

Background: AUG recognition is promoted by several initiation factors (eIFs).
Results: eIF5 interacts with the extreme N terminus of eIF3c/Nip1 to promote pre-initiation complex assembly, and eIF1 binds the region that immediately follows.
Conclusion: eIF1 binding to c/Nip1 is equally important for its 40 S ribosome recruitment and AUG selection.
Significance: Understanding start codon selection that sets the reading frame for decoding is key in gene expression studies.
Keywords: Gene Expression, Mutagenesis, Ribosomes, Translation Control, Translation Initiation Factors, AUG Recognition, eIF1, eIF2 Ternary Complex, eIF3, Translation Initiation
Abstract
In eukaryotes, for a protein to be synthesized, the 40 S subunit has to first scan the 5′-UTR of the mRNA until it has encountered the AUG start codon. Several initiation factors that ensure high fidelity of AUG recognition were identified previously, including eIF1A, eIF1, eIF2, and eIF5. In addition, eIF3 was proposed to coordinate their functions in this process as well as to promote their initial binding to 40 S subunits. Here we subjected several previously identified segments of the N-terminal domain (NTD) of the eIF3c/Nip1 subunit, which mediates eIF3 binding to eIF1 and eIF5, to semirandom mutagenesis to investigate the molecular mechanism of eIF3 involvement in these reactions. Three major classes of mutant substitutions or internal deletions were isolated that affect either the assembly of preinitiation complexes (PICs), scanning for AUG, or both. We show that eIF5 binds to the extreme c/Nip1-NTD (residues 1–45) and that impairing this interaction predominantly affects the PIC formation. eIF1 interacts with the region (60–137) that immediately follows, and altering this contact deregulates AUG recognition. Together, our data indicate that binding of eIF1 to the c/Nip1-NTD is equally important for its initial recruitment to PICs and for its proper functioning in selecting the translational start site.
Introduction
Translation is a fundamental process contributing to the regulation of gene expression, normal developmental processes, and occurrence of disease. It can be divided into initiation, elongation, termination, and ribosome recycling with the initiation phase serving as a target of the most regulatory pathways (for a review, see Ref. 1). Undoubtedly, the start codon selection is the key step of this phase and in fact of translation in general as it sets the reading frame for decoding. Initiation at an incorrect codon will produce a completely miscoded protein, wasting valuable resources of the cell and creating a potentially toxic peptide. In contrast to prokaryotic cells, the mRNAs of which posses a Shine-Dalgarno sequence that ensures a direct placement of the start codon into the ribosomal P-site, eukaryotic ribosomes have to search a 5′-untranslated region (UTR) of an mRNA for the start codon by a successive movement called scanning. During this process, ribosomes have to read and respond to a variety of integrated yet not well understood signals that orchestrate the AUG recognition. These signals originate from mutual molecular and functional interactions between mRNA and ribosomes with a number of proteins called eukaryotic translation initiation factors (eIFs) such as eIF1A, eIF1, eIF2 (in the form of the eIF2·Met-tRNAiMet·GTP ternary complex (TC)3), and eIF5.
Upon initial binding of the latter factors to the 40 S small ribosomal subunit stimulated by the multisubunit eIF3 complex, eIFs 1 and 1A serve to stabilize a specific conformation of the 40 S head relative to its body that opens the mRNA binding channel for mRNA recruitment to form the 48 S pre-initiation complex (PIC). That requires dissolving the latch formed by helices 18 and 34 of 18 S rRNA and establishing a new interaction between Rps3 and helix 16 (2). This so-called open/scanning-conducive conformation with the anticodon of Met-tRNAiMet not fully engaged in the ribosomal P-site to prevent premature engagement with putative start codons is then maintained during scanning for the AUG start codon in an ATP-dependent process (for reviews, see Refs. 3 and 4). During this search, eIF2 partially hydrolyzes its GTP with the help of the GTPase accelerating factor eIF5. Prior to start codon recognition, the “gate-keeping” function of eIF1 prevents the release of the resultant phosphate ion producing both GTP- and GDP·Pi-bound states of the factor possibly in equilibrium (5). Encounter of the AUG start codon induces a reciprocal conformational switch of the 48 S PIC to the closed/scanning-arrested form stabilized by a functional interaction between eIF1A and eIF5 (6) with the initiator Met-tRNA fully accommodated in the P-site (7). This irreversible reaction serves as the decisive step in stalling the entire machinery at the AUG start codon and is triggered by displacement or dissociation of eIF1 (8) possibly promoted by eIF1A and eIF5 and subsequent release of free Pi. In short, eIF1 and eIF1A (via its C-terminal tail) antagonize the codon-anticodon interactions in the P-site by blocking the full accommodation of initiator tRNA in the P-site in a manner that is overcome efficiently by the action of the N-terminal tail of eIF1A and eIF5 upon establishment of a perfect AUG-anticodon duplex in an optimal AUG context (for reviews, see Refs. 3 and 4).
Specific mutations that reduce the stringency of start codon recognition in budding yeast, allowing increased utilization of near-cognate codons (UUG or AUU), produce the Sui− phenotype (suppressor of initiation codon mutation). Mutations with the opposite effect of lowering UUG initiation in the presence of a given Sui− mutation impart the Ssu− phenotype (suppressor of Sui−). Defects in AUG selection can also be identified by measuring the efficiency of initiation at the AUG of uORF1 in GCN4 mRNA in a well established in vivo GCN4-lacZ reporter system (for example, see Refs. 9 and 10).
Besides the aforementioned factors, there is an increasing number of reports suggesting that the multifunctional eIF3 complex also significantly contributes to the regulation of AUG recognition (9–13). First of all, yeast eIF3 plays a critical role in productive mRNA recruitment (14, 15) and directly interacts with mRNA (16–18), suggesting that the way the mRNA interacts with the mRNA binding channel during scanning for AUG can be influenced by eIF3. In addition, yeast eIF3 is composed of six subunits (a/Tif32, b/Prt1, c/Nip1, i/Tif34, g/Tif35, and j/Hcr1), two of which directly interact with eIF1, TC, or eIF5 in the multifactor complex (MFC) (Fig. 1) (for a review, see Ref. 4). Particularly intriguing in this respect is the N-terminal domain (NTD) of c/Nip1 that makes direct contacts with eIFs 1 and 5 and via the latter also associates with the TC (19, 20). Indeed, several important segments (designated as Boxes) within the c/Nip1-NTD were identified previously; mutations of these segments impaired stringency of the start codon selection and produced either Sui− or Ssu− phenotypes in a manner intensified or suppressible by increased gene dosage of eIF5 or eIF1 (11). As could be expected, some of the identified mutations also affected assembly of the PICs. Furthermore, increased frequency of skipping the AUG of uORF1 in the GCN4-lacZ reporter (a leaky scanning phenotype) was observed with mutations disrupting the web of mutual interactions among the members of an eIF3 module composed of the RNA recognition motif in the NTD of b/Prt1 and j/Hcr1 and the C-terminal domain (CTD) of a/Tif32 (Fig. 1) (9, 10). The robust leaky scanning phenotype also accompanies perturbed interactions within the other eIF3 module formed by the extreme CTD of b/Prt1 and by i/Tif34 and g/Tif35 (Fig. 1) (13). Interestingly, both modules are thought to reside near the 40 S mRNA entry channel with g/Tif35 and the a/Tif32-CTD directly interacting with the Rps3 “latch” component (9, 10). Based on these observations, we recently proposed that the scaffold b/Prt1 subunit serves to connect both eIF3 modules at each of its termini to work together with c/Nip1 and other eIFs to fine-tune the AUG selection process (13).
FIGURE 1.

A three-dimensional model of eIF3 and its associated eIFs in the MFC (adapted from Ref. 13). ntd, N-terminal domain; ctd, C-terminal domain; hld, Hcr1-like domain; rrm, RNA recognition motif; pci, PCI domain. The NMR structure of the interaction between the RNA recognition motif of human eIF3b (green and light blue) and the N-terminal peptide of human eIF3j (yellow) (9), the NMR structure of the C-terminal RNA recognition motif of human eIF3g (red and sky blue) (18), the x-ray structure of the yeast i/Tif34·b/Prt1-CTD complex (13), and the three-dimensional homology model of the c/Nip1-CTD (31) were used to replace the original schematic representations of the corresponding molecules.
In this study, we subjected the aforementioned segments of the c/Nip1-NTD to semirandom mutagenesis to pinpoint critical residues that either promote the assembly of PICs (by screening for the Gcd− (general control derepressed) phenotype) or more importantly ensure stringent selection of the AUG start codon (by screening for the Sui− phenotype). Strikingly, we were able to separate the effects of distinct amino acid substitutions within a short 8-residue segment on the manifestation of either of the latter phenotypes, suggesting that the c/Nip1-NTD promotes both initiation reactions independently of each other at least to a certain degree. Based on our findings, we propose a model suggesting that not only the productive recruitment of eIF1 to 40 S ribosomes but also its proper functioning during the AUG recognition process depends on its contact with the stretch of amino acid residues 60–137 of the c/Nip1-NTD.
EXPERIMENTAL PROCEDURES
Construction of Yeast Strains and Plasmids
HMJ04 and HMJ06 were generated by introducing YCpMJ-MET-Nip1-W into HLV04 and HKN06 (11), respectively. The original pNIP1+ (c/NIP1 URA3) covering plasmid was evicted on SD plates containing 5-fluoro-orotic acid.
To create HMJ08, H2880 (21) was first transformed with YCpNIP1-His-L to cover for the deletion of NIP1 that was made in the next step by introducing the SacI-SphI fragment carrying the nip1Δ::hisG-URA3-hisG integration cassette from pLV10 (11). The uracil auxotrophy was regained by growing the cells on SD plates containing 5-fluoro-orotic acid. The resulting strain was subsequently transformed with YCpNIP1-His-U, and the leucine auxotrophy was regained by growing the cells in liquid medium containing leucine and selecting for those that lost the YCpNIP1-His-L plasmid on SD ± leucine plates producing HMJ08.
YCpMJ-MET-NIP1-W was constructed by inserting the 2618-bp BamHI-HindIII fragment from pGAD-NIP1 (22) into YCplac22MET-W (23) digested by BamHI-HindIII.
YCpNIP1-Box12-WW was made by fusion PCR using the template YCpNIP1-His-L and two sets of primers (JKNIP1Mut and MJreversV111WV112W; LV22ext and MJ111WW) in the first PCR. The PCR products thus obtained were mixed in 1:1 ratio, and the second PCR was performed using primers JKNIP1Mut and LV22ext. The resulting PCR product (631 bp) was cut with AvaI-XbaI and inserted into AvaI-XbaI-digested YCpNIP1-His-help3 (11).
The following plasmids were created essentially the same as those described above with the exception of the primer sets (containing the degenerate oligonucleotides) that were used in the first PCRs: YCpNIP1-Box12-SPW (JKNIP1Mut and LV12C-1XR; LV22ext and LVBOX12C-1X), YCpNIP1-Box12-WWPW (JKNIP1Mut and LV12C-3XR; LV22ext and LVBOX12C-3X), YCpNIP1-GAP85 (JKNIP1Mut and LV12C-2XR; LV22ext and LVBOX12C-2X), YCpNIP1-GAP92 (JKNIP1Mut and LV12C-4XR; LV22ext and LVBOX12C-4X), and YCpNIP1-Box15-2 (JKNIP1Mut and LV15C-2XR; LV22ext and LVBOX15C-2X).
YEpNIP1-GAP85, -GAP92, and -Box15-2 were constructed by inserting the 2887-bp HindIII-PstI fragments from the corresponding single copy plasmids described above into YEplac181 digested with HindIII-PstI. pT7-NIP1-N270, pT7-NIP1-N-Box12-SPW, pT7-NIP1-N-Box12-WWPW, pT7-NIP1-N-Box12-WW, pT7-NIP1-N-Box15-2, pT7-NIP1-N-GAP85, and pT7-NIP1-N-GAP92 were all made by inserting the NdeI-HindIII-digested PCR product obtained with primers JKNIP1Mut and LVRN270 using the templates YCpNIP1-His-L, YCpNIP1-Box12-SPW, YCpNIP1-Box12-WWPW, YCpNIP1-Box12-WW, YEpNIP1-Box15-2, YEpNIP1-GAP85, and YEpNIP1-GAP92, respectively, into NdeI-HindIII-digested pT7-7.
pT7-NIP1-N-Δ46/137 and pT7-NIP1-N-Δ60 were created by inserting the NdeI-BamHI-digested PCR product obtained using the template YCpNIP1-His-L and the set of primers MJNIP1_46–137 and MJNIP1_46–137rev or MJNIP1_61–205 and MJNIP1_61–205rev, respectively, into NdeI-BamHI-digested pT7-7. Lists of all yeast strains and plasmids can be found in Tables 1 and 2, respectively; a list of all PCR primers will be provided upon request.
TABLE 1.
Yeast strains used in this study
| Strain | Genotype | Source or Ref. |
|---|---|---|
| HMJ04 | MATα trp1 leu2-3,–112 ura3-52 his4-303[ATT] SUI1 nip1Δ GCN2 YCpMJ-Met-NIP1-W (MET3-NIP1 TRP1) | This study |
| HMJ06 | MATa trp1 leu2-3,–112 ura3-52 nip1Δ gcn2Δ YCpMJ-Met-NIP1-W (MET3-NIP1 TRP1) | This study |
| HMJ08 | MATa, trp1 leu2-3,–112 ura3-52 nip1Δ YCpNIP1-His-U (NIP1-His URA3) | This study |
| HLV04 | MATα trp1 leu2-3,–112 ura3-52 his4-303[ATT] SUI1 nip1Δ GCN2 pNIP1+ (NIP1 URA3) | 11 |
| HKN06 | MATa trp1 leu2-3,–112 ura3-52 nip1Δ gcn2Δ pNIP1+ (NIP1 URA3) | 11 |
| TD301-8D | MATα leu2-3,–112 ura3-52 his4-303[ATT] sui1-1 | 11 |
| H2880 | MATa trp1 leu2-3,–112 ura3-52 | 21 |
| H2881 | MATa trp1 leu2-3,–112 ura3-52 gcn2Δ | 21 |
TABLE 2.
Plasmids used in this study
| Plasmid | Description | Source of Ref. |
|---|---|---|
| YCplac111 | Single copy cloning vector, LEU2 | 43 |
| YEplac181 | High copy cloning vector, LEU2 | 43 |
| YEplac195 | High copy cloning vector, URA3 | 43 |
| YCplac22MET-W | Single copy cloning vector with conditional MET3 promoter, TRP1 plasmid from YCplac22 | K. Nasmyth |
| YCpMJ-MET-NIP1-W | Single copy NIP1 under MET3 promoter, TRP1 plasmid from YCplac22 | This study |
| YCpNIP1-His-U | Single copy NIP1-His, Ura3 plasmid from YCplac33 | 20 |
| YEpNIP1-His-U | High copy NIP1-His, Ura3 plasmid from YEplac195 | 20 |
| YCpNIP1-His-L | Single copy NIP1-His, Leu2 plasmid from YCplac111 | 20 |
| YEpNIP1-His-L | High copy NIP1-His, Ura3 plasmid from YEplac181 | 20 |
| YCpNIP1-Box12-SPW | Single copy NIP1-His containing K113S/K116P/K118W substitutions in Box12, LEU2 plasmid from YCplac111 | This study |
| YCpNIP1-Box12-WWPW | Single copy NIP1-His containing V111W/V112W/K116P K118W substitutions in Box12, LEU2 plasmid from YCplac111 | This study |
| YCpNIP1-Box12-WW | Single copy NIP1-His containing V111W/V112W substitutions in Box12, LEU2 plasmid from YCplac111 | This study |
| YCpNIP1-GAP85 | Single copy NIP1-His containing deletion of 85 residues (Val60-Asn144), LEU2 plasmid from YCplac111 | This study |
| YCpNIP1-GAP92 | Single copy NIP1-His containing deletion of 92 residues (Asp46-Asn137), LEU2 plasmid from YCplac111 | This study |
| YCpNIP1-Box15-2 | Single copy NIP1-His containing I142V/E145W/F146T/D147L/I149R substitutions in Box15, LEU2 plasmid from YCplac111 | This study |
| YEpNIP1-GAP85 | High copy NIP1-His containing deletion of 85 residues (Val60-Asn144), LEU2 plasmid from YEplac181 | This study |
| YEpNIP1-GAP92 | High copy NIP1-His containing deletion of 92 residues (Asp46-Asn137), LEU2 plasmid from YEplac181 | This study |
| YEpNIP1-Box15-2 | High copy NIP1-His containing I142V/E145W/F146T/D147L/I149R substitutions in Box15, LEU2 plasmid from YEplac181 | This study |
| pLV10 | nip1Δ::hisG::URA3::hisG (NIP1 disruption hisG cassette) | 11 |
| p367 | Low copy URA3 vector containing HIS4-ATG-lacZ fusion | 44 |
| p391 | Low copy URA3 vector containing HIS4-TTG-lacZ fusion | 44 |
| P2041 | Low copy URA3 vector containing HIS4-ATT-lacZ fusion, 3rd codon replaced with TTA | 44 |
| p180 (YCp50-GCN4-lacZ) | Low copy URA3 vector containing wild-type GCN4 leader | 45 |
| YEpTIF5-U | High copy TIF5-FLAG, URA3 plasmid from YEplac195 | 11 |
| YEpSUI1-U | High copy SUI1, URA3 plasmid from YEplac195 | 11 |
| p1780-IMT | High copy SUI2, SUI3, GCD11, IMT4, URA3 plasmid from YEp24 | 30 |
| pGEX-TIF5 | GST-TIF5 fusion plasmid from pGEX-4T-1 | 46 |
| pGEX-SUI1 | GST-SUI1 fusion plasmid from pGEX-5X-3 | 11 |
| pT7-NIP1-N270 | NIP1 (1–270) ORF under T7 promoter | This study |
| pT7-NIP1-N-Box12-SPW | NIP1 (1–270) ORF containing K113S/K116P K118W substitutions in Box12 under T7 promoter | This study |
| pT7-NIP1-N-Box12-WWPW | NIP1 (1–270) ORF containing V111W/V112W/K116P/K118W substitutions in Box12 under T7 promoter | This study |
| pT7-NIP1-N-Box12-WW | NIP1 (1–270) ORF containing V111W V112W substitutions in Box12 under T7 promoter | This study |
| pT7-NIP1-N-GAP85 | NIP1 (1–270) ORF containing deletion of 85 residues (Val60-Asn144) under T7 promoter | This study |
| pT7-NIP1-N-GAP92 | NIP1 (1–270) ORF containing deletion of 92 residues (Asp46-Asn137 under T7 promoter | This study |
| pT7-NIP1-N-Box15-2 | NIP1 (1–270) ORF containing I142V/E145W/F146T/D147L/I149R substitutions in Box15 under T7 promoter | This study |
| pT7-NIP1-N-Δ46/137 | NIP1 ORF (amino acid residues 46–137) under T7 promoter | This study |
| pT7-NIP1-N-Δ60 | NIP1 ORF (amino acid residues 61–205) under T7 promoter | This study |
Yeast Biochemical Methods
GST pulldown experiments with GST fusions and in vitro synthesized 35S-labeled polypeptides (see Table 2 for vector descriptions) were conducted as described in Fig. 5. Ni2+ chelation chromatography of eIF3 complexes containing His8-tagged c/Nip1 from yeast whole-cell extracts (WCEs) and Western blot analysis were conducted as described in detail previously (24). In short, WCEs were incubated at 4 °C overnight with 15 μl of 50% Ni2+-Sepharose 6 Fast Flow (GE Healthcare) suspended in 300 μl of buffer A (20 mm Tris-HCl (pH 7.5), 100 mm KCl, 5 mm MgCl2, 0.5 mm β-mercaptoethanol, 1 mm phenylmethylsulfonyl fluoride, 20 mm imidazole, 10% (v/v) glycerol, 1 EDTA-free complete Protease Inhibitor Mix tablets (Roche)) followed by washing and elution. Polysome profile analysis and 2% HCHO cross-linking followed by WCE preparation and fractionation of extracts for analysis of pre-initiation complexes were carried out as described previously (24). β-Galactosidase assays were conducted as described (25).
FIGURE 5.

The extreme N terminus of c/Nip1 interacts with eIF5, the segment that immediately follows binds eIF1, and nip1-Box12-SPW increases binding affinity of the c/Nip1-NTD for eIF1 in vitro. A, schematic as in Fig. 2B showing the minimal binding segments (as arrows) of the c/Nip1-NTD for eIFs 1 and 5. The lines beneath the arrows depict the 35S-labeled segments of the c/Nip1-NTD used in the binding assays of B with the amino acid end points and clone designations indicated. The binding of each c/Nip1 construct to GST-eIF1 or -eIF5 is summarized in the table on the right. Data from the WT ((N)*) construct were taken from Ref. 19 for comparison purposes only. B, eIF1 and eIF5 fused to GST (lanes 3 and 4) or GST alone (lane 2) was expressed in E. coli, immobilized on glutathione-Sepharose beads, and incubated with the 35S-labeled c/Nip1 segments indicated to the right of the lower panels at 4 °C for 2 h. The beads were washed three times with phosphate-buffered saline, and bound proteins were separated by SDS-PAGE. Gels were first stained with GelCode Blue Stain Reagent (Pierce) (top panel) followed by autoradiography (bottom panels). Lane 1 shows 10% of the input (In) amounts. C, same as in B except that the semirandom mutations of c/Nip1-Boxes are under study. The amount of each 35S-labeled c/Nip1 polypeptide bound to each GST fusion protein was quantified and is expressed above the corresponding panel as a percentage of the input (three independent measurements were made, and the outputs were averaged with the S.E. ranging between 3 and 11%).
RESULTS
Semirandom Mutagenesis of the Selected Segments of the N-terminal Domain of the c/Nip1 Subunit of eIF3
The c/Nip1-NTD mediates eIF3 interactions with eIFs 1 and 5 and indirectly with eIF2 (20). By subjecting the N-terminal 160 amino acids of c/Nip1 to the clustered 10-alanine mutagenesis, we showed previously that these c/Nip1 interactions stimulate the assembly of the 43 S PICs and somehow coordinate the functions of eIF1 and eIF5 in stringent AUG selection (11). To understand the molecular mechanism of the c/Nip1 involvement in these functions and also to identify specific residues that are critical for them, we randomly mutated selected groups of conserved residues within the previously identified clustered 10-alanine mutagenesis c/nip1-Box mutations 6R, 12, and 15 (Fig. 2A) (20) using degenerate oligonucleotides and the fusion PCR technique. Degenerate oligonucleotides were designed to substitute a given group of amino acid residues with degenerate codons NNN where the N nucleotide was synthesized with a mixture of A, C, G, and T, each at 25% of the total. This way we constructed nine mutant libraries containing random amino acid residues at selected sites within each group derived from the latter three Boxes (Fig. 2A). These mutant libraries were then separately introduced into two different yeast strains (both deleted for chromosomal NIP1 and carrying a wild-type (WT) NIP1 allele under the control of MET3 promoter) and screened for the Sui− and Gcd− phenotypes indicative of a relaxed stringency of AUG recognition (strain HMJ04) or of a defect in 43 S PIC assembly (strain HMJ06), respectively, in the presence of methionine, which shuts off expression of the WT gene.
FIGURE 2.

Semirandom mutagenesis of the preselected segments (Boxes) of the extreme NTD of c/Nip1 yields mutants displaying either Sui− or Gcd− phenotypes alone or in combination. A, amino acid sequence alignment of the Boxes of interest from the c/Nip1-NTD of Saccharomyces cerevisiae with that of other species. The amino acid sequence of S. cerevisiae c/Nip1 (accession number P32497.2 UniProtKB/Swiss-Prot) between residues 1 and 160 was aligned with its Caenorhabditis elegans (accession number A8WWU0.1 UniProtKB/Swiss-Prot), Homo sapiens (accession number Q99613.1 UniProtKB/Swiss-Prot), Arabidopsis thaliana (accession number AAC83464.1 GenBank), and Schizosaccharomyces pombe (accession number CAB11485.2 GenBank) homologues using ClustalW; only the sequences corresponding to the Boxes of interest are shown. Highly conserved residues are color-coded. Altogether, nine mutant libraries containing random amino acid residues at selected, color-coded sites marked with “X” and grouped together individually for each Box are indicated. B, schematic representation of the first 160 amino acid residues of c/Nip1 shown as numbered circles (Boxes 1–16), each of which is composed of 10 consecutive residues. Three Boxes of interest are color-coded, and the sequences and phenotypes of the mutants derived from the semirandom mutagenesis are given below the schematic. Two internal deletions that were unintentionally generated by this mutagenic procedure are also shown with the deleted residues indicated. A table to the right of the schematic summarizes phenotypes associated with individual mutations including Slg−, Sui−, Gcd−, and the polysome/monosome (P/M) ratios (averaged values of replicate experiments are given; S.E. values ranged between 5 and 15% and are not shown). C, the c/Nip1 mutants producing the Sui− phenotype indicative of a defect in AUG recognition. The HLV04 (nip1Δ his4-303) strain was transformed with the corresponding sc or hc plasmids carrying WT NIP1 and its mutant alleles, and the resident pNIP1+ (NIP1, URA3) covering plasmid was evicted on 5-fluoro-orotic acid. The resulting strains and the parental control strain TD301-8D (NIP1 sui1-1 his4-303) were then spotted in four serial 10-fold dilutions on SD medium containing histidine (upper panel) or lacking histidine (lower panel) and incubated at 30 °C for 2 (upper panel) or 7 days (lower panel). D, the c/Nip1 mutants producing the Gcd− phenotype indicative of a defect in the assembly of the PICs. The HKN06 (nip1Δ gcn2Δ) strain was transformed with the corresponding sc or hc plasmids carrying WT NIP1 and its mutant alleles, and the resident pNIP1+ (NIP1, URA3) covering plasmid was evicted on 5-fluoro-orotic acid. The resulting transformants and isogenic strains H2880 (GCN2) (lane 1) and H2881 (gcn2Δ) (lane 2) transformed with empty vector were spotted in four serial dilutions on SD (upper panel) or SD medium containing 30 mm 3-AT (lower panel) and incubated at 30 °C for 3 and 6 days, respectively. E, Western blot analysis of the WCEs derived from the indicated strains described in C using antibodies raised against c/Nip1 and Rps0A.
Mutations that relax the stringency of translational start site selection in yeast (so-called Sui− mutations) were isolated by selecting for growth on medium lacking histidine in a his4-303 genetic background where the start codon of the HIS4 gene has been mutated and instead its third coding triplet (UUG) is used to initiate HIS4 translation (26). In other words, such mutations suppress histidine auxotrophy of the WT his4-303 cells, turning their His− into a His+ phenotype. Mutations that affect formation of 43 S PICs by reducing the rate of the TC recruitment (so called Gcd− mutations) were isolated by their ability to constitutively derepress GCN4 translation in cells lacking the kinase Gcn2, which makes them resistant to the inhibitor of histidine biosynthesis, 3-aminotriazole (3-AT). Gcn4 is a transcriptional activator of amino acid biosynthetic genes whose expression is under tight translational control via four short upstream ORFs (uORFs) occurring in its leader mRNA. The gcn2Δ cells in which translational activation of GCN4 is eliminated fail to grow on 3-AT-containing medium (for more details, see Ref. 27).
Approximately 10,000 clones from each library were screened individually for the His+ and 3-ATRes phenotypes. Altogether, five mutants with strong phenotypes were isolated, two of which fall in the Box12 group and one that falls in the Box15 group (Fig. 2B). In addition to those, a few other mutations in the latter Boxes were also identified; however, given their less pronounced phenotypes, we did not characterize them any further. Similarly, no mutations with strong phenotypes were found in the Box6R group; hence, we stopped working with this group at this point. Although not programmed intentionally, two large internal deletions of 85 (GAP85) and 92 (GAP92) residues impinging into the region between Asp46 and Asn144 of the c/Nip1-NTD also resulted from this mutagenic procedure (Fig. 2B). All mutant phenotypes were corroborated to be plasmid-born by retransformation of isolated plasmid DNAs into the nip1Δ strains carrying the WT NIP1 URA3 covering plasmid that was subsequently evicted by plasmid shuffling. As shown in Fig. 2, C and D (summarized in Fig. 2B), three major classes of mutant substitutions were identified, imparting either (a) only the Sui− phenotype (c/nip1-Box12-SPW (Fig. 2C, lane 3)), (b) only the Gcd− phenotype (c/nip1-Box15-2 (Fig. 2D, lane 7)), or (c) both combined (c/nip1-Box12-WWPW and both GAP mutants (Fig. 2, C and D, lanes 4–6, respectively)). All of these mutations also resulted in a slow growth (Slg−) phenotype of different strengths as illustrated by their varying effects on translation initiation rates estimated by measuring the polysome:monosome ratios (summarized in Fig. 2B).
Our finding that no Sui− mutations were detected in the Box15 group is consistent with the fact that the original c/nip1-Box15 also displayed only the Gcd− phenotype (11). In contrast, whereas the original c/nip1-Box12 mutation produced merely the mild Sui− phenotype, our semirandom substitutions of Box12 impart strong Sui− as well as Gcd− phenotypes. Perhaps the most striking observation, however, is the fact that whereas the deletion of the first 160 residues is lethal (11) the internal deletions of 84 or 91 residues are not lethal and resulted “only” in severe Slg−, Sui−, and Gcd− phenotypes. Because the protein levels of Box15-2 and both GAP mutants in a single copy (sc) number were found partially reduced when compared with those of the WT c/Nip1, we generated high copy number versions of these three alleles. High copy (hc) expression increased their protein levels by ∼2-fold and more or less matched the sc expression of the WT c/Nip1 (Fig. 2E). Importantly, because there was no qualitative difference in the mutant phenotypes between the sc and hc expressions of the latter alleles (data not shown), we decided to use their hc versions throughout the rest of the study.
Dissection of Effects of the Specific c/Nip1-Box12 Residues on Phenotypes Indicating Impaired Formation of PICs Versus AUG Selection
We noted that substitution of three Box12 lysines (113, 116, and 118) by Ser, Pro, and Trp in c/nip1-Box12-SPW produced merely the Sui− phenotype, whereas substitution of two preceding highly conserved valines (111 and 112) by tryptophans in combination with K116P and K118W in c/nip1-Box12-WWPW produced both the Sui− and Gcd− phenotypes (Fig. 2, B–D). The fact that both mutants contain K116P and K118W substitutions might thus suggest that these two also highly conserved lysines specifically contribute to the stringency of AUG selection, whereas Val111 and Val112 participate in assembly of the PICs. To test that, we separated the V111W and V112W double substitution from K116P and K118W by producing two individual c/nip1-Box12-WW and c/nip1-Box12-PW mutant alleles, which were then tested for Sui− and Gcd− phenotypes as described above. Strikingly, in accord with our prediction, c/nip1-Box12-WW imparts the severe Gcd− phenotype but not Sui− (Fig. 2D, lane 8). At odds with our prediction, the c/nip1-Box12-PW mutant showed no apparent phenotype (data not shown). Thus, we propose that whereas the two consecutive valines (Val111 and Val112) somehow promote recruitment of the TC to the small ribosomal subunit all three consecutive lysines (Lys113, Lys116, and Lys118) are critically required for proper detection of the AUG start codon. In support, one of the “weaker” Box12 mutants that was isolated in our initial screen but not analyzed further carried substitutions of exactly these three lysines (K113Y, K116H, and K118H) and displayed the Sui− but not Gcd− phenotype (data not shown).
To demonstrate that the Sui− phenotype is indeed caused by increased selection of the UUG triplet as the start site and that our Sui− mutants increase initiation rates at near-cognate codons in general, we examined expression of matched HIS4-lacZ reporters containing UUG or AUU versus AUG start codons (Fig. 3A). Previously, we reported that the original c/nip1-Box12 10-Ala substitution increased the UUG/AUG and AUU/AUG initiation ratios by ∼3- and 5-fold, respectively (11). In comparison, all three Sui− mutants (12-SPW, -WWPW, and GAP85) generated here conferred a much more dramatic increase in the UUG/AUG initiation ratio (from 12- to 29-fold) and a similar increase in the AUU/AUG initiation ratio (∼6-fold), indicating a strong preference for selecting UUG over the AUU as the false start site. As expected, the “Gcd−-only” Box12-WW mutant showed little to no increase in both ratios.
FIGURE 3.
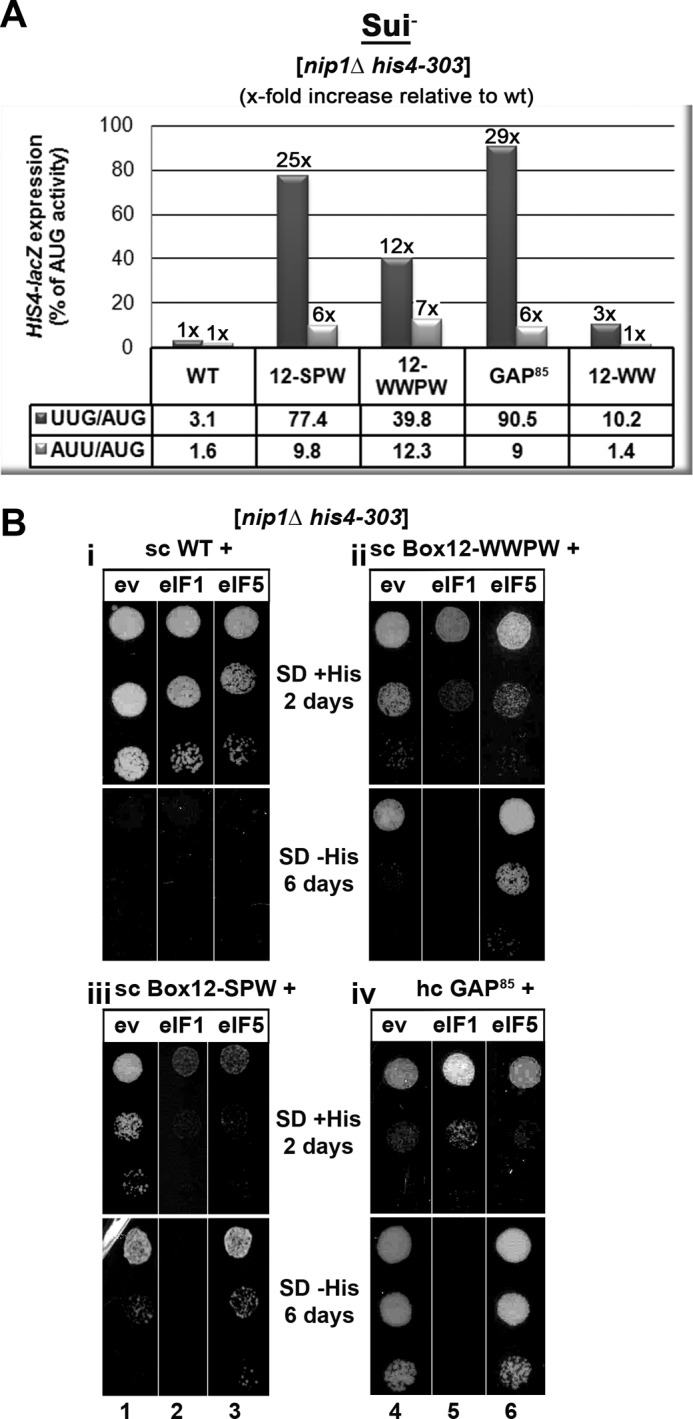
High dosage suppressor analysis of the c/Nip1-NTD Sui− mutants by the selected eIFs known to participate in the AUG recognition process. A, quantification of Sui− phenotypes. Strains from Fig. 2C were transformed with HIS4-lacZ reporters with AUG (p367), UUG (p391), or AUU (p2042), respectively, and grown in SD medium supplemented with His and Trp at 30 °C, and β-galactosidase activities were measured in WCEs. The mean percentages of the UUG or AUU initiation rates relative to those of AUG and an x-fold increase over the WT c/Nip1 were calculated from three experiments with six independent transformants. B, genetic effects of high copy expression of the indicated eIFs on selected Sui− mutants of c/Nip1. Strains from Fig. 2C were transformed with either empty vector (ev; lanes 1 and 4) or the corresponding plasmids overexpressing SUI1 (eIF1; lanes 2 and 5) or TIF5 (eIF5; lanes 3 and 6), respectively, and the resulting transformants were spotted in three serial dilutions on SD medium containing histidine (upper panels i and ii) or lacking histidine (lower panels iii and iv) and incubated at 30 °C for 2 (upper panels i and ii) or 6 days (lower panels iii and iv).
High Dosage Suppressor Analysis of the c/Nip1-NTD Sui− Mutants by Selected eIFs Known to Participate in the AUG Recognition Process
We showed previously that the Sui− phenotype of the original Box12 mutation was suppressed by overexpressing WT eIF1 despite the fact that at the same time its Slg− phenotype was exacerbated (11). In contrast, overproduction of eIF5 had an intensifying effect on both Sui− and Slg− phenotypes of nip1-Box12. To examine genetic interactions between our new Sui− mutants and these key players in the AUG recognition pathway, we transformed the nip1-Box12 mutants and nip1-GAP85 with hc vectors overexpressing eIF1 or eIF5 and scored the resulting transformants for growth in the presence or absence of histidine (Fig. 3B).
In analogy with our original data, overexpressing eIF1 reduced the growth rate of both Box12 mutants in the presence of histidine and suppressed their Sui− phenotype in its absence (Fig. 3B, panels ii and iii, lanes 5 and 2). Because the reduced growth rate of both mutants on His+ medium somewhat complicates our conclusion regarding the suppression of the Sui− defect, we measured the UUG/AUG and AUU/AUG ratios in WWPW and SPW mutants bearing either empty vector or hc eIF1. We found that overexpression of eIF1 reduced the latter ratios in the SPW mutant by ∼44 and 40%, respectively, and in the WWPW mutant by ∼30 and 33%, respectively. These findings clarify that the eIF1-mediated suppression is significant but not full. Interestingly, high dosage eIF1 also suppressed the Sui− phenotype of nip1-GAP85; however, it did not worsen but improved its Slg− phenotype (panel iv, lane 5), indicating that in this particular mutant the eIF1 suppression effect is more potent. Overexpression of eIF5 surprisingly had no significant effect on the GAP85 mutant (panel iv, lane 6), whereas it exacerbated the Sui− but not Slg− phenotype of Box12-WWPW (panel ii, lane 6). It is worth reiterating that these two mutants are not only Sui− but also display the Gcd− phenotype, which may contribute to the observed genetic interactions that differ from the original “Sui−-only” Box12 mutant. Consistently, overexpressing eIF5 in the Sui−-only nip1-Box12-SPW allele had an intensifying effect on both its Slg− and Sui− phenotypes (panel iii, lane 3). The intensifying effect on the Sui− phenotype is inferred from the fact that it reduced growth on His+ but not on His− medium. It should be mentioned that neither of these Sui− mutants conferred a significant Ssu− phenotype when combined with dominant negative Sui− alleles of eIF5 (in SUI5G31R) or SUI3 (a subunit of eIF2; in SUI3S264Y) (data not shown). Taken together, it is evident that each of these three Sui− mutants impairs the AUG selection process in a more or less different way (see “Discussion”).
The nip1-Box15-2, -Box12-WW, and -Box12-WWPW Mutants Affect Recruitment of the TC to the PICs in a Manner Partially Reversible by Overexpressing TC
The NTD of c/Nip1 indirectly interacts with the TC via their common binding partner eIF5 and thus promotes its recruitment to the 40 S ribosomes (11, 19, 20). As hinted above, an inability to recruit and/or stably anchor the TC to the PICs constitutively derepresses the otherwise tightly controlled expression of GCN4 under non-starvation conditions, producing the Gcd− phenotype even in the absence of the Gcn2 kinase. Mutants having such a mechanistic defect in the gcn2Δ background are commonly characterized by having their Gcd− defect suppressible by overexpression of the TC as an increased cellular concentration of TC offsets the defect in its recruitment. The failure of hc TC to suppress the Gcd− phenotype suggests a more complex defect(s) in the GCN4 regulation, the nature of which has not been understood so far.
To determine which of our newly generated NIP1 mutants affect the TC loading to the PICs as the rate-limiting defect, we introduced them with either empty vector or hc TC and tested the resulting transformants for growth in the presence of 3-AT. As shown in Fig. 4A, hc TC significantly suppressed the Gcd− phenotypes of the nip1-Box12-WW and -Box15-2 mutants and to a smaller degree perhaps that of -Box12-WWPW but not that of the GAP85 mutant, although it did partially suppress its slow growth defect. Importantly, increased TC dosage also suppressed, in fact almost fully, the Slg− phenotype of Box15-2, strongly indicating that the TC recruitment is the dominating defect in this mutant.
FIGURE 4.

The c/nip1-Boxes 12-WWPW, 12-WW, and 15-2 impair GCN4 translational control (produce the Gcd− phenotype) in the canonical manner suppressible by overexpressing the TC. A, strains from Fig. 2D were transformed with either empty vector or all three subunits of eIF2 + IMT4 (TC) on a high copy plasmid. The resulting transformants and isogenic strains H2880 (GCN2) and H2881 (gcn2Δ) transformed with empty vector were spotted in four serial dilutions on SD medium (upper panel) or SD medium containing 30 mm 3-AT (lower panel) and incubated at 30 °C for 2 and 7 days, respectively. B, quantification of the Gcd− phenotypes with or without the hc TC effect. Selected nip1Δ strains from A were further transformed with p180 containing the GCN4-lacZ fusion with all four uORFs present (shown as a schematic above the plot) and grown in minimal medium for 6 h, and the β-galactosidase activities were measured in the WCEs and expressed in units of nmol of o-nitrophenyl-β-d-galactopyranoside hydrolyzed/min/mg of protein. The mean values and standard deviations obtained from at least six independent measurements with three independent transformants, the x-fold increases in activities of the mutant strains relative to the corresponding WT expressing either empty vector (EV) or hc TC, and percentages of hc TC activities relative to empty vector activities in each individual strain (indicating the extent of the hc TC suppression effect) are given in the histogram. nd, not done.
To quantify the Gcd− phenotypes of all Gcd− mutants and to confirm or exclude its suppression by hc TC in WWPW and GAP85, we assayed expression of a GCN4-lacZ reporter containing all four uORFs in the mRNA leader under non-starvation conditions (Fig. 4B). As expected, the WT gcn2Δ NIP1+ strain showed constitutively low GCN4-lacZ expression irrespective of the presence of either empty vector or hc TC, whereas all four mutations bearing an empty vector displayed significantly increased derepression of GCN4-lacZ expression in the gcn2Δ background after normalizing for their effects on the expression of an incontrollable GCN4-lacZ construct lacking all four uORFs (Fig. 4B). These results thus confirm that all of our Gcd− mutants do diminish translational repression of GCN4 imposed by its uORFs as expected. Measuring the β-galactosidase activities in the presence of hc TC also confirmed moderate (by ∼30%) suppression of the WWPW Gcd− defect and no suppression of that displayed by the GAP85 mutant (Fig. 4B). Together, these results clearly imply that the impaired TC recruitment and thus the proper assembly of the PICs are to a varying degree one of the rate-limiting defects in all but one of our Gcd− mutants.
The Extreme N Terminus of c/Nip1 Interacts with eIF5, the Segment That Immediately Follows Binds eIF1, and nip1-Box12-SPW Increases Binding Affinity of the c/Nip1-NTD for eIF1 in Vitro
Having analyzed all newly generated mutations by genetic means, we next sought to investigate molecular effects of the semirandom substitutions as well as internal deletions of the c/Nip1-NTD on binding affinities toward its direct binding partners in eIF1 and eIF5. We began by examining the effects of both GAP deletions on in vitro binding of 35S-labeled c/Nip1-NTD (residues 1–270) to GST-eIF5 or GST-eIF1 fusions produced in Escherichia coli (Fig. 5A). Although this assay is more qualitative than quantitative in its nature, it has been reliably used in the past to provide rough estimates of altered binding affinities of mutant proteins (for example, see Refs. 9–13, 20, 24, and 28–30). In agreement with previous results, the WT c/Nip1-NTD polypeptide bound specifically to GST-eIF5 and GST-eIF1 but not to GST alone (Fig. 5B). Strikingly, both GAP deletions (Δ60–144 and Δ46–137) completely eliminated binding to eIF1 but not to eIF5 (lane 3 versus lane 4). In fact, binding to eIF5 was even strengthened perhaps because of the fact that the eIF5-binding site in the remaining c/Nip1 polypeptide was more ideally exposed for the interaction. In any case, given that the minimal c/Nip1 fragment required and sufficient for WT binding to both eIF1 and eIF5 is 156 residues (19), these results strongly suggest that the extreme N-terminal 45 residues constitute the binding site for eIF5, whereas the segment encompassing residues 60–137 represents the eIF1-binding site (Fig. 5A). In support, removal of the first 60 residues (in Δ60) or expressing the internal “GAP92” segment (in Δ45/137) had the opposite effect; i.e. it eliminated binding to eIF5 but not to eIF1 (lane 4 versus lane 3). In an effort to directly demonstrate that the first 45 residues are sufficient for the interaction with eIF5, we created pT7 constructs expressing only the first 45 or 60 N-terminal residues of c/Nip1; however, they did not yield stable proteins. Taken together, these data suggest that the molecular nature of defects displayed by both GAPs is associated with the loss of eIF1 binding to the c/Nip1-NTD. This is consistent with the fact that hc eIF1 suppressed the Sui− phenotype and partially suppressed Slg− phenotype of GAP85 (Fig. 3B, panel iv). Further analysis of binding affinities of semirandom Box mutants did not reveal any specific changes with the exception of Box12-SPW, which showed a reproducible ∼2-fold increase in binding to eIF1 (Fig. 5C, third panel, lane 3 with asterisk) (see also below). Note that Box12 falls in the eIF1 binding region, whereas Box15 lies further downstream.
nip1-GAP85 Completely Diminishes Whereas nip1-Box12-SPW Modestly Increases the Amount of eIF1 Associated with the MFC in Vivo
To test whether our mutations affect binding of c/Nip1 to eIFs 1 and 5 and the TC in the context of the entire eIF3, we analyzed the in vivo composition of the MFC in cells expressing the individual His8-tagged NIP1 mutant alleles by Ni2+ chelation chromatography. As reported previously (11), a fraction of a/Tif32, i/Tif34, g/Tif35, eIF2, eIF5, and eIF1 co-purified specifically with WT c/Nip1-His but not with its untagged version (Fig. 6A, lane 5 versus lane 2). Although neither of the mutations affects the extreme N-terminal binding site for eIF5, Box15-2 and GAP85 reduced in vivo association of mutant eIF3 with eIF5 by more than 60% (lanes 17 and 20). In addition, Box12-WWPW also lowered the eIF3 binding affinity for eIF5 but to a smaller extent (by ∼30%) (lane 11). This is most probably an indirect effect arising from the overall changes of the c/Nip1-NTD fold in the context of the entire eIF3/MFC. As eIF5 mediates an indirect contact between eIF2 and the c/Nip1-NTD (19, 20), dramatic reductions in eIF2 association with the rest of MFC were also observed in these three mutant strains in a remarkable proportional accord with the extent of the eIF5 loss. In perfect agreement, all these NIP1 mutant alleles impart severe Gcd− phenotypes (Fig. 2D), strongly suggesting that impaired TC loading to the PICs represents one of their functional defects in translation. Interestingly, another Gcd− mutant, Box12-WW, showed virtually WT levels of eIF5 associated with the rest of eIF3 but significantly reduced amounts of eIF2 (lane 14). These results suggest that its TC binding defect mechanistically differs from those of the other three mutants (see “Discussion” for more details). In contrast, Box12-SPW, which is the only mutant allele that does not produce the Gcd− phenotype, showed WT levels of both eIF5 and eIF2 (Fig. 6A, lane 8). This Sui− mutant, however, did display a modest increase in the eIF1 amounts associated with the MFC, consistent with our in vitro binding data presented in Fig. 5C. It should be noted here that despite numerous repetitions the experimental error for eIF1 remained relatively high in this particular experiment. Nevertheless, because the increase in eIF1 recovery averaged out to ∼20% relative to WT, we propose that Box12-SPW slightly but specifically increases the binding affinity of the c/Nip1-NTD for eIF1 both in vitro and in vivo. Conversely, eIF1 binding was completely diminished by GAP85 (Fig. 6A, lane 20) as expected because this mutant has the eIF1-binding site deleted. Finally, eIF1 binding in the remaining three Box mutants (12-WWPW, 12-WW, and 15-2) was also reduced but to a much smaller degree than in the case of the GAP85 mutant. Taking our in vitro binding data into account (Fig. 5C), we think that these reductions are not a result of impaired direct binding between eIF1 and the NTD of c/Nip1 but rather an indirect consequence of changes in the arrangement of the assembly of the eIFs around the mutant c/Nip1-NTD.
FIGURE 6.

The semirandom mutations of c/Nip1-Boxes selectively affect composition of the MFC in vivo. A, WCEs prepared from the selected strains described in Fig. 2C were incubated with Ni2+-Sepharose, and the bound proteins were eluted and subjected to Western blot analysis with the antibodies indicated in each row. In, lanes contained 5% of the input WCEs; E, lanes contained 100% of the eluate from the resin (a typical recovery is ∼10% of the input for the WT); FT, lanes contained 5% of the flow-through from 100% input that did not bind to the resin (the limiting factor in this experiment). B, the Western signals for the indicated proteins in the eluate fractions of the WT NIP1-His and its mutants were quantified, normalized for the amounts of the WT c/Nip1 in these fractions, and plotted in the histogram as percentages of the corresponding values calculated for the WT c/Nip1; S.D. values are given.
The c/Nip1-NTD Mutants Generally Reduce 40 S Subunit Association of eIF3 and eIF5 in Vivo but Have Varying Effects on eIF1 and TC Recruitment to the 43–48 S PICs
All results described so far suggest that the three identified classes of the c/Nip1-NTD mutants that we generated have a varying impact on the composition of the 43–48 S PICs, the analysis of which would greatly help with the explanation of their molecular defects. To address this issue, we measured binding of selected eIF3 subunits and other MFC components to 40 S subunits by formaldehyde cross-linking followed by high velocity sedimentation in sucrose gradients (24). This method provides the best available approximation of the native composition of 43–48 S pre-initiation complexes in vivo. As shown in Fig. 7, A–C (left panels), we observed the expected co-sedimentation of a proportion of eIF3, eIF2, eIF5, and eIF1 with the 40 S species in the WCEs derived from WT cells. In the case of the class “a” Box12-SPW mutant, we observed a relative ∼50% decrease in the amounts of selected eIF3 subunits (c/Nip1, i/Tif34, and g/Tif35) and eIF5 associated with 40 S subunits (Fig. 7A, right panel), indicating that this mutation destabilizes eIF3 and its tightly bound partner eIF5 (28) on the ribosome in vivo perhaps by increasing their dissociation rates as suggested previously for some other eIF3 mutants (13, 31). In contrast, eIF2 levels remained practically unchanged, which nicely correlates with the fact that this mutant does not impart the Gcd− phenotype. Importantly and in accord with our binding data mentioned above, we reproducibly detected significantly increased amounts of eIF1 in the 40 S subunit-containing fractions (note the peak of the eIF1 signal in the 40S subunit-containing fractions that is apparent only in the panel showing the SPW mutant). Hence, we propose that tighter binding of eIF1 to the c/Nip1-NTD somehow prevents eIF1 from properly functioning in the AUG start codon selection process, producing the severe Sui− phenotype (see “Discussion” for our model).
FIGURE 7.

The c/Nip1-NTD mutants generally reduce 40 S subunit association of eIF3 and eIF5 in vivo but have varying effects on eIF1 and TC recruitment to the 43–48 S PICs. A–D, selected strains described in Fig. 2, C and D, were grown in YPD medium at 30 °C to an A600 of ∼1.5 and cross-linked with 2% HCHO prior to harvesting. WCEs were prepared, separated on a 7.5–30% sucrose gradient by centrifugation at 41,000 rpm for 5 h, and subjected to Western blot analysis. Fractions 1–4, 5–9, and 10–12 (43–48 S) were pooled, and 5% of each pooled sample was loaded on the gel; lane “In” shows 5% input. Proportions of the 40 S subunit-bound proteins relative to the amount of 40 S subunits were calculated using Quantity One software (Bio-Rad) from at least three independent experiments. The resulting values obtained with the WT strain were set to 100%, and those obtained with mutant strains were expressed as percentages of the WT in the histograms on the right (S.D. values are given).
The class “b” WWPW mutant imparting both the Sui− and Gcd− phenotypes reduced 40 S subunit binding of eIF3 to a similar extent and that of eIF5 to an even greater extent than did Box12-SPW (Fig. 7B, right panel). Here, the reduction in eIF5 on the 40 S ribosome might be attributed either to the reduced amount of eIF5 in the affinity-purified MFC and/or to the decrease in 40 S subunit-associated eIF3; we cannot rule out either of these possibilities. In accord with its Gcd− phenotype and in contrast to Box12-SPW, it also significantly reduced the eIF2 levels on the ribosome. Another striking difference between WWPW and SPW mutants is that the former also markedly reduced the 40 S subunit-associated amounts of eIF1. These results thus imply that (i) the nature of the Sui− defect differs between these two mutants and (ii) the character of the WWPW Sui− defect conspicuously resembles so-called class “i” mutations in SUI1 encoding eIF1 (3). Class i Sui− mutations weaken eIF1 association with PICs, and their Sui− phenotype can be partially suppressed by their own overexpression; in accord, the Sui− defect of WWPW is also partially suppressible by hc eIF1 (Fig. 3). Intuitively, the same results could be also expected with another class b mutant, GAP85, which completely removes the eIF1-binding site (Fig. 5) and practically eliminates eIF1 from the MFC in vivo (Fig. 6). Despite numerous attempts, however, the results were not satisfactorily conclusive because they suffered from huge experimental error. We think the most probable reason for this is the extremely long doubling time of the large internal deletion of this mutant that makes it very hard to work with when compared with other, not so slow growing mutants. Nevertheless, our in vitro and in vivo binding as well as hc eIF1 suppression data strongly suggest that GAP85 also causes the Sui− phenotype by weakening the eIF1 binding to PICs in a manner partially suppressible by hc eIF1.
The class “c” WW mutant, which still falls in the original Box12 segment, imparts only the Gcd− phenotype in a manner partially suppressible by hc TC (Fig. 4) and as such was expected to reduce mainly the eIF2 amounts on the 40 S ribosome. As shown in Fig. 7C, besides eIF2, we also found decreased amounts of eIF5 but not eIF3 and eIF1. Interestingly and in accord with our binding data showing a significant loss of eIF2 but not eIF5 from the MFC in vivo (Fig. 6), 40 S ribosome binding of eIF2 was more severely impaired than that of eIF5. This is significant because in all other Gcd− mutants examined in this work eIF5 association with PICs was always more impaired than that of eIF2. These results seem to indicate that the WW mutant impairs eIF2 recruitment by a different mechanism than via the destabilization of eIF5 binding to the c/Nip1-NTD in the MFC, which is the only way that we have seen so far (see “Discussion” for further details). These results also suggest that the “PW” but not the “WW” part of the WWPW mutant is responsible for the weakened eIF3 binding to PICs in vivo.
Finally, another representative class c mutant, Box15-2, reduced binding of eIF3 and eIF5 with 40 S species (Fig. 7D, right panel), indicating that this part of the NTD of c/Nip1 also is required for stable incorporation of eIF3 and eIF5 into the PICs. Importantly, consistent with the Gcd− only phenotype of this mutant, 40 S subunit-associated amounts of eIF2 (TC) were also significantly reduced in contrast to eIF1, the binding of which was affected only modestly.
DISCUSSION
In this report, we added great detail into our understanding of the dual role of the NTD of c/Nip1 in TC recruitment and AUG selection, delineated its binding sites for eIF1 and eIF5, and pinpointed specific residues involved in both of these functions. First, we showed that the extreme N-terminal 45 residues of the c/Nip1-NTD in the GST-GAP92 construct, which practically lacks the rest of the originally defined minimal eIF5-binding segment (in residues 1–156; Ref. 19), are sufficient for binding to eIF5 in vitro, whereas their removal completely abrogates it (Fig. 5). These findings clearly imply that the tip of the c/Nip1 protein is the direct mediator of eIF3 control over the eIF5 function(s) on the ribosome. This is consistent with our earlier observations that two 10-Ala substitution mutants (Box2 and Box4) falling directly in this region preferentially reduced association of the c/Nip1-NTD with eIF5 versus eIF1 in vitro and accordingly lowered eIF5 occupancy in native PICs (11). Interestingly, similar in vivo but not in vitro effects were also observed with the original Box6R mutation, although the mutated region lies outside of the minimal eIF5-binding domain defined here (11). Likewise, neither of our newly generated mutations occurs in this domain and thus shows no impairment of eIF5 binding in vitro (Fig. 5), but most of mutations reduce eIF5 amounts in the MFC in vivo (Fig. 6). In addition, we observed that the minimal eIF5-binding domain lacking the eIF1-binding site that immediately follows shows much stronger affinity for eIF5 than the entire N-terminal segment (Fig. 5). To accommodate these findings, we propose that the c/Nip1-NTD, which is not involved in any eIF3 intersubunit interactions, adopts a dynamic fold that undergoes a relatively large restructuring when eIF3 associates with other eIFs in the MFC. This could play an important role when the MFC contacts the 40 S subunit and eIF3 and its other components (all in contact with the c/Nip1-NTD) must find their attachment sites on both sides of the ribosome (see our model below). If true, one can envisage that even mutations lying outside of the defined c/Nip1-binding sites for eIFs 1 and 5 might have a marked impact on the folding of the rest of the NTD and could prevent association of eIF1 and/or eIF5 with the MFC indirectly, for example due to spatial constraints.
Also, the c/Nip1-NTD internal deletions (Δ60–144 in GAP85 and Δ46–137 in GAP92) completely eliminated association of eIF1 with the NTD in vitro and with eIF3 in the MFC in vivo (Figs. 5 and 6 and data not shown), and the missing fragments showed WT binding to eIF1 in vitro (Fig. 5 and data not shown). This clearly suggests that the segment spanning residues 60–137 is responsible for eIF1 recruitment to the MFC and via MFC to the 40 S ribosome. Interestingly, neither of our Box12 mutations reduced eIF1 binding in vitro, indicating that the region from 111 to 120 is not the most critical determinant of the c/Nip1-eIF1 interaction. The absolute loss of contact between eIF1 and the c/Nip1-NTD that dominates in the GAP mutants produced very severe Slg− and Sui− phenotypes partially and fully suppressible, respectively, by high copy eIF1 (Fig. 3). Conversely, overexpressing eIF5 showed no effect on either phenotype. Intuitively, these results suggest that the rate-limiting defect in both GAPs should be the loading of eIF1 to the PIC and/or the lack of the eIF1-c/Nip1-NTD contact therein. The former would be reminiscent of the class i sui1 mutations that specifically reduce the eIF1/Sui1 amounts in the PICs and produce the Sui− phenotype in a manner partially suppressible by their own overexpression (3, 8). Although the aforementioned technical difficulties prevented us from showing this directly, we posit that the eIF1 recruitment to the ribosome is one of the major defects displayed by this mutant in addition to its failure to properly promote TC assembly with the PICs (in a manner not suppressible by hc TC, which we cannot explain). In many ways, a similar but certainly not completely identical molecular effect also underlies a Sui− defect of the largest semirandom Box12-WWPW mutant. It markedly reduced amounts of the MFC- and PIC-bound eIF1 in vivo but not in vitro (Figs. 5–7), and its Sui− but not Slg− phenotype was partially suppressible by high copy eIF1 by the principle of mass action (Fig. 3).
The class a (Sui−-only) mutant Box12-SPW is a variant of the WWPW mutation within Box12 lacking the first two Trp substitutions of Val111 and Val112 (shown to produce the canonical Gcd− phenotype on their own; see below) but having an extra substitution of Lys113 by serine. It is intriguing that such a short stretch of residues, apparently not even directly contacting eIF1, contains two distinct sets of residues that are involved in two mutually distinguishable processes. Although the Sui− phenotype of the SPW mutant was also partially suppressible by hc eIF1 as in the case of GAP85 and WWPW (Fig. 3), our biochemical experiments clearly suggest that the molecular effects of its Sui− defect significantly differ from the latter two. We think that it is because GAP85 and WWPW also impair a step preceding the AUG selection, in particular the TC recruitment, which must undoubtedly greatly influence the normal progress of all following steps. In this respect, the SPW effect on AUG selection can be considered the most direct.
The SPW mutation surprisingly increased the affinity of the c/Nip1-NTD for eIF1 in vitro (Fig. 5) and to a certain degree in vivo in the context of the entire MFC (Fig. 6). In addition, perhaps as a result of this, it also markedly increased the steady state levels of eIF1 in the PICs while reducing the overall amounts of eIF3 and eIF5 (Fig. 7). To explain these effects, we propose the following model. If we presume that the c/Nip1-NTD delivers eIF1 to the back side of the 40 S ribosome from which it has to relocate to the P-site area on the interface side (32) (see our model below), tighter binding of eIF1 to the SPW mutant could shift the equilibrium between eIF1 bound near the P-site toward eIF1 bound to eIF3, which is known to reside on the back side of the 40 S ribosome (4). (The existence of two eIF1-binding sites on the ribosome (one on eIF3 and the other near the P-site) was also recently proposed by A. Hinnebusch (3).) This could at least partially account for the Sui− phenotype of SPW by altering the proper positioning of eIF1 for stringent AUG selection. In other words, because of the increased affinity of the mutant c/Nip1-NTD for eIF1, the SPW mutation would interfere with the eIF1 gatekeeping role at the P-site either by preventing its stable binding therein and/or by evoking its premature dissociation/displacement at near-cognate start codons. Both defects would be mitigated by eIF1 overexpression as was observed. However, this “altered distribution/improper positioning” model would still not explain the fact that the occupancy of eIF1 is increased in native PICs. One way to account for this elevated eIF1 occupancy would be to propose that a second consequence of the SPW mutation is to delay eIF1 dissociation from the 40 S ribosome specifically at AUG codons in the manner described previously for the “class ii” Sui− mutant in SUI1 represented by sui1-G107R, which produces the Sui− defect by slowing not accelerating the release of eIF1 from 48 S PICs (33). Indeed, HCHO cross-linking analysis of G107R showed that this mutation also reduces the amounts of eIF3 and eIF5 associated with the PICs in vivo while producing a slight increase in the eIF1 amounts. In fact, this second defect would not only explain the observed higher occupancy of eIF1 in bulk PICs (which are dominated by AUG initiation events), but in addition, it would also be expected to contribute to the SPW Sui− phenotype by reducing initiation specifically at AUG codons.
Finally, the three mutations Box12-WW, 12-WWPW, and Box15-2 produced canonical Gcd− phenotypes suppressible by overexpressing the TC in cells lacking protein kinase Gcn2 (Fig. 4), providing strong genetic evidence that they decrease the rate of TC binding to 40 S ribosomes and thus deregulate GCN4 translational control. Accordingly, they significantly decreased the eIF2/TC levels in both the MFC (Fig. 6) and the PIC in vivo (Fig. 7). Increased dosage of the TC also fully suppressed the Slg− phenotype of the Box15-2 mutant, suggesting that the TC recruitment is the rate-limiting defect in these cells and residues Ile142, Glu145, Phe146, Asp147, and Ile149 are critically required for this c/Nip1 role. Although we did not attempt to dissect the individual effects of these five residues any further, we assume that they are all required because of the following facts. Two mutant libraries of the original Box15, each containing ∼10,000 clones, were screened. One library contained mutations in only two of these five residues (E145X and D147X), and no Gcd− mutants (not even weak ones) were identified. The other library contained mutations in all five residues, and only one of ∼10,000 transformants showed a strong Gcd− phenotype. In contrast to Box15-2, the Slg− phenotype of the WW mutant was not suppressed, indicating that the molecular nature of its defect differs to some degree. Indeed, whereas Box15-2 also markedly decreased the eIF5 levels in the MFC, the WW mutation had a negligible effect if any (Fig. 6). To explain this difference, we propose that although Box15-2 affects the c/Nip1-NTD fold in such a way that primarily impedes the eIF5 attachment to its tip in the context of the MFC Box12-WW allows Nip1-NTD-eIF5-CTD binding but changes the overall orientation of the eIF5-CTD in the MFC so that it can no longer contact the β-subunit of eIF2 to promote efficient TC recruitment to 40 S ribosomes. It is also noteworthy that in contrast to all other mutants the WW mutation did not destabilize eIF3 binding to the ribosome. Hence, it seems that the former effect and the latter lack of effect are the unique characteristics of the “V111W/V112W” substitutions because the “parental” WWPW mutant showed biochemical effects similar to those of Box15-2.
Based on these results, we propose the following model. The fact that the NTD of c/Nip1 interacts simultaneously with eIF1 and the CTD of eIF5 (19, 20) and via the latter with the β-subunit of the TC makes the c/Nip1-NTD one of the most critical domains ensuring the functions of the MFC in recruiting the latter factors to the 43 S PIC and regulating their contributions to the start codon selection process thereon. It has been shown that disrupting mutual interactions among individual components of the yeast MFC significantly reduces translation initiation rates by affecting not only the steps of the PIC assembly but also the subsequent postassembly events (9–12, 14, 15, 18, 20, 21, 29). This not only suggests that the MFC-driven pathway of PIC assembly ensures the efficiency of the whole initiation process but also that the postassembly persistence of at least some contacts among the MFC components is required for smooth scanning through the 5′-UTR of the mRNA and proper AUG recognition. There is increasing evidence that the major eIF3 body contacts the solvent side of the 40 S subunit in proximity to mRNA at both the entry and exit channel pores (9, 10, 13, 18, 22, 34). However, some of its domains, including the c/Nip1-NTD, were proposed to reach out under the 40 S beak toward the ribosomal A-site (11, 22). Although it is not known where the eIF5-CTD resides, the very recent analysis of 40 S ribosome-associated eIF2 suggested that the eIF2γ-bound eIF2β faces the A-site (35), indicating that the CTD of eIF5 also occurs somewhere in this area. The modeled eIF2β location would be consistent with the proposed placement of the CTD of the a/Tif32 subunit of eIF3 (22) that directly interacts with this eIF2 subunit in the MFC (20). If true, this could mean that the MFC-established c/Nip1-NTD-eIF5-CTD and a/Tif32-CTD-eIF2β contacts also remain preserved in the scanning PICs. In such a case, the latter two eIF3 domains would be ideally positioned to actively contribute to the regulation of AUG selection via their contacts with eIF2 and eIF5. Indeed, mutations in the a/Tif32-CTD and its interacting partner j/Hcr1 were shown to markedly increase the frequency of skipping the AUG start site, producing the leaky scanning phenotype (9, 10). In contrast, the binding site of eIF1 in the eIF1·40 S ribosome complex lacking all other eIFs was mapped close to the ribosomal P-site in the interface platform area (32, 36). In addition, eIF1 appears to bind to the ribosome via the same surface that also contacts the c/Nip1-NTD (36, 37). Hence, unlike the eIF2β and the eIF5-CTD interactions, the eIF1 contact with the c/Nip1-NTD on the ribosome must be undoubtedly given up at a certain point of initiation.
Taken together, we propose that upon MFC binding to the ribosome the c/Nip1-NTD together with the a/Tif32-CTD accommodate eIF1, the eIF5-CTD, and the eIF2β-NTD near the A-site. Whereas the latter two domains remain bound in this area, eIF1 is subsequently transferred to its “scanning-competent” position near the P-site. This transfer could be a part of a large conformational rearrangement of the 40 S head that is triggered by eIFs 1 and 1A and that opens up the mRNA binding channel for mRNA recruitment (2). We think that this particular process is slowed down in our gain-of-function SPW mutant because of its increased affinity for eIF1, resulting in the severe Sui− defect.
Based on biophysical studies conducted with a yeast in vitro reconstituted system using eIFs 1, 1A, and 5 and TC but not eIF3, eIF1 was proposed to be ejected from PICs upon AUG recognition to “open the gate” for the subsequent Pi release, resulting in scanning arrest (38). Premature loss of eIF1 from the scanning PICs in numerous mutants is also generally considered to be the major cause of the Sui− phenotype (8). An interesting alternative scenario to the “ejection model” is that eIF1 upon AUG recognition triggers a reciprocal conformational rearrangement from the open to closed states, and instead of being directly ejected, it drifts back to the c/Nip1-NTD in the A-site. Although there is no direct evidence for this option with the exception of the fact that addition of eIF3 to reconstituted PICs in the system of Lorsch and co-workers (38), while having no effect on the rate constant for eIF1 dissociation, reduces the extent of eIF1 dissociation from reconstituted PICs, we think it has an important physiological relevance as follows. i) The c/Nip1-NTD could control timing and dynamics of the eIF1 shuffling between its two positions on the ribosome depending on the immediate scanning status, i.e. on a conformational state of the ribosome. ii) Holding on to eIF1 by eIF3 post-initiation could speed up the reinitiation process after translation of short uORFs. eIF3 critically promotes reinitiation by staying 80 S ribosome-bound during the first elongation cycles to stabilize the 40 S subunit on mRNA after termination on a short uORF (17, 39, 40). In the next step, the 40 S subunit must resume scanning to locate the next AUG of a downstream ORF. Intuitively, to be able to resume scanning, it is very likely that conformational changes similar to those occurring in the newly formed 48 S PIC also must occur in the mRNA-bound post-termination 40 S complex (39). Hence, having eIF1 already present could make the transition from post-termination to scanning complex faster and thus more efficient. iii) eIF3 together with eIFs 1 and 1A was proposed to greatly stimulate a ribosomal recycling step following termination (41). If eIF1 is ejected in a complex with eIF3 post-initiation as eIF5 is with eIF2 prior to subunit joining (42), the eIF3·eIF1 complex could in turn directly participate in recycling at one end of the translational cycle and serve as a nucleation center for the new round of MFC formation at the other end. Certainly, more experiments are needed to support or exclude these possibilities.
Acknowledgments
We are thankful to Alan G. Hinnebusch and the members of the Valášek and Krásný laboratories for helpful comments, to Jan Kosla for help with preparation of the c/Nip1 mutagenesis, and to Olga Krydová for technical and administrative assistance. We are also indebted to an unknown reviewer whose insightful comments greatly improved the quality of this work.
This research was supported by The Wellcome Trust Grants 076456/Z/05/Z and 090812/B/09/Z and Czech Science Foundation Grant 305/10/0335.
- TC
- ternary complex
- NTD
- N-terminal domain
- PIC
- pre-initiation complex
- MFC
- multifactor complex
- CTD
- C-terminal domain
- SD
- synthetic defined medium
- YPD
- yeast extract-peptone-dextrose medium
- WCE
- whole-cell extract
- 3-AT
- 3-aminotriazole
- uORF
- upstream ORF
- sc
- single copy
- hc
- high copy.
REFERENCES
- 1. Sonenberg N., Hinnebusch A. G. (2009) Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136, 731–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Passmore L. A., Schmeing T. M., Maag D., Applefield D. J., Acker M. G., Algire M. A., Lorsch J. R., Ramakrishnan V. (2007) The eukaryotic translation initiation factors eIF1 and eIF1A induce an open conformation of the 40S ribosome. Mol. Cell 26, 41–50 [DOI] [PubMed] [Google Scholar]
- 3. Hinnebusch A. G. (2011) Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol. Mol. Biol. Rev. 75, 434–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Valášek L. S. (2012) 'Ribozoomin'—translation initiation from the perspective of the ribosome-bound eukaryotic initiation factors (eIFs). Curr. Protein Pept. Sci. 13, 305–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Algire M. A., Maag D., Lorsch J. R. (2005) Pi release from eIF2, not GTP hydrolysis, is the step controlled by start-site selection during eukaryotic translation initiation. Mol. Cell 20, 251–262 [DOI] [PubMed] [Google Scholar]
- 6. Maag D., Algire M. A., Lorsch J. R. (2006) Communication between eukaryotic translation initiation factors 5 and 1A within the ribosomal pre-initiation complex plays a role in start site selection. J. Mol. Biol. 356, 724–737 [DOI] [PubMed] [Google Scholar]
- 7. Saini A. K., Nanda J. S., Lorsch J. R., Hinnebusch A. G. (2010) Regulatory elements in eIF1A control the fidelity of start codon selection by modulating tRNAiMet binding to the ribosome. Genes Dev. 24, 97–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheung Y. N., Maag D., Mitchell S. F., Fekete C. A., Algire M. A., Takacs J. E., Shirokikh N., Pestova T., Lorsch J. R., Hinnebusch A. G. (2007) Dissociation of eIF1 from the 40S ribosomal subunit is a key step in start codon selection in vivo. Genes Dev. 21, 1217–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Elantak L., Wagner S., Herrmannová A., Karásková M., Rutkai E., Lukavsky P. J., Valásek L. (2010) The indispensable N-terminal half of eIF3j/HCR1 cooperates with its structurally conserved binding partner eIF3b/PRT1-RRM and with eIF1A in stringent AUG selection. J. Mol. Biol. 396, 1097–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chiu W. L., Wagner S., Herrmannová A., Burela L., Zhang F., Saini A. K., Valásek L., Hinnebusch A. G. (2010) The C-terminal region of eukaryotic translation initiation factor 3a (eIF3a) promotes mRNA recruitment, scanning, and, together with eIF3j and the eIF3b RNA recognition motif, selection of AUG start codons. Mol. Cell. Biol. 30, 4415–4434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Valásek L., Nielsen K. H., Zhang F., Fekete C. A., Hinnebusch A. G. (2004) Interactions of eukaryotic translation initiation factor 3 (eIF3) subunit NIP1/c with eIF1 and eIF5 promote preinitiation complex assembly and regulate start codon selection. Mol. Cell. Biol. 24, 9437–9455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nielsen K. H., Valásek L., Sykes C., Jivotovskaya A., Hinnebusch A. G. (2006) Interaction of the RNP1 motif in PRT1 with HCR1 promotes 40S binding of eukaryotic initiation factor 3 in yeast. Mol. Cell. Biol. 26, 2984–2998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Herrmannová A., Daujotyte D., Yang J. C., Cuchalová L., Gorrec F., Wagner S., Dányi I., Lukavsky P. J., Valásek L. S. (2012) Structural analysis of an eIF3 subcomplex reveals conserved interactions required for a stable and proper translation pre-initiation complex assembly. Nucleic Acids Res. 40, 2294–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mitchell S. F., Walker S. E., Algire M. A., Park E. H., Hinnebusch A. G., Lorsch J. R. (2010) The 5′-7-methylguanosine cap on eukaryotic mRNAs serves both to stimulate canonical translation initiation and to block an alternative pathway. Mol. Cell 39, 950–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jivotovskaya A. V., Valásek L., Hinnebusch A. G., Nielsen K. H. (2006) Eukaryotic translation initiation factor 3 (eIF3) and eIF2 can promote mRNA binding to 40S subunits independently of eIF4G in yeast. Mol. Cell. Biol. 26, 1355–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pisarev A. V., Kolupaeva V. G., Yusupov M. M., Hellen C. U., Pestova T. V. (2008) Ribosomal position and contacts of mRNA in eukaryotic translation initiation complexes. EMBO J. 27, 1609–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Szamecz B., Rutkai E., Cuchalová L., Munzarová V., Herrmannová A., Nielsen K. H., Burela L., Hinnebusch A. G., Valásek L. (2008) eIF3a cooperates with sequences 5′ of uORF1 to promote resumption of scanning by post-termination ribosomes for reinitiation on GCN4 mRNA. Genes Dev. 22, 2414–2425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cuchalová L., Kouba T., Herrmannová A., Dányi I., Chiu W. L., Valásek L. (2010) The RNA recognition motif of eukaryotic translation initiation factor 3g (eIF3g) is required for resumption of scanning of posttermination ribosomes for reinitiation on GCN4 and together with eIF3i stimulates linear scanning. Mol. Cell. Biol. 30, 4671–4686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Asano K., Clayton J., Shalev A., Hinnebusch A. G. (2000) A multifactor complex of eukaryotic initiation factors, eIF1, eIF2, eIF3, eIF5, and initiator tRNAMet is an important translation initiation intermediate in vivo. Genes Dev. 14, 2534–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Valásek L., Nielsen K. H., Hinnebusch A. G. (2002) Direct eIF2-eIF3 contact in the multifactor complex is important for translation initiation in vivo. EMBO J. 21, 5886–5898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nielsen K. H., Szamecz B., Valásek L., Jivotovskaya A., Shin B. S., Hinnebusch A. G. (2004) Functions of eIF3 downstream of 48S assembly impact AUG recognition and GCN4 translational control. EMBO J. 23, 1166–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Valásek L., Mathew A. A., Shin B. S., Nielsen K. H., Szamecz B., Hinnebusch A. G. (2003) The yeast eIF3 subunits TIF32/a, NIP1/c, and eIF5 make critical connections with the 40S ribosome in vivo. Genes Dev. 17, 786–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Valásek L., Trachsel H., Hasek J., Ruis H. (1998) Rpg1, the Saccharomyces cerevisiae homologue of the largest subunit of mammalian translation initiation factor 3, is required for translational activity. J. Biol. Chem. 273, 21253–21260 [DOI] [PubMed] [Google Scholar]
- 24. Nielsen K. H., Valásek L. (2007) In vivo deletion analysis of the architecture of a multiprotein complex of translation initiation factors. Methods Enzymol. 431, 15–32 [DOI] [PubMed] [Google Scholar]
- 25. Grant C. M., Hinnebusch A. G. (1994) Effect of sequence context at stop codons on efficiency of reinitiation in GCN4 translational control. Mol. Cell. Biol. 14, 606–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Donahue T. (2000) in Translational Control of Gene Expression (Sonenberg N., Hershey J. W. B., Mathews M. B., eds) pp. 487–502, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 27. Hinnebusch A. G. (2005) Translational regulation of GCN4 and the general amino acid control of yeast. Annu. Rev. Microbiol. 59, 407–450 [DOI] [PubMed] [Google Scholar]
- 28. Phan L., Schoenfeld L. W., Valásek L., Nielsen K. H., Hinnebusch A. G. (2001) A subcomplex of three eIF3 subunits binds eIF1 and eIF5 and stimulates ribosome binding of mRNA and tRNAiMet. EMBO J. 20, 2954–2965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yamamoto Y., Singh C. R., Marintchev A., Hall N. S., Hannig E. M., Wagner G., Asano K. (2005) The eukaryotic initiation factor (eIF) 5 HEAT domain mediates multifactor assembly and scanning with distinct interfaces to eIF1, eIF2, eIF3, and eIF4G. Proc. Natl. Acad. Sci. U.S.A. 102, 16164–16169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Asano K., Krishnamoorthy T., Phan L., Pavitt G. D., Hinnebusch A. G. (1999) Conserved bipartite motifs in yeast eIF5 and eIF2Bepsilon, GTPase-activating and GDP-GTP exchange factors in translation initiation, mediate binding to their common substrate eIF2. EMBO J. 18, 1673–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kouba T., Rutkai E., Karásková M., Valášek L. S. (2012) The eIF3c/NIP1 PCI domain interacts with RNA and RACK1/ASC1 and promotes assembly of translation preinitiation complexes. Nucleic Acids Res. 40, 2683–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rabl J., Leibundgut M., Ataide S. F., Haag A., Ban N. (2011) Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiation factor 1. Science 331, 730–736 [DOI] [PubMed] [Google Scholar]
- 33. Nanda J. S., Cheung Y. N., Takacs J. E., Martin-Marcos P., Saini A. K., Hinnebusch A. G., Lorsch J. R. (2009) eIF1 controls multiple steps in start codon recognition during eukaryotic translation initiation. J. Mol. Biol. 394, 268–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kouba T., Danyi I., Munzarová V., Vlčková V., Cuchalová L., Neueder A., Milkereit P., Valášek L. S. (2012) Small ribosomal protein RPS0 stimulates translation initiation by mediating 40S-binding of eIF3 via its direct contact with the eIF3a/TIF32 subunit. PLoS One, 7, e40464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shin B. S., Kim J. R., Walker S. E., Dong J., Lorsch J. R., Dever T. E. (2011) Initiation factor eIF2γ promotes eIF2-GTP-Met-tRNAiMet ternary complex binding to the 40S ribosome. Nat. Struct. Mol. Biol. 18, 1227–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lomakin I. B., Kolupaeva V. G., Marintchev A., Wagner G., Pestova T. V. (2003) Position of eukaryotic initiation factor eIF1 on the 40S ribosomal subunit determined by directed hydroxyl radical probing. Genes Dev. 17, 2786–2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reibarkh M., Yamamoto Y., Singh C. R., del Rio F., Fahmy A., Lee B., Luna R. E., Ii M., Wagner G., Asano K. (2008) Eukaryotic initiation factor (eIF) 1 carries two distinct eIF5-binding faces important for multifactor assembly and AUG selection. J. Biol. Chem. 283, 1094–1103 [DOI] [PubMed] [Google Scholar]
- 38. Maag D., Fekete C. A., Gryczynski Z., Lorsch J. R. (2005) A conformational change in the eukaryotic translation preinitiation complex and release of eIF1 signal recognition of the start codon. Mol. Cell 17, 265–275 [DOI] [PubMed] [Google Scholar]
- 39. Munzarová V., Pánek J., Gunišová S., Dányi I., Szamecz B., Valášek L. S. (2011) Translation reinitiation relies on the interaction between eIF3a/TIF32 and progressively folded cis-acting mRNA elements preceding short uORFs. PLoS Genet. 7, e1002137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pöyry T. A., Kaminski A., Jackson R. J. (2004) What determines whether mammalian ribosomes resume scanning after translation of a short upstream open reading frame? Genes Dev. 18, 62–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pisarev A. V., Hellen C. U., Pestova T. V. (2007) Recycling of eukaryotic posttermination ribosomal complexes. Cell 131, 286–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jennings M. D., Pavitt G. D. (2010) eIF5 has GDI activity necessary for translational control by eIF2 phosphorylation. Nature 465, 378–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gietz R. D., Sugino A. (1988) New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74, 527–534 [DOI] [PubMed] [Google Scholar]
- 44. Cigan A. M., Pabich E. K., Donahue T. F. (1988) Mutational analysis of the HIS4 translational initiator region in Saccharomyces cerevisiae. Mol. Cell. Biol. 8, 2964–2975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mueller P. P., Hinnebusch A. G. (1986) Multiple upstream AUG codons mediate translational control of GCN4. Cell 45, 201–207 [DOI] [PubMed] [Google Scholar]
- 46. Phan L., Zhang X., Asano K., Anderson J., Vornlocher H. P., Greenberg J. R., Qin J., Hinnebusch A. G. (1998) Identification of a translation initiation factor 3 (eIF3) core complex, conserved in yeast and mammals, that interacts with eIF5. Mol. Cell. Biol. 18, 4935–4946 [DOI] [PMC free article] [PubMed] [Google Scholar]