

Abstract
Bioluminescence methodologies have been extraordinarily useful due to their high sensitivity, broad dynamic range, and operational simplicity. These capabilities have been realized largely through incremental adaptations of native enzymes and substrates, originating from luminous organisms of diverse evolutionary lineages. We engineered both an enzyme and substrate in combination to create a novel bioluminescence system capable of more efficient light emission with superior biochemical and physical characteristics. Using a small luciferase subunit (19 kDa) from the deep sea shrimp Oplophorus gracilirostris, we have improved luminescence expression in mammalian cells ∼2.5 million-fold by merging optimization of protein structure with development of a novel imidazopyrazinone substrate (furimazine). The new luciferase, NanoLuc, produces glow-type luminescence (signal half-life >2 h) with a specific activity ∼150-fold greater than that of either firefly (Photinus pyralis) or Renilla luciferases similarly configured for glow-type assays. In mammalian cells, NanoLuc shows no evidence of post-translational modifications or subcellular partitioning. The enzyme exhibits high physical stability, retaining activity with incubation up to 55 °C or in culture medium for >15 h at 37 °C. As a genetic reporter, NanoLuc may be configured for high sensitivity or for response dynamics by appending a degradation sequence to reduce intracellular accumulation. Appending a signal sequence allows NanoLuc to be exported to the culture medium, where reporter expression can be measured without cell lysis. Fusion onto other proteins allows luminescent assays of their metabolism or localization within cells. Reporter quantitation is achievable even at very low expression levels to facilitate more reliable coupling with endogenous cellular processes.
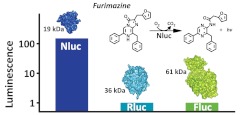
Bioluminescence is found across a diversity of life that includes bacteria, insects, fungi, and an abundance of marine organisms.1 It occurs when a photon-emitting substrate (luciferin) is oxidized by a generic class of enzymes called luciferases. These enzymes have been popular as reporters of cellular physiology because of their ability to provide highly sensitive quantitation with broad linearity. Firefly (Fluc, 61 kDa) and Renilla (Rluc, 36 kDa) luciferases have accounted for the majority of such applications, particularly for elucidating molecular processes coupled to gene expression. More recently, bioluminescence has been applied to other aspects of cellular analysis. Fluc has been configured into assay reagents for quantitation of cell viability, apoptosis, and various processes linked to cellular metabolism.2,3 Luciferases have been fused to other proteins to monitor their metabolism4 and interactions,5 circularly permuted to create intracellular biosensors,6 and split into fragments to monitor protein interactions in living cells.7
The widely recognized utility of bioluminescence has spurred investigation of alternative luciferases, predominantly from marine organisms. Luciferase genes have been derived from the copepods Gaussia princeps (20 kDa)8 and Metridia longa (24 kDa),9 the ostracod Cypridina noctiluca (61 kDa),10 the dinoflagellate Pyrocystis lunula (40 kDa),11 and the deep sea shrimp, Oplophorus gracilirostris (106 kDa).12Gaussia luciferase in particular has been used as a secreted reporter in mammalian cells,13 reportedly providing increased assay sensitivity owing to its bright luminescence and accumulation in the cell culture medium.8 However, the light intensity decays rapidly under most conditions, thus requiring luminometers equipped with injectors to measure the transient peak luminescence. Furthermore, the coelenterazine substrate is prone to chemical instability and high autoluminescence background,14 properties that make sample handling difficult and decrease assay sensitivity.
Although bright luminescence is generally desirable, a sustained signal with low background is necessary to enable efficient assay methods with high sensitivity. Preferably the luciferase should be small, monomeric, and structurally stable to environmental conditions. The luciferase from the deep sea shrimp, Oplophorus, suggested a route for achieving these capabilities.15 The native luciferase is secreted by the shrimp in brilliant luminous clouds as a defense mechanism against predation. Like many marine luciferases, it utilizes coelenterazine in an ATP-independent reaction to produce blue light (spectral maximum 454 nm). The native enzyme was found to be structurally stable with a high specific activity and quantum yield.15 Although it has a heteromeric structure consisting of two 35 kDa subunits and two 19 kDa subunits, cDNA clones revealed that bioluminescence activity was associated only with the smaller subunit (Oluc-19).12 Unfortunately, this smaller recombinant subunit does not retain many of the desirable features evident in the native enzyme, as it is unstable and poorly expressed in the absence of the 35 kDa partner.12
Our examination of the Oluc-19 amino acid sequence revealed an association with the family of intracellular lipid binding proteins (iLBPs), indicating that the underlying protein architecture should support development of a stable structure. A program to achieve a more optimal enzyme structure was undertaken, which also afforded an opportunity to explore variants of the luminogenic substrate. This resulted in a novel bioluminescent system consisting of an engineered luciferase called NanoLuc (Nluc) coupled with a novel coelenterazine analogue called furimazine. The combination of these generates much brighter luminescence than Fluc or Rluc, provides improved physical and chemical characteristics, and is generally compatible with mammalian cells. Overall, we found that Nluc performed exceptionally well as a reporter and anticipate that it will further advance the use of bioluminescence for cellular analysis.
Results and Discussion
Prediction of Stabilizing Amino Acids
There is no experimentally determined structure for Oluc-19, and standard sequence search methods failed to uncover significant similarities with known proteins.12 Using fold-recognition,16 we identified remote similarities between Oluc-19 and the well-characterized family of iLBPs. This protein family exhibits a strongly conserved structural motif, where an Arg or Lys of strand 10 hydrogen bonds with strand 1 and packs against a conserved Trp residue.17 This stabilizing interaction is only partially retained in Oluc-19, where the conserved Arg/Lys is replaced by Asn (Supplementary Figure s1). We hypothesized that a more stable variant of Oluc-19 could be created by mutating Asn to either Arg or Lys. Both substitutions produced higher enzyme stability and improved luminescence output in bacterial lysates, further substantiating the structural similarity to iLBPs. The Arg variant (Oluc-N166R) was most improved (∼50% increased stability at 37 °C and ∼3-fold higher luminescence intensity) and was used as the template for directed evolution.
Structural Optimization and Screening of Novel Coelenterazine Substrates
The luminescence expression of Oluc-N166R was optimized in E. coli in three phases. The first phase utilized a single round of random mutagenesis and screening in bacterial lysates with coelenterazine for brighter luminescence. Eight beneficial mutations (A4E, Q11R, A33K, V44I, A54F, P115E, Q124K, and Y138I) were combined to produce the variant C1A4E. When analyzed in HEK293 cell lysates, C1A4E was approximately 29,000-fold brighter than Oluc-N166R (Table 1). Western blot analysis indicated C1A4E was produced more efficiently than Oluc-N166R in cells (Supplementary Figure s2), accounting for much of the increased luminescence. The increased expression is consistent with improved enzyme stability of C1A4E at 37 °C, where the half-life of activity retention was increased 65-fold over that of Oluc-N166R (Table 2). Despite the increased stability, gel permeation chromatography revealed that the purified protein was largely aggregated (Supplementary Figure s3).
Table 1. Luminescence in HEK293 Lysates.
| luciferase | coelenterazine signal intensitya,b,c | furimazine signal intensitya,b,d | ||
|---|---|---|---|---|
| Oluc-N166R | 0.000089 ± 0.000007 | (3) | 0.0023 ± 0.0002 | (78) |
| C1A4E | 2.6 ± 0.2 | (88,000) | 16 ± 1 | (540,000) |
| Nluc | 2.4 ± 0.3 | (81,000) | 75 ± 9 | (2,500,000) |
| Rluc | 0.51 ± 0.02 | (17,000) | 0.00045 ± 0.00003 | (15) |
N = 4.
Normalized to Fluc/ONE-Glo.
10 μM coelenterazine.
50 μM furimazine. Values normalized to Oluc-19/coelenterazine are shown in parentheses.
Table 2. Signal and Enzyme Stability in HEK293 Lysates.
| luciferase | enzyme stabilitya,b (37 °C; t1/2, min) |
signal durationa,b (22 °C; t1/2, min) |
|---|---|---|
| Oluc-N166R | 5.1 ± 0.4 | NDc |
| C1A4E | 330 ± 17(5.5 h) | 92 ± 5 |
| Nluc | 11,000 ± 220(7.7 days) | 160 ± 18 |
| Rluc | 99 ± 2(1.7 h) | 86 ± 5 |
| Fluc | 7.3 ± 0.3 | 62 ± 5 |
N = 4.
Oluc-N166R, C1A4E, and Nluc measured using assay buffer/50 μM furimazine; Rluc measured using Renilla-Glo buffer/10 μM coelenterazine; Fluc measured using ONE-Glo.
ND = not determined.
The second phase of optimization focused on identification of a superior substrate and screening for further increases in luminescence. Twenty-four novel coelenterazine analogues were synthesized (Supplementary Figure s4) containing different motifs at positions 2, 6, and 8 of the imidazopyrazinone core (Figure 1). A preferred substrate would yield brighter luminescence while also having greater chemical stability and lower background autoluminescence. We anticipated that efficient catalytic utilization of a novel substrate may require corresponding modifications to the enzyme structure. Thus, together with native coelenterazine, 11 representative analogues were used to screen a random library of C1A4E mutants for brighter luminescence. Variants exhibiting brighter luminescence were then screened again with the entire panel of compounds.
Figure 1.

Chemical structures. (a) Coelenterazine. (b) Coelenterazine imidazopyrazinone core (with numbering scheme). (c) Furimazine and presumed reaction products.
Although some of the library mutants revealed improvements specific to particular substrate analogues, many exhibited increased luminescence across multiple substrates. These mutations (Q18L, F54I, F68Y, L72Q, M75K, and I90V) were apparently generally stabilizing to the enzyme structure, and their combination further enhanced enzyme stability (∼10-fold over C1A4E at 37 °C) and luminescence expression. The substrate producing the brightest luminescence signal with this enzyme variant (∼25-fold over coelenterazine) was 2-furanylmethyl-deoxy-coelenterazine (furimazine; Figure 1). Furimazine was found to be more stable in cell culture media and produce lower autoluminescence (Supplementary Figure s5a,b) than coelenterazine or coelenterazine h (R2 = benzyl; Figure 1b).
The final phase of optimization focused on maximizing luminescence with the furimazine substrate. Furimazine was used to screen another random library for brighter luminescence. Beneficial mutations were identified (L27V, K33N, K43R, and Y68D) and combined to produce NanoLuc (Nluc). In total, 16 amino acid substitutions were identified over the wild-type Oluc-19, constituting alteration to about 10% of the amino acid sequence (Supplementary Figure s6). An amended gene sequence encoding Nluc was designed by optimizing codon usage for expression in mammalian cells (www.kazusa.or.jp/codon), removing potentially strong mRNA secondary structure (http://mfold.rna.albany.edu), and removing consensus promoter sequences, other transcription factor binding sites, and potential eukaryotic mRNA splice sites.
Enzymological and physical attributes resulting from this development process are summarized in Tables 1 and 2 relative to Fluc and Rluc. Although several variants of Fluc and Rluc have been described,18−20 we chose forms that have been routinely used as benchmarks in comparative studies. Comparisons to these reporters were done using glow-type assay formats, which are generally preferred for achieving reproducible measurements across multiple samples.
Nluc paired with furimazine produced 2.5 million-fold brighter luminescence in mammalian cells relative to Oluc-19 with coelenterazine (Table 1). Because light intensity is typically correlated inversely with signal duration, these assay characteristics generally should be considered together. The luminescence produced by Nluc decayed with a half-life >2 h, significantly longer than for C1A4E. This is also longer than in the glow-type assays used for Fluc and Rluc, yet Nluc produced 75- and 89-fold more luminescence in mammalian cells, respectively.
Light intensity produced by Rluc was affected only modestly by increasing the substrate concentrations to 50 μM (Km = 15 μM for both coelenterazine and coelenterazine h). Increased coelenterazine raised the light intensity by 1.6-fold; however, the signal decayed twice as fast (Supplementary Figure s7a,b). Coelenterazine h was less efficient, raising light intensity by 1.3-fold but with similarly rapid signal decay (Supplementary Figure s7a,b).
The dramatic improvement in light output for Nluc was due in part to the change of substrate, as Nluc, C1A4E, and Oluc-N166R all produced more luminescence with furimazine compared to coelenterazine. Nluc in particular was ∼30-fold brighter with furimazine, although for Rluc it produced >1,000-fold less luminescence relative to that of coelenterazine. Nonetheless, the increased luminescence was gained mostly through improvements in protein stability, where Nluc showed markedly greater retention of activity in lysates following incubation at 37 °C (Table 2). Moreover, in contrast to C1A4E, purified Nluc appeared only as a monomer by gel permeation chromatography (Supplementary Figure s8).
NanoLuc Characterization and Comparison to Fluc and Rluc
The assay conditions for Nluc (see Methods for buffer composition) were optimized for high luminescence intensity, sustained signal duration, and good working stability under typical laboratory conditions. The reagent is added in equal volume directly to cells in culture medium to elicit steady-state luminescence with a half-life routinely >2 h. Upon combining furimazine with the assay buffer, the working solution at ambient temperature loses potency with a half-life of ∼2 days.
The apparent Km for purified Nluc using either furimazine or coelenterazine was ∼10 μM (Figure 2a), while the maximum luminescence (i.e., apparent Vmax) was ∼30-fold higher for furimazine than for native coelenterazine. This difference in brightness is consistent with the data from lysates of cells expressing Nluc (Table 1). Purified Nluc produced ∼150-fold more luminescence than either Fluc or Rluc on a per mole basis under these glow-type assay conditions (Figure 2b). Quantum yield alone cannot account for this increased luminescence, as the values for Rluc (0.05–0.1)21,18 and Fluc (0.4)22 are already relatively high. Accordingly, the higher light intensity of Nluc is more likely caused by increased catalytic turnover. Strong linearity was evident for all of the luciferases, although non-linearity can occur for Nluc at higher concentrations because the higher turnover rate can deplete available substrate. However, this is a minor practical impediment since it occurs near the saturation limit of common laboratory luminometers.
Figure 2.

(a) Furimazine and coelenterazine titrations using Nluc for determining relative signal intensities and Km (n = 3). Note the left and right axes have different scales. (b) Comparison of luminescence intensity (at 10 min) for purified Nluc, Fluc, and Rluc. (c) Spectral profiles for Nluc (furimazine), Rluc (coelenterazine), Fluc (d-luciferin), and click beetle red luciferase (CBR) (d-luciferin). Emission peaks: Nluc (460 nm), Rluc (480 nm), Fluc (565 nm), and CBR (605 nm). RLU = relative luminescence units.
The spectral profile of Nluc revealed an emission maximum of 460 nm, consistent with the reported spectrum of the wild-type luciferase.15 The spectrum is 20 nm blue-shifted relative to that of Rluc and about 20% narrower (Figure 2c). As a consequence, Nluc should be well suited for applications involving multiplexing with longer wavelength luminescence reporters,23 providing dual luciferase assays with well separated spectra to support greater composite dynamic range and sensitivity. It may be particularly beneficial to pair Nluc with a beetle luciferase having an emission maximum greater than 600 nm.24,20 Furthermore, given the widespread use of Rluc in bioluminescence resonance energy transfer (BRET), Nluc may be preferable due to its brighter luminescence and narrow spectrum. In contrast, the blue-shifted emission may hinder usage of Nluc in animal models, where shorter wavelengths do not readily penetrate mammalian tissues.
The enzymatic activity of Nluc was found to be more robust than that of Fluc when compared under a variety of environmental conditions. Nluc showed greater thermal stability, retaining activity following incubation at 55 °C for 30 min, whereas Fluc began losing activity below 30 °C (Figure 3a). Using buffers ranging from pH 5 to 9 (Figure 3b), Nluc displayed a broader optimal range between pH 7 and 9 and retained significant activity under more acidic conditions. In contrast, Fluc showed a narrower pH profile with activity dropping sharply below pH 8. Urea sensitivity (Figure 3c) was compared by treatment for 30 min followed by 20,000-fold dilution into assay buffer containing 50 μM furimazine for Nluc or into ONE-Glo for Fluc. Nluc maintained its activity following exposure up to 8 M, whereas Fluc lost activity above 2 M. Both Nluc and Fluc tolerated high concentrations of NaCl, but Nluc showed less inhibition at 10 M (Figure 3d).
Figure 3.

Comparison between purified Nluc and Fluc for sensitivity to (a) elevated temperature (n = 4), (b) pH (n = 3), (c) urea (n = 3), and (d) NaCl (n = 3).
Performance as a Genetic Reporter
In addition to producing bright and robust luminescence, it is important that reporters be expressed without bias in their experimental hosts. By originating from a marine invertebrate and having modifications in both the enzyme and gene, Nluc is unlikely to exhibit biases unique to mammalian cells. Nonetheless, the possibility of spurious interactions was examined. Immunodetection (Figure 4a) and luminescence imaging (Figure 4b,c) both suggested that Nluc was expressed uniformly in mammalian cells, including the nuclei. Moreover, no morphological differences were evident between cells expressing Nluc and control cells (not shown). Western blots prepared from cells expressing Nluc revealed only a single band of the expected molecular weight (Supplementary Figures s2, s11). Total mass analysis by mass spectrometry (LC–MS), using purified enzyme from E. coli and mammalian cells, indicated the absence of post-translational modifications. The proteins from both sources revealed identical molecular masses, matching the calculated mass for the expected unmodified protein.
Figure 4.

Intracellular distribution of Nluc determined by (a) confocal imaging/ICC of transient expression in U2OS cells fixed and processed with anti-Nluc IgG/Alexa488-conjugated secondary IgG (left panel = fluorescence; right panel = DIC); scale bar = 20 μm. (b) BLI of transient expression in U2OS cells; scale bar = 40 μm. (c) BLI of stable expression in Hela cells; scale bar = 40 μm. BLI was performed on an Olympus LV200 Bioluminescence Imager using a single addition of furimazine.
The intracellular lifetime of a genetic reporter can substantially shape its expression characteristics. A stable reporter can persist longer in cells and thus accumulate to greater levels, allowing greater assay sensitivity and reduced variability from aberrant fluctuations in gene expression. However, by this same consideration, a stable reporter has diminished ability to detect changes in transcriptional rate. Reduced lifetime will yield less signal but may provide better response dynamics.25 Which scenario is appropriate depends on the experimental objectives.
To achieve better response dynamics, Fluc having an appended 41-amino-acid PEST sequence is often used to shorten its intracellular lifetime.25 We evaluated this approach by making a similar fusion to Nluc (NlucP; Supplementary Figure s6) and inserted both Nluc and NlucP into expression plasmids containing tandem cAMP-response elements (CRE). These plasmids, along with analogous plasmids containing Fluc or FlucP, were introduced into mammalian cells, and gene expression was induced by titrated addition of forskolin (FSK). FSK activates adenylate cyclase, causing an increase of intracellular cAMP to stimulate the CRE sites. All four reporters yielded similar response profiles and showed identical EC50 values (Figure 5a). As expected, NlucP was dimmer than Nluc but showed a faster rate of signal increase and greater overall response (2,000-fold versus 750-fold) following pathway activation. When compared to their Fluc counterparts over a range of similar experiments, Nluc was generally brighter than Fluc (on average 80-fold), and NlucP was brighter than FlucP (on average 10-fold).
Figure 5.

(a) Reporter induction by tandem cAMP response elements (CRE). Nluc, Fluc, NlucP, and FlucP were transiently expressed in HEK293 cells under multiple CRE linked to a minimal promoter; luminescence measured 5 h after adding varying concentrations of FSK (n = 3). (b) Intracellular lifetime of reporters following treatment with cycloheximide. Remaining luminescence was monitored over time (n = 3) for Nluc, NlucP, Fluc, and FlucP transiently expressed in HEK293 under a constitutive promoter. (c) Reporter induction by tandem NFκB-response elements. Nluc, Fluc, NlucP, and FlucP were transiently expressed in HEK293 cells under multiple response elements linked to a minimal promoter; fold induction determined after adding recombinant, human TNFα (100 ng/mL) by comparison of treated relative to untreated samples for each time point (n = 3). (d) Assay of reporter secreted to the culture medium. HEK293 cells transiently expressing secNluc under tandem CRE were treated with FSK (10 μM) or vehicle alone; luminescence measured periodically from aliquots of culture medium (n = 3).
It was surprising to find that the PEST sequence had a larger effect on brightness and response dynamics when combined with Nluc rather than Fluc. We estimated intracellular lifetime by adding cycloheximide to block protein synthesis and measuring the decline of residual activity in the cells (Figure 5b). Both Nluc and Fluc were stable for at least 6 h. From multiple experiments, Nluc appears to have a longer lifetime than Fluc, which is consistent with the increased physical stability of Nluc. However, quantitative assessments by this method are difficult for half-lives beyond 6 h due to the toxicity of cycloheximide. As expected, FlucP had a shorter half-life (2 h) due to degradation induced by the PEST sequence. Yet, despite the high physical stability of Nluc, the intracellular half-life of NlucP (20 min) was 6-fold shorter than for FlucP.
It can be expected, therefore, that NlucP may provide better coupling to transcriptional dynamics while still providing good assay sensitivity. The influence of the intracellular lifetime is evident by the relative response of different reporters coupled to an NFκB-response element (Figure 5c). Upon stimulation with TNFα, the least relative response was achieved by Nluc, although it produced the most luminescence. The greatest relative response was achieved by NlucP, where the signal was produced faster and with higher signal over background. Both Fluc and FlucP produced intermediate responses. The greater responsiveness of NlucP as a reporter may be particularly important when the underlying genetic response of interest is subtle (Supplementary Figure s9).
Transcriptional reporters are commonly used for the high-throughput screening (HTS) of diverse compound libraries, where Fluc in particular has gained widespread use. However, the influence of chemical compounds on reporter activity, such as by inhibition or stabilization, can lead to false hits in the screening results.26 The performance of Nluc in the context of a diverse chemical collection was assessed by screening purified enzyme against the LOPAC1280 library (Supplementary Figure s10a). Relative to a parallel screen using Fluc (Supplementary Figure s10b), the data for Nluc showed a tighter distribution with lower inhibitor potency. As a percentage of total activity, 1.2% of the library compounds inhibited Nluc by >10%, 0.5% of these inhibited by >20%, and no compounds inhibited by >30%. For Fluc, 1.9% of compounds inhibited by >10%, where 0.8% inhibited by >20% and 0.5% inhibited by >30%. We speculate that greater structural rigidity associated with the increased thermal stability of Nluc may reduce its potential to bind nonspecifically to small molecules.
The secretion of bioluminescence by the Oplophorus shrimp suggested that Nluc might retain the capacity for efficient secretion from mammalian cells. Like other secreted proteins, the Oplophorus luciferase contains multiple cysteine residues, which are thought generally to provide stability in extracellular environments through disulfide bonds.27 For instance, Gaussia luciferase is a secreted enzyme that contains 11 cysteines. These cysteines all contribute to enzymatic activity but can hinder expression of functional enzyme within the reducing environment of the cell interior.28 Although the Oplophorus luciferase contains 12 cysteines, all but one are located in the larger 35 kDa subunit.12 Nluc, derived from the smaller subunit, contains only one non-essential29 cysteine and thus has no disulfide bonds. Hence, it is suited for both intracellular and secreted reporter configurations.
Initial reports of the recombinant Oluc-19 suggested that the signal sequence may not work effectively in mammalian cells.12 Thus we evaluated the capacity for secretion by appending both the native Oluc-19 secretion peptide and the secretion signal from human IL630 to the N-terminus of Nluc. Although both peptides caused Nluc to accumulate in the media, the IL6 construct (secNluc; Supplementary Figure s6) produced a brighter signal when transiently expressed in HEK293 and Hela cells, with ∼99% of the total luminescence localized to the cell culture medium. The half-life of secreted Nluc in the culture medium at 37 °C was 4.2 ± 0.2 days for HEK293 cells (DMEM + 10% FBS) and 7.2 ± 0.3 days for CHO cells (F12 + 10% FBS). Thus, secreted reporter activity can be retained in culture conditions with negligible loss (<10%) for over 15 h. These values are comparable to what has been observed for secreted Gaussia luciferase.13
Western blot analysis indicated the presence of processed secNluc in the medium, while Nluc without a secretion sequence was detected only in cell lysates (Supplementary Figure s11). The blot also indicates a similar size for secNluc and Nluc in cell lysates, consistent with simultaneous translation and signal peptide processing of secNluc. Images of U2OS cells by immunocytochemistry show a punctate staining pattern consistent with vesicular transport through the secretory pathway31 (Supplementary Figure s12). The performance of secNluc as a transcriptional reporter was evaluated using a plasmid as before containing multiple CRE sites. Following a medium change to remove secNluc activity resulting from basal expression, cells were treated with FSK to observe both the kinetics of signal accumulation (Figure 5d) and a dose response (Supplementary Figure s13). As expected, FSK caused a steady accumulation of secNluc in the medium at levels significantly higher than cells treated with vehicle alone.
Performance as a Fusion Reporter
The enhanced luminescence and small size of Nluc make it an attractive candidate as a protein fusion tag for a variety of applications.32 Luciferase fusions have become increasingly popular for measuring regulated changes in intracellular protein lifetime via the ubiquitin/proteasome system (UPS).33,4 For instance, such fusions have been used to monitor stress response pathways, where the activity of a transcription factor is typically regulated by altering intracellular abundance.34 We applied this approach by fusing Nluc to the C-terminus of p53, which is known to degrade rapidly in unstressed cells. However, DNA damaging agents (e.g., etoposide) cause the transcription factor to accumulate within cells by decoupling this degradation. Upon treatment with etoposide we observed a dose-dependent increase in signal (15-fold response) for cells expressing the p53 fusion, while no effect was evident for cells expressing Nluc alone (Figure 6). This result indicates the potential for Nluc as a fusion tag for monitoring intracellular protein lifetimes as indicators of cellular stress.
Figure 6.

Use of Nluc for monitoring regulated changes in p53 stability. HEK293 cells transiently expressing p53-Nluc or Nluc were treated with etoposide for 6 h (n = 5). Response was calculated by comparing treated samples to untreated controls.
We also examined the ability to monitor subcellular localization by bioluminescence imaging (BLI) using Nluc fusions to proteins with distinct static and dynamic localization patterns. We observed that a Histone H3-Nluc fusion was appropriately localized to the nucleus (Supplementary Figure s14a–c). When fused (with a secretion signal) to the N-terminus of the β2-adrenergic receptor (β2-AR), luminescence was localized to the plasma membrane, indicating proper trafficking through the secretory pathway (Supplementary Figure s14d). BLI has the potential for revealing protein dynamics in living cells without the need for repeated sample excitation, a cytotoxic artifact associated with use of fluorescent proteins. However, the limited brightness of existing luciferase enzymes has hindered the common use of BLI for real-time measurements. Using a Nluc-glucocorticoid receptor (GR) fusion, we monitored the expected translocation from the cytosol to the nucleus upon treatment with dexamethasone (Figure 7a,b). Finally, a fusion of Nluc to protein kinase C alpha (PKCα) was properly recruited to the plasma membrane following treatment with phorbol-12-myristate-13-acetate (PMA) (Figure 7c,d).
Figure 7.

Monitoring translocation of Nluc fusion proteins using BLI. Hela cells transiently expressing Nluc-GR fusions show (a) cytosolic localization and (b) nuclear accumulation after 15 min of dexamethasone (500 nM) treatment. U2OS cells transiently expressing Nluc-PKCα fusions show (c) cytosolic localization and (d) plasma membrane accumulation after 20 min of PMA (100 nM) treatment. Scale bar = 40 μm.
The behavior of the PKCα and GR fusion proteins was consistent with the subcellular localization dynamics of endogenous PKCα35 and GR,36 suggesting that Nluc did not significantly perturb the functionality of the fusion partner. Of significant note, the required sample exposure times in these experiments ranged from 1 to 5 s, in contrast to the 3–10 min previously reported when using an enhanced beetle luciferase adapted specifically for BLI.37 Exposure times exceeding 1 min would make video-rate analysis of rapid (<20 min) translocation events prohibitively difficult, whereas the short exposure times used for the Nluc fusions enabled continuous monitoring of both Nluc-GR (Supporting Video 1) and Nluc-PKCα (Supporting Video 2). Gaussia luciferase has been used for high frame-rate BLI of insulin secretion;38 however, its reduced intracellular activity may limit its general use for this type of application.
Bioluminescent images were acquired immediately following addition of 20 μM furimazine to the culture medium. No significant morphological changes were evident under these conditions (Figure 4b,c; Supplementary Figure s15a–f). Quantitation of cell viability following 2 h of exposure showed that furimazine was tolerated up to about 20 μM (Supplementary Figure s16a). Similar viability profiles were observed for both native coelenterazine and coelenterazine h. Measurement of luminescence intensity from cells expressing Nluc revealed that maximal signal was attained at about 10–20 μM furimazine (Supplementary Figure s16b). These results indicate that high light intensity can be achieved from Nluc in living cells with minimal perturbance to their normal physiology.
Summary
Bioluminescence has been associated primarily with quantifying genetic processes, although increasingly it is proving valuable to other aspects of cellular analysis. The potential for achieving new capabilities has motivated the search for new luminescent chemistries able to deliver greater sensitivity and adaptability to experimental programs. We recognized that the Oplophorus luciferase offered such an opportunity by the inherent luminescent efficiency of the native enzyme combined with the discovery that the small catalytic subunit was structurally related to iLBPs. Because these proteins exhibit well-behaved structural properties and are ubiquitously expressed in vertebrates and invertebrates, this protein scaffold provided a good candidate for development of a robust luminescent reporter. The reportedly broad substrate specificity of Oplophorus luciferase also afforded the opportunity to design an improved luminogenic substrate.21
To achieve this, we combined structural optimization of the small catalytic subunit using various schemes of mutagenesis with the organic synthesis of a panel of novel substrate analogues. By folding these aspects together into an integrated process, we created a small luciferase called NanoLuc (Nluc; 19 kDa) capable of producing very bright and sustained luminescence. While elevated light intensity in short bursts has been possible with other luciferases, achieving sustained luminescence at these high levels has been elusive. Sustained luminescence greatly simplifies the instrumentation and processing requirements for sample quantification, and is essential for analysis using laboratory automation.
Nluc exhibits high physical stability despite being much smaller than Fluc, showing much greater tolerance to temperature, pH, and urea. In cells, Nluc is present as a single molecular species devoid of post-translational modifications and is uniformly distributed without apparent compartmental biases. The novel substrate, furimazine, yields light intensity higher than that of native coelenterazine and is more stable with lower background autoluminescence. The combination of these characteristics positions Nluc to be generally useful as a cellular reporter, generating a highly sensitive signal with a physical constitution that is robust to environmental influences. Nluc is particularly suited for high-throughput screening, where the bright luminescence facilitates measurements in very small sample volumes and the assay is resistant to interference from library compounds.
The structure of Nluc can be readily adapted to meet different experimental needs. It may be appended with a degradation signal (e.g., PEST) to allow expression levels to change rapidly in response to transcriptional dynamics or appended with a secretion sequence to allow export into the culture medium. The stability of Nluc does not rely on having disulfide bonds, so the enzyme may be efficiently expressed either inside or outside of cells. The small size of Nluc makes it well suited as a protein fusion tag, allowing luminescence to be associated with the physiological dynamics of specific intracellular proteins. For example, changes in luminescence can be correlated to the regulated degradation associated with many transcription factors. Luminescence from Nluc can be generated within living cells, providing sufficient signal intensity for imaging the subcellular location of fusion proteins. Further investigation will determine how broad the application space is for Nluc. The properties described here are expected to enable successful use in BRET, where the enhanced brightness over Rluc should improve assay sensitivity. Nluc may also provide unique opportunities for the development of protein complementation assays, where small size, brightness, and structural stability may offer advantages over existing approaches.
Methods
Synthesis of Coelenterazine Analogues
Details on the synthesis and characterization of furimazine and the other 23 analogues (Supplementary Figure s4) can be found in the Supporting Information.
Variant Enzyme Screening
Random libraries were generated by error-prone PCR (average of 2–3 mutations per clone). Library 1 (phase 1; template = Oluc-N166R) was screened (4,400 variants) with coelenterazine. Library 2 (phase 2; template = C1A4E) was screened (4,400 variants) with 11 novel coelenterazine analogues: 3840, 3841, 3842, 3857, 3880, 3881, 3886, 3887, 3889, 3897, and 3900 (Supplementary Figure s4). The 11 analogues represented substitutions at positions 2, 6, and 8 and were considered to be representative of the entire set of 24 compounds; 2,200 variants were screened with compounds 3896 and 3894 (Supplementary Figure s4). All hits (improved luminescence) were screened again with the remaining coelenterazine analogues. Library 3 (phase 3; template = C1A4E + Q18L/K33N/F54I/F68Y/L72Q/M75K/I90V) was screened in the context of a mouse Id-X-HaloTag (where X = library) using coelenterazine and furimazine (Figure 1c). Library screens were performed on a Freedom robotic workstation (Tecan) as follows: induced bacterial cultures (in 96-well microtiter plates) were lysed with a buffer containing 300 mM HEPES pH 8, 200 mM thiourea, 0.3X Passive Lysis Buffer (PLB, Promega), 0.3 mg mL–1 lysozyme, and 0.002 units of RQ1 DNase (Promega). Assay reagent containing 1 mM CDTA, 150 mM KCl, 10 mM DTT, 0.5% (v/v) Tergitol, and 20 μM substrate was then added to equal volumes of lysate. Samples were measured on a GENios Pro luminometer (Tecan). Secondary screening to confirm hits (defined as those variants producing greater luminescence compared to that of the parental clone) and to test combination sequences was completed using a similar protocol but in manual fashion and in triplicate.
NanoLuc Assay buffer
The buffer for Nluc reactions consisted of 100 mM MES pH 6.0, 1 mM CDTA, 0.5% (v/v) Tergitol, 0.05% (v/v) Mazu DF 204, 150 mM KCl, 1 mM DTT, and 35 mM thiourea. Furimazine substrate was added to give a working reagent that was then added in equal volume directly to assay samples (final concentration of furimazine in the assay was commonly between 10 and 50 μM).
Complete methods and additional details can be found in the Supporting Information.
Acknowledgments
We thank M. Scurria, R. Arbit, H. Wang, L. Bernad, D. Simpson, R. Hurst, S. Saveliev, A. Niles, M. O’Brien, E. Strauss, J. Wilkinson, and T. Lubben for technical expertise and insightful discussions. We also thank G. Colwell at Gene Dynamics, LLC for help with vector constructions and J. Bujnicki at the IIMCB in Warsaw, Poland for assistance with fold-recognition analysis.
Supporting Information Available
This material is available free of charge via the Internet at http://pubs.acs.org
The authors declare no competing financial interest.
This paper was published ASAP on August 30, 2012. Additions were made to the Variant Enzyme Screening of the Methods section. The revised version was posted on October 24, 2012.
Supplementary Material
References
- Widder E. A. (2010) Bioluminescence in the ocean: origins of biological, chemical, and ecological diversity. Science 328, 704–708. [DOI] [PubMed] [Google Scholar]
- Melnick J. S.; Janes J.; Kim S.; Chang J. Y.; Sipes D. G.; Gunderson D.; Jarnes L.; Matzen J. T.; Garcia M. E.; Hood T. L.; Beigi R.; Xia G.; Harig R. A.; Asatryan H.; Yan S. F.; Zhou Y.; Gu X. J.; Saadat A.; Zhou V.; King F. J.; Shaw C. M.; Su A. I.; Downs R.; Gray N. S.; Schultz P. G.; Warmuth M.; Caldwell J. S. (2006) An efficient rapid system for profiling the cellular activities of molecular libraries. Proc. Natl. Acad. Sci. U.S.A. 103, 3153–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doshi U.; Li A. P. (2011) Luciferin IPA-based higher throughput human hepatocyte screening assays for CYP3A4 inhibition and induction. J. Biomol. Screen. 16, 903–909. [DOI] [PubMed] [Google Scholar]
- Smirnova N. A.; Haskew-Layton R. E.; Basso M.; Hushpulian D. M.; Payappilly J. B.; Speer R. E.; Ahn Y. H.; Rakhman I.; Cole P. A.; Pinto J. T.; Ratan R. R.; Gazaryan I. G. (2011) Development of Neh2-luciferase reporter and its application for high throughput screening and real-time monitoring of Nrf2 activators. Chem. Biol. 18, 752–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perroy J.; Pontier S.; Charest P. G.; Aubry M.; Bouvier M. (2004) Real-time monitoring of ubiquitination in living cells by BRET. Nat. Methods 1, 203–208. [DOI] [PubMed] [Google Scholar]
- Fan F.; Binkowski B. F.; Butler B. L.; Stecha P. F.; Lewis M. K.; Wood K. V. (2008) Novel genetically encoded biosensors using firefly luciferase. ACS Chem. Biol. 3, 346–351. [DOI] [PubMed] [Google Scholar]
- Remy I.; Michnick S. W. (2006) A highly sensitive protein-protein interaction assay based on Gaussia luciferase. Nat. Methods 3, 977–979. [DOI] [PubMed] [Google Scholar]
- Tannous B. A.; Kim D. E.; Fernandez J. L.; Weissleder R.; Breakefield X. O. (2005) Codon-optimized Gaussia luciferase cDNA for mammalian gene expression in culture and in vivo. Mol. Ther. 11, 435–443. [DOI] [PubMed] [Google Scholar]
- Markova S. V.; Golz S.; Frank L. A.; Kalthof B.; Vysotski E. S. (2004) Cloning and expression of cDNA for a luciferase from the marine copepod Metridia longa. A novel secreted bioluminescent reporter enzyme. J. Biol. Chem. 279, 3212–3217. [DOI] [PubMed] [Google Scholar]
- Nakajima Y.; Kobayashi K.; Yamagishi K.; Enomoto T.; Ohmiya Y. (2004) cDNA cloning and characterization of a secreted luciferase from the luminous Japanese ostracod, Cypridina noctiluca. Biosci. Biotechnol. Biochem. 68, 565–570. [DOI] [PubMed] [Google Scholar]
- Suzuki C.; Nakajima Y.; Akimoto H.; Wu C.; Ohmiya Y. (2005) A new additional reporter enzyme, dinoflagellate luciferase, for monitoring of gene expression in mammalian cells. Gene 344, 61–66. [DOI] [PubMed] [Google Scholar]
- Inouye S.; Watanabe K.; Nakamura H.; Shimomura O. (2000) Secretional luciferase of the luminous shrimp Oplophorus gracilirostris: cDNA cloning of a novel imidazopyrazinone luciferase. FEBS Lett. 481, 19–25. [DOI] [PubMed] [Google Scholar]
- Wurdinger T.; Badr C.; Pike L.; de Kleine R.; Weissleder R.; Breakefield X. O.; Tannous B. A. (2008) A secreted luciferase for ex vivo monitoring of in vivo processes. Nat. Methods 5, 171–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreu N.; Zelmer A.; Fletcher T.; Elkington P. T.; Ward T. H.; Ripoll J.; Parish T.; Bancroft G. J.; Schaible U.; Robertson B. D.; Wiles S. (2010) Optimisation of bioluminescent reporters for use with mycobacteria. PLoS One 5, e10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura O.; Masugi T.; Johnson F. H.; Haneda Y. (1978) Properties and reaction mechanism of the bioluminescence system of the deep-sea shrimp Oplophorus gracilirostris. Biochemistry 17, 994–998. [DOI] [PubMed] [Google Scholar]
- Kurowski M. A.; Bujnicki J. M. (2003) GeneSilico protein structure prediction meta-server. Nucleic Acids Res. 31, 3305–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flower D. R.; North A. C.; Sansom C. E. (2000) The lipocalin protein family: structural and sequence overview. Biochim. Biophys. Acta 1482, 9–24. [DOI] [PubMed] [Google Scholar]
- Loening A. M.; Fenn T. D.; Wu A. M.; Gambhir S. S. (2006) Consensus guided mutagenesis of Renilla luciferase yields enhanced stability and light output. Protein Eng., Des. Sel. 19, 391–400. [DOI] [PubMed] [Google Scholar]
- Woo J.; von Arnim A. G. (2008) Mutational optimization of the coelenterazine-dependent luciferase from Renilla. Plant Methods 4, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branchini B. R.; Ablamsky D. M.; Davis A. L.; Southworth T. L.; Butler B.; Fan F.; Jathoul A. P.; Pule M. A. (2010) Red-emitting luciferases for bioluminescence reporter and imaging applications. Anal. Biochem. 396, 290–297. [DOI] [PubMed] [Google Scholar]
- Inouye S.; Shimomura O. (1997) The use of Renilla luciferase, Oplophorus luciferase, and Apoaequorin as bioluminescent reporter protein in the presence of coelenterazine analogues as substrate. Biochem. Biophys. Res. Commun. 233, 349–353. [DOI] [PubMed] [Google Scholar]
- Ando Y.; Niwa K.; Yamada N.; Enomoto T.; Irie T.; Kubota H.; Ohmiya Y.; Akiyama H. (2008) Firefly bioluminescence quantum yield and colour change by pH-sensitive green emission. Nat. Photonics 2, 44–47. [Google Scholar]
- Davis R. E.; Zhang Y. Q.; Southall N.; Staudt L. M.; Austin C. P.; Inglese J.; Auld D. S. (2007) A cell-based assay for IκBα stabilization using a two-color dual luciferase-based sensor. Assay Drug Dev. Technol. 5, 85–103. [DOI] [PubMed] [Google Scholar]
- Almond B.; Hawkins E.; Stecha P.; Garvin D.; Paguio A.; Butler B. L.; Beck M.; Wood M.; Wood K. (2003) Introducing ChromaLuc technology. Promega Notes 85, 11–14. [Google Scholar]
- Swanson B.; Fan F.; Wood K. V. (2007) Enhanced response dynamics for transcription analysis using new pGL4 luciferase reporter vectors. Cell Notes 17, 3–5. [Google Scholar]
- Auld D. S.; Zhang Y. Q.; Southall N. T.; Rai G.; Landsman M.; MacLure J.; Langevin D.; Thomas C. J.; Austin C. P.; Inglese J. (2009) A basis for reduced chemical library inhibition of firefly luciferase obtained from directed evolution. J. Med. Chem. 52, 1450–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahey R. C.; Hunt J. S.; Windham G. C. (1977) On the cysteine and cystine content of proteins. Differences between intracellular and extracellular proteins. J. Mol. Evol. 10, 155–160. [DOI] [PubMed] [Google Scholar]
- Goerke A. R.; Loening A. M.; Gambhir S. S.; Swartz J. R. (2008) Cell-free metabolic engineering promotes high-level production of bioactive Gaussia princeps luciferase. Metab. Eng. 10, 187–200. [DOI] [PubMed] [Google Scholar]
- Inouye S.; Sasaki S. (2007) Overexpression, purification and characterization of the catalytic component of Oplophorus luciferase in the deep-sea shrimp, Oplophorus gracilirostris. Protein Expression Purif. 56, 261–268. [DOI] [PubMed] [Google Scholar]
- Wong G. G.; Witek-Giannotti J.; Hewick R. M.; Clark S. C.; Ogawa M. (1988) Interleukin 6: identification as a hematopoietic colony-stimulating factor. Behring Inst. Mitt. 83, 40–47. [PubMed] [Google Scholar]
- Lippincott-Schwartz J.; Roberts T. H.; Hirschberg K. (2000) Secretory protein trafficking and organelle dynamics in living cells. Annu. Rev. Cell Dev. Biol. 16, 557–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons S. O. (2011) Fireflies in the coalmine: luciferase technologies in next-generation toxicity testing. Comb. Chem. High Throughput Screening 14, 688–702. [DOI] [PubMed] [Google Scholar]
- Rehemtulla A.; Taneja N.; Ross B. D. (2004) Bioluminescence detection of cells having stabilized p53 in response to a genotoxic event. Mol. Imaging 3, 63–68. [DOI] [PubMed] [Google Scholar]
- Horn H. F.; Vousden K. H. (2007) Coping with stress: multiple ways to activate p53. Oncogene 26, 1306–1316. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y. (1984) The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature 308, 693–698. [DOI] [PubMed] [Google Scholar]
- Htun H.; Barsony J.; Renyi I.; Gould D. L.; Hager G. L. (1996) Visualization of glucocorticoid receptor translocation and intranuclear organization in living cells with a green fluorescent protein chimera. Proc. Natl. Acad. Sci. U.S.A. 93, 4845–4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima Y.; Yamazaki T.; Nishii S.; Noguchi T.; Hoshino H.; Niwa K.; Viviani V. R.; Ohmiya Y. (2010) Enhanced beetle luciferase for high-resolution bioluminescence imaging. PLoS One 5, e10011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T.; Kondo C.; Kanamori T.; Inouye S. (2011) Video rate bioluminescence imaging of secretory proteins in living cells: localization, secretory frequency, and quantification. Anal. Biochem. 415, 182–189. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.