


Abstract
MicroRNAs (miRNAs) negatively regulate the levels of messenger RNA (mRNA) post-transcriptionally. Recent advances in CLIP (cross-linking immunoprecipitation) technology allowed capturing miRNAs with their cognate mRNAs. Consequently, thousands of validated mRNA–miRNA pairs have been revealed. Herein, we present a comprehensive outline for the combinatorial regulation by miRNAs. We implemented combinatorial and statistical constraints in the miRror2.0 algorithm. miRror estimates the likelihood of combinatorial miRNA activity in explaining the observed data. We tested the success of miRror in recovering the correct miRNA from 30 transcriptomic profiles of cells overexpressing a miRNA, and to identify hundreds of genes from miRNA sets, which are observed in CLIP experiments. We show that the success of miRror in recovering the miRNA regulation from overexpression experiments and CLIP data is superior in respect to a dozen leading miRNA-target prediction algorithms. We further described the balance between alternative modes of joint regulation that are executed by pairs of miRNAs. Finally, manipulated cells were tested for the possible involvement of miRNA in shaping their transcriptomes. We identified instances in which the observed transcriptome can be explained by a combinatorial regulation of miRNA pairs. We conclude that the joint operation of miRNAs is an attractive strategy to maintain cell homeostasis and overcoming the low specificity inherent in individual miRNA–mRNA interaction.
INTRODUCTION
MicroRNAs (miRNAs) are short non-coding RNAs (ncRNAs) that negatively regulate gene expression post-transcriptionally (1). Recent miRNA detection techniques confirmed the presence of hundreds of miRNAs in healthy and diseased tissues (2,3). An estimate across animal genomes suggests that up to 1–2% of the genes in human and Caenorhabditis elegans consist of miRNAs. These estimates are derived from a combination of computational and experimental methods (4,5). Deep sequencing datasets have led to an expansion in numbers of known miRNAs. Currently, miRBase is the most exhaustive collection of miRNAs, IsomiRs, miRNA families and experimental evidence (6).
In humans and other metazoa, miRNAs play a role as an additional layer of post-transcriptional regulators (7). miRNAs are best known for the regulation of stem cell differentiation, immunological cell function, organogenesis, cell identity, apoptosis and more. The study of miRNAs in the context of cancer biology shows that disruption in miRNA biogenesis leads to tumorigenesis and to a drastic change in the relative expression of a large number of mRNAs (8). Furthermore, several miRNAs directly regulate cell cycle genes and thus induce oncogenic activity (3,9). In other instances, the activity of miRNA resembles tumor suppression (10).
Mechanistically, miRNAs exert their function via base-pair complementarity with mRNA. This interaction occurs within the RNA-induces silencing complex (RISC) (11,12). The binding of miRNA to mRNA occurs mainly at the 3′-untranslated region (3′-UTR). However, binding sites at the coding region or the 5′-UTR were reported (13). Binding of the miRNA to the mRNA leads to gene silencing due to changes in mRNA stability, enhanced degradation and to some extent, translational arrest (14,15). A coherent picture of miRNA regulation remains elusive, mainly due to gaps in knowledge of miRNA modes of action in vivo (16).
The attenuation of target expression by individual miRNAs is usually quite modest (17). Consequently, a definitive mapping of miRNAs to their genuine targets is inherently challenging (18). Numerous databases, algorithms and resources provide predictions for the matching of miRNAs with their direct targets (19). Although all resources use the knowledge of the minimal requirement for sequence complementarity (called seed), some algorithms account for imperfect hybridization, context-dependent features (e.g. accessibility of binding sites, GC content), inter-species conservation, thermodynamic stability of the miRNA–mRNA duplex and combinations of these constraints. Additional descriptive features associated with miRNAs include the distribution of miRNA binding sites, positioning of the binding sites on the mRNAs, transcript length and energetic favorability of the transcript secondary structures (20,21). Although each of these features is founded on biological insight, poor coherence among miRNA-target prediction resources was reported (22–24). The main source for inconsistency is attributed to the many false positives across all prediction methods (25). In addition, the number of false negatives is still unknown (26,27), especially as each miRNA is assumed to attenuate tens to hundreds of targets. Importantly, distinguishing between a direct and indirect miRNA–target interaction remains challenging.
Regulation at the levels of gene promoters and transcripts are interconnected (28). With almost 2000 reported miRNAs in human (29), and about 10 000 candidate genes (excluding alternative splicing variants), the network of interactions is extremely complex. The concentration of individual miRNAs in cells may vary by four orders of magnitude (30) and thus the effectiveness of miRNA regulation also depends on the stoichiometry with competing factors (31). The degree of miRNA regulation results from the integration of: (i) accessibility of miRNAs to their cognate targets; (ii) miRNA turnover; (iii) transcript stability; and (iv) the concentration of targeted genes (both genuine and off-targets) (32,33). Furthermore, the effective modeling of off-target effects, local concentration, saturation of binding and competition on binding sites is still in its infancy (34).
A coordinated action of miRNAs on their targets was proposed (see Discussion (35,36)). For example, for the known targets of miR-375, a combined addition of miR-124 and let-7b led to a synergy in target inhibition (35). Similarly, the expression of miR-16, miR-34a and miR-106b altered the cell cycle. Combining these miRNAs resulted in cell cycle arrest that was stronger than for each of the miRNAs alone (36). The regulation of the tumor suppressor FUS1 in cancer cells depends on the presence of at least three miRNAs (miR-93, miR-98, miR-197 and additional unidentified miRNAs) (37). The potential of miRNA pairs or triplets to alter the integrity of pathways was systematically studied (38). The generality of these observations is not yet established, mostly because accurate information is missing on the stability, local concentration and competition among miRNAs in living cells.
In this study, we focus on the rich set of human miRNAs while seeking trends that are best explained by a large collection of experimental data. In addition to miRNA over-expressed experiments in cell-line, HITS-CLIP (high-throughput sequencing of RNAs isolated by cross-linking immunoprecipitation) technology provides an in vivo collection of the miRNAs that were hybridized with their targets at the RISC complex (27). We show that the success of miRror to recover the miRNA regulation from over-expression experiments and CLIP data is superior in respect to a dozen leading miRNA-target prediction databases (MDBs). Moreover, we show that the incorporation of statistical and combinatorial considerations empowered the systems’ predictive success in view of leading miRNA-target prediction resources. Large-scale data from CLIP technologies were used for testing the validity and quality of our combinatorial-statistical analysis. We provide evidence for the concept of ‘miRNAs working together’ for an individual targeted gene and a large set of genes. We offer miRror2.0 as a computational, statistical platform that incorporates the notion of a collective mode of action, in view of the experimental results. We conclude by formulating general trends on the design principles for cellular regulation by multiple miRNAs. We discuss the notion of a miRNA-Duo (pair) as a basic regulatory element. We assess the validity of miRNA-Duos in governing the profile of gene expression in manipulated cells. We discuss several instances that support the involvement of a set of miRNAs in governing the observed gene expression. We conclude that the miRror2.0 platform is useful in reducing the prediction noise and in capturing the underlying combinatorial modus operandi of miRNA in a wide range of cellular conditions.
MATERIALS AND METHODS
Prediction databases
Database files that are included in miRror encompass most of the available miRNA-target prediction tools covering human miRNAs (and several other species). The resources used are collectively called MDBs: (i) TargetScan database (39); (ii) microCosm that is based on the miRanda algorithm (40); (iii) PicTar, according to species conservation scheme (35); (iv) DIANA–MicroT (41); (v) PITA (42); (vi) MirZ (43); (vii) microRNA.org that allows analysis on multiple miRNA acting on the same gene-target, based on miRanda (44); (viii) miRDB resource (45); (ix) TargetRank (46); (x) RNA22 (47); (xi) MAMI (48); and (xii) miRNAMap2 (49). We used the latest versions available (February 2012). miRNAMap2 is based on RNAhybrid, miRanda and TargetScan, while requiring a minimal score from miRanda. Reducing false positives is achieved by increasing the numbers of binding sites and their accessibility. We included a resource that applied statistical machine learning algorithms (i.e. miRDB, (22)) and a meta-server that integrates some of the major resources (i.e. MAMI (48)). The number of candidate genes and the number of miRNAs are indicated for each of the major MDBs (Supplementary Figure S1A, Supplementary Table S1). The union of the miRNAs and genes for all the reported MDBs is ∼2500 and ∼47 000, respectively. These numbers exceed the number of known miRNAs due to inconsistency in naming by the different MDBs. For this study, MDB refers to the entire collection of miRNA—gene matches that result from the underlying prediction algorithm.
Gene expression data
The complete list of experiments was extracted from the GEO (50) and ArrayExpress (51). The platforms that we considered are from Affymetrix (HG-U133 Plus 2.0, 22K probes, 14.5K genes) and Agilent spotted oligonucleotides (Human 3.0 A1, 23K genes). Data were collected from the SOFT files. See Supplementary Figure S1 and Supplementary Table S2 for a complete list.
Prediction performance
The MDBs are not designed to deal with hundreds and thousands of genes as input. In order to assess the performance of each MDB, we score each miRNA by counting the number of down-regulated genes that are predicted as targeted by it. The miRNA that is ranked first is the miRNA that is associated with the highest number of predictions from the down-regulated genes that were tested.
CLIP data were retrieved from StarBase v2.1 (52). The identification of miRNAs from the CLIP data is based on predictions from TargetScan, PicTar, RNA22, PITA and miRanda. We confirmed that 98% of the unified list of miRNAs derived from these 5 MDBs. We exported and analyzed all documented miRNA–target interactions that concerned human cells. For the CLIP data, the MDBs’ prediction accuracy was estimated by counting the number of cases the appropriate gene was predicted from the large number of its regulating miRNAs.
miRror application for miRNA cooperative regulation
The miRror platform (53) was used to match between the input gene list and the miRNAs that best explain such input. The statistical basis of miRror is implemented in the miRTegrate algorithm (53) that calculates the probability of matching a gene list while considering the coverage of the miRNA-targets for each of the prediction databases. Formally, the probability of the miRNA interaction with the input gene set as opposed to the rest of the genes in that MDB is calculated. We used a statistical threshold to ensure that the contribution of any MDB that covers only a small number of miRNAs with high specificity remains significant. On the other extreme, the impact of an MDB that over predicts (i.e. each miRNA matches almost any gene) to miRror results should be minimal. Calculating the P-value for genes as input was performed according to the hypergeometric distribution.
Where N is the total number of genes in the database, n is the number of genes predicted for the specific miRNA, m is the number of genes in the input set and k is the number of genes that appear in both the input set and the predicted miRNA targets, the probability of such k is:
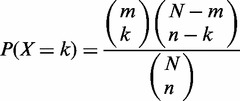 |
P-value is obtained by summing over all X ≥ k (53). A correction for multiple testing was included using the false discovery rate (FDR) procedure. The P-value threshold that was used in this study is 0.05 (unless mentioned otherwise).
In this study, we used the miRror2.0 that includes number of new capabilities. Most notably, implementing an optional analysis on the basis of miRNA families, incorporating miRIS (miRror Internal Score) as a quality score, adding several MDBs (RNA22, MAMI and miRNAMap2) and updating the new scoring method presented by miRanda-microRNA.org.
Mapping genes to pairs of miRNAs
For each prediction results, we considered the 5 top predictions and analyzed by miRror the success of each pair (10 pairs). The ranking of miRror is according to miRIS. This score balances between (i) the proportions of predicting MDBs out of all tested MDBs and (ii) the fraction of the potentially regulated genes from the entire input genes. The 10 pairs are tested by their ability to increase the miRIS score beyond the miRIS obtained from the individual miRNA. Each pair is tested for the degree of complementing each of the individual miRNA and the degree of overlapping, redundant mode. These values range from 0 to 1. Formally:
An increase in miRIS to the level that was considered a standard deviation apart from the calculated value is considered a successful recovery of miRNA.
An inherent redundancy in miRNAs is evident in miRNA families (as defined by miRBase). The redundancy is based on target binding site and seed overlap. Still, several MDBs do not cover the same miRNA or the same targets, thus small differences are still found. These cases are often identified by high redundancy and low complementation. The consideration of pairs of miRNAs provides the notion of joint miRIS (JM), an intuitive generalization of miRIS:
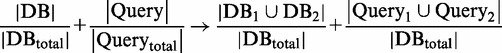 |
The best five ranked candidates are chosen. We tested the joint miRIS values and the Jaccard Index (JI) to identify candidates of miRNA-Duos that explain best the gene expression profiles. Analyzing all combinations of the top 5 miRNAs provides 10 pairs per for each experiment.
miRNA family unification
A source for inconsistency and thus a reduced quantity of prediction is attributed to the use of slightly different names for the same miRNAs. A compilation of miRNAs to their families was applied according to the miRBase notations. Compressing the miRNAs to their families reduced the number of entries by 2.7-folds (Supplementary Table S3). Another conservative and systematic approach assigned miRNAs to their seed families. We cluster miRNAs into the same seed family if nucleotides 2–7 of the mature miRNA are identical. Seed families compress the number of miRNAs by 1.6-folds (on average).
RESULTS
Limited success in identifying the underlying miRNA from over-expression experiments
With the goal of revealing global trends in miRNA regulation, we considered the major miRNA resources. Data from MDBs that comprise the most stable resources were collected. We designed a systematic analysis of each MDB from an unbiased list of transcripts. Several criteria were used in order to select experimental data: (i) Experiments are conducted on human cell-lines that are transfected with a single miRNA. (ii) The entire cell transcriptome is compared with controlled cells (often introduced with a scrambled or mutated sequence). (iii) The platforms used for the gene expression profiling cover most known genes (See Materials and Methods). We tested the potential of recovering the appropriate miRNA by each MDB from the unbiased transcriptomic data. We selected 12 of the leading resources (PITA-Top, DIANA-microT3.0, MiRanda-microRNA.org, MicroCosm, TargetScan, TargetRank, PicTar-4ways, MirZ-ElMMo, RNA22, MAMI, miRDB and MAP2) for the rest of this study (Supplementary Figure S1).
The transcriptome from hsa-miR-124 overexpression experiment (54) (GSE6207, HepG2 cells, time series for 4–120 h after transfection) was used as a test case. We found that the success in predicting the subjected miRNA is restricted to data collected from ≥24 h after transfection. A maximal level of expression for the functional miRNA is reached between 24 and 48 h after transfection. Data from earlier time points (4–16 h after transfection) were uninformative and have not been further analyzed. We focused on the down-regulated genes at a permissive repression level of 1.2-fold relative to control cells (Supplementary Table S4). Different names for miR-124 (i.e. miR-124a and miR-124-3p) were unified to limit the inconsistency for these results. The 100 most significant down-regulated genes were used as the input for the 12 MDBs (see Materials and Methods). We found that 75% of the MDBs identified hsa-miR-124 as their top prediction and the rest of MDBs predicted hsa-miR-124 among the top 9 predictions. Notably, the actual degree (e.g. fold change) by which miRNA reduced the amount of its candidate targets is a poor indicator for a successful recovery (t-test, P-value = 0.8).
We collected raw transcriptomic data from additional 30 large-scale experiments that cover 26 different miRNAs from a variety of cell-lines (Supplementary Figure S1). Figure 1 illustrates the workflow for the miRNA-gene prediction task. All together we produced over 10 000 prediction results (30 experiments, 12 MDBs, 8 input sets, 4 selections of MDB internal scores). A common protocol for the 12 MDBs (Figure 1, right) and miRror (53) as a unified-predictor (Figure 1, Left) is shown. The miRror platform examines the combinatorial nature of miRNA regulation. miRror seeks the miRNAs that are the most likely explanation for the observed gene expression profile. For this study we applied miRror2.0, an improved version of the original platform (see Materials and Methods).
Figure 1.

A workflow for assessing the performance of miRNA predictions. Analysis of miRror2.0 is illustrated for genes as an input (Gene2miR mode, default parameters). A filter for the collection of the top predictions according to internal scores was activated. Stringency refers to parameters including the choice of P-value threshold. Additional parameters that provide fine-tuning for the platform are ignored (tissue of interest, highly expressed subset, MDB choices). By changing these parameters, a relaxed or a strict search protocol can be activated. We assigned several categories for success according to the rank of the prediction (prediction ranked 1, 2, 3–5, 6–9, ≥10).
Figure 2 shows the performance for each MDB in view of the miRror2.0 results. We marked the success in predicting the appropriate miRNA at various levels of accuracy (ranked 1, 2 and 3–9). Figure 2A shows the results from 30 experiments and across 12 MDBs. Most MDBs fail to recover over 50% of the subjected miRNAs. miRror (Figure 1A, purple bar) outperformed all MDBs with 60% success (for the 1–9 ranked predictions, Supplementary Table S5).
Figure 2.

Success rates of MDB’s prediction. (A) 10% of the down-regulated genes from 30 experiments were subjected to miRNA prediction by the 12 MDBs and miRror. The color intensity is according to the indicated predictions’ quality. miRror results are shown in purple (Supplementary Table S5). (B) Reanalysis of the same input as in (A) with top predictions (25%) by the internal scoring system for each MDB. Only 10/12 MDBs provide an internal score (Supplementary Table S5). For example, PicTar-4 performs poorly when the top 25% of its internal score is applied. However, without such filtration, a good performance of PicTar-4 is recorded (compare Figure 4A with Figure 4B). (C) Average success rates based on the 12 MDBs for each of the 30 transcriptomics datasets. A miRNA overexpression experiment that is marked as 99% (green line) means that the rank across the 12 MDB is in the top 1% predictions. The colors mark the prediction success as very high (green), high (blue), moderate (orange) and poor (red).
Internal prediction scores fail to improve the quality of predictions
Most MDBs provide internal scores for ranking their prediction results. Such scoring is presented in order to reduce the inherent numbers of false predictions (discussed in (55)). We tested whether the miRNA predictions can be improved by applying the internal scoring as provided by each MDB (Figure 2B). The internal scores for the MDBs differ by their underlying principles, their distribution and range. We thus used percentiles (i.e. top 10, 25 and 50%) to select the suggested top predictions as defined by each MDB. Figure 2B shows prediction results, using the top 25% according to the internal scores (only 10 of the 12 MDBs support internal scorings). Surprisingly, only a few of the MDBs (microCosm, TargetScan and Miranda-microRNA.org) improved their success when the filter of the internal score was activated. For the other MDBs, the prediction success was reduced. Consequently, a slight reduction in the number of successful prediction is recorded for miRror (Figure 2B, dark purple, Supplementary Table S5).
An experiment-centric view shows that only some experiments are associated with a successful recovery of the relevant miRNA (Figure 2C, green line). The average number of miRNA predictions for the analyzed MDB is 720 (Supplementary Figure S1 and Supplementary Table S1). For about half of the experiments the average success by the MDBs is very poor (Figure 2C, orange and red lines).
The high performance of miRror in view of each MDB is intriguing (Figure 2). We thus assessed the consistency of the results by combining a large number of prediction tests (Supplementary Figure S2). The results confirmed the robustness of miRror predictions in view of the individual MDBs. The miRror2.0 protocol consistently outperformed the 12 other MDBs with 55–60% success. The performance of miRror is robust in view of thousands of predictions across a wide range of parameters (Supplementary Table S6).
miRror score unifies the combinatorial nature of the input and the coherence of predictions
Based on the reported results, we developed a scoring method that benefits from the individual MDBs. The score, called miRIS, balances between (i) the proportions of predicting MDBs out of all tested MDBs and (ii) the fraction of the potentially regulated genes from the entire input genes. A default of two successful MDBs is a minimal demand for consistency, as well as at least two genes from the input set—a minimal demand for cooperativity.
Figure 3A shows the contribution of the two components that comprise miRIS. The analysis is based on hsa-miR-124 (GSE6207, HepG2 cells, 24 h after transfection). We analyzed 240 genes as input (10% down-regulated genes, P-value = 0.05, Supplementary Table S4). Figure 3A shows the contribution of the fraction of MDBs and the fraction of hits from the input list. The two measures are not replaceable and only weakly correlate (r = 0.3–0.5). miRIS balances between these two complementary components (Figure 3A).
Figure 3.

miRror internal scoring. (A) The overexpression of hsa-miR-124 transcriptomic data (10% of the down-regulated genes) is used to predict the top 100 miRNAs. (A) The fraction of MDBs (from 12) that agree on each of the miRNA prediction (orange color), and the fraction of genes from the input genes that are explained by the miRNA prediction (blue color). A minimal agreement of two MDBs and of two genes from the input is requested. The unified miRIS is the averaged of these components. The 100 predicted miRNAs are sorted according to miRIS. (B) The prediction list, ranked by miRIS for hsa-miR-7 (GSE14507). The highest miRIS identified hsa-miR-7 whereas the other MDBs failed to predict it. The average rank for hsa-miR-7 by all the MDBs is 38.4 ± 30.6. (C) Predictions of hsa-miR-124 (GSE6207). The miRIS indicated hsa-miR-124 as the second prediction and hsa-miR-506 as the first one. These two miRNAs belong to the same family according to TargetScan. Insets display a zoomed view on the top 10 predictions. Note the abrupt drop in score among the top 10 predictions.
An additional statistical prerequisite is the P-value for the miRTegrate algorithm (P-value ≤ 0.05, see Materials and Methods). This threshold remains a useful parameter for tuning the stringency by changing the statistical significance of the analyses. The application of a stringent P-value tends to increase the prediction success (not shown).
Presenting the entire list of miRror predictions revealed that remarkably, only few miRNAs are associated with the highest miRIS values (Figure 3B and C). We illustrate the miRIS ranking for hsa-miR-7 (Figure 3B) and hsa-miR-124 (Figure 3C). Notably, all other 12 MDBs failed to recover the hsa-miR-7 (Supplementary Tables S5). For 75% of the successful predictions miRIS values drops after 1–3 predictions. This trend is not dependent on the size of the input or the length of the predicted list (compare Figure 3B with Figure 3C). Although the absolute miRIS values might be relatively low, an abrupt drop in miRIS often signifies a successful prediction.
Combinatorial nature of miRNA regulation is reflected by CLIP-Seq data
We further tested the combinatorial nature of miRNAs in human cells. The discussed miRNA experiments (Supplementary Figure S1) represent non-physiological settings that are prone to off-target noise (56). Therefore, we tested the combinatorial concept on CLIP technology experiments (24,27). Specifically, validated data for ∼6500 genes that are regulated by 653 miRNAs are compiled in StarBase (52). Note that based on the CLIP technology, the actual sequence from the mRNA that is bound to miRNA is identified by sequencing. Thus, it eliminates the largest source of error (e.g. the label of a site as true of false).
Figure 4A shows a cumulative representation of miRNA regulation according to the number of miRNAs per gene. Only 10% of the genes are regulated by a single miRNA. We noted that 50% of the targeted genes are associated with up to 8 miRNAs. About 90% of the genes are targeted by up to 35 miRNAs, with a few genes that are regulated by as many as >80 miRNAs (Supplementary Figure S3). The analysis includes the task of recovering the human genes by an input of the set of miRNAs that jointly regulate it (Figure 4B). We use the miRror procedure in a miR2Gene mode (Figure 4B). Figure 4C shows the analysis for the 12 MDBs and miRror (ranked by miRIS) for the genes that are regulated by 30–40 miRNAs (362 genes, Supplementary Table S7). The results show that for 33% of the genes, miRror correctly determined the subjected gene whereas the other MDBs failed to reach such success (success is considered a correct prediction among the top ranked 20 results). miRror also outperforms the other methods for recovering the genes at the top 1% of the predictions (Figure 4C, inset). Note that half of the MDBs failed to recover 90% of the relevant genes (Figure 4C).
Figure 4.

CLIP data analysis. (A) A cumulative view of the number of miRNAs regulating each gene, from the CLIP experiments in StarBase (52). The cumulative view demonstrates the prevalent regulation of genes by multiple miRNAs. The Total number of genes in the analysis is 6287. (B) A scheme for miRror interpretation of the CLIP data. Using miR2Gene mode to assess the recovery of a gene from the collection of miRNAs that are based on CLIP experimental data. (C) Sets of miRNAs that are regulated by 30–40 miRNAs per gene are the input for miRror application. 362 such genes were analyzed and success is categorized as two levels (top 1–9 and top 10–20 genes). Y-axis is defined as the fraction of success prediction from all selected targeted genes (total 362). The prediction success of miRror (purple) and the 12 MDBs are shown (Supplementary Table S7). Inset, the fraction of identified genes in the entire prediction list. miRror outperformed the other 12 MDBs in both success measurements.
Evidence for miRNA pairs governing cellular regulation
The tools and methodologies developed for the experimental data from miRNA overexpression (Figures 2 and 3) and CLIP datasets (Figure 4) are used to explore the concept of ‘miRNA-Duos’. A Duo (pair) of miRNAs is the simplest form of coordinated co-regulation by miRNAs and thus a starting point for the assessment. The number of human genes that are reported by the numerous CLIP data is ∼6500. A vast majority of them (90%) are regulated by multiple miRNAs (Figure 4A).
For a quantitative view of all possible pairs of miRNAs, we used the human CLIP data as an in vivo evidence for miRNA-Duos. There are 212 878 human miRNA-pairs in StarBase. Regulation by a miRNA-Duo can be generally described as one of two modes: (i) a miRNA-Duo that expands the set of gene-targets, thus allowing a higher coverage of the genes; and (ii) a miRNA-Duo with overlapping targets that can be considered as a backup mode (Figure 5A). The two extreme scenarios are formulated using the JI. Intuitively, JI is a straightforward measure for comparing the correspondence (intersection) and expansion (union) of two sets. Thus, a low JI value is indicative of the expansion mode whereas high JI indicates dominance by a backup mode (Figure 5A). We noted that the CLIP data have exceptionally low JI (average = 0.020, Supplementary Table S8).
Figure 5.

Assessment of the combinatorial nature of miRNAs. (A) miRNA regulation of miRNA-Duos according to the JI. The JI combines the intersection and the union of the targeted genes by the miRNA-Duos. Data on the miRNA-pairs are extracted from the CLIP-based experiments. The average JI value for several of the most successful MDBs is shown in view of the observations from the CLIP experiments (from StarBase). Average JI calculated for all 12 MDBs are shown in Supplementary Table S8. (B) The distribution of miRNA-Duos target overlap for several of the most successful MDBs is shown. Note that each MDB is characterized by a unique distribution. The CLIP data shows 17.5% of miRNA-Duos as having no joint targets, in contrast with the MDBs who show no such pairs. Still, a generally low overlap dominates both the MDBs and the experimental data. (C) An illustration of a scheme for the intersection of any six pairs of miRNA (for miRNAs that are marked miR-a, miR-b, miR-c and miR-d). In this scheme, the interesting candidate pair that reaches a maximal coverage (measured by JM) is the combination of miR-a and miR-b. Note that miR-c and miR-d are actually included in the predicted list of genes of miR-a and miR-b. (D) Global analysis of 20 experiments in manipulated cell-lines (Supplementary Table S9). The top 5 miRNA predictions are listed as 10 pairs (total of 200 data-points). The joint miRIS is plotted in view of the JI. miRNA pairs that are within the statistical variance of the data are highlighted in light yellow. Several extreme cases are emphasized. Note the 4 pairs that involve miR-218. The combination of some miRNAs with miR-218 led to a substantial increase in the number of predictions relative to the input list.
The miRNA pairs (from the CLIP data) are tested by JI (Figure 5A). Each MDB is centered at a characteristic range of JI. This is strongly affected by the number of shared targets between the pair (Figure 5B). The average JI is 0.032, 0.027 and 0.096 for PITA-Top, TargetScan-conserved and Miranda-microRNA.org, respectively (Supplementary Table S8). The tendency of each MDB toward a restricted range of JI exposes the inherent bias of each MDB.
Inspecting transcriptome profiles for evidence of a combinatorial miRNA regulation mode
We tested the validity of miRNA-Duo regulation in a wide range of cellular contexts. As miRror provides a robust approach for a combinatorial regulation by miRNA, we tested miRror’s capacity toward miRNA-Duos. Figure 5C illustrates a simplified test case. Each miRNA is assigned with its calculated miRIS. An edge connecting two nodes is marked by JM value which is associated with a miRNA pair. Specifically, the JM considers the miRIS of each of the miRNAs under the assumption that the miRNA pair is a new entity. For example, a miRNA that covers 50% of the genes query and 50% of the MDBs will have a miRIS of 0.5. Similarly, the miRIS of its pair miRNA is also 0.5. For this example, the JM must range from 0.5 to 1.0. The JM is thus an indirect measure of the intersection and union (measured by JI) of the miRNA in the pair. We illustrate miRNA-Duos in which the overlapping, backup mode dominates (miR-a and miR-d; miR-b and miR-c). Other miRNAs adopt an expanding, complementation mode (miR-a and miR-c; miR-c and miR-d).
As miRNAs that belong to the same family regulate a similar list of genes, a bias toward a backup mode of regulation is expected (Figure 5A). Thus, we integrated a compression of miRNAs into their families (Supplementary Table S3) for eliminating miRNA-Duos that trivially overlap their gene lists. We selected 20 large-scale gene expression experiments (Supplementary Table S9), and set to evaluate the potential of miRNA-pairs to explain the observed transcriptomic profile. We focused on experiments that were conducted on human cell-lines that were exposed to a variety of stress conditions, molecular and pharmacological manipulations. Importantly, in none of these experiments the involvement of miRNAs regulation was proposed.
Figure 5D shows the relationship of two essential features for miRNA-Duos regulation: the joint miRIS (JM, Materials and Methods) and the JI. For each of the 20 experiments, the 5 top ranked predictions (by miRIS) were considered as miRNA-pairs. A correlation between the Joint miRIS and the JI is substantial (R2 = 0.245). We focused on pairs that were specified by unusually high JI and low Joint miRIS. Such pairs are miR-25 and miR-32; miR-363 and miR-367. We found that these miRNAs share the same 6-mer seed and thus represent cases of overlapping, backup regulatory mode. Note that the family relations were not reported by miRBase or any other resource. The same is valid for the miRNA-Duo of miR-26 and miR-1297 whose overlap in the seed sequence is overlooked (Figure 5D).
The regulatory strategy of expanding, complementation mode dominates the CLIP data (Figure 5B, CLIP). We search pairs that are characterized by a low JI and a significant Joint miRIS. Such instances can be explained by cooperative activity by a small set of miRNAs. miRNAs that interact with hsa-miR-218 (Figure 5D, orange symbols) are promising candidates. The miRNA-Duos that pair with hsa-miR-218 maximized the likelihood of the observed profile of down-regulation genes. These pairs were predicted for the transcriptome of 293T cells treated with camptothecin (GSE2451). We found no evidence to support miRNA-Duos as a hidden layer of regulation for most (85%) of the tested experiments, as expected (Supplementary Table S10). However, for 3 experiments a significant coverage of the input genes can be explained by miRNA-pair acting together: (i) the RPTEC cells that were exposed to hypoxia (GSE12792); (ii) ultrasound treatment in human leukemia U937 cells (GSE10212); and (iii) 293T cells treated with camptothecin (GSE2451). Camptothecin is a natural cytotoxic quinoline alkaloid drug that inhibits DNA topoisomerase I and is used in cancer chemotherapy. Many of the down-regulated genes from this experiment are associated with cell-cycle regulation genes that are subjected to miRNA regulation. We anticipate that irrespectively to the primary target of the drug, the camptothecin treated cell reached homeostasis through the involvement of miRNA regulation. Indeed, several recent studies confirmed the participation of miRNAs in camptothecin treated cells (57).
DISCUSSION
The concept of combinatorial miRNA regulation is not new. It was discuss regarding an individual target (58), a combination of miRNAs (35,59) and as part of a network (60) with transcription regulation (61). At the cell phenotype level, a synergism in miRNA action was studied in the context of human diseases (62) and the disruption of human pathways (38). In this study, we present experimental evidence that emphasize the importance of a combinatorial mode for analyzing cell regulation under changing conditions. We present miRror2.0 as a tool that implemented the concept of ‘miRNAs working together’ into a meta-prediction platform. We show that miRror can be applied to input consisting of either a gene set or a collection of miRNAs. As such, it supports the experimental biologists in gaining insights from a broad range of experimental protocols, under a wide range of parameters. The predictive power of miRror relative to the other 12 relevant MDBs strengthens the notion of a combinatorial miRNA control mode, particularly in the form of miRNA-pairs. The MDBs do not carry the capacity to analyze a large number of genes or miRNAs. These MDBs are designed to match a miRNA with its direct targets or a specific gene to its regulating miRNAs. The success of miRror2.0 in explaining the cell’s transcriptomic signature and the CLIP data should be considered as a proof of concept. Accordingly, a transcriptomic signature can be applied as input for miRror to assess the contribution of miRNA regulation. We leverage miRror’s predictive capabilities to successfully identify a hidden layer of regulation in manipulated cells (Figure 5D).
Coping with noisy predictions by miRror
An estimation of prediction accuracy by established MDBs is reported in (21). The miRror2.0 system outperformed other MDBs, with the success attributable to: (i) the incorporation of statistical considerations for predictions above a predetermined threshold; and (ii) the concept that is applied to genes or miRNAs sets as input. We demonstrate that our protocol (Figure 1) successfully overcomes the noisy predictions of existing MDBs. Surprisingly, we found that the use of the internal scoring methods had a negative impact on miRror’s success rate (Figure 2B), dropping from 60% to 53% (success being defined as a correct entry in the top 9 predictions). Only Miranda-microRNA.org and microT3.0 improved their success (from 40% to 47% and from 40% to 50%, respectively). Both MDBs have recently refined their internal scoring methods (55,63). MAMI and TargetScan maintained their success rate. The success of the other MDBs had actually deteriorated. For PicTar-4 the success was reduced from 40% to 10% using its suggested internal score. Thus, it seems that the value of the scoring methods for some of the MDBs should be revisited.
A known pitfall in the field of miRNA prediction concerns the inconsistency in miRNA notation schemes. The use of 12 difference resources led to an artificial expansion of the miRNA input set (e.g. hsa-miR-19 is sometimes denoted as hsa-mir-19a, hsa-mir-19b or hsa-mir-19b-2). Furthermore, the hsa-mir-19a* and hsa-mir-19b* are disputably grouped as part of the has-miR-19 equivalence class. Due to the inconsistency in miRNA nomenclature, the union of miRNAs and genes submitted to miRror reached an unrealistic number of 2528 and 46 921, respectively (Supplementary Figure S1). Therefore, we applied the miRNA family compression scheme. The family representation according to miRBase definition compresses the MDBs by 2–4-folds. However, a family assignment that is based on seed sequence identity reduces the number of miRNAs by only 38% (Supplementary Table S3). Such representation improved our prediction success, with only a minimal reduction in sensitivity. Furthermore, the prevalence of the overlapping mode of miRNA regulation was reduced to a minimum following the compressing of miRNAs to their respective families.
Interestingly, many variants of miRNAs (called isomiRs) were found by deep-sequencing studies. Experimental analysis showed that isomiRs and their canonical counterparts act coordinately to target functionally related genes and pathways (64). Thus, in addition to the seed dependent family assignment (Supplementary Figure S3) isomiRs function as an additional mode for increasing the specificity of miRNA regulation.
Exposing a combinatorial mode of action from raw data
miRNAs constitute an additional layer of post-transcriptional regulation. Several studies aim to unveil the design principle underlying miRNA regulation (14). Systems biology has identified small miRNA modules that by simultaneous regulation alter a functional module (e.g. EGFR-driven cell-cycle pathway (65)). The hierarchy among miRNAs suggests that some miRNAs act as pathway hubs whereas others are interconnected with transcription factors to provide small functional modules (60). In this study, we assessed the capacity and limitations of the miRror platform in exposing miRNA cell regulation in healthy human cells. We applied the Joint miRIS (JM) as a measure for the validity of regulation by miRNA-Duos. The assumption being that a small number of coordinated miRNAs act to maintain robust homeostasis. Under this assumption, even a modest modulation executed in a combinatorial fashion can yield to a substantial change in cell phenotype. In our analysis we had not considered the spacing of the binding sites along the transcripts. It was shown that a combinatorial regulation at the level of one transcript is not necessarily cooperative but may be competing. Thus, the outcome of a combined action of miRNAs may be a lower efficacy than a single site (66).
An intriguing observation from the analyzed CLIP data is the dominant appearance of expanding, complementation mode. We confirm an identical distribution for simulated data (randomizing the pairs of miRNA and genes in a two-sided graph) and the observed CLIP data. Thus, we reject the possibility of a biased sampling of the in vivo situation (see Discussion in (24)). Instead, a strong preference in the CLIP data toward the expanded mode suggests that accessibility and competition with RNA binding proteins drives the preferential binding of miRNA on their targets (14). However, a test for each of the MDBs (e.g. TargetScan, Supplementary Figure S3) and the union of the MDBs differ from the properties of the CLIP data (24,27,67,68) (Supplementary Figure S3). Whether the expanding mode is the preferred mode of action in vivo under stress and extreme conditions in yet to be determined.
We anticipate that the miRror platform will be used successfully for screening for overlooked miRNA regulation (in either Gene2miR or miR2Gene modes). In this regards we intentionally excluded from our analysis any transcriptomic data from cancer tissues and transformed cells as many of these datasets are dominated overwhelmingly by cancer-related miRNAs (69). Notably, the majority of the transcriptomic studies (20 experiments, Supplementary Table S10) do not claim any miRNA regulation process. However, for 15% of cell manipulated experiments (3/20), a regulation by miRNAs remains a plausible hypothesis (Figure 5D).
The expression levels of miRNAs in cell-lines (70) and normal tissues are accumulating (2,71). In this view, the expression of hsa-miR-218 and hsa-miR-374 (Figure 5D) co-occur in most independent experiments. Actually, the miRNA-Duo of miR-218 and miR-374 doubled the number of successful hits from 27 to 54 genes (Supplementary Table S10). We expect to identify sets of miRNAs that have the potential to govern the observed transcriptomic profile in view of the co-occurrence in miRNAs expression.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–10 and Supplementary Figures 1–3.
FUNDING
Funding for open access charge: EU Framework VII—Prospects consortium; ISF 592/07 and BSF 2007219.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Guy Naamati whose contribution to the miRror platform development was seminal. We thank Nati Linial for insightful ideas throughout this research. We thank Manor Askenazi for his comments and reading the manuscript. Solange Karsenty supports the miRror website. A student fellowship (O.B.) is awarded by the SCCB, the Sudarsky Center for Computational Biology.
REFERENCES
- 1.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 2.Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calin GA, Ferracin M, Cimmino A, Di Leva G, Shimizu M, Wojcik SE, Iorio MV, Visone R, Sever NI, Fabbri M, et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N. Engl. J. Med. 2005;353:1793–1801. doi: 10.1056/NEJMoa050995. [DOI] [PubMed] [Google Scholar]
- 4.Grad Y, Aach J, Hayes GD, Reinhart BJ, Church GM, Ruvkun G, Kim J. Computational and experimental identification of C. elegans microRNAs. Mol. Cell. 2003;11:1253–1263. doi: 10.1016/s1097-2765(03)00153-9. [DOI] [PubMed] [Google Scholar]
- 5.Friedlander MR, Chen W, Adamidi C, Maaskola J, Einspanier R, Knespel S, Rajewsky N. Discovering microRNAs from deep sequencing data using miRDeep. Nat. Biotechnol. 2008;26:407–415. doi: 10.1038/nbt1394. [DOI] [PubMed] [Google Scholar]
- 6.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2010;39:D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bushati N, Cohen SM. microRNA functions. Annu. Rev. Cell Dev. Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- 8.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl Acad. Sci. USA. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee YS, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007;21:1025–1030. doi: 10.1101/gad.1540407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomson DW, Bracken CP, Goodall GJ. Experimental strategies for microRNA target identification. Nucleic Acids Res. 2011;39:6845–6853. doi: 10.1093/nar/gkr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forman JJ, Legesse-Miller A, Coller HA. A search for conserved sequences in coding regions reveals that the let-7 microRNA targets Dicer within its coding sequence. Proc. Natl Acad. Sci. USA. 2008;105:14879–14884. doi: 10.1073/pnas.0803230105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brodersen P, Voinnet O. Revisiting the principles of microRNA target recognition and mode of action. Nat. Rev. Mol. Cell Biol. 2009;10:141–148. doi: 10.1038/nrm2619. [DOI] [PubMed] [Google Scholar]
- 15.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat. Rev. Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 17.Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 18.Doench JG, Sharp PA. Specificity of microRNA target selection in translational repression. Genes Dev. 2004;18:504–511. doi: 10.1101/gad.1184404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rajewsky N. microRNA target predictions in animals. Nat. Genet. 2006;38:S8–S13. doi: 10.1038/ng1798. [DOI] [PubMed] [Google Scholar]
- 20.Long D, Lee R, Williams P, Chan CY, Ambros V, Ding Y. Potent effect of target structure on microRNA function. Nat. Struct. Mol. Biol. 2007;14:287–294. doi: 10.1038/nsmb1226. [DOI] [PubMed] [Google Scholar]
- 21.Shirdel EA, Xie W, Mak TW, Jurisica I. NAViGaTing the micronome–using multiple microRNA prediction databases to identify signalling pathway-associated microRNAs. PLoS ONE. 2011;6:e17429. doi: 10.1371/journal.pone.0017429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alexiou P, Maragkakis M, Papadopoulos GL, Reczko M, Hatzigeorgiou AG. Lost in translation: an assessment and perspective for computational microRNA target identification. Bioinformatics. 2009;25:3049–3055. doi: 10.1093/bioinformatics/btp565. [DOI] [PubMed] [Google Scholar]
- 23.Mendes ND, Freitas AT, Sagot MF. Current tools for the identification of miRNA genes and their targets. Nucleic Acids Res. 2009;37:2419–2433. doi: 10.1093/nar/gkp145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wen J, Parker BJ, Jacobsen A, Krogh A. MicroRNA transfection and AGO-bound CLIP-seq data sets reveal distinct determinants of miRNA action. RNA. 2011;17:820–834. doi: 10.1261/rna.2387911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin RC, Liu PP, Goloviznina NA, Nonogaki H. microRNA, seeds, and Darwin?: diverse function of miRNA in seed biology and plant responses to stress. J. Exp. Bot. 2010;61:2229–2234. doi: 10.1093/jxb/erq063. [DOI] [PubMed] [Google Scholar]
- 26.Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol. 2003;5:R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen K, Rajewsky N. The evolution of gene regulation by transcription factors and microRNAs. Nat. Rev. Genet. 2007;8:93–103. doi: 10.1038/nrg1990. [DOI] [PubMed] [Google Scholar]
- 29.Griffiths-Jones S. miRBase: the microRNA sequence database. Methods Mol. Biol. 2006;342:129–138. doi: 10.1385/1-59745-123-1:129. [DOI] [PubMed] [Google Scholar]
- 30.Willenbrock H, Salomon J, Sokilde R, Barken KB, Hansen TN, Nielsen FC, Moller S, Litman T. Quantitative miRNA expression analysis: comparing microarrays with next-generation sequencing. RNA. 2009;15:2028–2034. doi: 10.1261/rna.1699809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larsson E, Sander C, Marks D. mRNA turnover rate limits siRNA and microRNA efficacy. Mol. Syst. Biol. 2010;6:433. doi: 10.1038/msb.2010.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arvey A, Larsson E, Sander C, Leslie CS, Marks DS. Target mRNA abundance dilutes microRNA and siRNA activity. Mol. Syst. Biol. 2010;6:363. doi: 10.1038/msb.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Linsley PS, Schelter J, Burchard J, Kibukawa M, Martin MM, Bartz SR, Johnson JM, Cummins JM, Raymond CK, Dai H, et al. Transcripts targeted by the microRNA-16 family cooperatively regulate cell cycle progression. Mol. Cell Biol. 2007;27:2240–2252. doi: 10.1128/MCB.02005-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, et al. Combinatorial microRNA target predictions. Nat. Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 36.Ivanovska I, Cleary MA. Combinatorial microRNAs working together to make a difference. Cell Cycle. 2008;7:3137–3142. doi: 10.4161/cc.7.20.6923. [DOI] [PubMed] [Google Scholar]
- 37.Du L, Schageman JJ, Subauste MC, Saber B, Hammond SM, Prudkin L, Wistuba, Ji L, Roth JA, Minna JD, et al. miR-93, miR-98, and miR-197 regulate expression of tumor suppressor gene FUS1. Mol. Cancer Res. 2009;7:1234–1243. doi: 10.1158/1541-7786.MCR-08-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Naamati G, Friedman Y, Balaga O, Linial M. Susceptibility of the human pathways graphs to fragmentation by small sets of microRNAs. Bioinformatics. 2012;28:983–990. doi: 10.1093/bioinformatics/bts077. [DOI] [PubMed] [Google Scholar]
- 39.Lewis BP, Shih I-h, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 40.John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maragkakis M, Alexiou P, Papadopoulos GL, Reczko M, Dalamagas T, Giannopoulos G, Goumas G, Koukis E, Kourtis K, Simossis VA, et al. Accurate microRNA target prediction correlates with protein repression levels. BMC Bioinform. 2009;10:295. doi: 10.1186/1471-2105-10-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007;39:1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- 43.Hausser J, Berninger P, Rodak C, Jantscher Y, Wirth S, Zavolan M. MirZ: an integrated microRNA expression atlas and target prediction resource. Nucleic Acids Res. 2009;37:W266–W272. doi: 10.1093/nar/gkp412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Betel D, Wilson M, Gabow A, Marks DS, Sander C. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36:D149–D153. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X. miRDB: a microRNA target prediction and functional annotation database with a wiki interface. RNA. 2008;14:1012–1017. doi: 10.1261/rna.965408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nielsen CB, Shomron N, Sandberg R, Hornstein E, Kitzman J, Burge CB. Determinants of targeting by endogenous and exogenous microRNAs and siRNAs. RNA. 2007;13:1894–1910. doi: 10.1261/rna.768207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, Lim B, Rigoutsos I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203–1217. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 48.Sethupathy P, Megraw M, Hatzigeorgiou AG. A guide through present computational approaches for the identification of mammalian microRNA targets. Nat. Methods. 2006;3:881–886. doi: 10.1038/nmeth954. [DOI] [PubMed] [Google Scholar]
- 49.Hsu SD, Chu CH, Tsou AP, Chen SJ, Chen HC, Hsu PW, Wong YH, Chen YH, Chen GH, Huang HD. miRNAMap 2.0: genomic maps of microRNAs in metazoan genomes. Nucleic Acids Res. 2008;36:D165–D169. doi: 10.1093/nar/gkm1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barrett T, Edgar R. Mining microarray data at NCBI's Gene Expression Omnibus (GEO)*. Methods Mol. Biol. 2006;338:175–190. doi: 10.1385/1-59745-097-9:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parkinson H, Sarkans U, Kolesnikov N, Abeygunawardena N, Burdett T, Dylag M, Emam I, Farne A, Hastings E, Holloway E, et al. ArrayExpress update–an archive of microarray and high-throughput sequencing-based functional genomics experiments. Nucleic Acids Res. 2011;39:D1002–D1004. doi: 10.1093/nar/gkq1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang JH, Li JH, Shao P, Zhou H, Chen YQ, Qu LH. starBase: a database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res. 2011;39:D202–D209. doi: 10.1093/nar/gkq1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Friedman Y, Naamati G, Linial M. MiRror: a combinatorial analysis web tool for ensembles of microRNAs and their targets. Bioinformatics. 2010;26:1920–1921. doi: 10.1093/bioinformatics/btq298. [DOI] [PubMed] [Google Scholar]
- 54.Wang X. Systematic identification of microRNA functions by combining target prediction and expression profiling. Nucleic Acids Res. 2006;34:1646–1652. doi: 10.1093/nar/gkl068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Betel D, Koppal A, Agius P, Sander C, Leslie C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010;11:R90. doi: 10.1186/gb-2010-11-8-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Dongen S, Abreu-Goodger C, Enright AJ. Detecting microRNA binding and siRNA off-target effects from expression data. Nat. Methods. 2008;5:1023–1025. doi: 10.1038/nmeth.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zeng CW, Zhang XJ, Lin KY, Ye H, Feng SY, Zhang H, Chen YQ. Camptothecin induces apoptosis in cancer cells via microRNA-125b-mediated mitochondrial pathways. Mol. Pharmacol. 2012;81:578–586. doi: 10.1124/mol.111.076794. [DOI] [PubMed] [Google Scholar]
- 58.Hon LS, Zhang Z. The roles of binding site arrangement and combinatorial targeting in microRNA repression of gene expression. Genome Biol. 2007;8:R166. doi: 10.1186/gb-2007-8-8-r166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu S, Huang S, Ding J, Zhao Y, Liang L, Liu T, Zhan R, He X. Multiple microRNAs modulate p21Cip1/Waf1 expression by directly targeting its 3' untranslated region. Oncogene. 2010;29:2302–2308. doi: 10.1038/onc.2010.34. [DOI] [PubMed] [Google Scholar]
- 60.Shalgi R, Brosh R, Oren M, Pilpel Y, Rotter V. Coupling transcriptional and post-transcriptional miRNA regulation in the control of cell fate. Aging. 2009;1:762–770. doi: 10.18632/aging.100085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou YM, Ferguson J, Chang JT, Kluger Y. Inter-and intra-combinatorial regulation by transcription factors and microRNAs. BMC Genomics. 2007;8:396. doi: 10.1186/1471-2164-8-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu J, Li CX, Li YS, Lv JY, Ma Y, Shao TT, Xu LD, Wang YY, Du L, Zhang YP, et al. MiRNA-miRNA synergistic network: construction via co-regulating functional modules and disease miRNA topological features. Nucleic Acids Res. 2010;39:825–836. doi: 10.1093/nar/gkq832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maragkakis M, Reczko M, Simossis VA, Alexiou P, Papadopoulos GL, Dalamagas T, Giannopoulos G, Goumas G, Koukis E, Kourtis K, et al. DIANA-microT web server: elucidating microRNA functions through target prediction. Nucleic Acids Res. 2009;37:W273–W276. doi: 10.1093/nar/gkp292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cloonan N, Wani S, Xu Q, Gu J, Lea K, Heater S, Barbacioru C, Steptoe AL, Martin HC, Nourbakhsh E, et al. MicroRNAs and their isomiRs function cooperatively to target common biological pathways. Genome Biol. 2011;12:R126. doi: 10.1186/gb-2011-12-12-r126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Uhlmann S, Mannsperger H, Zhang JD, Horvat EA, Schmidt C, Kublbeck M, Henjes F, Ward A, Tschulena U, Zweig K, et al. Global microRNA level regulation of EGFR-driven cell-cycle protein network in breast cancer. Mol. Syst. Biol. 2012;8:570. doi: 10.1038/msb.2011.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Saetrom P, Heale BS, Snove O, Jr, Aagaard L, Alluin J, Rossi JJ. Distance constraints between microRNA target sites dictate efficacy and cooperativity. Nucleic Acids Res. 2007;35:2333–2342. doi: 10.1093/nar/gkm133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, Clark TA, Schweitzer AC, Blume JE, Wang X, et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456:464–469. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Darnell RB. HITS-CLIP: panoramic views of protein-RNA regulation in living cells. Wiley Interdiscip. Rev. RNA. 2011;1:266–286. doi: 10.1002/wrna.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat. Rev. Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 70.Liu H, D'Andrade P, Fulmer-Smentek S, Lorenzi P, Kohn KW, Weinstein JN, Pommier Y, Reinhold WC. mRNA and microRNA expression profiles of the NCI-60 integrated with drug activities. Mol. Cancer. Ther. 2010;9:1080–1091. doi: 10.1158/1535-7163.MCT-09-0965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liang Y, Ridzon D, Wong L, Chen C. Characterization of microRNA expression profiles in normal human tissues. BMC Genomics. 2007;8:166. doi: 10.1186/1471-2164-8-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.