 10.1128/jvi.71.7.5560-5569.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orvedahl A, Alexander D, Tallóczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B. HSV-1 ICP34. 5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host and Microbe. 2007;1:23–45. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 31.Coscoy L. Immune evasion by Kaposi’s sarcoma-associated herpesvirus. Nat Rev Immunol. 2007;7:391–401. doi: 10.1038/nri2076. [DOI] [PubMed] [Google Scholar]
- 32.Pattingre S, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 33.Virgin HWt, et al. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J Virol. 1997;71:5894–904. doi: 10.1128/jvi.71.8.5894-5904.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liang C, et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–99. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 35.Liang XH, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 36.Qu X, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–20. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA. 2003;100:15077–82. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levine B. Cell biology: Autophagy and cancer. Nature. 2007;446:745–7. doi: 10.1038/446745a. [DOI] [PubMed] [Google Scholar]
- 39.Wei Y, Pattingre S, Sinha S, Levine B. JNK-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. 2007 doi: 10.1016/j.molcel.2008.06.001. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He C, Orvedahl A. 2007 Keystone symposium on autophagy in health and disease. Autophagy. 2007;3:527–36. doi: 10.4161/auto.4595. [DOI] [PubMed] [Google Scholar]
- 41.Katze MG, He Y, Gale M. Viruses and interferon: A fight for supremacy. Nat Rev Immunol. 2002;2:675–87. doi: 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- 42.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008 doi: 10.1016/j.cell.2007.12.018. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu L, et al. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci USA. 2006;103:4952–7. doi: 10.1073/pnas.0511288103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–53. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klionsky DJ. How selective is autophagy? Autophagy. 2006;2:151–2. [Google Scholar]
- 46.Xiao G. Autophagy and NF-κB: Fight for fate. Cytokine Growth Factor Rev. 2007;18:233–43. doi: 10.1016/j.cytogfr.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]