

Abstract
Severe and fatal viral infections remain common after hematopoietic stem cell transplantation. Adoptive transfer of cytotoxic T lymphocytes (CTLs) specific for Epstein–Barr virus (EBV), cytomegalovirus (CMV), and adenoviral antigens can treat infections that are impervious to conventional therapies, but broader implementation and extension to additional viruses is limited by competition between virus-derived antigens and time-consuming and laborious manufacturing procedures. We now describe a system that rapidly generates a single preparation of polyclonal (CD4+ and CD8+) CTLs that is consistently specific for 15 immunodominant and subdominant antigens derived from 7 viruses (EBV, CMV, Adenovirus (Adv), BK, human herpes virus (HHV)-6, respiratory syncytial virus (RSV), and Influenza) that commonly cause post-transplant morbidity and mortality. CTLs can be rapidly produced (10 days) by a single stimulation of donor peripheral blood mononuclear cells (PBMCs) with a peptide mixture spanning the target antigens in the presence of the potent prosurvival cytokines interleukin-4 (IL4) and IL7. This approach reduces the impact of antigenic competition with a consequent increase in the antigenic repertoire and frequency of virus-specific T cells. Our approach can be readily introduced into clinical practice and should be a cost-effective alternative to common antiviral prophylactic agents for allogeneic hematopoietic stem cell transplant (HSCT) recipients.
Introduction
Although hematopoietic stem cell transplant (HSCT) may cure hematological malignancies and genetic disorders, extension to donors other than HLA-matched siblings has resulted in the emergence of viral infections as major contributors to post-transplant morbidity and mortality.1,2,3,4 With the advent of more intensive viral screening and improved detection, increasing numbers of viral pathogens have been implicated in these complications, expanding from cytomegalovirus (CMV), Epstein–Barr virus (EBV), herpes-simplex virus, Adenovirus (Adv), and BK to include human herpes virus (HHV)-6, respiratory syncytial virus (RSV), parainfluenza, and influenza.2 While pharmacological agents are standard therapy for some, they have substantial toxicities, generate resistant variants, are frequently ineffective and do not provide long-term protection.5,6
Restoration of virus-specific immunity offers an attractive alternative to conventional drugs. We have shown that in vitro expanded virus-specific cytotoxic T lymphocytes (CTL) generated from stem cell donors with specificity for one (EBV), two (EBV and Adv), or three (EBV, CMV, and Adv) viruses are safe and effectively prevent and treat viral infection or disease in the HSCT setting.7,8,9 More recently, banked, partially HLA-matched virus-specific CTL (3rd party CTLs) are showing promise in allograft recipients with advanced viral disease.10
Despite these encouraging clinical results broader implementation of T cell therapy is restricted by (i) the limited spectrum of viruses that can be effectively targeted in a single T cell line and (ii) the logistics of manufacture. Antigenic competition between high and low frequency T cells as well as between multiple antigens expressed at different levels and competing for presentation on shared antigen-presenting cells (APCs) may favor generation of lines dominated by responses to a single virus or to a restricted spectrum of viral antigens,11,12 thus limiting the antiviral coverage provided by a single T cell product. In addition, our current manufacturing process is complex, requiring infectious virus material (EBV/Adv), production of a clinical grade vector, and prolonged (10–12 weeks) in vitro culture.8,9 To address this latter problem some groups have evaluated more rapid approaches for producing T cell products for adoptive transfer. These include multimer selection to directly isolate virus-specific CD8+ T cells from peripheral blood,13 as well as the selection of cells based on cytokine production [interferon γ (IFNγ)] or expression of activation markers (e.g., CD154) following antigen exposure.14,15,16 However, these approaches are expensive, require a large starting blood volume, which is not always available, particularly in the matched unrelated donor setting, and cannot be applied to viruses with low circulating T cell precursor frequencies.
We now describe a mechanism by which we can rapidly generate a single preparation of polyclonal (CD4+ and CD8+) CTLs, which is consistently specific for 15 immunodominant and subdominant antigens derived from seven viruses (EBV, CMV, Adv, BK, HHV6, RSV, and Influenza) that are frequent causes of post-transplant disease or death. Our approach, which uses standardized (synthetic) peptides as a stimulus and enhancing cytokines to promote the survival and expansion of T cells, is readily adaptable to clinical implementation and may thus be used as a safe and effective broad spectrum antiviral agent for all high risk transplant recipients.
Results
IL2, IL15, and IL4+7 promote the expansion of peptide-activated T cells in vitro
To increase the range of viral antigens that could be recognized by a single CTL line and to mitigate the impact of antigenic competition in order to retain both high and low frequency T cells, we stimulated peripheral blood mononuclear cells (PBMCs) in the presence of different Th1, proproliferative and prosurvival cytokines. We then compared the frequency and repertoire of responding cells to those generated by conventional activation in the absence of cytokines. In exploratory experiments PBMCs were simulated with a pepmix (peptide library of overlapping 15mers) spanning the immunodominant CMV-pp65 antigen, then expanded without cytokines, or with media supplemented with (i) interleukin-15 (IL15) (5 ng/ml), (ii) IL2 (20 U/ml), or (iii) IL4 (1,666 U/ml) + IL7 (10 ng/ml). After 9–12 days we assessed cell expansion, phenotype, specificity, and function.
Cultures supplemented with IL15 or IL4+7 showed the greatest overall expansion (5 ± 0.6 and 3.7 ± 0.5-fold increase, respectively) over 9 days (n = 5). Cultures that were stimulated in the absence of cytokines did not expand (0.6 ± 0.04), while the IL2 condition was intermediate (2.7 ± 0.1) (Figure 1a). To determine whether the superior cell numbers were a consequence of improved T cell proliferation, enhanced survival, or the combination, cells were labeled with CFSE on day 0 and then analyzed every 2–3 days to measure cell doubling, while live and apoptotic/necrotic cells were distinguished by Annexin-PI staining (data not shown). Flow cytometric analysis demonstrated no difference in the number of cell divisions from day 0–5. However, from day 5 onward cells cultured in cytokines continued to divide, whereas in their absence, cell division was reduced and viability was consistently lower (Figure 1b). These data suggest that the improved survival of proliferating cells made the primary contribution to the observed increase in cell numbers in cytokine-supplemented cultures.
Figure 1.

Growth-promoting cytokines enhance the activation and expansion of antigen-specific cytotoxic T lymphocytes (CTLs). Peripheral blood mononuclear cells (PBMC) were stimulated with pp65 pepmix in the presence of IL2, IL15, IL4+7 or without exogenous cytokines. Cell expansion was evaluated after 9–11 days of culture by cell counting using trypan blue exclusion (n = 5). Results are shown as mean cell numbers ± SEM. (a). Panel b CD3+ T cell proliferation in the different culture conditions as evaluated by CFSE dilution. M1 shows the percentage of cells that underwent at least seven cell doublings on day 10 after stimulation. Bulk cultures were analyzed for T and NK-cell marker expression on day 10 after activation. Mean expression ± SEM in CTL lines generated from five donors are shown in c. (d) cytokine production from CD3/CD4+ (helper) and CD3/CD8+ (cytotoxic) CTLs on day 9 after initiation in one representative donor (dot plots shown were gated on CD3+ cells). Summary intracellular cytokine production results from three donors (mean ± SD) are shown in (e). Finally the cytokine production profile of pp65-specific CTL initiated with or without cytokines was evaluated by multiplex assay using supernatant harvested 18 hours after antigenic restimulation (n = 4). Th1 cytokines are shown in the left panel while prototypic Th2 cytokines are shown in the right panel (f). Presence of regulatory T cells were evaluated by FoxP3 staining. Plots shown are gated on CD3+/CD4+ CTLs (g).
IL4+7 support the selective expansion of polyclonal, Th1-polarized T cells
Optimal in vivo T cell persistence and activity requires both helper (CD4+) and cytotoxic (CD8+) T cells. We therefore used phenotypic analyses to determine that the cells in the cytokine-supplemented cultures reflected the selective expansion of polyclonal T cells. We found the lowest frequency of CD3+ T cells in cultures supplemented with IL2 or IL15 (72.8 ± 2.1% and 61.3 ± 3.7%, respectively), which instead contained significantly higher numbers of CD56+ NK cells than other conditions (27.1 ± 2.3% and 37.7 ± 3.7%, respectively) (n = 5). By contrast, IL4+7 cultures were comprised almost entirely of CD3+ T cells (92.6 ± 0.4%), with both CD8+ T cells and significantly more CD4+ T cells (61 ± 2.7%) than the other cytokine-supplemented conditions (IL2 26 ± 4%, IL15 17.6 ± 4.3%, P = 0.024, P = 0.004, respectively) (Figure 1c). To confirm that both CD8+ and CD4+ T cells were antigen-specific and produced effector cytokines we performed intracellular cytokine staining (ICS) for IFNγ. Figure 1d shows representative results from 1 donor, while Figure 1e shows summary results for three donors. Our data confirm that IL4+7-supplemented cultures contained antigen-specific IFNγ-producing T cells in both compartments (CD4+ 39.3% ± 16.4%, CD8+ 22.2% ± 2.2%), at levels substantially higher than in other conditions (no cytokine: CD4+ 2.3% ± 3.9%, CD8+ 0.8% ± 0.5%; IL15: CD4+ 1.7% ± 1.4%, CD8+ 13.9% ± 2.8% and IL2: CD4+ 2.2% ± 3.1%, CD8+ 12.6% ± 2.6%, n = 3). Similar results were obtained using pepmixes from subdominant Adv (Penton) and EBV (LMP2) viral antigens; indeed outgrowth of NK cells was even more evident in the IL2 and IL15-supplemented conditions (Supplementary Figure S1).
IL4 is a prototypic Th2 cytokine, therefore to more comprehensively evaluate the cytokine profile of the induced CTLs the supernatant of antigen-activated T cells was assessed using luminex array. Figure 1f shows that, in addition to IFNγ, the IL4+7-supplemented lines produced the prototypic Th1 cytokines granulocyte-macrophage colony-stimulating factor, IL2 and tumor necrosis factor α (TNFα), at levels similar to that of IL15-induced CTLs. In addition, levels of Th2 cytokines (IL5 and IL13) were not substantially different and there was no evidence of regulatory T cell outgrowth, as assessed by CD4/CD25/FoxP3+ staining (Figure 1g). Thus, IL4, in combination with IL7, induces selective expansion of polyclonal, Th1-polarized T cells that produce multiple effector cytokines upon stimulation (Supplementary Figure S2).
Overlapping 15mer peptide libraries activate T cells with similar specificity and avidity to those generated using endogenously processed full-length antigen
To address concerns that pepmixes might reactivate low avidity T cells unable to recognize antigens that are naturally processed and presented by virus-infected cells we compared pp65 pepmix-activated CTLs with those generated using dendritic cells (DCs) nucleofected with a DNA plasmid encoding the same antigen. After activation, each set of cells was expanded in IL4+7. Expansion was similar between the groups, with 107 ± 23.4 × 106 cells generated using pepmix-pulsed PBMCs (7.2-fold expansion) versus 130.3 ± 46.9 × 106 cells in the DC-stimulated cultures (8.7-fold expansion) (Figure 2a) (n = 3). Phenotypic analysis demonstrated that the pepmix-activated CTLs were predominantly CD4+ (74.3 ± 19.3%), with a minor CD8+ component (22.8 ± 19.2%), as were the plasmid-activated CTLs (CD4+ 70.6 ± 14.2% and CD8+ 26.5 ± 13.4%) and both expressed similar levels of the memory and activation markers CD62L, CD28, and CD45RO (61 ± 46.7%, 86.5 ± 3.5%, 92 ± 7.1% pepmix versus 77 ± 28.3%, 85.5 ± 0.7%, 87.5 ± 13.4% plasmid) (Figure 2b). We next compared the breadth of epitopes recognized by measuring responses to 110 20mer peptides (overlapping by 15aa) spanning CMV-pp65 and arranged into 22 pools such that each peptide was represented in 2 pools. Figure 2c shows that both the recognition of a given peptide and the magnitude of the response thereto were little changed by the antigen source. Finally, we compared the functional avidity of our polyclonal CTL lines by IFNγ enzyme-linked immunosorbent spot (ELISpot) using log dilutions of the pp65 pepmix or epitope peptides (A2-NLV and A24-QYD) as a stimulus. As shown in Figure 2d, there was no significant difference in the avidity of the CTLs. This data was confirmed for other viral antigens using Adv-Hexon pepmix and viral antigen-encoding plasmid as a stimulus (Supplementary Figure S3).
Figure 2.

Peptide-stimulated and plasmid-activated cytotoxic T lymphocytes (CTLs) share similar phenotypic and functional characteristics. (a) CTLs were stimulated either directly with a pp65 pepmix or using DCs nucleofected with a DNA plasmid encoding the same antigen. Cell expansion was evaluated by counting using trypan blue exclusion (n = 4). (b) The expression of cell surface markers (average ± SD expression) on CTLs 11 days after stimulation (n = 4). The breadth of T cell reactivity in plasmid and pepmix-activated pp65-specific CTLs was evaluated by IFNγ ELISpot on day 9 using a total of 22 mini peptide pools representing all pp65 peptides. Data were normalized to 100% for maximum number of SFC per 1 × 105 CTL. Data from 3 donors screened is shown in (c). (d) The T-cell receptor (TCR) avidity of plasmid versus pepmix-activated CTL generated from 2 representative donors. To assess avidity pp65-CTLs were stimulated bulk CTL cultures with serial dilutions of pp65 pepmix (pp65) or relevant (HLA-matched) epitope peptides (NLV, QAD). IFNγ release of stimulated CTLs was evaluated by ELISpot assay and maximum SFC/1 × 105 cells was normalized to 100% for comparison purposes.
15mer peptides activate CD4+ and CD8+ T cells as efficiently as long (20mer or 30mer) peptides
Since CD4+ epitopes (>20aa) may be longer that CD8 epitopes (8–10aa) we next determined whether longer peptides would induce higher frequencies of antigen-specific CD4+ T cells. We obtained three overlapping peptide libraries (#1–15mers overlapping by 11, #2–20mers overlapping by 15, and #3–30mers overlapping by 15) spanning the C-terminus (aa539-953) of Adv-Hexon; a region rich in both CD4+ and CD8+ epitopes17,18 (Figure 3a). We directly stimulated PBMCs with each of the libraries and evaluated the phenotype, epitope specificity and breadth of the lines.
Figure 3.

Peptide length does not affect breadth of reactivity. (a) A schematic of three peptide libraries spanning a portion of Adv-Hexon which were used for CTL initiation. Peptide libraries consisted of 15aa, 20aa, or 30aa peptides covering the immunogenic C-terminal 414aa of Adv-Hexon. (b) Phenotypic analysis of cytotoxic T lymphocytes (CTLs) performed on day 10 after stimulation (n = 6). Results are shown as mean ± SEM. Breadth of reactivity was tested using interferon γ (IFNγ) ELISpot as a readout, with the 15mer Hexon overlapping peptide library divided into minipools such that each pool contained 5–6 contiguous peptides, as a stimulus.
Phenotypically the lines were comparable, with a predominance of CD4+ cells (mean 56 ± 5.5% versus 59 ± 5.8% versus 60 ± 6%) and a minor CD8+ component (mean 21 ± 0.2% versus 20 ± 0.1% versus 16 ± 0.2%), and similar levels of the memory and activation markers CD62L, CD28, and CD45RO (CD62L – 60 ± 1.9% versus 57 ± 1.9% versus 51 ± 1.6%, CD28 – 88 ± 0.6% versus 84 ± 2.1%, versus 89 ± 0.6%, and CD45RO – 58 ± 1.7% versus 60 ± 1.6% versus 60 ± 1.2%) (15mer versus 20mer versus 30mer) (n = 6). To learn whether the spectrum of epitopes recognized differed based on the stimulating library; we rechallenged the induced CTLs with subpools of peptides from each library and found no consistent or statistically significant differences in the breadth of peptides recognized. Results for the 15mer minipool rechallenge are shown in Figure 3c. Since 15mer pepmixes are readily available as both research and clinical products we performed all subsequent experiments with this antigen source.
Generation of a single T cell culture with simultaneous specificity for Adv, EBV, and CMV
After successfully generating CTLs using peptides derived from a single viral antigen and culture in IL4+7, we next prepared a single culture of CTLs simultaneously recognizing CMV, EBV, and Adv. For each virus we targeted immunogenic antigens; CMV – IE-1 and pp65, Adv – Hexon and Penton, and EBV – EBNA1 (an immunodominant CD4+ T cell target antigen expressed in all EBV-associated malignancies and in normal EBV-infected B cells), LMP2 (an antigen that is immunogenic across multiple HLA types and expressed in most EBV malignancies) and BZLF1 (an immediate early lytic cycle antigen that stimulates both CD4+ and CD8+ T cells19 and pulsed PBMCs with the relevant pepmixes before culture in IL4+7. After 9–12 days we compared the antiviral reactivity of the resulting CTLs with those generated using our current clinical trivirus CTL protocol which uses DCs nucleofected with plasmids encoding the same antigens as a stimulus (Figure 4). IFNγ ELISpot confirmed that pepmix-generated CTLs from four donors had antiviral activity against all three viruses and seven stimulating antigens. The frequency of T cells reactive against EBV (EBNA1, LMP2, BZLF1) and CMV (IE-1, pp65) was comparable irrespective of the stimulus. In contrast, all 4 donors had significantly more Adv-reactive T cells (Hexon and Penton) in pepmix-stimulated cultures (Hexon—median 462.3, range 373–572.5 versus median 112, range 53–421.5 SFC/2 × 105 CTL; P = 0.01, Penton—median 317, range 105.5–345 versus median 51.25, range 4–134 SFC/2 × 105 CTL, P = 0.02, pepmix versus plasmid, respectively).
Figure 4.

Pepmix-activated trivirus-specific CTL lines show similar specificity to plasmid-activated T cells. CTL lines were generated using DCs nucleofected with DNA plasmids encoding EBNA1, LMP2, BZLF1 (EBV), Hexon, Penton (adenovirus), IE-1, and pp65 [cytomegalovirus (CMV)] or direct peripheral blood mononuclear cells (PBMC) stimulation with the corresponding pepmixes. Specificity was determined 10 days after initiation with interferon γ (IFNγ) ELISpot as readout. Results are expressed as SFC/1 × 105 input cells. Control was IFNγ release in response to stimulation with irrelevant pepmix.
Extension to additional viruses
To determine whether our direct pepmix stimulation approach could be extended to generate multivirus-specific CTL lines targeting a broader spectrum of different clinically relevant viruses we stimulated PBMCs with pepmixes spanning 2 or 3 T cell immunogenic antigens from CMV, Adv, EBV, BK, Influenza, RSV, and HHV6 (Table 1). To determine whether antigenic competition would preclude pooling we segregated the pepmixes and stimulated PBMCs with minipools-containing pepmixes from (i) each virus; (ii) immunodominant (CMV, RSV, Flu, HHV6) and subdominant (Adv, EBV, BK) viruses; (iii) lytic (Adv, RSV, Flu) and latent (EBV, CMV, HHV6, BK) viruses, or (iv) a mastermix of all pepmixes (Figure 5a). There was no difference in either the rate of expansion (Supplementary Figure S4), the overall specificity or magnitude of the response directed against each antigen, irrespective of the composition of the stimulating pepmix pool (Figure 5b). Thus, all further studies used the mastermix (condition D). Figure 5c shows eight additional CTL lines with consistent multivirus specificity. The highest responses were seen against CMV-pp65 and Adv-Hexon (951.6 ± 82.1 and 461.4 ± 19.2 SFC/1 × 105 CTL) whereas activity against HHV6-U90, EBV-BZLF1 and EBV-LMP2 was weakest (26.9 ± 4.2, 35.6 ± 5, 39.6 ± 2.6 SFC/1 × 105 CTL). Adv-Penton, Influenza-MP1, and RSV-F demonstrated intermediate response rates (191 ± 13.7, 117.6 ± 8.6, 90.1 ± 10.3 SFC/1 × 105 CTL, respectively) (Figure 5c). The lines were polyclonal and polyfunctional with activity against the stimulating viruses detectable in both CD4+ and CD8+ fractions (Figure 5d), and reactive cells produced both IFNγ and TNFα after stimulation, a profile associated with superior in vivo activity.20,21 Figure 5e shows the results for one representative donor in whom 63% of all Adv, 55% of CMV, 40% of EBV, 46% of RSV, 36% of Influenza, and 28% of HHV6-specific CTLs produced both IFNγ and TNFα after antigenic stimulation. Intracellular cytokine staining for IFNγ and/or TNFα in a total of six CTL lines generated and screened showed that 67.7 ± 13.3% of all T cells in multivirus cultures were antigen-specific (data not shown). This percentage is likely an underestimate since some virus-specific CTLs do not produce cytokines or produce effector cytokines other than IFNγ and TNFα. Finally, even though these CTLs had received only a single stimulation there was no evidence of alloreactivity, assessed by Cr51 release assay using HLA-mismatched PHA blasts as targets (Supplementary Figure S5), an important consideration if these cells are to be used for the treatment of allogeneic HSCT recipients.
Table 1. Clinically relevant viruses and equivalent antigens used for T cell stimulation.
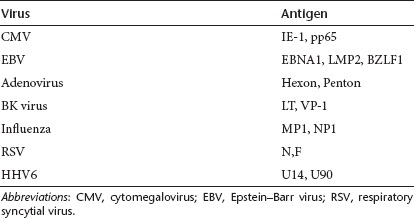
Figure 5.

Generation of multivirus-specific cytotoxic T lymphocytes (CTLs). (a) A schematic of antigen pooling strategy for CTL initiation. Peripheral blood mononuclear cells (PBMCs) were stimulated with pepmixes pooled by virus (condition A), divided into subpools—immunodominant and subdominant (condition B), divided into subpools encompassing antigens from latent or lytic viruses (condition C), and finally all antigens were pooled together in a mastermix (condition D). After activation PBMCs were pooled and transferred to the G-Rex10 (15 × 106/G-Rex). After 10 days the specificity of the CTL lines generated using these 4 pooling strategies were analyzed using interferon γ (IFNγ) ELISpot assay as readout and individual pepmixes as a stimulus. Results from two representative donors are presented in (b) showing no difference in the specificity of lines. (c) Confirms that multivirus CTL can be reproducibly generated by pooling all pepmixes into one mastermix for activation (n = 8). Results are expressed as SFC/1 × 105 input cells ± SEM. Control was IFNγ release in response to stimulation with an irrelevant pepmix. Antigen specificity of CD3/CD8+ (cytotoxic) and CD3+CD8- (helper) T cells was evaluated by intracellular IFNγ staining after overnight stimulation with the equivalent antigens. Results from one representative donor are shown in (d). (e) The lines are polyfunctional as assessed using intracellular cytokine staining (ICS) for IFNγ and TNFα in one representative donor.
Multivirus-specific CTL can be expanded in vitro
To discover whether multivirus-specific CTLs could be further expanded to provide numbers suited for third party or “off-the-shelf” use, we restimulated the cells with autologous PHA blasts pulsed with the same mastermix of pepmixes. Secondary expansion of a mean of 8.4 ± 2-fold was obtained over 7 days, to a final cell number of 604.6 ± 23.7 × 106 (Figure 6a). Figure 6b shows that the expanded CTLs remained polyclonal, with activity detected in both CD4+ and CD8+ compartments. Expansion was associated with an overall increase in the magnitude of the response directed against all of the stimulating antigens on day 16 relative to day 9 (Figure 6c) so that >80% of cells in the restimulated cultures produced IFNγ and/or TNFα. Similarly, these expanded cultures had greater cytolytic activity, ranging from >60% (CMV) to 14% (BK), demonstrating retained specificity for both subdominant and immunodominant antigens/viruses (Figure 6d) without alloreactivity (Supplementary Figure S5).
Figure 6.

Multivirus-specific cytotoxic T lymphocytes (CTLs) can be expanded in vitro. On day 9 after initial stimulation CTLs were restimulated using pepmix-pulsed PHA blasts. (a) shows the expansion of CTLs from initiation (day 0) to day 16, following a 2nd stimulation on day 9/10 (n = 4). CTL expansion was evaluated using trypan blue exclusion and results are shown as mean cell numbers ± SD. (b) Results from 1 representative donor illustrating the antigen specificity of CD3/CD8+ and CD3/CD8– (CD4+) CTLs after the 2nd round of stimulation using interferon γ (IFNγ) intracellular cytokine staining (ICS). (c) Summary results from six donors after the 1st (day 9) and 2nd (day 16) stimulation, using IFNγ ELISpot as a readout. Results are expressed as SFC/1 × 105 input cells ± SD and the control was IFNγ release in response to stimulation with irrelevant pepmix. The cytotoxic abilities of the generated CTLs were evaluated by standard 4–6 hours Cr51 release assay using pepmix-pulsed PHA blasts as targets. Specific lysis after the 1st and 2nd stimulation from two representative donors are shown in (d).
Discussion
We have shown that we can rapidly generate polyclonal, CD4+ and CD8+ T cells with specificities directed to a wide range of lytic and latent viruses responsible for infection in the immunocompromised host and after HSCT. These cells were Th1-polarized, had high avidity for a multiplicity of individual viral antigens, produced multiple effector cytokines upon stimulation, and killed virus-infected targets without alloreactivity. Because we generated these T cells using combinations of clinically available peptide libraries and prosurvival cytokines, our approach should be well suited to clinical application.
While CMV, EBV, and Adv are the most frequently detected viral infections following allogeneic HSCT, recipients are also susceptible to numerous other viruses, including BK, JC, HHV6, HHV7, influenza, parainfluenza, coronavirus, and RSV, all of which may cause severe morbidity and mortality.1,2 Several of these viruses are usually seasonally detected (e.g., influenza, RSV) while others, such as HHV7, JC, and coronavirus, are infrequent, so that it is impracticable to cover all these pathogens post-transplant by generating individualized patient and single virus-specific T cell products. Hence, we sought to develop a strategy that would enable the production of a single CTL line with simultaneous specificity for a multiplicity of antigens, which were chosen based on their T cell stimulatory capacity and protective nature in our own studies (refs. 8,9,18,22,23 and U. Gerdemann, L. Keukens, J.H. Keirnan, U.L. Katari, C.T.Q. Ngugen, A.P. de Pagtee et al., manuscript submitted) and in those from other groups.13,15,19,24,25,26,27,28,29,30,31,32
In our present clinical trials of virus-specific T cells, we have used EBV-LCL, adenovectors and/or viral antigen-encoding DNA plasmids to generate virus-directed T cells.8,9,33 The use of full-length antigen ensures that CTL can be generated from all donors, irrespective of HLA, and that the antigen is physiologically processed by APCs and produces CTLs that recognize multiple CD4+ and CD8+ T cell epitopes and have sufficient avidity to kill virus-infected targets. The induction of lines that recognize multiple epitopes also minimizes virus escape due to epitope loss and produces potent and sustained antiviral activity in vivo.34 However, the requirements for live virus/vectors are barriers to broader and late phase clinical studies, and also limit the number of pathogens to which a single T cell line can be directed.8,9 We therefore evaluated whether clinically applicable pepmixes could be used as an alternative. Though clinical studies using minimal epitope peptides as vaccines have resulted in immune tolerance or the activation of low avidity T cells, Melief and colleagues recently demonstrated improved results with long (22-45aa) peptides containing both CD4+ and CD8+ epitope sequences.35 They observed that these long peptides were processed endogenously, presented to T cells by APCs, and induced both helper and cytotoxic T cells, resulting in robust and effective CTL responses.35 Based on these data, we chose to use a whole antigen source in the form of overlapping peptide libraries, but for optimal induction of polyclonal CTL we compared peptides of different lengths (15mers, 20mers, and 30mers) for stimulation. When using Hexon, which is predominantly recognized by CD4+ T cells, as a model antigen we saw no difference in the phenotype, specificity or epitope breadth of our lines. However, Maecker and colleagues found that 20mers were suboptimal for activating pp65-directed CD8+ T cells.36 Thus, though still rather controversial, the optimal length of peptide for the activation of T cell lines containing both CD4+ and CD8+ cells appears to be 15aa. This analysis also highlights the differences between delivering peptides as a vaccine, where one relies on endogenous APCs to take up and process antigen versus in vitro T cell activation using professional APCs within PBMCs at optimal effector:target ratios, where peptides are either endogenously processed or edited by exoproteases following binding to cell surface HLA molecules.37,38,39,40 Given the ready clinical availability of pepmixes containing 15mer peptides that cover all possible CD8+ and the majority of CD4+ epitopes, we substituted this antigen source and were able to demonstrate equivalency to “conventionally generated” CTLs with respect to both epitope specificity and avidity.
We next addressed how best to extend the breadth of antigen/epitope specificities that could be accommodated within a single CTL line. Physiologically, T cells are activated when they receive signals from T-cell receptor stimulation (signal 1), costimulation (signal 2), and cytokines (signal 3). Our “conventional CTLs” are activated in the absence of exogenous cytokines, a deficit that appears to adversely affect their proliferative capacity in vitro and also increases their susceptibility to activation-induced cell death, likely resulting in a more restricted repertoire of epitope recognition. Consistent with this possibility, we found that both the frequency and breadth of cells with viral specificity could be increased by supplementing cultures with inflammatory and prosurvival cytokines at initiation. We chose to test cytokines that support cell proliferation in vitro and in vivo (IL2, IL15),41,42 as well as combinations (IL4+7) that also support the retention of a central memory phenotype, and promote the survival of activated T cells by upregulation of anti-apoptotic molecules e.g., Bcl-2.43,44,45,46 Only lines supplemented with IL4+7 selectively promoted the expansion and survival of both CD4+ and CD8+ virus-specific T cells recognizing multiple epitopes, a combination that should favor the subsequent sustained expansion of transferred cells in vivo.34 Of note, the induced cells were Th1-polarized despite exposure to IL4, a prototypic Th2 cytokine. Given the clinical availability of both cytokines and their safety in human clinical trials,47,48 IL4+7 fulfilled the requirements of the current study, however, other proinflammatory cytokines capable of mimicking the milieu present during viral infection may produce similar benefits. For example, von Rossum and colleagues recently reported that CD3/28-activated CD8+ T cells cultured in an inflammatory cocktail consisting of IL1+IL6+IL23 underwent significantly less cell death after activation as compared with cells activated in any of the cytokines alone or activated in the presence of IL12.49
The direct stimulation of PBMCs with pepmixes and culture in cytokine-supplemented conditions also allowed us to overcome a second major barrier to increasing the spectrum of viruses targeted in a single CTL line, namely antigenic competition resulting from the use of a common APC to simultaneously present multiple antigenic components from different viruses.8,9 Antigenic competition results both from limited access of peptides to HLA molecules and physical constraints on the simultaneous stimulation of both high and low frequency T cells.11,12 To overcome these issues, investigators have used artificial APCs that are engineered with molecules to provide the necessary T-cell receptor and co-stimulatory events required for immune synapse formation.50 However, to avoid the inevitable complexities and costs of introducing a gene-modified cellular product into the manufacturing process, we evaluated whether patient PBMCs themselves could act as both a source of antigen-presenting and responding cells. B cells, monocytes and macrophages may all have the capacity to present antigen to T cells and these APCs can utilize endo- and exopeptidases to liberate class I or class II epitopes from 15mer peptides. By taking advantage of these properties, we can avoid reliance on a single APC endogenously expressing multiple antigens at different levels as a shared T cell stimulator, and instead have a diverse group of APCs in which each cell has the potential to display a diverse repertoire of peptides, allowing sufficient access for both high and low frequency T cells. Thus, antigenic competition both within the APC and between T cells could be alleviated. As proof of principal we generated a single culture of T cells with reactivity for 15 antigens derived from 7 latent and lytic viruses (EBV, CMV, BK, HHV6, Adv, Flu, and RSV) using pooled pepmixes as a stimulus and saw no evidence of competition. Additional pathogens may be included in this platform, although ultimately APC numbers will likely eventually become limiting, thus additions must be performed in a stepwise manner and one must evaluate changes in the frequency and breadth of T cell recognition of all peptides in the mix.
Critically for clinical feasibility, our approach was able to produce large numbers of virus-specific T cells. By seeding just 1.5 × 107 PBMCs in the G-Rex and a single in vitro stimulation we could regularly manufacture 1 × 108 CTLs within 10 days, with a >10-fold enrichment in virus-specific cells and a corresponding reduction in alloreactive T cells to levels observed in repetitively stimulated conventional CTLs, which have a proven safety record in vivo. Thus, using our new manufacturing technology we predict that multivirus-specific CTL will be safe for infusion after a single exposure to pepmixes and will provide broad spectrum antiviral protection without GvHD. Should additional cells be required, for example if banked virus-specific CTLs are established for 3rd party recipients, a second stimulation using pepmix-pulsed PHA blasts can expand the total number of CTLs without impairing their epitope specificity or breadth.
Materials and Methods
Donors and cell lines. PBMCs were obtained from healthy volunteers with informed consent using a Baylor College of Medicine institutional review board-approved protocol. The donors tested all had pre-existing immunity to the target viruses. PBMCs were used to generate DCs, CTL lines, and PHA blasts. PHA blasts were generated from PBMC (2 × 106/ml) using PHA (5 µg/ml) and maintained in CTL media [RPMI 1640, 45% Click's (Irvine Scientific, Santa Ana, CA), 2 mmol/l GlutaMAX TM-I, and 5% Human AB Serum] supplemented with IL2 (100 U/ml; NIH, Bethesda, VA), which was replenished every 3 days.
CTL generation—peptide stimulation
Peptides/pepmixes. For PBMC stimulation we used commercially available pepmixes (15mers overlapping by 11aa) spanning EBV-LMP2, BZLF1, EBNA1; Adv-Penton, Hexon; CMV-pp65, IE-1; BKV-VP1, large T; Influenza A-MP1 (H3N2), NP (H3N2); RSV-F, N; JPT Technology, Berlin, Germany. Pepmixes spanning HHV6 U14 and U90 were synthesized by Genemed Synthesis, San Antonio, TX. Peptide libraries spanning the 414aa C-terminus of Adv-Hexon were synthesized by Proimmune, Oxford, UK or Alta Bioscience, University of Birmingham, Edgbaston, Birmingham, UK. Lyophilized peptides were reconstituted at 5 mg/ml in DMSO.
PBMC stimulation. 15 × 106 fresh/frozen PBMCs were pelleted in a 15-ml tube and pulsed for 30–60 minutes at 37 °C with peptide libraries/pepmixes, either singly or pooled, at a concentration of 100 ng/peptide/15 × 106 PBMCs. After incubation cells were resuspended in CTL media alone or supplemented with cytokines (as outlined below) and transferred to a G-Rex10 (Wilson Wolf Manufacturing, New Brighton, MN) (15 × 106/G-Rex10) or plated out in a 24-well plate (2 × 106/well). Media and cytokines were replenished on day 5, and cultures were split when they reached a density >50 × 106/G-Rex10 or >3 × 106 cell/24-well. On day 9–12 CTLs were harvested, counted and used for phenotypic and functional studies.
Cytokines for promoting CTL activation and expansion. We compared four conditions; (i) no cytokine, (ii) IL7 (10 ng/ml) + IL4 (1,666 U/ml), (iii) IL15 (5 ng/ml) (R&D Systems, Minneapolis, MN), and (iv) IL2 (20 U/ml). Cytokines were added to CTLs at day 0 and replenished on day 5.
CTL expansion. For expansion CTLs were restimulated at a S:R ratio of 1:1 with irradiated (30Gy) pepmix-pulsed autologous PHA blasts in CTL media with IL4+7 and IL15 (5 ng/ml) on the day of restimulation and fed with IL15 twice weekly. Seven days later CTLs were harvested, and used for further studies.
CTL generation using plasmid-nucleofected DCs
Plasmids. DNA plasmids expressing the viral antigens (i) Hexon and Penton, (ii) IE-1 and pp65, and (iii) EBNA1ΔLMP2 and BZLF1 were generated by NTC (Lincoln, NE).33
CTL activation. Monocyte-derived DCs were matured on day 5 using a cytokine cocktail containing 100 ng IL6, 10 ng/ml IL-1β, 10 ng/ml TNF-α (R&D Systems), 1 µg/ml PGE2 (Sigma, St Louis, MO), 800 U/ml granulocyte-macrophage colony-stimulating factor (Immunex, Seattle, WA) and 1,000 U/ml IL4 for 48 hours. Twenty four hours after maturation 0.5–1 × 106 DCs were nucleofected with 5 µg/plasmid using program U2 (Lonza Nucleofector IV) and subsequently incubated overnight in DC media (CellGenix, 2 mmol/l GlutaMAX TM-I) (CellGenix, Antioch, IL) with the cytokine maturation cocktail and then used for T cell stimulation. For CTL initiation CD14-ve 15 × 106 PBMCs were stimulated with nucleofected DCs at a S:R ratio of 1:20 in the G-Rex10 in CTL media supplemented with IL4+7.33
Flow cytometry
Immunophenotyping. CTLs were surface-stained with monoclonal antibodies to: CD3, CD4, CD8, CD16, CD56, CD28, CD45RO, and CD62L (Becton Dickinson BD, Franklin Lakes, NJ). Cells were washed once with phosphate-buffered saline (PBS) (Sigma) containing 2% fetal bovine serum (FBS) (HyClone, Thermo Fisher Scientific, Logan, UT), pelleted, and antibodies added in saturating amounts (10 µl). After 15 minutes at 4 °C in the dark, cells were washed twice and analyzed. Approximately 20,000 live cells were acquired using a FACS Calibur equipped with Cell Quest software.
CFSE. To measure cell proliferation PBMCs were isolated, pelleted and pulsed with pp65 pepmix (100 ng/15 × 106 PBMC) for 30–60 minutes. Next PBMCs were washed twice using PBS+0.1% FBS and incubated for 10 minutes with 150 µl/20 × 106 PBMC 10 µmol/l CSFE. Subsequently FBS was added at a 1:1 ratio and incubated for 10 minutes at 37 °C. After CFSE labeling PBMCs were washed twice using PBS+2% FBS and plated at a concentration of 1 × 106/ml in CTL media with cytokines. Dilution of CFSE was examined every 2–3 days by flow after surface staining with CD3, CD4, CD8, and CD56.
FoxP3 staining. To measure regulatory T cells Foxp3 staining was performed using the e-Bioscience FoxP3 staining kit. Briefly, CTLs were rested in CTL media for 48 hours, then 1 × 106 CTLs were resuspended in PBS+2% FBS and surface stained for CD3, CD25 and CD4. After washing the cells were resuspended in 1 ml Fixation/Permeabilization solution and incubated for 1 hour at 4 °C, then washed, resuspended in permeabilization buffer and incubated with 0.2 µl isotype or 10 µl FoxP3 antibody (Clone PCH101) for 30 minutes at 4 °C. After a final wash cells were acquired using a FACSCalibur equipped with Cell Quest software.
Intracellular cytokine staining. CTLs were harvested, resuspended at a concentration of 5 × 106/ml in CTL media and plated at 200 µl/well in a 96 well plate. The cells were then stimulated with 100 ng of test or control pepmix in the presence of Brefeldin A (1 µg/ml), (BD) CD28 and CD49d (1 µg/ml) for 5–7 hours. Subsequently, CTLs were washed with PBS+2% FBS, pelleted, and surface stained with CD8, CD4, and CD3 (10 µl/antibody/tube). After 15 minutes, cells were washed twice, pelleted, fixed, and permeabilized with Cytofix/Cytoperm solution (BD) for 20 minutes at 4 °C in the dark. After washing twice with PBS/2%FBS containing 0.1% saponin (Calbiochem, EMD Chemicals, Gibbstown, NJ) cells were incubated with 20 µl IFNγ and/or TNFα antibodies (BD) for 30 minutes at 4 °C in the dark. Cells were then washed twice with cold PBS/2%FBS containing 0.1% saponin and at least 200,000 live cells from each population were analyzed with a FACSCalibur equipped with Cell Quest software (BD).
Functional studies
Multiplex assay. To assess cytokine production we used a multiplex assays. 1 × 105 pp65-CTLs were restimulated using 500 ng/ml pp65 or control pepmix. After 16 hours supernatant was collected and the cytokine profile assessed using the MILLIPLEX High Sensitivity Human Cytokine Magnetic Bead Panel (Millipore, Billerica, MA). Specifically, 50 µl supernatant was incubated overnight at 4 °C with cytokine antibody beads. After incubation, samples were washed and incubated for 1 hour at room temperature with the biotinylated detection antibody. Finally Streptavidin–Phycoerythrin was added for 30 minutes at room temperature, then samples were washed and analyzed using the Luminex 200 instrument. Samples were run in duplicate.
Enzyme-linked immunospot assay. We used ELISpot to quantify IFNγ -producing T cells and assess the breadth of reactivity in our CTL lines. The populations were serially diluted from 4-1 × 105 cells/well, and antigen-specific activity measured after direct pepmix or peptide minipool stimulation. Each condition was run in triplicate. After 20 hours, plates were developed, dried overnight at room temperature, then sent to Zellnet Consulting, New York, NY for quantification. SFC and input cell numbers were plotted, and a linear regression calculated after excluding plateau data points.
T-cell receptor avidity assessment. T-cell receptor avidity was assessed by IFNγ ELISpot. 2 × 105 CTLs were stimulated with serial dilutions of pepmixes (pp65, Hexon) or 9mer peptides (NLV-pp65: NLVPMVATV HLA-A2 restricted, QYD-pp65: QYDPVAALF HLA-A24 restricted; TDL-Hexon: TDLGQNLLY HLA-A1 restricted). The frequency of T cells specific for each antigen/peptide was expressed as a percentage of the maximal SFC/input cell number.
Chromium release assay. We measured the cytotoxic specificity in a standard 4 hours Cr51 release assay, using E:T ratios of 40:1, 20:1, 10:1, and 5:1. CTLs were used as effectors and the targets were PHA blasts pulsed with pepmixes. Autologous and allogeneic PHA blasts alone or loaded with an irrelevant pepmix were used as specificity and alloreactivity controls. The percentage of specific lysis was calculated as [(experimental release – spontaneous release)/(maximum release – spontaneous release)] × 100.
SUPPLEMENTARY MATERIAL Figure S1. Phenotype and specificity of Penton and LMP2-specific CTLs generated in the presence of different growth-promoting cytokines. Figure S2. CD3+ T cell expansion after addition of growth-promoting cytokines. Figure S3. TCR avidity is comparable in Hexon DC plasmid-activated and pepmix-stimulated PBMCs. Figure S4. Comparable expansion of CTLs stimulated with pooled versus single pepmixes. Figure S5. Lack of alloreactivity in pepmix-stimulated PBMCs.
Acknowledgments
The authors were supported by NIH grants U54 HL081007, N01-HB-10-03, and the NHLBI Production Assistance for Cellular Therapies (PACT). A.M.L. was supported by the National Marrow Donor Program through funding from the Amy Strelzer Manasevit Research Program and a Texas Children's Hospital Research Pilot award. U.G. was funded by a Leukemia and Lymphoma Society Special Fellow in Clinical Research Award, an ASBMT Young Investigator Award and the HHV6 Foundation. H.E.H. is supported by a Dan L. Duncan Chair and M.K.B. by a Fayez Sarofim Chair. The authors declared no conflict of interest.
Supplementary Material
Phenotype and specificity of Penton and LMP2-specific CTLs generated in the presence of different growth-promoting cytokines.
CD3+ T cell expansion after addition of growth-promoting cytokines.
TCR avidity is comparable in Hexon DC plasmid-activated and pepmix-stimulated PBMCs.
Comparable expansion of CTLs stimulated with pooled versus single pepmixes.
Lack of alloreactivity in pepmix-stimulated PBMCs.
REFERENCES
- Schönberger S, Meisel R, Adams O, Pufal Y, Laws HJ, Enczmann J.et al. (2010Prospective, comprehensive, and effective viral monitoring in children undergoing allogeneic hematopoietic stem cell transplantation Biol Blood Marrow Transplant 161428–1435. [DOI] [PubMed] [Google Scholar]
- Verdeguer A, de Heredia CD, González M, Martínez AM, Fernández-Navarro JM, Pérez-Hurtado JM, GETMON: Spanish Working Party for Blood and Marrow Transplantation in Children et al. Observational prospective study of viral infections in children undergoing allogeneic hematopoietic cell transplantation: a 3-year GETMON experience. Bone Marrow Transplant. 2011;46:119–124. doi: 10.1038/bmt.2010.52. [DOI] [PubMed] [Google Scholar]
- Lang P., and, Handgretinger R. Haploidentical SCT in children: an update and future perspectives. Bone Marrow Transplant. 2008;42 Suppl 2:S54–S59. doi: 10.1038/bmt.2008.285. [DOI] [PubMed] [Google Scholar]
- Sauter C, Abboud M, Jia X, Heller G, Gonzales AM, Lubin M.et al. (2011Serious infection risk and immune recovery after double-unit cord blood transplantation without antithymocyte globulin Biol Blood Marrow Transplant 171460–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantz S, Garnier-Geoffroy F, Mazeron MC, Garrigue I, Merville P, Mengelle C, French CMV Resistance Survey Study Group et al. Drug-resistant cytomegalovirus in transplant recipients: a French cohort study. J Antimicrob Chemother. 2010;65:2628–2640. doi: 10.1093/jac/dkq368. [DOI] [PubMed] [Google Scholar]
- Ljungman P, Ribaud P, Eyrich M, Matthes-Martin S, Einsele H, Bleakley M, Infectious Diseases Working Party of the European Group for Blood and Marrow Transplantation et al. Cidofovir for adenovirus infections after allogeneic hematopoietic stem cell transplantation: a survey by the Infectious Diseases Working Party of the European Group for Blood and Marrow Transplantation. Bone Marrow Transplant. 2003;31:481–486. doi: 10.1038/sj.bmt.1703798. [DOI] [PubMed] [Google Scholar]
- Heslop HE, Brenner MK., and, Rooney CM. Donor T cells to treat EBV-associated lymphoma. N Engl J Med. 1994;331:679–680. doi: 10.1056/NEJM199409083311017. [DOI] [PubMed] [Google Scholar]
- Leen AM, Myers GD, Sili U, Huls MH, Weiss H, Leung KS.et al. (2006Monoculture-derived T lymphocytes specific for multiple viruses expand and produce clinically relevant effects in immunocompromised individuals Nat Med 121160–1166. [DOI] [PubMed] [Google Scholar]
- Leen AM, Christin A, Myers GD, Liu H, Cruz CR, Hanley PJ.et al. (2009Cytotoxic T lymphocyte therapy with donor T cells prevents and treats adenovirus and Epstein-Barr virus infections after haploidentical and matched unrelated stem cell transplantation Blood 1144283–4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque T, Wilkie GM, Jones MM, Higgins CD, Urquhart G, Wingate P.et al. (2007Allogeneic cytotoxic T-cell therapy for EBV-positive posttransplantation lymphoproliferative disease: results of a phase 2 multicenter clinical trial Blood 1101123–1131. [DOI] [PubMed] [Google Scholar]
- Kedl RM, Rees WA, Hildeman DA, Schaefer B, Mitchell T, Kappler J.et al. (2000T cells compete for access to antigen-bearing antigen-presenting cells J Exp Med 1921105–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedl RM, Schaefer BC, Kappler JW., and, Marrack P. T cells down-modulate peptide-MHC complexes on APCs in vivo. Nat Immunol. 2002;3:27–32. doi: 10.1038/ni742. [DOI] [PubMed] [Google Scholar]
- Cobbold M, Khan N, Pourgheysari B, Tauro S, McDonald D, Osman H.et al. (2005Adoptive transfer of cytomegalovirus-specific CTL to stem cell transplant patients after selection by HLA-peptide tetramers J Exp Med 202379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuchtinger T, Matthes-Martin S, Richard C, Lion T, Fuhrer M, Hamprecht K.et al. (2006Safe adoptive transfer of virus-specific T-cell immunity for the treatment of systemic adenovirus infection after allogeneic stem cell transplantation Br J Haematol 13464–76. [DOI] [PubMed] [Google Scholar]
- Feuchtinger T, Opherk K, Bethge WA, Topp MS, Schuster FR, Weissinger EM.et al. (2010Adoptive transfer of pp65-specific T cells for the treatment of chemorefractory cytomegalovirus disease or reactivation after haploidentical and matched unrelated stem cell transplantation Blood 1164360–4367. [DOI] [PubMed] [Google Scholar]
- Khanna N, Stuehler C, Conrad B, Lurati S, Krappmann S, Einsele H.et al. (2011Generation of a multipathogen-specific T-cell product for adoptive immunotherapy based on activation-dependent expression of CD154 Blood 1181121–1131. [DOI] [PubMed] [Google Scholar]
- Leen AM, Sili U, Savoldo B, Jewell AM, Piedra PA, Brenner MK.et al. (2004Fiber-modified adenoviruses generate subgroup cross-reactive, adenovirus-specific cytotoxic T lymphocytes for therapeutic applications Blood 1031011–1019. [DOI] [PubMed] [Google Scholar]
- Leen AM, Christin A, Khalil M, Weiss H, Gee AP, Brenner MK.et al. (2008Identification of hexon-specific CD4 and CD8 T-cell epitopes for vaccine and immunotherapy J Virol 82546–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hislop AD, Taylor GS, Sauce D., and, Rickinson AB. Cellular responses to viral infection in humans: lessons from Epstein-Barr virus. Annu Rev Immunol. 2007;25:587–617. doi: 10.1146/annurev.immunol.25.022106.141553. [DOI] [PubMed] [Google Scholar]
- Badr G, Bédard N, Abdel-Hakeem MS, Trautmann L, Willems B, Villeneuve JP.et al. (2008Early interferon therapy for hepatitis C virus infection rescues polyfunctional, long-lived CD8+ memory T cells J Virol 8210017–10031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannanganat S, Ibegbu C, Chennareddi L, Robinson HL., and, Amara RR. Multiple-cytokine-producing antiviral CD4 T cells are functionally superior to single-cytokine-producing cells. J Virol. 2007;81:8468–8476. doi: 10.1128/JVI.00228-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leen A, Meij P, Redchenko I, Middeldorp J, Bloemena E, Rickinson A.et al. (2001Differential immunogenicity of Epstein-Barr virus latent-cycle proteins for human CD4(+) T-helper 1 responses J Virol 758649–8659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heslop HE, Ng CY, Li C, Smith CA, Loftin SK, Krance RA.et al. (1996Long-term restoration of immunity against Epstein-Barr virus infection by adoptive transfer of gene-modified virus-specific T lymphocytes Nat Med 2551–555. [DOI] [PubMed] [Google Scholar]
- Gotch F, McMichael A, Smith G., and, Moss B. Identification of viral molecules recognized by influenza-specific human cytotoxic T lymphocytes. J Exp Med. 1987;165:408–416. doi: 10.1084/jem.165.2.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMichael AJ, Gotch F, Cullen P, Askonas B., and, Webster RG. The human cytotoxic T cell response to influenza A vaccination. Clin Exp Immunol. 1981;43:276–284. [PMC free article] [PubMed] [Google Scholar]
- Bunde T, Kirchner A, Hoffmeister B, Habedank D, Hetzer R, Cherepnev G.et al. (2005Protection from cytomegalovirus after transplantation is correlated with immediate early 1-specific CD8 T cells J Exp Med 2011031–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickham K, Münz C, Tsang ML, Larsson M, Fonteneau JF, Bhardwaj N.et al. (2001EBNA1-specific CD4+ T cells in healthy carriers of Epstein-Barr virus are primarily Th1 in function J Clin Invest 107121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münz C, Bickham KL, Subklewe M, Tsang ML, Chahroudi A, Kurilla MG.et al. (2000Human CD4(+) T lymphocytes consistently respond to the latent Epstein-Barr virus nuclear antigen EBNA1 J Exp Med 1911649–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balduzzi A, Lucchini G, Hirsch HH, Basso S, Cioni M, Rovelli A.et al. (2011Polyomavirus JC-targeted T-cell therapy for progressive multiple leukoencephalopathy in a hematopoietic cell transplantation recipient Bone Marrow Transplant 46987–992. [DOI] [PubMed] [Google Scholar]
- Rock MT, McKinney BA, Yoder SM, Prudom CE, Wright DW., and, Crowe JE., Jr Identification of potential human respiratory syncytial virus and metapneumovirus T cell epitopes using computational prediction and MHC binding assays. J Immunol Methods. 2011;374:13–17. doi: 10.1016/j.jim.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao HY, Lin YW, Yu SL, Lin HY, Chitra E, Chang YC.et al. (2011Immunoprotectivity of HLA-A2 CTL peptides derived from respiratory syncytial virus fusion protein in HLA-A2 transgenic mouse PLoS ONE 6e25500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuchtinger T, Richard C, Joachim S, Scheible MH, Schumm M, Hamprecht K.et al. (2008Clinical grade generation of hexon-specific T cells for adoptive T-cell transfer as a treatment of adenovirus infection after allogeneic stem cell transplantation J Immunother 31199–206. [DOI] [PubMed] [Google Scholar]
- Gerdemann U, Christin AS, Vera JF, Ramos CA, Fujita Y, Liu H.et al. (2009Nucleofection of DCs to generate Multivirus-specific T cells for prevention or treatment of viral infections in the immunocompromised host Mol Ther 171616–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA.et al. (2010Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients Blood 115925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenter GG, Welters MJ, Valentijn AR, Lowik MJ, Berends-van der Meer DM, Vloon AP.et al. (2009Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia N Engl J Med 3611838–1847. [DOI] [PubMed] [Google Scholar]
- Maecker HT, Dunn HS, Suni MA, Khatamzas E, Pitcher CJ, Bunde T.et al. (2001Use of overlapping peptide mixtures as antigens for cytokine flow cytometry J Immunol Methods 25527–40. [DOI] [PubMed] [Google Scholar]
- Kozlowski S, Corr M, Shirai M, Boyd LF, Pendleton CD, Berzofsky JA.et al. (1993Multiple pathways are involved in the extracellular processing of MHC class I-restricted peptides J Immunol 1514033–4044. [PubMed] [Google Scholar]
- Toes RE, van der Voort EI, Schoenberger SP, Drijfhout JW, van Bloois L, Storm G.et al. (1998Enhancement of tumor outgrowth through CTL tolerization after peptide vaccination is avoided by peptide presentation on dendritic cells J Immunol 1604449–4456. [PubMed] [Google Scholar]
- Sherman LA, Burke TA., and, Biggs JA. Extracellular processing of peptide antigens that bind class I major histocompatibility molecules. J Exp Med. 1992;175:1221–1226. doi: 10.1084/jem.175.5.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen SL, Pedersen LO, Buus S., and, Stryhn A. T cell responses affected by aminopeptidase N (CD13)-mediated trimming of major histocompatibility complex class II-bound peptides. J Exp Med. 1996;184:183–189. doi: 10.1084/jem.184.1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, Yannelli JR, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS.et al. (1994Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2 J Natl Cancer Inst 861159–1166. [DOI] [PubMed] [Google Scholar]
- Becker TC, Wherry EJ, Boone D, Murali-Krishna K, Antia R, Ma A.et al. (2002Interleukin 15 is required for proliferative renewal of virus-specific memory CD8 T cells J Exp Med 1951541–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vella AT, Dow S, Potter TA, Kappler J., and, Marrack P. Cytokine-induced survival of activated T cells in vitro and in vivo. Proc Natl Acad Sci USA. 1998;95:3810–3815. doi: 10.1073/pnas.95.7.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JT, Ernst B, Kieper WC, LeRoy E, Sprent J., and, Surh CD. Interleukin (IL)-15 and IL-7 jointly regulate homeostatic proliferation of memory phenotype CD8+ cells but are not required for memory phenotype CD4+ cells. J Exp Med. 2002;195:1523–1532. doi: 10.1084/jem.20020066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchionda F, Fry TJ, Milliron MJ, McKirdy MA, Tagaya Y., and, Mackall CL. Adjuvant IL-7 or IL-15 overcomes immunodominance and improves survival of the CD8+ memory cell pool. J Clin Invest. 2005;115:1177–1187. doi: 10.1172/JCI23134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetoui N, Boisvert M, Gendron S., and, Aoudjit F. Interleukin-7 promotes the survival of human CD4+ effector/memory T cells by up-regulating Bcl-2 proteins and activating the JAK/STAT signalling pathway. Immunology. 2010;130:418–426. doi: 10.1111/j.1365-2567.2009.03244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sportès C, Babb RR, Krumlauf MC, Hakim FT, Steinberg SM, Chow CK.et al. (2010Phase I study of recombinant human interleukin-7 administration in subjects with refractory malignancy Clin Cancer Res 16727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majhail NS, Hussein M, Olencki TE, Budd GT, Wood L, Elson P.et al. (2004Phase I trial of continuous infusion recombinant human interleukin-4 in patients with cancer Invest New Drugs 22421–426. [DOI] [PubMed] [Google Scholar]
- von Rossum A, Krall R, Escalante NK., and, Choy JC. Inflammatory cytokines determine the susceptibility of human CD8 T cells to Fas-mediated activation-induced cell death through modulation of FasL and c-FLIP(S) expression. J Biol Chem. 2011;286:21137–21144. doi: 10.1074/jbc.M110.197657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maus MV, Thomas AK, Leonard DG, Allman D, Addya K, Schlienger K.et al. (2002Ex vivo expansion of polyclonal and antigen-specific cytotoxic T lymphocytes by artificial APCs expressing ligands for the T-cell receptor, CD28 and 4-1BB Nat Biotechnol 20143–148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phenotype and specificity of Penton and LMP2-specific CTLs generated in the presence of different growth-promoting cytokines.
CD3+ T cell expansion after addition of growth-promoting cytokines.
TCR avidity is comparable in Hexon DC plasmid-activated and pepmix-stimulated PBMCs.
Comparable expansion of CTLs stimulated with pooled versus single pepmixes.
Lack of alloreactivity in pepmix-stimulated PBMCs.