

Abstract
Porcine circovirus type 2 (PCV2) uses autophagy machinery to enhance its replication in PK-15 cells. However, the underlying mechanisms are unknown. By the use of specific inhibitors, RNA interference, and coimmunoprecipitation, we show that PCV2 induces autophagy in PK-15 cells through a pathway involving the kinases AMP-activated protein kinase (AMPK) and extracellular signal-regulated kinase 1/2 (ERK1/2), the tumor suppressor protein TSC2, and the mammalian target of rapamycin (mTOR). AMPK and ERK1/2 positively regulate autophagy through negative control of the mTOR pathway by phosphorylating TSC2 in PCV2-infected PK-15 cells. Thus, PCV2 might induce autophagy via the AMPK/ERK/TSC2/mTOR signaling pathway in the host cells, representing a pivotal mechanism for PCV2 pathogenesis.
INTRODUCTION
Porcine circovirus type 2 (PCV2) is a small, nonenveloped, and single-stranded DNA virus (58) that is widespread in pig populations. It is recognized as the causative agent of a number of diseases in pigs collectively referred to as PCVAD (porcine circovirus-associated diseases) (42), including postweaning multisystemic wasting syndrome (PMWS) (3). Severe clinical PCVAD usually result from coinfection of PCV2 with other swine pathogens such as reproductive and respiratory syndrome virus, Mycoplasma hyopneumoniae, etc. (42). Lymphoid depletion is characteristic of PMWS (45), and the architecture of secondary lymphoid organs with depletion of B and T cells is lost during the development of PMWS (9). Current commercial vaccines based on the PCV2a genotype are effective. However, with the predominant prevalence of PCV2b in the field, efforts are being made to develop future generations of vaccines targeting the PCV2b genotype, including compliance markers and safer modified attenuated vaccines that would enhance humoral and cell-mediated immunity with reduced cost (2). PCV2 contains at least three open reading frames (ORFs). ORF1 encodes the replication proteins (Rep and Rep′) involved in virus replication (35). ORF2 encodes the capsid protein (Cap) (41) that is associated with development of neutralizing antibodies (36, 43) and with autophagy induction (68). The ORF3 protein is reported to induce apoptosis in virus-infected cells (31), although its role in pathogenicity remains to be verified (20). Thus, the pathogenic mechanisms of PCV2 remain poorly understood.
Autophagy is a catabolic cellular process conserved in all eukaryotes that involves the degradation and turnover of cytoplasmic components by lysosomal fusion (15, 24). Recent research reveals that autophagy may act as a host defense mechanism against bacterial and viral infections (52, 65). Several signaling pathways have been reported to regulate autophagy, such as the mammalian target of rapamycin (mTOR) pathway (13, 15, 57, 64) and the extracellular signal-regulated kinase 1/2 (ERK1/2) pathway (15, 46). A number of cellular signals converge to mTOR complex 1 (mTORC1) to regulate autophagy upstream of the core machinery. Molecules upstream of mTOR, such as AMPK (AMP-activated protein kinase), ERK1/2, TSC2 (tuberous sclerosis protein 2), and PTEN (phosphatase and tensin homolog), have been shown to inhibit mTORC1 and lead to activation of autophagic process, while tumor promoter pathways initiated by class I phosphoinositide 3-kinase (PI3K), Akt, IGF receptors, and Rheb (Ras homolog enriched in brain) activate mTORC1 and therefore inhibit autophagy (4, 13, 16, 40, 62). In mammalian cells, AMPK negatively regulates mTOR either by directly inhibiting mTOR or by activating TSC2 protein, an upstream negative effector of mTOR (19, 64). In addition, AMPK might regulate mTOR signaling by directly phosphorylating the mTORC1 subunit Raptor (17).
Viruses could regulate autophagy in infected cells, but detailed mechanisms are elusive and may vary with virus types (8, 28, 44, 67, 68). Tobacco mosaic virus and hepatitis C virus (HCV) can induce autophagosome formation by inducing endoplasmic reticulum stress (29, 53). Some viruses regulate mTOR signaling to control autophagy (6, 25, 38, 56). HCV induces autophagy through the Rab5 and class III PI3K Vps34 pathway (55). Human immunodeficiency virus inhibits autophagy through the Src-Akt and STAT3 pathway (59). We have found that PCV2 induces autophagy in PK-15 cells (68). In the present study, we further investigated the signaling pathway of autophagy induction by PCV2 infection. Our results demonstrate that PCV2 induces autophagy in PK-15 cells by repressing mTOR in a cascade of phosphorylated proteins involving TSC2, ERK1/2, and AMPK.
MATERIALS AND METHODS
Cells culture, treatments with chemicals and virus infection.
PK-15 cells were maintained in complete minimal essential medium (HyClone, South Logan, UT) supplemented with 10% heat-inactivated newborn calf serum (Gibco, Grand Island, NY), 1% l-glutamine, 1% nonessential amino acids, 100 U of penicillin G/ml, and 100 μg of streptomycin/ml. PK-15 cells stably expressing enhanced green fluorescent protein (EGFP)-LC3 (referred to here as PK-15/EGFP-LC3 cells) (68) were maintained in complete medium containing 0.2 mg of G418/ml. All cell monolayers were incubated at 37°C and 5% CO2.
A PCV2 strain SY4 (PCV2b; GenBank accession no. GU325754) isolated from a diseased pig (68) was propagated in PK-15 cells. Cells were infected with PCV2 at a multiplicity of infection of one 50% tissue culture infective dose for autophagy induction. After a 1-h absorption, the supernatant was removed, and the cells were rinsed with sterile phosphate-buffered saline at pH 7.2 (PBS) to remove unabsorbed viruses. The infected cells were grown in fresh medium at 37°C and 5% CO2 for the indicated times.
The chemicals used to treat or pretreat PK-15 cells included 0.5 μM rapamycin (Merck, Whitehouse Station, NJ), 20 μM UO126 (Sigma, St. Louis, MO), 5 μM compound C (Sigma), and 1 mM 5-aminoimidazole-4-carboxamide riboside (AICAR; Sigma). The cells were then infected as described above and cultured in fresh medium in the absence or presence of the same drug (at the same concentrations for pretreatments), or the corresponding level of the solvent dimethyl sulfoxide (DMSO) as a control, unless otherwise specified in the figure legends.
Plasmid construction and small interfering RNA (siRNA) transfection.
Rheb (Rheb Q64L) is reported to positively regulate mTOR (30, 63). To construct pcDNA3.1-flag-Rheb Q64L, the porcine Rheb gene was PCR amplified from porcine liver cDNA by using the primers 5′-AAGGATCCATGCCGCAGTCCAAGTCC-3′ and 5′-CCCTCGAGTCACATCACCGAGCAGGA-3′. PCR-amplified Rheb was subsequently cloned into pcDNA3.1 (Invitrogen, Eugene, OR) fused to an N-terminal flag. A mutant construct with a Q64L substitution was generated by site-directed mutagenesis using the primers 5′-ACACAGCTGGGCTGGATGAATATT-3′ and 5′-AATATTCATCCAGCCCAGCTGTGT-3′. All constructs were confirmed by DNA sequencing. The construction of pcDNA3.1-Cap has been described previously (68).
The following ERK- and TSC2-specific siRNAs were purchased from Genepharma (Shanghai, China): ERK sense (5′-CUCCAAAGCUCUGGAUUUAtt-3′) and ERK antisense (5′-UAAAUCCAGAGCUUUGGAGtt-3′) and TSC2 sense (5′-CGAGCUCG UUGAGAGAUAUtt-3′) and TSC2 antisense (5′-AUAUCUCUCAACGAGCUCGtt-3′) (deoxyribonucleotides are shown as lowercase letters). For the delivery of siRNAs into PK-15 cells, 90 nM nontargeting siRNA (NS) and siRNAs targeting ERK and TSC2 were transfected with SuperFectin II reagent (Qiagen, Valencia, CA) according to the manufacturer's instructions. Cells were infected with PCV2 at 24 h posttransfection and subjected to further incubation for 36 h. Infected cells were then harvested for further analyses. The ERK and TSC2 knockdown efficiency was evaluated by Western blotting with rabbit anti-ERK or TSC2 monoclonal antibodies (Epitomics, Burlingame, CA).
Immunoblotting.
Immunoblotting (IB) was performed as described previously (68). Briefly, cells were lysed in lysis buffer after infection or chemical treatments for indicated times. The lysates were collected and protein concentration quantified by using a bicinchoninic acid assay kit. Proteins were separated in standard SDS-PAGE gels and electrotransferred onto a polyvinylidene difluoride membrane (Millipore, Billerica, MA). The membranes were blocked for 2 h in Tris-buffered saline containing 0.05% Tween 20 and 5% nonfat milk and then probed for 1 h with the following primary antibodies: mouse monoclonal anti-Cap IgG (produced in our laboratory), rabbit polyclonal anti-LC3B (Sigma), mouse monoclonal anti-β-actin (MultiSciences, Hangzhou, China), and rabbit monoclonal antibodies to Ras, Raf1, mTOR, ERK1/2, AMPK, and TSC2, as well as to phosphorylated proteins of mTOR, ERK1/2, AMPK, and TSC2 (Epitomics). Blots were washed, incubated for another hour with goat anti-rabbit or anti-mouse horseradish peroxidase-labeled antibodies (KPL, Gaithersburg, MD), and visualized using SuperSignal West Pico chemiluminescent substrate (Thermo, Marina, CA) under the conditions recommended by the manufacturer. Images were captured in a Gel 3100 chemiluminescent imaging system (Sagecreation, Beijing, China), and the densities of the protein bands were normalized to the β-actin signal and quantified using Quantity One software (Bio-Rad, Hercules, CA). The abundance of interested proteins in various treatments was expressed relative to those under mock conditions.
Coimmunoprecipitation.
Cells were lysed with immunoprecipitation lysis buffer (IPLB; 50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, and 1% Triton X-100) in the presence of protease cocktail inhibitor (Roche, Mannheim, Germany). After centrifugation at 12,000 × g for 15 min, supernatant samples of the lysates were pretreated with protein A/G (Beyotime, Haimen, China) for 30 min at 4°C to eliminate nonspecific binding to the agarose gel. Antibodies against phospho-ERK1/2, phospho-AMPK, or phospho-TSC2 (1 μg) were used to pull down the protein complexes (500 μg) overnight at 4°C. The complexes were then immobilized with 20 μl of protein A/G-coated beads, followed by incubation overnight at 4°C on a rocker. The beads were spun down and washed three times with IPLB, and the proteins were eluted in SDS loading buffer by heating at 100°C for 5 min. SDS-PAGE and immunoblotting were carried out as previously described. The blots were probed with antibodies against phospho-ERK1/2, phospho-AMPK, phospho-TSC2, or Raf1 and visualized as described above. The lysates of mock-infected PK-15 cells were used as controls.
Confocal microscopy.
For the detection of autophagosomes, PK-15/EGFP-LC3 cells cultured in a petri dish with a glass bottom (10 mm in diameter) were infected with PCV2 or treated with 20 μM UO126 or transfected with pcDNA3.1, pcDNA3.1-flag-Rheb Q64L, or siERK for the indicated times. The cells were washed with PBS, fixed, and permeabilized with 80% cold acetone in PBS at −20°C for 20 min and then washed again with PBS. The cell samples were then incubated with rabbit polyclonal anti-flag for 1 h at room temperature, washed with PBS, and incubated with DyLight 549-conjugated goat anti-rabbit IgG (MultiSciences) for 1 h at room temperature. Nuclei were counterstained with DAPI (4′,6′-diamidino-2-phenylindole; Invitrogen). The fluorescence of EGFP-LC3 was observed under a laser scanning confocal microscope (TCS SP5; Leica Munich, Germany). The average number of EGFP-LC3 punctae per cell from at least 60 to 80 cells per sample was counted (39). Only cells with at least five dots were scored as positive.
MTT assay.
MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] was performed as described previously (68). In brief, PK-15 cells were seeded in a 96-well plate, followed by incubation at 37°C for 24 h. The medium was replaced with fresh complete medium containing DMSO, 20 μM UO126, or 5 μM compound C or 1 mM AICAR or transfected with siERK or siTSC2, and the plates were incubated for 36 h. MTT (10 μl) was added to each well, and the cells were further incubated for 4 h. Then, 100 μl of acidified isopropanol was added to each well to dissolve the precipitates completely. The optical density was measured at 570 nm using the SpectraMax M2 spectrophotometer. The viability of treated cells was expressed as a percentage relative to untreated cells.
Statistical analysis.
All results in figures were presented, where appropriate, as means ± the standard deviations from three independent experiments and analyzed by using the Student t tests.
RESULTS
PCV2 induces autophagy by inhibiting mTOR signaling.
Previously, we found that PCV2 infection activated autophagy (68). However, the underlying mechanism remains elusive. Since mTOR is a major signaling pathway that inhibits autophagy (12, 21), we investigated whether PCV2 infection inhibited mTOR by infecting PK-15 cells with PCV2 or treating with rapamycin, an mTOR inhibitor. Figure 1 shows that PCV2 or rapamycin caused significant reduction in phosphor-mTOR and increased the LC3-II/β-actin ratio compared to uninfected control cells (Fig. 1A and B, P < 0.05) (no change was detected in the levels of total mTOR), suggesting that mTOR signaling is involved in PCV2-mediated autophagy in PK-15 cells.
Fig 1.

PCV2 induces autophagy by inhibiting the mTOR signaling pathway. (A and B) LC3-II, PCV2 Cap expression, phosphorylated mTOR (p-mTOR), and total mTOR in PK-15 cells infected with PCV2, mock infected (Ctrl), or treated with 0.5 μM rapamycin (Rapa) for 36 h. Their ratios to β-actin were normalized to mock infection, set at 1.0. (C) PK-15 cells were first transfected with control vector or the vector expressing active (Q64L) Rheb mutant. After 24 h, the cells were further infected with PCV2 or mock infected for 36 h. Immunoblotting results are presented as in panel A. (D) Ratios of p-mTOR and LC3-II to β-actin normalized to the control vector, set at 1.0. (E) Formation of autophagosomes, shown as green punctae in PK-15/EGFP-LC3 cells that were treated as described in panel C and analyzed by fluorescence microscopy (scale bar, 10 μm). (F) Average number of punctae in each cell from 60 to 80 cells in each treatment. All data are reported as means ± the standard errors of the mean of three independent experiments (*, P < 0.05; **, P < 0.01) here and in all subsequent figures.
We then investigated whether inhibition of mTOR is an essential step in autophagy induction by PCV2. For this purpose, we overexpressed a constitutive active form of Rheb (Rheb Q64L) which positively regulates mTOR (30, 63). As shown in Fig. 1C and D, the expression of Rheb Q64L restored phospho-mTOR in PCV2-infected PK-15 cells and suppressed LC3 lipidation. The expression of Rheb Q64L also reduced the number of cells containing punctae of EGFP-LC3 (i.e., autophagosome-like vesicles) in PK-15/EGFP-LC3 cells infected with PCV2 (Fig. 1E and F, P < 0.01). Collectively, these observations indicate that PCV2 induces autophagy by inhibiting mTOR signaling.
PCV2 induces autophagy by activating ERK1/2 signaling.
The ERK1/2 pathway is reported to play a key role in regulating autophagy (5, 10), and PCV2 infection activates ERK1/2 signaling (61). Thus, we examined whether ERK1/2 is involved in autophagy induction in PCV2-infected PK-15 cells. Figure 2A and B show that PCV2-infected cells had an increased ratio of LC3-II or phospho-ERK1/2 to β-actin relative to uninfected cells (P < 0.05), whereas no change was detected in the levels of total ERK1/2. Inhibition of ERK1/2 phosphorylation with UO126 suppressed PCV2-induced conversion of LC3-I to LC3-II (Fig. 2C and D, P < 0.01). In PK-15/EGFP-LC3 cells, UO126 decreased the number of cells containing EGFP-LC3 punctae (Fig. 2E and F, P < 0.01). Similar findings were seen with knockdown of ERK1/2 by siRNA. LC3-II levels were significantly decreased in PK-15 cells transfected with siRNA targeting ERK1/2 compared to control siRNA (Fig. 2G and H, P < 0.05). In PK-15/EGFP-LC3 cells, ERK1/2-targeted siRNA diminished the number of cells containing EGFP-LC3 punctae (Fig. 2I and J, P < 0.01). Therefore, it is apparent that PCV2 infection triggers autophagy in PK-15 cells through the ERK1/2 signaling pathway.
Fig 2.

PCV2 induces autophagy by activating ERK1/2 signaling pathway. (A and B) LC3-II, PCV2 Cap expression, phosphorylated ERK1/2 (p-ERK1/2), and total ERK1/2 in PK-15 cells infected with PCV2 or mock infected (Ctrl) for 36 h. Their ratios to β-actin were normalized to mock infection, set at 1.0. (C and D) Effect of UO126 treatment on endogenous LC3-II, p-ERK1/2, and total ERK1/2 of PK-15 cells infected with PCV2 for 36 h. Their ratios to β-actin were normalized to mock infection, set at 1.0. (E) Formation of autophagosomes, shown as green punctae in PK-15/EGFP-LC3 cells that were treated as described in panel C. (F) Average number of punctae in each cell from 60 to 80 cells in each treatment. (G and H) PK-15 cells were mock treated (NS) or treated with ERK1/2-specific siRNA for 24 h. The cells were then further infected with PCV2 or mock infected for 36 h, and p-ERK and LC3-II ratios to β-actin were normalized to mock-infected cells, set at 1.0. (I) Formation of autophagosomes, shown as green punctae in PK-15/EGFP-LC3 cells that were treated as described in panel G. (J) Average number of punctae in each cell from 60 to 80 cells in each treatment.
PCV2 infection activates ERK1/2 with subsequent downregulation of mTOR.
The mTOR function is tightly regulated by ERK1/2 signaling through tuberous sclerosis complex 2 (TSC2) (1, 33, 60). To characterize the molecular mechanisms underlying reduced mTOR activation and autophagy induction by PCV2 infection, UO126 was used to block the ERK1/2 pathway. As shown in Fig. 3A and B, inhibition of ERK1/2 increased phosphorylation of mTOR in PCV2-infected cells (P < 0.05), indicating that ERK1/2 was required for PCV2 to inhibit mTOR signaling. To further confirm the role of ERK1/2 in mTOR inhibition during PCV2 infection, we suppressed ERK1/2 by using siRNA. Knockdown of ERK1/2 downregulated phospho-ERK1/2 (Fig. 3C and D). The levels of mTOR phosphorylation were decreased in PCV2-infected cells with ERK1/2-specific RNA silencing compared to control siRNA (Fig. 3C and D, P < 0.05). Based on these findings, we conclude that PCV2 infection in PK-15 cells might activate the ERK1/2 pathway, which downregulates mTOR in turn with subsequent induction of autophagy.
Fig 3.

Downregulation of mTOR signaling is associated with ERK1/2 pathway activation after PCV2 infection. (A) PK-15 cells were treated as described in Fig. 2C, and whole-cell extracts were subjected to immunoblot analysis for LC3-II, p-mTOR, total mTOR, p-ERK1/2, and total ERK1/2. (B) Ratios of p-mTOR, p-ERK1/2, and LC3-II to β-actin were normalized to mock infection, set at 1.0. (C and D) PK-15 cells were treated as in Fig. 2G, and their ratios to β-actin were normalized to mock infection, set at 1.0.
AMPK is the upstream regulator of ERK1/2 in PCV2-mediated autophagy.
ERK1/2 activation has been shown to occur via the Ras/Raf1/ERK1/2 pathway (54). However, AMPK has recently been reported to be a positive regulator of autophagy (14, 23, 26, 48, 49). Also, AMPK could positively regulate ERK1/2 activity (7, 22, 47, 60). To determine whether ERK1/2 activation occurs downstream of the Ras/Raf1 pathway or AMPK, we analyzed whether Ras, Raf1, or AMPK was activated by PCV2 infection. Figure 4A shows that PCV2 infection did not have significant effects on expression of Ras or Raf1, suggesting that the Ras/Raf1 pathway does not seem to contribute to PCV2-induced autophagy. In contrast, the starvation medium (i.e., Earle's balanced salt solution [EBSS]; Gibco), which is known to activate the Ras/Raf1/ERK1/2 pathway (18), increased expression of both Ras and Raf1 (Fig. 4A).
Fig 4.

AMPK is the upstream regulator of ERK1/2 in PCV2-mediated autophagy. (A) Immunoblotting of Ras and Raf1 in PK-15 cells infected with PCV2 or mock infected for 36 h or treated with EBSS for 2 h. (B) Immunoblotting of phosphorylated AMPK (p-AMPK) and total AMPK in PK-15 cells infected with PCV2, mock infected (Ctrl), or treated with AICAR for 36 h. (C) Immunoblotting of LC3-II, p-AMPK, and total AMPK in PK-15 cells transfected with pcDNA3.1 (vector) or pcDNA3.1-Cap for 24 h. (D and E) Effect of compound C treatment on LC3-II, phosphorylation, and total levels of mTOR, ERK1/2, and AMPK of PK-15 cells infected with PCV2 for 36 h. Their ratios to β-actin were normalized to mock-infected cells, set at 1.0. (F) Effect of UO126 treatment on p-ERK1/2, total ERK1/2, p-AMPK, and total AMPK of PK-15 cells infected with PCV2 for 36 h. (G and H) PK-15 cells were infected with PCV2 or mock infected (Ctrl), and the interactions between ERK1/2 and AMPK, between AMPK and ERK1/2, or between ERK1/2 and Raf1 were determined by using the indicated phospho-specific antibodies. EBSS treatment served as a positive control for Ras/Raf1 activation in PK-15 cells. IB, immunoblot; IP, immunoprecipitation; TCL, total cell lysates.
We then examined whether PCV2 infection activated AMPK. PK-15 cells were infected with PCV2 or treated with AICAR (an AMPK activator) (27, 34) for 36 h. Figure 4B shows that PCV2 infection or AICAR treatment significantly increased phosphorylation of AMPK, whereas no change was detected in the levels of total AMPK. Since we found that PCV2 capsid protein induces autophagosome formation (68). We attempted to examine whether Cap is the protein that activates AMPK. By transfecting PK-15 cells with pcDNA3.1-Cap, we found that Cap expression did indeed increase phosphorylation of AMPK (Fig. 4C). Moreover, PCV2-induced activation of ERK1/2 and AMPK or LC3 lipidation was inhibitable by compound C, an agent known to inhibit AMPK (37) (Fig. 4D and E, P < 0.05), whereas the inhibition of ERK1/2 by UO126 did not inhibit AMPK activation in response to PCV2 infection (Fig. 4F), suggesting that AMPK might be an upstream regulator of ERK1/2 in PCV2-mediated autophagy. To understand how ERK1/2 is linked to AMPK, PK-15 cells were infected with PCV2 or mock-infected for 36 h, and the protein lysates were analyzed by reciprocal coimmunoprecipitation (Co-IP). Clearly, PCV2 infection activated both ERK1/2 and AMPK, and both proteins were interacted with each other (Fig. 4G). However, Raf1 did not seem to interact with ERK1/2 during PCV2 infection (Fig. 4H). Therefore, we propose that ERK1/2 interacts with its upstream regulator AMPK, but not with Raf1 in response to PCV2 infection. Since we did not see direct interaction between AMPK and Cap in Co-IP analyses (data not shown), there might be some other component upstream of AMPK that could interact with the viral protein in initiating the signaling cascade.
ERK1/2 inhibits mTOR by activating TSC2 in PCV2-induced autophagy.
ERK1/2 could activate TSC2 (60). AMPK is also involved in phosphorylation of TSC2 and enhances its activity (11, 19). We found that PCV2 infection induced activation of TSC2 and reduced mTOR activity (Fig. 5A and B). Suppression of TSC2 by siRNA recovered mTOR phosphorylation and downregulated LC3-II (Fig. 5A and B, P < 0.05), suggesting that TSC2 regulates autophagy via mTOR. To examine whether ERK1/2 inhibits mTOR via activating TSC2 after PCV2 infection or whether AMPK exerts its direct effect on TSC2 or indirectly via ERK1/2, PK-15 cells were infected with PCV2 with or without compound C or UO126. We found that inhibition of AMPK with compound C repressed ERK1/2 and TSC2, and recovered mTOR activity (Fig. 5C and 5D, P < 0.05). Inhibition of ERK1/2 also suppressed TSC2 activation (Fig. 5E and F, P < 0.05). However, TSC2 remained repressed irrespective of the high level of p-AMPK when p-ERK1/2 was inhibited by UO126. Co-IP analysis suggests that PCV2 infection activated both TSC2 and ERK1/2 and that both proteins interacted with each other (Fig. 5G). These results indicate that TSC2 is the downstream effector of ERK1/2, and AMPK seems to affect TSC2 via ERK1/2 in regulation of autophagy during PCV2 infection.
Fig 5.

ERK1/2 downregulates mTOR activity by upregulating TSC2 after PCV2 infection. (A) PK-15 cells were mock treated (NS) or treated with TSC2-specific siRNA for 24 h. The cells were then further infected with PCV2 or mock infected for 36 h, and whole-cell extracts were subjected to immunoblot analysis for LC3-II, phosphorylation, and total levels of mTOR, TSC2, ERK1/2, and AMPK. (B) Ratios of the molecules in panel A to β-actin, which was normalized to mock infection, set at 1.0. (C) Effect of compound C treatment on LC3-II, phosphorylation, and the total levels of mTOR, TSC2, ERK1/2, and AMPK of PK-15 cells infected with PCV2 for 36 h. (D) Ratios of the molecules in panel C to β-actin. (E) Effect of UO126 treatment on LC3-II, phosphorylation, and the total levels of mTOR, TSC2, ERK1/2, and AMPK of PK-15 cells infected with PCV2 for 36 h. (F) Ratios of the molecules in panel E to β-actin. (G) PK-15 cells were infected with PCV2 or mock infected for 36 h, and the interactions between ERK1/2 and TSC2 or between TSC2 and ERK1/2 were determined by using the indicated phospho-specific antibodies. IB, immunoblot; IP, immunoprecipitation; TCL, total cell lysates.
Pharmacological treatments and RNA interference do not affect cell viability.
To assess whether treatments with UO126, AICAR or compound C and siRNA targeting ERK1/2 or TSC2 transfection affected cell viability, which could ultimately affect experimental results in host cells, we used the MTT assay to analyze their effects on cell viability. There were virtually no significant effects on the viability of PK-15 cells treated with the chemicals or siRNA transfection (Fig. 6).
Fig 6.
Pharmacological or siRNA alterations of autophagy do not affect cell viability. Cell viability was determined by MTT assay after treatment with 20 μM UO126, 1 mM AICAR, or 5 μM compound C or transfection with siERK1/2 or TSC2 for 36 h. Percent relative cell viabilities are expressed as means ± the SEM (n = 3).
DISCUSSION
PCV2, an important pathogen of pigs, causes lymphoid depletion in infected tissues, most probably by inducing apoptosis and/or autophagy, although the precise pathogenesis of PCVAD remains largely unknown. In our previous work, PCV2 was found to explore the autophagic machinery to enhance its replication in PK-15 cells (68). Here, we further show that PCV2 induces autophagy by repressing mTOR in a cascade of phosphorylated proteins involving TSC2, ERK1/2, and AMPK (Fig. 7). This signaling cascade has not been seen with other viruses that regulate autophagy, such as vesicular stomatitis virus (Akt signaling) (51) and avian influenza virus (Akt/TSC2/mTOR) (56). Encephalomyocarditis virus, as well as porcine reproductive and respiratory syndrome virus, also induces autophagy with enhanced viral replication; however, the signaling pathways involved remain unknown (32, 67). Simian virus 40 small T antigen induces autophagy by activating AMPK, with a concomitant decrease in phospho-mTOR and an increase in phospho-raptor, mTOR binding protein, and mTOR negative regulator under a glucose-deficient condition (25). However, it remains uncertain whether AMPK has direct interaction with mTOR or raptor or acts indirectly via other signaling molecules.
Fig 7.
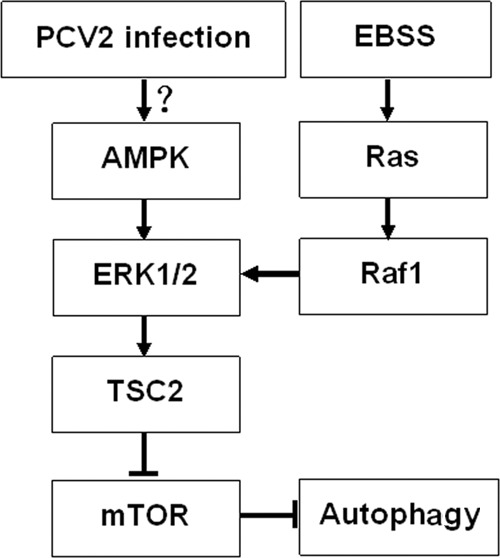
Hypothetical model of AMPK/ERK/TSC2/mTOR signaling in PCV2-induced autophagy in PK-15 cells. PCV2 infection activated AMPK and then activated ERK1/2 and TSC2 and suppressed mTOR signaling, thereby inducing autophagy in PK-15 cells.
AMPK may positively regulate ERK1/2 (22, 47, 60). It is also involved in phosphorylation of TSC2 and enhances its activity (11, 19). ERK1/2 has been found to inhibit TSC2 in vitro and in vivo (33, 50). However, ERK1/2 has recently been shown to inhibit mTOR via activating TSC2, thus enhancing Beclin1-mediated autophagy (60). These opposite effects reflect the complexity of the signaling cross talk that exists either among AMPK, ERK1/2, and mTOR or even other signaling pathways of the autophagic processes induced by different factors.
With PCV2-induced autophagy, the activation of ERK1/2 and TSC2, as well increased LC3 lipidation, was inhibitable by compound C (Fig. 4C and D and Fig. 5C and D), suggesting that AMPK might be an upstream regulator of ERK1/2. TSC2 was suppressed and remained repressed irrespective of high level of phospho-AMPK when p-ERK1/2 was inhibited by UO126 (Fig. 5E and F). Co-IP analysis reveals that PCV2 infection activated both TSC2 and ERK1/2, and both proteins interacted with each other (Fig. 5G). These results indicate that TSC2 is the downstream regulator of ERK1/2, and AMPK affects TSC2 via ERK1/2 in autophagy regulation during PCV2 infection (Fig. 7). This is consistent with a previous study demonstrating that AMPK upregulates ERK1/2 activity via activating TSC2 (60). We further confirm that Cap is the protein of PCV2 that activates AMPK. Because we did not see direct interaction between AMPK and Cap in Co-IP, the signaling upstream of AMPK with PCV2-induced autophagy remains to be studied.
With starvation-induced autophagy, ERK1/2 activation has been shown to occur via the Ras/Raf1/ERK1/2 signaling pathway (54). HCV infection could also activate this pathway, leading to enhanced viral replication. Further investigation indicates that the Ras/Raf/ERK pathway could disrupt JAK-STAT signaling by reducing the expression of its upstream IFNAR1 and IFNAR2 (66). However, PCV2-induced autophagy did not seem to involve Ras/Raf1 signaling (Fig. 4 and 5).
We have established here the molecular connections among AMPK, ERK1/2, TSC2, and mTOR in PCV2-regulated autophagy. These findings may lead to a better understanding of PCV2 pathogenesis.
ACKNOWLEDGMENTS
This study was supported by grants from the National Natural Science Foundation of China (31272534) and the Hangzhou Science and Technology Development Project (20102012A05).
We thank J. P. Li for confocal microscopic analysis at the Zhejiang University Core Facility Consortium.
Footnotes
Published ahead of print 22 August 2012
REFERENCES
- 1. Ballif BA, et al. 2005. Quantitative phosphorylation profiling of the ERK/p90 ribosomal S6 kinase-signaling cassette and its targets, the tuberous sclerosis tumor suppressors. Proc. Natl. Acad. Sci. U. S. A. 102:667–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beach NM, Meng XJ. 2012. Efficacy and future prospects of commercially available and experimental vaccines against porcine circovirus type 2 (PCV2). Virus Res. 164:33–42 [DOI] [PubMed] [Google Scholar]
- 3. Bolin SR, Stoffregen WC, Nayar GP, Hamel AL. 2001. Postweaning multisystemic wasting syndrome induced after experimental inoculation of cesarean-derived, colostrum-deprived piglets with type 2 porcine circovirus. J. Vet. Diagn. Invest. 13:185–194 [DOI] [PubMed] [Google Scholar]
- 4. Botti J, Djavaheri-Mergny M, Pilatte Y, Codogno P. 2006. Autophagy signaling and the cogwheels of cancer. Autophagy 2:67–73 [DOI] [PubMed] [Google Scholar]
- 5. Cagnol S, Chambard JC. 2010. ERK and cell death: mechanisms of ERK-induced cell death—apoptosis, autophagy, and senescence. FEBS J. 277:2–21 [DOI] [PubMed] [Google Scholar]
- 6. Chaumorcel M, Souquère S, Pierron G, Codogno P, Esclatine A. 2008. Human cytomegalovirus controls a new autophagy-dependent cellular antiviral defense mechanism. Autophagy 4:46–53 [DOI] [PubMed] [Google Scholar]
- 7. Chen HC, et al. 2002. Activation of the ERK pathway and atypical protein kinase C isoforms in exercise-and aminoimidazole-4-carboxamide-1-β-d-riboside (AICAR)-stimulated glucose transport. J. Biol. Chem. 277:23554–23562 [DOI] [PubMed] [Google Scholar]
- 8. Chen Q, et al. 2012. Induction of autophagy enhances porcine reproductive and respiratory syndrome virus replication. Virus Res. 163:650–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chianini F, Majó N, Segalés J, Domínguez J, Domingo M. 2003. Immunohistochemical characterisation of PCV2 associate lesions in lymphoid and non-lymphoid tissues of pigs with natural postweaning multisystemic wasting syndrome (PMWS). Vet. Immunol. Immunopathol. 94:63–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Corcelle E, et al. 2007. Control of the autophagy maturation step by the MAPK ERK and p38: lessons from environmental carcinogens. Autophagy 3:57–59 [DOI] [PubMed] [Google Scholar]
- 11. Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan KL. 2004. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 18:1533–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Diaz-Troya S, Perez-Perez ME, Florencio FJ, Crespo JL. 2008. The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy 4:851–865 [DOI] [PubMed] [Google Scholar]
- 13. Djavaheri-Mergny M, et al. 2007. Regulation of autophagy by NF-κB transcription factor and reactive oxygen species. Autophagy 3:390–392 [DOI] [PubMed] [Google Scholar]
- 14. Egan DF, et al. 2011. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331:456–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Esclatine A, Chaumorcel M, Codogno P. 2009. Macroautophagy signaling and regulation. Curr. Top. Microbiol. Immunol. 335:33–70 [DOI] [PubMed] [Google Scholar]
- 16. Fleming A, Noda T, Yoshimori T, Rubinsztein DC. 2011. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat. Chem. Biol. 7:9–17 [DOI] [PubMed] [Google Scholar]
- 17. Gwinn DM, et al. 2008. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30:214–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He C, Klionsky DJ. 2009. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43:67–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Inoki K, Zhu T, Guan KL. 2003. TSC2 mediates cellular energy response to control cell growth and survival. Cell 115:577–590 [DOI] [PubMed] [Google Scholar]
- 20. Juhan NM, LeRoith T, Opriessnig T, Meng XJ. 2010. The open reading frame 3 (ORF3) of porcine circovirus type 2 (PCV2) is dispensable for virus infection but evidence of reduced pathogenicity is limited in pigs infected by a ORF3-null PCV2 mutant. Virus Res. 147:60–66 [DOI] [PubMed] [Google Scholar]
- 21. Jung CH, Ro SH, Cao J, Otto NM, Kim DH. 2010. mTOR regulation of autophagy. FEBS Lett. 584:1287–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim EK, et al. 2012. Human mesenchymal stem cell differentiation to the osteogenic or adipogenic lineage is regulated by AMP-activated protein kinase. J. Cell Physiol. 227:1680–1687 [DOI] [PubMed] [Google Scholar]
- 23. Kim J, Kundu M, Viollet B, Guan KL. 2011. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13:132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Klionsky DJ. 2005. The molecular machinery of autophagy: unanswered questions. J. Cell Sci. 118:7–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kumar SH, Rangarajan A. 2009. Simian virus 40 small T antigen activates AMPK and triggers autophagy to protect cancer cells from nutrient deprivation. J. Virol. 83:8565–8574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee JW, Park S, Takahashi Y, Wang HG. 2010. The association of AMPK with ULK1 regulates autophagy. PLoS One 5:e15394 doi:10.1371/journal.pone.0015394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lempiäinen J, Finckenberg P, Levijoki J, Mervaala E. 2012. AMPK activator AICAR ameliorates ischemia reperfusion injury in the rat kidney. Br. J. Pharmacol. doi:10.1111/j.1476-5381.2012.01895.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li J, et al. 2011. Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. J. Virol. 85:6319–6333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li L, et al. 2012. The invasion of tobacco mosaic virus RNA induces endoplasmic reticulum stress-related autophagy in HeLa cells. Biosci. Rep. 32:171–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li Y, Inoki K, Guan KL. 2004. Biochemical and functional characterizations of small GTPase Rheb and TSC2 GAP activity. Mol. Cell. Biol. 24:7965–7975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu J, Chen I, Du Q, Chua H, Kwang J. 2006. The ORF3 protein of porcine circovirus type 2 is involved in viral pathogenesis in vivo. J. Virol. 80:5065–5073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu Q, et al. 2012. Autophagy sustains the replication of porcine reproductive and respiratory virus in host cells. Virology doi:org/10.1016/j.virol.2012.03.022 [DOI] [PMC free article] [PubMed]
- 33. Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. 2005. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 121:179–193 [DOI] [PubMed] [Google Scholar]
- 34. Magnoni LJ, Vraskou Y, Palstra AP, Planas JV. 2012. AMP-activated protein kinase plays an important evolutionary conserved role in the regulation of glucose metabolism in fish skeletal muscle cells. PLoS One 7:e31219 doi:10.1371/journal.pone.0031219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mankertz A, Mankertz J, Wolf K, Buhk HJ. 1998. Identification of a protein essential for replication of porcine circovirus. J. Gen. Virol. 79(Pt 2):381–384 [DOI] [PubMed] [Google Scholar]
- 36. McNeilly F, et al. 2001. Production, characterization and application of monoclonal antibodies to porcine circovirus 2. Arch. Virol. 146:909–922 [DOI] [PubMed] [Google Scholar]
- 37. Meley D, et al. 2006. AMP-activated protein kinase and the regulation of autophagic proteolysis. J. Biol. Chem. 281:34870–34879 [DOI] [PubMed] [Google Scholar]
- 38. Meng S, et al. 2012. Avian reovirus triggers autophagy in primary chicken fibroblast cells and Vero cells to promote virus production. Arch. Virol. doi:10.1007/s00705-012-1226-x [DOI] [PubMed] [Google Scholar]
- 39. Mizushima N, Yoshimori T, Levine B. 2010. Methods in mammalian autophagy research. Cell 140:313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Morselli E, et al. 2009. Anti- and pro-tumor functions of autophagy. Biochim. Biophys. Acta 1793:1524–1532 [DOI] [PubMed] [Google Scholar]
- 41. Nawagitgul P, et al. 2000. Open reading frame 2 of porcine circovirus type 2 encodes a major capsid protein. J. Gen. Virol. 81(Pt 9):2281–2287 [DOI] [PubMed] [Google Scholar]
- 42. Opriessnig T, Meng MJ, Halbur PG. 2007. Porcine circovirus type 2 associated disease: update on current terminology, clinical manifestations, pathogenesis, diagnosis, and intervention strategies. J. Vet. Diagn. Invest. 19:591–615 [DOI] [PubMed] [Google Scholar]
- 43. Pogranichnyy RM, et al. 2000. Characterization of immune response of young pigs to porcine circovirus type 2 infection. Viral Immunol. 13:143–153 [DOI] [PubMed] [Google Scholar]
- 44. Rodriguez-Rocha H, et al. 2011. Adenoviruses induce autophagy to promote virus replication and oncolysis. Virology 416:9–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rosell C, et al. 2000. Identification of porcine circovirus in tissues of pigs with porcine dermatitis and nephropathy syndrome. Vet. Rec. 146:40–43 [DOI] [PubMed] [Google Scholar]
- 46. Saiki S, et al. 2011. Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy 7:176–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sajan MP, et al. 2010. AICAR and metformin, but not exercise, increase muscle glucose transport through AMPK-, ERK-, and PDK1-dependent activation of atypical PKC. Am. J. Physiol. Endocrinol. Metab. 298:E179–E192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shang L, Wang X. 2011. AMPK and mTOR coordinate the regulation of Ulk1 and mammalian autophagy initiation. Autophagy 7:924–926 [DOI] [PubMed] [Google Scholar]
- 49. Shang L, et al. 2011. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. U. S. A. 108:4788–4793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shaw RJ, Cantley LC. 2006. Ras, PI(3)K, and mTOR signalling controls tumour cell growth. Nature 441:424–430 [DOI] [PubMed] [Google Scholar]
- 51. Shelly S, Lukinova N, Bambina S, Berman A, Cherry S. 2009. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity 30:588–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sir D, Ou JH. 2010. Autophagy in viral replication and pathogenesis. Mol. Cells 29:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sir D, et al. 2008. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 48:1054–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Steelman LS, et al. 2011. Roles of the Raf/MEK/ERK and PI3K/ PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging 3:192–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Su WC, et al. 2011. Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J. Virol. 85:10561–10571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sun Y, et al. 2012. Inhibition of autophagy ameliorates acute lung injury caused by avian influenza A H5N1 infection. Sci. Signal. 5:ra16. [DOI] [PubMed] [Google Scholar]
- 57. Tanida I. 2011. Autophagosome formation and molecular mechanism of autophagy. Antioxid. Redox Signal. 14:2201–2214 [DOI] [PubMed] [Google Scholar]
- 58. Todd D, et al. 2005. Circoviridae, p 327–334 In Fauquet CM, et al. (ed), Virus taxonomy: VIIIth report of the International Committee on Taxonomy of Viruses, 2nd ed Elsevier Academic Press, San Diego, CA [Google Scholar]
- 59. Van Grol J, et al. 2005. HIV-1 inhibits autophagy in bystander macrophage/monocytic cells through Src-Akt and STAT3. PLoS One 5:e11733 doi:10.1371/journal.pone.0011733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang J, et al. 2009. A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin1. J. Biol. Chem. 284:21412–21424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wei L, Liu J. 2009. Porcine circovirus type 2 replication is impaired by inhibition of the extracellular signal-regulated kinase (ERK) signaling pathway. Virology 386:203–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Xiao G. 2007. Autophagy and NF-κB: fight for fate. Cytokine Growth Factor Rev. 18:233–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yang J, et al. 2011. Deficiency of hepatocystin induces autophagy through an mTOR-dependent pathway. Autophagy 7:748–759 [DOI] [PubMed] [Google Scholar]
- 64. Yang ZF, Klionsky DJ. 2010. Mammalian autophagy: core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 22:124–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yuk JM, Yoshimori T, Jo EK. 2012. Autophagy and bacterial infectious diseases. Exp. Mol. Med. 44:99–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang Q, et al. 2012. Activation of the Ras/Raf/MEK pathway facilitates hepatitis C virus replication via attenuation of the interferon-JAK-STAT pathway. J. Virol. 86:1544–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang YN, Li ZC, Ge XN, Guo X, Yang HC. 2011. Autophagy promotes the replication of encephalomyocarditis virus in host cells. Autophagy 7:1–16 [DOI] [PubMed] [Google Scholar]
- 68. Zhu BL, et al. 2012. Porcine circovirus type 2 explores the autophagic machinery for replication in PK-15 cells. Virus Res. 163:476–485 [DOI] [PubMed] [Google Scholar]