

Abstract
Most known proteins have at least one local Hsp70 chaperone binding site. Does this mean that all proteins interact with Hsp70 as they fold? This study makes an initial step to address the above question by examining the interaction of the E.coli Hsp70 chaperone (known as DnaK) and its co-chaperones DnaJ and GrpE with a slow-folding E.coli substrate, RNase HD. Importantly, this protein is a nonobligatory client, and it is able to fold in vitro even in the absence of chaperones. We employ stopped-flow mixing, chromatography, and activity assays to analyze the kinetic perturbations induced by DnaK/DnaJ/GrpE (K/J/E) on the folding of RNase HD. We find that K/J/E slows down RNase HD's apparent folding, consistent with the presence of transient chaperone-substrate interactions. However, kinetic retardation is moderate for this slow-folding client and it is expected to be even smaller for faster-folding substrates. Given that the interaction of folding-competent substrates such as RNase HD with the K/J/E chaperones is relatively short-lived, it does not significantly interfere with the timely production of folded biologically active substrate. The above mode of action is important because it preserves K/J/E bioavailability, enabling this chaperone system to act primarily by assisting the folding of other misfolded and (or) aggregation-prone cellular proteins that are unable to fold independently. When refolding is carried out in the presence of K/J and absence of the nucleotide exchange factor GrpE, some of the substrate population becomes trapped as a chaperone-bound partially unfolded state.
Keywords: protein folding, molecular chaperones, DnaK, RNase H, protein-chaperone complex
Introduction
The ubiquitous Hsp70 chaperone family is involved in preventing protein aggregation and assisting co- and post-translational protein folding.1–3 The ATP-dependent interaction of Hsp70 with its client proteins is critical for the function of the Hsp70 machinery. DnaK, the cytosolic E.coli Hsp70, is the best-studied member of the Hsp70 family.4 DnaK interacts with its substrates via the sequence-specific recognition of the DnaK-binding motif, which consists of a core of approximately five nonpolar amino acids flanked by basic residues.5 This motif is very common in proteins and is predicted to occur with a frequency of about 1 in 40 residues,5 implying that most proteins have at least one local DnaK binding site.
DnaK is an ATP-dependent chaperone and functions in concert with the two co-chaperones DnaJ and GrpE.6 The kinetics and thermodynamics of the interaction of DnaK with its co-chaperones and with peptide substrates has been extensively characterized.4,7–11 Substrates enter the Hsp70 chaperone cycle by binding either DnaJ12 or the ATP-bound state of DnaK.13 ATP hydrolysis, stimulated by binding of both DnaJ and the substrate,14 locks the substrate into a high-affinity ADP-DnaK-bound state. GrpE, the nucleotide exchange factor, then releases the bound ADP.15 The latter process facilitates the binding of ATP, resulting in substrate release. The chaperone cycle is thus reset, and ready for additional rounds of substrate binding and release.
Fewer investigations have been carried out with larger protein substrates,10,16 which are believed to bind the chaperone in a globally unfolded conformation.17–19 The DnaK/DnaJ/GrpE (K/J/E) chaperone system is known to significantly accelerate the in vitro refolding of large proteins incapable of independent folding, for example, firefly luciferase,12,20 by actively unfolding the misfolded substrate.12,20 Luciferase requires chaperones for efficient in vitro refolding and tends to aggregate, except at extremely low concentrations.21,22 However, the generality of the active refolding assistance by K/J/E has not been addressed in the literature. In particular, it is not understood whether K/J/E catalyzes the refolding of all slow-folding proteins, including those that are able to fold even in the absence of chaperones. In summary, the extent of interaction between folding-competent substrates and the K/J/E chaperone network remains poorly characterized.
In this work, we employ a two-state slow-folding variant of E.coli RNase H (RNase HD)23 as a model client protein for the K/J/E chaperone system. We investigate RNase HD folding kinetics in the presence of various combinations of DnaK, DnaJ, and GrpE. Given that RNase HD has a strong local binding site for DnaK (Fig. 1) and is known to fold slowly on the typical timescale of chaperone binding, one would expect that RNase HD may significantly interact with K/J/E during folding. Such interaction could lead to either acceleration or retardation of the client protein folding.
Figure 1.
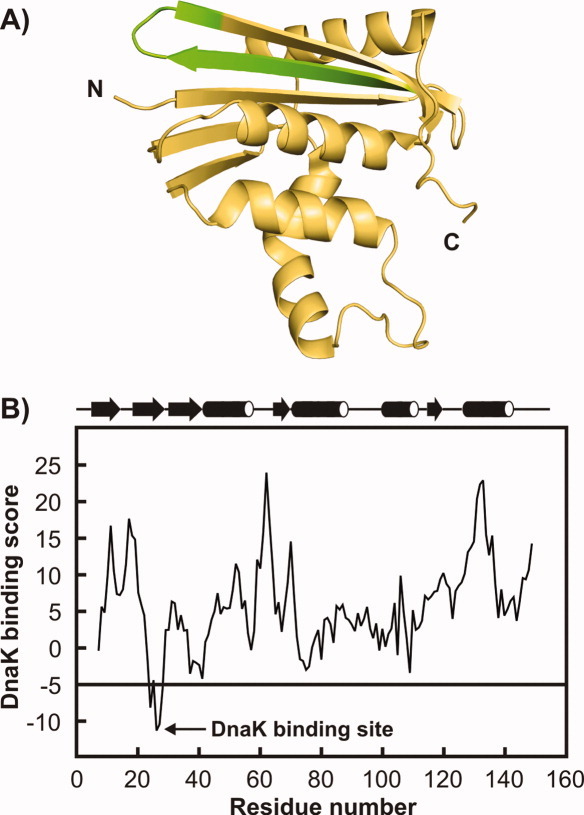
A: Three-dimensional structure of E.coli RNase H (PDB code: 1F21). The N and C termini are labeled. The unique DnaK binding site is shown in green. B: Predicted local DnaK binding scores5 of RNase HD.23 Regions with a score lower than −5 are potential strong DnaK binding sites. The secondary structure of wild type E.coli RNase H derived from the protein crystal structure is mapped above the graph.
We find a small but significant decrease in RNase HD refolding rate, in the presence of K/J/E. This result shows that catalytic acceleration of protein folding by K/J/E, observed for some large multidomain proteins, is not a universal feature of this chaperone system. Our data also support the presence of transient interactions of K/J/E with the folding-competent substrate RNase HD. We propose that the physiological role of such contacts may be to decrease any likelihood of misfolding/aggregation while, at the same time, avoiding significant delays in the production of bioactive substrate.
Results
RNase HD is a good slow-folding model substrate to monitor interactions with the K/J/E chaperone system
To explore the influence of the E.coli Hsp70 chaperone system in the folding pathway of a protein substrate, we chose RNase H* I53D23 as a model client protein. E.coli Ribonuclease HI (RNase H) is a 155 amino acid endoribonuclease that catalyzes the hydrolysis of RNA in RNA-DNA hybrids. The wild type enzyme lacks disulphide bonds in the native state. The mutant construct RNase H*, which has all three Cys replaced by Ala is folded and enzymatically active.24 The RNase H variant that we employ in our study, RNase H* I53D, here denoted as RNase HD, is the triple Cys-to-Ala mutant containing an additional point mutation at residue 53, with Ile53 replaced by Asp.23 In the native state, I53 is buried in the hydrophobic core at the interface between helices A and D. The Ile53 to Asp mutation results in a destabilization of the RNase H kinetic intermediate and switches the folding mechanism from three-state to two-state, while retaining the RNase H enzymatic activity.23 RNase HD is thus a slow two-state-folding protein with a folding rate constant of 0.03 s−1. It is among the slowest known two-state folders in vitro25 at 25°C.
A widely used computational algorithm that scans protein primary structure for potential local DnaK interaction sites5 predicts a strong DnaK binding site near the N-terminus of RNase HD, as shown in Figure 1. Since it is known that RNase HD folds slowly, it is possible that this hydrophobic DnaK binding motif remains solvent-exposed for a sufficiently long time to interact with the Hsp70 chaperone system. The above considerations make E.coli RNase HD an attractive model protein for studying the time-course of the interaction of folding-competent substrates with the E.coli K/J/E chaperone machinery.
The apparent folding rate of RNase HD decreases in the presence of K/J/E
To probe the kinetics of RNase HD refolding in the absence and presence of K/J/E, we employed stopped-flow fast mixing in conjunction with far-UV circular dichroism (CD) detection. The CD signal at 222 nm is a measure of the α-helical secondary structure in a protein. RNase HD was unfolded in 5M urea pH 6 and allowed to refold in the absence or presence of various combinations of DnaK, DnaJ, and GrpE. Chaperone concentrations were chosen to reflect the physiologically relevant ratio of K:J:E = 5:1:2 in E.coli cells.16 The concentration ratio of substrate:DnaJ was ∼1.6:1, which is nearly optimal for the in vitro K/J/E-mediated refolding of luciferase.14,26
Stopped-flow refolding traces of RNase HD are shown in Figure 2. All kinetic traces were fit to a single exponential (see Materials and Methods) to extract rate constants, and burst-phase and final CD amplitudes. Comparative histograms showing apparent refolding rate constants of RNase HD in buffer containing or lacking DnaJ, DnaK/ATP, DnaK/DnaJ/ATP, and DnaK/DnaJ/GrpE/ATP are reported in Figure 3(A).
Figure 2.
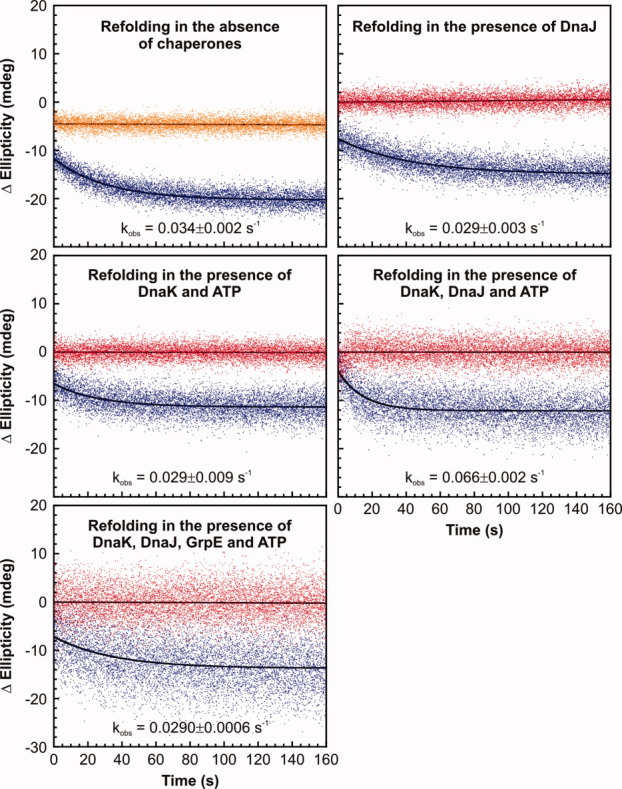
Experimental far-UV CD-detected stopped-flow kinetic traces for the refolding of RNase HD (blue) in the absence and presence of various combinations of DnaK, DnaJ, and GrpE at 30°C at 222 nm. Time-dependent variations in ellipticity of refolding buffer and urea-unfolded RNase HD are shown in red and orange, respectively. The ellipticity of the refolding buffer subtracted from the ellipticity of the refolding trace, denoted as Δellipticity, is plotted on the y-axis. Hence, the ellipticity of the refolding solution, containing contributions from chaperones, has been subtracted out. The observed time-dependent change in ellipticity thus arises from the substrate protein alone, assuming that the secondary structure of the chaperones is not significantly perturbed. Final concentrations of RNase HD, DnaK, DnaJ, and GrpE and ATP in the refolding mixture were 5, 15, 3, and 6 μM and 1 mM, respectively. Least squares fits of the kinetic traces to linear or single-exponential functions are in black. Average rate constants derived from curve fitting are listed close to each corresponding refolding trace. Error bars represent 1 standard error calculated from 2 to 8 independent repeats, with each repeat comprising 4–6 replicate measurements.
Figure 3.
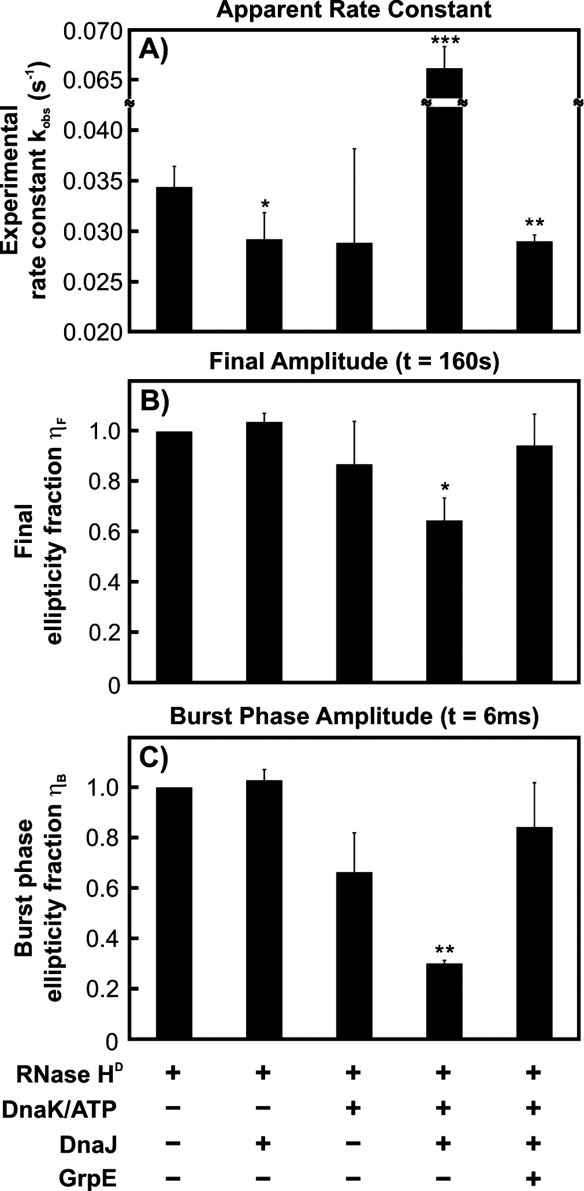
Stopped-flow refolding data analysis. Bar diagram reporting the (A) rate constants, (B) final (ηF) ellipticity fractions [see Eqs. (1) and (2)], and (C) burst phase amplitudes (ηB), and obtained from stopped-flow data analysis. Chaperone combinations pertinent to each experimental condition are listed below the corresponding bar. Error bars represent 1 standard error calculated from 2 to 8 independent measurements, with each measurement consisting of 4–6 independent runs. Statistically significant differences (relative to the RNase HD-only experiment) are shown in the graph by asterisks (*P ≤ 0.1, **P ≤ 0.05, ***P ≤ 0.001).
An initial important result illustrated in Figure 3(A) is that the refolding of RNase HD is not accelerated by the presence of the entire K/J/E chaperone system. Instead, there is a small but statistically significant (P < 0.05) apparent deceleration in RNase HD refolding. It is thus clear that the catalytic refolding activity of the Hsp70 chaperone system, observed in the case of the folding-incompetent client luciferase,12 is substrate dependent. Hence, catalytic acceleration of substrate folding is not a general feature of Hsp70. In the case of RNase HD, which is a slow two-state folder capable of achieving its native state even in the absence of chaperones, the folding process is effectively slowed down when K/J/E are present.
In contrast, the apparent RNase HD refolding rate constant in the presence of DnaK, DnaJ, and ATP, but no GrpE, is about two-fold larger than in the absence of the whole chaperone system. In the absence of GrpE, client proteins are known to become kinetically trapped in the DnaK substrate-binding pocket in the ADP-bound state of DnaK.27 As supported by additional evidence from gel filtration, HPLC, SDS-PAGE and RNase H activity assays (see below), the large observed rate constant in the presence of DnaK/DnaJ/ATP reflects the generation of a kinetically stable RNase HD-chaperone complex by some of the substrate molecules. Formation of this complex is dramatically enhanced by the presence of DnaJ, which stimulates the basal ATP hydrolysis of DnaK in conjunction with the substrate.14 Persistent substrate-DnaK complex formation accounts for the fact that a faster apparent rate is only detected when the refolding buffer contains both DnaK and DnaJ, but not when only DnaK or DnaJ is present.
From the average observed rate constants in Figure 3(A), one may infer that DnaJ or ATP-DnaK independently retard the folding of RNase HD to a similar extent as K/J/E. However, the observed refolding rate constants in the presence of either DnaJ or ATP-DnaK are not sufficiently statistically different from the chaperone-free case, due to a P value >0.05. Interestingly, numerical simulations on the folding of RNase HD in the presence of DnaJ, ATP-DnaK, or the entire K/J/E system (Sekhar A, Lam HN, and Cavagnero S, submitted) predict a small kinetic retardation due to transient client binding and release by DnaJ, ATP-DnaK, or K/J/E.
Stopped-flow CD is consistent with the generation of a partially unfolded K/J-bound substrate, in the absence of GrpE
To evaluate the extent of folding occurring in the presence of chaperones and the formation of any chaperone-substrate complexes, we computed the final ellipticity fraction (ηF) of RNase HD by far-UV CD. The ηF parameter is a measure of the change in final ellipticity due to the client protein, in the presence of chaperones. Since the CD signal at 222 nm is related to the degree of α-helical secondary structure, ηF is a probe of the final (F) fraction of native RNase HD formed 160 s after refolding initiation in the presence of chaperones. If RNase HD has folded completely to its native state, ηF is expected to be 1. The ηF parameter is defined as
| 1 |
Here, [θ]R,F and [θ]RC,F correspond to the ellipticity at 222 nm of the RNase HD refolding (R) traces in the absence and presence of chaperones (C), respectively, at the end of the stopped-flow measurements (t = 160 s). [θ]C,F is the ellipticity arising from the chaperone-containing solution in the absence of RNase HD after 160 s from refolding initiation. All [θ]F values were obtained from least-squares fitting of the corresponding kinetic traces either to a linear ([θ]C,F) or a single exponential relation ([θ]R,F and [θ]RC,F), as detailed in the Materials and Methods.
As shown in Figure 3(B), in the presence of ATP-DnaK and DnaJ and in the absence of GrpE, ηF is ∼0.65. This means that the CD signal intensity at the end of the kinetic run is only about 65% of the expected value for fully folded substrate.
This reduction in ellipticity may arise from a smaller α-helical content of RNase HD or, alternatively, it may also be due to conformational changes within the chaperones (DnaK and DnaJ) upon interaction with the substrate. Published studies of DnaK and DnaJ in the absence and presence of various substrates revealed no significant perturbations in the secondary or tertiary structure of these chaperones upon substrate binding.28–30 Hence, it is more likely that the observed decrease in ellipticity can be ascribed entirely to RNase HD.
Gel filtration, HPLC, SDS-PAGE, and substrate activity assays collectively support formation of about 35% complex between RNase HD and K/J after a few minutes from mixing unfolded protein and chaperone-containing solution (see sections below). Given that the stopped-flow experiment was run at concentrations of substrate and chaperones slightly different from those used in the chromatographic and activity assays, we conclude that some chaperone-substrate complex is also present at the end of the stopped-flow experiment. However, we cannot precisely quantify its population due to the difference in experimental conditions. Hence, the stopped-flow ηF value of ∼0.65 [Fig. 3(B)] shows that the smaller α-helical content of RNase HD detected at the end of the kinetic run must be because the chaperone-bound substrate population is partially or completely unfolded. This intriguing result is in agreement with previous experimental observations with other substrates, indicating that DnaK binds polypeptide and protein clients in a globally unfolded conformation.18,19,28
Figure 3(B) also shows that ηF in the presence of only DnaJ or ATP-DnaK, and in the presence of the complete K/J/E chaperone system, is statistically indistinguishable from 1 (P > 0.1). This result confirms that the RNase HD folds to completion under these conditions. The mere presence of ATP-DnaK or DnaJ in the folding environment of RNase HD is not sufficient to create a stable chaperone-substrate complex, in agreement with the notion that the synergistic stimulation of ATP hydrolysis by the substrate and DnaJ is necessary for trapping substrates as complexes with ADP-DnaK.14 In contrast, GrpE in the folding buffer reverses the kinetic trapping seen with K/J by facilitating the exchange of bound nucleotide, allowing ATP to bind DnaK and dissociate the chaperone-substrate complex.
Burst phase CD amplitudes support the existence of transient interactions between RNaseHD and chaperones
We computed burst phase CD signal amplitudes to gauge the extent of substrate secondary structure at 6 ms from refolding initiation (i.e., within the dead time of our stopped-flow measurement). The burst phase (denoted by the subscript B) amplitude of the far-UV CD signal was quantified in a similar fashion to the final amplitude, via a parameter denoted as burst ellipticity fraction (ηB), defined as:
| 2 |
According to the above formula, ηB is an indication of the fraction of the substrate's native helicity after 6 ms from stopped-flow experiment initiation.
As illustrated in Figure 3(C), ηB is statistically different from 1 (P < 0.1) only in the case of RNase HD folding in the presence of ATP-DnaK and DnaJ. This result stems from the fact that the burst phase amplitude is dramatically smaller, than for the refolding of RNase HD alone. The small value of ηB (equal to ca. 0.3) therefore shows that the addition of K/J to the refolding buffer causes a global decrease in substrate structure at 6 ms, relative to the chaperone-free process. The simplest explanation for the origin of this effect is the presence of some interactions between the substrate and the K/J chaperone system. The latter conclusion is in qualitative agreement with evidence from gel filtration, HPLC and activity assays (see below), which support the presence of a persistent substrate-chaperone complex after a few minutes from refolding initiation.
The ηB values in the presence of ATP-DnaK (P = 0.14) and DnaJ alone (P = 0.28), as well as in the presence of the entire K/J/E chaperone system (P = 0.22), are statistically indistinguishable from 1. This result likely reflects our inability to assess transient chaperone-substrate interactions above noise level. In the case of folding in the presence of DnaJ, however, the observed ηB value of 1 may also be a result of a DnaJ-RNase HD complex possessing the same helical content as the native unfolded state of RNase HD. This explanation is in agreement with the fact that DnaJ binds protein surfaces,31 thereby allowing the development of helical secondary structure within the bound polypeptide.
Analytical size-exclusion, reverse-phase HPLC and SDS-PAGE confirm the existence of a kinetically trapped RNase HD-chaperone complex in the presence of K/J
Stopped-flow kinetic rate constants and final ellipticity fractions observed in the absence of GrpE suggest that RNase HD forms a kinetically trapped complex with chaperones. To obtain direct evidence for RNase HD-chaperone complex formation, we carried out analytical size exclusion HPLC experiments, which discriminate between molecules based on their hydrodynamic radii. Hence, this method is a sensitive reporter of complex formation under refolding conditions. Unfolded RNase HD was allowed to refold in buffer containing DnaK, DnaJ, and ATP for 5 min, and the mixture was then injected into the size exclusion column. Samples were eluted from the column immediately using refolding buffer lacking chaperones and ATP. A five-fold higher concentration of chaperones and RNase HD was employed in these experiments while maintaining the same substrate:chaperone ratio, to obtain good signal-to-noise.
Figure 4 shows gel filtration chromatograms of RNase HD refolded in the absence (panel A) and presence (panel C) of ATP-DnaK and DnaJ. There is a significant reduction in the intensity of the native RNase HD peak in the presence of DnaK and DnaJ, indicating that a fraction of RNase HD molecules form complexes with chaperones.
Figure 4.
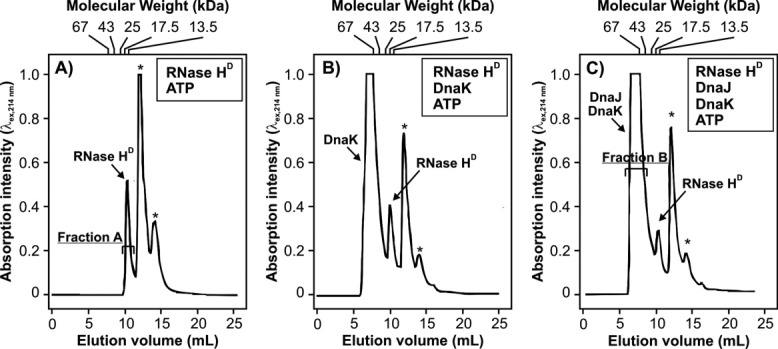
Analytical gel filtration chromatograms, followed by electronic absorption at 214 nm, of RNase HD (A) in the absence of chaperones, (B) in the presence of DnaK/ATP, and (C) in the presence of DnaK, DnaJ, and ATP. Urea-unfolded RNase HD was refolded in 20 mM sodium acetate pH 6.0 containing 100 mM KCl, 5 mM MgCl2, and 1 mM ATP in the absence (A) and presence (B and C) of chaperones before injecting into the size-exclusion column. Peaks denoted with asterisks arise from buffer components (i.e., ATP and ADP). The elution volume corresponding to each molecular-weight standard used to calibrate the column is indicated at the top of each panel (see Materials and Methods for names of standard proteins). In all chromatograms, absorption intensities are reported on the same absolute scale.
To confirm the presence of a complex of RNase HD and K/J on the size exclusion column, we collected elution fractions A and B (Fig. 4) and analyzed the contents using analytical reverse phase HPLC. Panels A and B of Figure 5 show the reverse phase HPLC chromatograms of fractions A and B, respectively. Pure RNase HD elutes at 8 minutes [Fig. 5(A)]. The gel filtration DnaK peak (fraction B) for the Figure 4(C) sample, containing RNase HD refolded in ATP-DnaK and DnaJ, shows a significant amount of RNase HD in the HPLC chromatogram [Fig. 5(B)]. This observation confirms that RNase HD and DnaK coelute on the gel filtration column, providing firm evidence for the physical interaction of RNase HD and DnaK under these conditions, in the absence of GrpE. Similar results are also obtained by SDS-PAGE, as shown in Figure 6.
Figure 5.
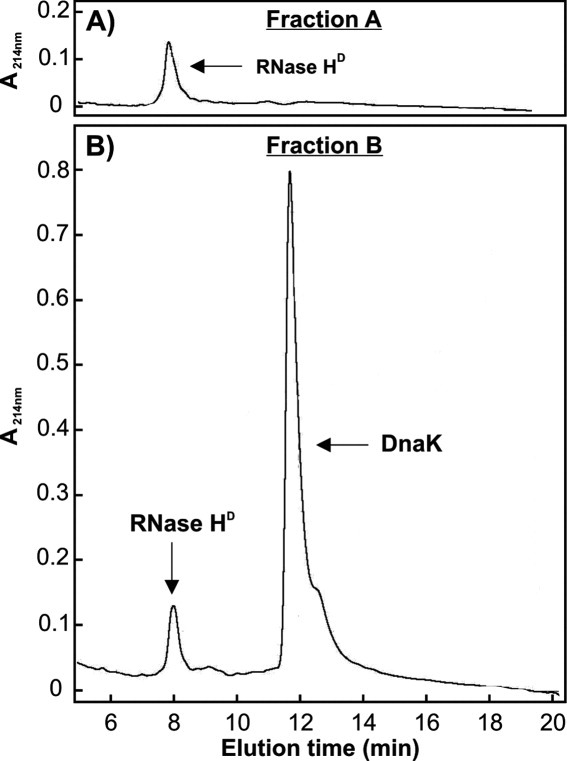
Analytical reverse-phase HPLC chromatograms of gel filtration fractions A and B [see Fig. 4(A,C)].
Figure 6.
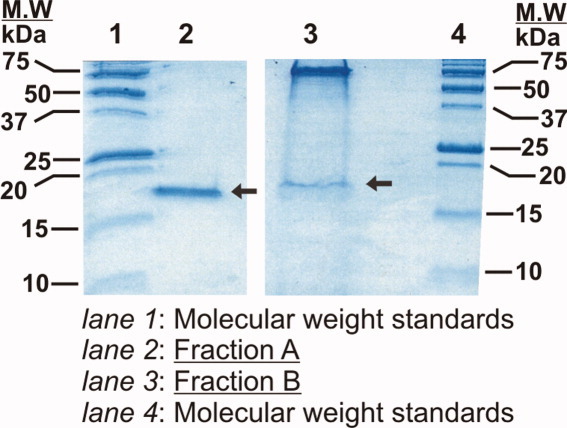
SDS-PAGE gel analysis of gel filtration fractions A and B [see Fig. 4(A,C)]. Prior to running each of the gel filtration fractions through the gel (16.5% Tris-tricine), precipitation with trichloroacetic acid (TCA) was carried out to effectively concentrate the samples for SDS-PAGE analysis. The arrows indicate bands corresponding to RNase HD.
To provide further evidence on chaperone-substrate association in the presence of various chaperone combinations, Figure 7 illustrates the reduction in native unbound RNase HD gel filtration peak intensities. In the presence of only ATP/DnaK or DnaJ, small amounts of complexes are formed. As expected, the extent of complex formation increases significantly in the presence of both ATP-DnaK and DnaJ.
Figure 7.
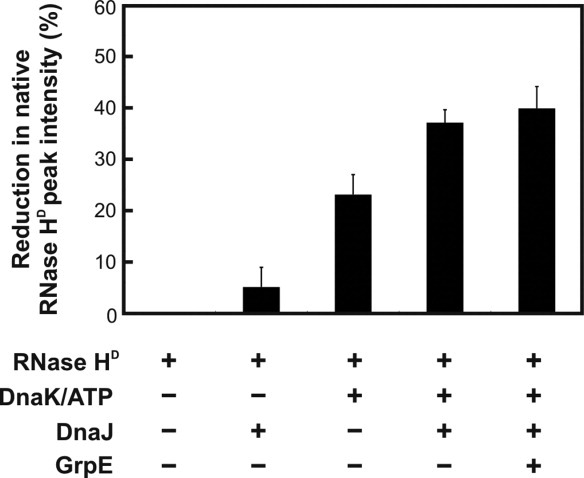
Extent of complex formation between RNase HD and the DnaK chaperone measured by gel filtration as the reduction in native RNase HD peak intensity. Error bars correspond to ±1 standard error, derived from 2 to 7 independent measurements.
While DnaJ is essential for significant complex formation in the presence of DnaK/ATP, as evidenced by stopped-flow and gel filtration, one may wonder why the DnaJ chaperone is not explicitly observed in the HPLC and SDS-PAGE analysis of fraction B. At least two reasons contribute to this observation. First, the concentration of DnaJ injected on the column is five-fold smaller than that of DnaK. Second, free DnaJ is known to exist in solution as a heterogeneous oligomeric mixture32; hence, it elutes from the size-exclusion column as a broad peak. Now, HPLC and SDS-PAGE (Figs. 5 and 6) show that DnaJ in fraction B falls below the detection threshold of these techniques, suggesting that DnaJ, whose presence enhances complex formation with DnaK, does not actually participate in a stable ternary complex between DnaK and RNase HD. The simplest interpretation of this result is that DnaJ dissociates from the ternary complex once RNase HD has achieved stable binding to DnaK. This observation is entirely consistent with the known catalytic role of DnaJ in the Hsp70 chaperone cycle.33
Gel filtration reveals the presence of DnaK-RNase HD interactions even in the presence of GrpE (Fig. 7). This is likely an artifact resulting from ATP depletion in the refolding buffer prior to complete release of all chaperone-bound RNase HD, which occurs rapidly because of the high chaperone concentrations in the gel filtration sample. This idea is supported by computer simulations (see below). In addition, the spatio-temporal separation of DnaK-RNase HD complexes from both GrpE and ATP on the gel filtration column may render GrpE unable to release the bound substrate during the course of the gel filtration run, contributing to the observed result.
RNase activity assays further support the presence of a substrate complex with K/J, upon refolding RNase HD in the absence of GrpE
To assess the effect of molecular chaperones on the production of free, bioactive RNase HD directly, we performed RNase HD activity assays23 immediately after refolding in the absence or presence of different molecular chaperones [Fig. 8(A)]. This assay is based on the spectrophotometric monitoring of the cleavage of RNA-DNA hybrids by native RNase HD. Activity assays were carried out in the presence of excess RNA-DNA hybrid to ensure that the initial reaction rate is directly proportional to the amount of native RNase HD. Figure 8(A) shows the enzymatic activity of RNase HD refolded in the absence/presence of chaperones, expressed as a percent of the activity observed when chaperones are absent. Hence, the y-axis of Figure 8(A) is a direct measure of the fraction of bioactive RNase HD under different chaperone environments.
Figure 8.
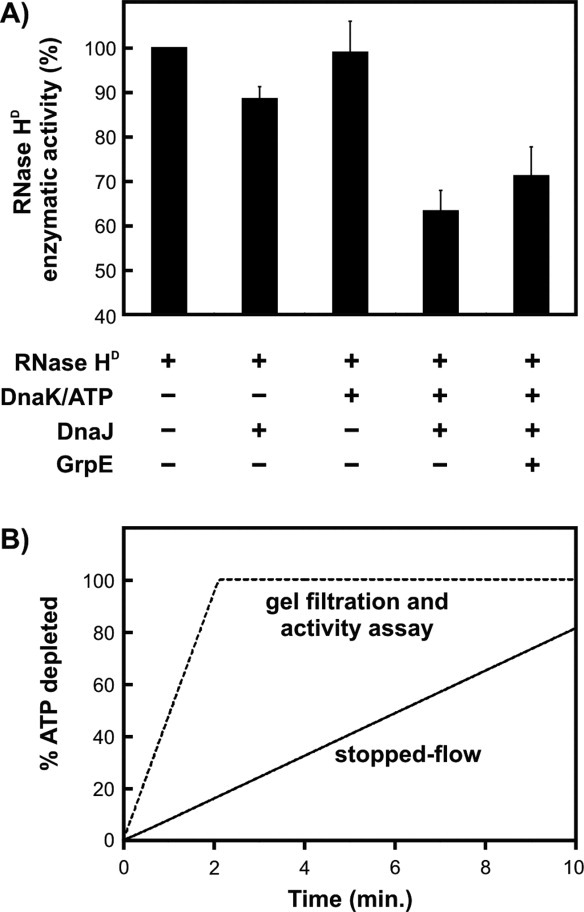
A: Enzymatic activity of RNase HD measured after 10 min from refolding initiation, in the presence of different combinations of chaperones. Data are shown as percent of the activity of a control RNase HD sample refolded in the absence of chaperones. Error bars represent ± 1 standard error of the mean, calculated from three independent measurements. B: Computer simulations predicting the expected ATP depletion as a function of time under the stopped-flow and gel filtration/activity assay experimental conditions. The refolding time in the stopped-flow experiments is 160 s, while the refolding time in the gel filtration and activity assays is 10 min. The spatial separation of the solution components during gel filtration experiments may introduce additional effects (not taken into account in this simulation).
In the presence of only DnaJ or DnaK/ATP, RNase HD essentially folds to completion as observed from the >90% enzymatic activity. Hence there is no chaperone-substrate complex at equilibrium, after refolding is complete. As discussed above, the observed small kinetic retardation under these conditions is consistent with the presence of only rapid and transient interactions between substrate and chaperones, upon substrate refolding under these conditions.
However, when both DnaK and DnaJ are present, only 65% of the RNase HD molecules fold to the bioactive native state. This result is consistent with the fact that the remaining 35% of RNase HD molecules are sequestered as a chaperone complex, consistent with the gel-filtration and HPLC analysis. Hence, several internally consistent experimental observations concur in showing that there is a substantial extent of persistent complex formation when RNase HD folds in the presence of DnaK, DnaJ, and ATP, in the absence of GrpE.
Addition of GrpE to the chaperone mixture releases a part of the DnaK-substrate complexes, resulting in an increase of enzymatic activity to about 70%. The fact that substrate release is incomplete under these conditions is likely due to the depletion of ATP during the 10 minute refolding time used in the assay. Once ATP is hydrolyzed to ADP, GrpE loses its ability to release DnaK-bound RNase HD, explaining the lower-than-expected enzymatic activity. This concept is supported by the computer simulation of ATP consumption shown in Figure 8(B), carried out with a recently developed computational model for protein folding in the presence of the K/J/E chaperone system (Sekhar A, Lam HN, and Cavagnero S, submitted). This simulation shows that virtually all the ATP is hydrolyzed within the first three minutes of preincubation, under the experimental conditions used in the gel filtration and activity assay experiments. In contrast, most of the ATP is available in the stopped-flow experiments during the 160 s refolding time, enabling complete substrate release and folding. The key difference between the stopped-flow and gel filtration/activity assay experimental conditions, responsible for the faster ATP hydrolysis in the case of gel filtration/activity assays, is that gel filtration and activity assays were run with approximately five-fold larger chaperone concentration than the stopped-flow experiments.
Discussion
The effect of the K/J/E chaperones on protein folding kinetics is highly substrate-dependent
RNase HD is a 2-state slow-folding protein capable of attaining its native state independently, that is, in the absence of molecular chaperones. This study compares the apparent folding rates of RNase HD in the absence and presence of the K/J/E molecular chaperones, to gain insights into the potential role of these chaperones in the cellular environment. We show that the K/J/E chaperone system interacts transiently with the E.coli single-domain protein RNase HD while it folds to the native state. This interaction slightly decreases the observed folding rate of this protein. Our observation is consistent with a prior report showing retardation in the folding of a large multidomain maltose binding protein (MBP) variant in the presence of K/J/E.34 However, while MBP is a large multidomain protein, RNase HD is a smaller client composed of only one domain. Our studies, which demonstrate transient interactions between K/J/E and a single-domain protein on its way to the native state, are important because single-domain proteins account for ∼70% of the E.coli proteome.35
Taken together, our experimental results on RNase HD and previously published data on MBP and luciferase establish that the kinetic effect of K/J/E on protein folding is highly substrate-dependent. Not all slow-folding client proteins are accelerated by K/J/E, even though they may extensively interact with the chaperone system before folding to the native state.
It remains unclear what specific substrate characteristics (primary/secondary structure, tertiary fold, folding pathways, etc.) prompt the K/J/E catalytic refolding activity. It is possible that K/J/E is particularly efficient at rescuing substrate proteins from kinetic traps in their folding energy landscape, as in the case of firefly luciferase22 by actively unfolding misfolded substrates and allowing them additional chances to undergo folding. Acceleration in protein folding may also depend on the extent of conformational search that a substrate can accomplish while bound to DnaK. In this context, proteins with several properly positioned hydrophobic residues may fold faster in the presence of K/J/E because they undergo a more rapid diffusion across the folding energy landscape while bound to the DnaK chaperone.
The DnaK chaperone interacts transiently with partially unfolded RNase HD, under substrate refolding conditions
Intriguingly, the burst-phase fraction ηB data indicate that interaction with chaperone is accompanied by smaller client secondary structure (at comparable time-points) than client folding in the absence of chaperones. This result suggests that the transient interaction with chaperone effectively retards structure formation and, in so doing, it maintains the substrate in a conformational state that may be particularly appropriate to support proper folding.
The existence of a predominantly unfolded DnaK-bound substrate conformation is entirely consistent with prior studies at equilibrium focusing on complexes between N-terminal protein fragments and the substrate-binding domain of DnaK.19,36 However, no prior investigations addressed the transiently chaperone-bound conformation of folding-competent protein clients on their way to the native state.
Transient substrate-chaperone interactions may serve important biological roles
Our stopped-flow, gel filtration and activity assay experiments show significant interactions between folding RNase HD and chaperones under conditions where the complex is kinetically trapped (i.e., K/J conditions). Our results also show that in the presence of the complete chaperone system (K/J/E experiment), that is, under the most physiologically relevant conditions for a healthy cell, folding is slowed down only moderately [Fig. 3(A)] and there is no significant substrate-chaperone complex at the end of the folding process. This result is consistent with the presence of only transient interactions between substrate and K/J/E, under physiological, non-heat-shock, conditions.
The most important biological consequence of a transient association with K/J/E is that the chaperone-substrate interaction does not slow down protein folding significantly.
Folding to the native conformation is usually a prerequisite for the biological function of a protein. From a cellular perspective, it is thus disadvantageous for K/J/E to significantly slow down protein folding, particularly for proteins that can fold efficiently even in the absence of interaction with chaperones. From our results, it appears that the kinetics of the K/J/E chaperone system is tuned to maximize chaperone-substrate interactions while maintaining a reasonable rate of native protein production.
The transient nature of chaperone-substrate interactions also facilitates the recycling of DnaK, DnaJ, and GrpE, likely preserving the bioavailability of these chaperones in the cellular context. On the other hand, transient chaperone-substrate interactions may be important for the ability of Hsp70 to prevent the aggregation that has been observed in previous studies on a different substrate.36
The transient association observed under physiological conditions is in interesting contrast with the biological scenario under heat-shock conditions. At 42°C, GrpE is inactivated by reversible unfolding,37 generating effective K/J-only conditions and leading to the kinetic trapping of the substrate within the DnaK binding pocket. Chaperone-substrate interactions are known to be more persistent under heat shock conditions, explaining the need to upregulate the DnaK concentration at 42°C, given the higher propensity for misfolding and aggregation at this temperature. This scenario is consistent with our experiments in the presence of K/J, though heat shock conditions with K/J/E were not explicitly tested here.
Mechanistic considerations on the interaction of the K/J/E chaperone system with slow-folding proteins such as RNase HD
Interaction with the K/J/E chaperone system slows down the apparent refolding of the RNase HD. Therefore, the catalytic acceleration of folding detected for some large multidomain proteins unable to fold independently is not a general feature of the K/J/E chaperone system. The measurements with RNase HD suggest that a simple kinetic partitioning model38,39 for protein folding in the presence of the K/J/E chaperone system is sufficient to account for our experimental findings. We are currently carrying out additional studies to address specifically the role of kinetic partitioning for the K/J/E interaction with two-state-folding proteins exhibiting different intrinsic folding rates and thermodynamic stability.
Consistent with this hypothesis, low resolution EPR distance measurements40 showed that substrate conformation is identical when bound to ATP-, ADP-, and NF-DnaK. Thus, ATP-binding and hydrolysis do not appear to be coupled to conformational work performed on the chaperone-associated client substrate at the DnaK binding site. The authors of this study argued that the above observation provides support for a kinetic partitioning mechanism.40 Additional studies are clearly necessary to provide more direct evidence for kinetic partitioning of client proteins between folding and chaperone-binding routes.
From a kinetic standpoint, our measurements are consistent with a simple holdase41 mechanism for K/J/E activity in the case of RNase HD. However, our results do not exclude that the folding of a fraction of the substrate molecules may be accelerated by K/J/E. However, either the fraction of molecules or the relative increase in folding rate (or both) is not sufficiently large to be detected as a separate stopped-flow kinetic phase. The consistency of our results with a kinetic partitioning mechanism does not conflict with K/J/E-induced conformational changes in the substrate at the molecular level. The final (ηF) ellipticity fraction, together with evidence from activity assays, indicate that RNase HD binds chaperones in a predominantly unfolded conformation. This observation is compatible with the unfoldase activity41 of K/J/E observed in its interaction with a luciferase variant.42 However, given that we are unable to assess the conformation of the unfolded substrate under refolding conditions before it binds the chaperones, our data alone are unable to either support or disprove any unfoldase activity by the K/J/E chaperone system.
Conclusions
In summary, this study shows that RNase HD interacts significantly with DnaK (and its co-chaperones DnaJ and GrpE) on its way to the native state, that RNase HD is at least partially unfolded upon its interaction with DnaK, and that the chaperone-substrate interaction retards the timescale of RNase HD folding only slightly, under physiologically relevant (nonheat-shock) conditions. Our findings have three major biological implications.
First, the transient, nature of chaperone-substrate interactions ensures that the production of a bioactive native protein like RNase HD, which does not need the chaperone to fold, is not substantially slowed down by the interaction with the K/J/E chaperones. This result suggests that the cellular function of slow-folding nonobligatory clients, binding K/J/E on their way to the native state, is not significantly perturbed by interaction with the K/J/E chaperone system. It is conceivable that this conclusion also applies to the folding of faster-folding substrates, which are likely to interact with the K/J/E system even less.
Second, the existence of transient substrate-chaperone association ensures that the effective concentration of chaperone-free unfolded or partially folded substrate is substantially decreased by the K/J/E chaperone system during protein folding, thereby reducing opportunities for misfolding and aggregation.
Third, the fact that K/J/E slows down the apparent folding of RNase HD, while it speeds up the folding of multidomain kinetically trapped misfolded proteins (e.g., firefly luciferase), shows that K/J/E-induced folding catalysis is not a general feature of the K/J/E chaperone system.
Materials and Methods
Protein Expression and Purification
RNase HD
RNase HD is a mutant of E.coli RNase H with all three native Cys replaced by Ala, in addition to the I53D mutation.23 The pSM101 plasmid encoding RNase HD was transformed into E. coli cells, and the protein was overexpressed as described.24 Cell pellets were thawed and resuspended in ice-cold lysis buffer (50 mM Tris, pH 8.0, 1 mM EDTA, and 10 mM β-mercaptoethanol), and lysed with lysozyme followed by mild sonication. RNase HD present in the inclusion bodies was extracted from the pellet with 50% acetonitrile containing 0.1% TFA, followed by a 0.5M urea wash.23 The protein was purified by reverse-phase HPLC on a Vydac C4 preparative scale column (Grace, Deerfield, IL). ESI-MS analysis of purified RNaseHD yielded a molecular weight of 17,502 Da, in agreement with the expected average mass of 17,503 Da.
DnaK
Overexpression and purification of DnaK was carried out according to published procedures43–45 with minor modifications. E.coli DnaK was overexpressed in BL21(DE3) cells (Novagen, EMD Chemicals, Gibbstown, NJ) carrying a pET-11a vector encoding the dnaK gene. Cells were grown in standard Luria broth containing 50 μg/mL carbenicillin at 30°C. Overexpression of the protein was induced by addition of isopropyl-β-thiogalactoside (1 mM) at an OD600 of 0.8. Cells were harvested after reaching an OD600 of 1.9–2.0. The cell pellet was resupsended in lysis buffer (25 mM Hepes, pH 7.5, 50 mM KCl, 5 mM MgCl2, 1 mM EDTA, and 10 mM BME) and lysed with lysozyme in the presence of 2 mM PMSF as a protease inhibitor, followed by mild sonication.
DnaK was initially purified on a 300 mL anion exchange Q-Sepharose column (GE Healthcare, Piscataway, NJ) equilibrated with lysis buffer. The protein was eluted from the column using a linear KCl gradient (0–1M). Fractions containing DnaK were pooled and concentrated to 5 mL with Amicon centrifugal concentrators (10 MWCO, Millipore, Billerica, MA). The concentrated DnaK solution was then purified on a Superdex 200 gel filtration column (GE Healthcare) equilibrated with lysis buffer. Pure fractions were pooled, concentrated to ∼20 mL and dialyzed against 25 mM HEPES (pH 7.5) buffer containing 1 mM EDTA and 10 mM BME in 10 MWCO dialysis cassettes (12–30 mL, Thermo Scientific, Rockford, IL).
DnaK-bound nucleotide was removed by treatment with alkaline phosphatase.44 2 units of alkaline phosphatase (Sigma-Aldrich Corp., St. Louis, MO) were added per mg of DnaK and the solution was stirred at 4°C with simultaneous dialysis to remove AMP. The A260/A280 ratio was monitored until the value went below 0.7. This typically takes about 5 h. Alkaline phosphatase was subsequently removed by purification on the Superdex 200 gel filtration column (GE Healthcare). The pure nucleotide-free DnaK was finally concentrated, dialyzed against storage buffer (25 mM HEPES pH 7.5, 5 mM MgCl2, and 50 mM KCl), flash frozen in liquid N2 and stored at −80°C.
DnaJ
A plasmid containing the full-length E. coli DnaJ gene was overexpressed from a pUHE21Δ12 plasmid in E. coli BL21 cells (Novagen) harboring a placIq plasmid. DnaJ was purified according to a published protocol26,46–48 with minor modifications. Detergents were not used in any step of the purification. The first chromatographic step was changed from the S-sepharose column to the SP-sepharose cation exchange column (GE Healthcare), and the last step involving Q-sepharose chromatography was removed.
GrpE
The expression plasmid pUHE21Δ12 containing the full-length E. coli GrpE gene was overexpressed in E. coli BL21 (Novagen) cells containing a placIq plasmid. The protein was purified according to established procedures.26,48,49 Briefly, cells were lysed with lysozyme and mild sonication. GrpE was then successively purified on the HiTrap Q-sepharose anion exchange column (GE Healthcare), the HiTrap Blue Sepharose column (GE Healthcare) and a hydroxyapatite column (Bio-Rad Laboratories, Hercules, CA). Pure fractions were flash frozen and stored at −80°C.
The activity of purified DnaK, DnaJ, and GrpE was assessed via a firefly luciferase reactivation assay.12 Concentrations of all proteins were estimated by UV-visible absorption spectrophotometry (extinction coefficients: 35,469,50 15,800,51 14,000,52 and 1,40052 M−1cm−1 for RNase HD, DnaK, DnaJ, and GrpE, respectively).
Stopped-flow kinetics
RNase HD refolding time courses were monitored with an SFM-400 MOS-450/AF-CD stopped-flow spectrophotometer equipped with an HDS mixer (Bio-Logic, Claix, France). The experimental dead time was 6 ms. A Xe-Hg lamp (Hamamatsu Photonics, Hamamatsu, Japan) and an 8-mm excitation slit width were used. Stopped-flow experiments were carried out with an FC-20 cuvette (2 × 2 mm2). For each kinetic trace, 8000 data points were collected. All stopped-flow experiments were performed at 30°C. Far-UV CD signals were monitored at 222 nm.
RNase HD was unfolded in 20 mM sodium acetate (pH 6.0) containing 5M urea (MP Biomedicals, Solon, OH). Unfolded protein samples were incubated at 4°C overnight prior to data collection to ensure complete unfolding. The urea-unfolded protein was refolded by 10-fold dilution into buffer containing 20 mM sodium acetate, 50 mM KCl, 1 mM ATP, and 5 mM MgCl2 in the absence or presence of chaperones. Unfolded controls consisted of unfolded protein diluted into 5M urea. Final protein concentrations of RNase HD, DnaK, DnaJ, and GrpE were 5, 15, 3 and 6 μM, respectively. All kinetics data were background-corrected against appropriate buffer blanks.
All stopped-flow CD-detected kinetic traces were fit to single exponential relations of the form
| 3 |
where y(t) is the time-dependent ellipticity, a is the ellipticity at time t = 0 and b is the total change in ellipticity upon refolding.
Size-exclusion chromatography
Analytical size-exclusion chromatography was performed on a TSK-GEL G2000SW (Tosoh Bioscience LLC, King of Prussia, PA) column connected to an HPLC (Shimadzu, Columbia, MD) equipped with a SPD-6AV detector monitoring absorbance at 214 nm. Flow rates were 1 mL/min. RNase HD was unfolded under the same conditions as in the stopped-flow experiments and refolded by diluting 10-fold into elution buffer (20 mM sodium acetate pH 6.0, 100 mM KCl, and 5 mM MgCl2) containing 1 mM ATP in the absence or presence of molecular chaperones. Samples were incubated at room temperature for 5 min before loading onto the column. The elution buffer did not contain any ATP since the absorbance was too high for detection at 214 nm. The gel filtration column was calibrated using the following molecular weight standards: bovine serum albumin (67 kDa), ovalbumin (43 kDa), chymotrypsinogen A (25 kDa), horse heart myoglobin (17.6 kDa), and ribonuclease A (13.7 kDa). Blue dextran was used to determine the column void volume.
Gel filtration chromatograms were digitized with the Bytescout Graph Digitizer (Vancouver, Canada). Peaks arising from RNase HD, chaperones and buffer components were deconvoluted by fitting each peak to a log-normal function using the Levenberg Marquardt algorithm53–55 within the Fityk software package.56 The deconvoluted native RNase HD peak intensity was used to calculate the extent of peak intensity reduction in the presence of various combinations of chaperones.
Reverse-phase chromatography
Protein fractions collected from the gel filtration experiments were lyophilized and resuspended in 35% acetonitrile containing 0.1% trifluoroacetic acid (Fisher Scientific, Fair Lawn, NJ). Samples were analyzed on an analytical reverse-phase Vydac C18 HPLC column (Grace). A flow rate of 1 mL/min was maintained and protein elution was monitored using absorbance at 214 and 280 nm.
RNase HD activity assays
RNase HD activity assays were performed on the protein after its refolding in the absence or presence of chaperones, to test the extent of its bioactivity after refolding and interaction with the K/J/E chaperones (Fig. 8). RNase HD was first unfolded in urea and refolded (final protein concentration: 27 μM, for 10 min) in the absence or presence of pertinent molecular chaperones: DnaK (70 μM), DnaJ (12 μM), GrpE (30 μM) and, whenever appropriate, ATP (1 mM). Refolded RNase HD was then diluted 1000-fold in the assay buffer (50 mM Tris, 50 mM NaCl, and 10 mM MgCl2 pH 8.0) containing 25 mg/mL rA/dT20 RNA-DNA hybrid to initiate the activity assay. RNase H activity assays were performed as described57 except that the timecourse of cleavage of RNA-DNA hybrids was followed via the increase in absorbance at 260 nm. Absorbance is quenched by base stacking in the DNA-RNA hybrid (hypochromic effect). The quenching vanishes upon cleavage, which eliminates base stacking.
Statistical analysis of stopped-flow refolding
The statistical significance of the differences between the observed stopped-flow rate constants and burst and final CD ellipticity amplitudes was determined via the one-tailed Student's t-test.58 For the apparent rate constants, the t-test was carried out on two means as described,58 to check the significance of differences between rate constants in the absence and presence of various chaperone combinations. In the case of CD signal amplitudes (ηB and ηF), the t-tests were carried out on a single mean, to evaluate the significance of the difference between the mean and the expected value in the absence of any change induced by K/J/E. The P values obtained from the Student's t-test are discussed in the legend of Figure 3.
Acknowledgments
The authors thank Bernd Bukau and Matthias Mayer for chaperone cell lines, plasmids, the luciferase refolding assay protocol, and for helpful discussions. In addition, they are grateful to Jochen Reinstein for assistance with the DnaK purification procedure, and to Susan Marqusee for providing the RNase HD-encoding plasmid and the RNase HD purification protocol. Finally, they thank Anders Knight, Neşe Kurt and Rudy Clausen for a critical reading of the manuscript.
References
- 1.Mayer MP, Brehmer D, Gässler CS, Bukau B. Hsp70 chaperone machines. Adv Protein Chem. 2001;59:1–12. doi: 10.1016/s0065-3233(01)59001-4. [DOI] [PubMed] [Google Scholar]
- 2.Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- 3.Frydman J. Folding of newly translated proteins in vivo: the role of molecular chaperones. Annu Rev Biochem. 2001;70:603–647. doi: 10.1146/annurev.biochem.70.1.603. [DOI] [PubMed] [Google Scholar]
- 4.Mayer MP, Rudiger S, Bukau B. Molecular basis for interactions of the DnaK chaperone with substrates. Biol Chem. 2000;381:877–885. doi: 10.1515/BC.2000.109. [DOI] [PubMed] [Google Scholar]
- 5.Rüdiger S, Germeroth L, Schneider-Mergener J, Bukau B. Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J. 1997;16:1501–1507. doi: 10.1093/emboj/16.7.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mayer MP, Bukau B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmid D, Baici A, Gehring H, Christen P. Kinetics of molecular chaperone action. Science. 1994;263:971–973. doi: 10.1126/science.8310296. [DOI] [PubMed] [Google Scholar]
- 8.Chesnokova LS, Slepenkov SV, Protasevich II, Sehorn MG, Brouillette CG, Witt SN. Deletion of DnaK's lid strengthens binding to the nucleotide exchange factor, GrpE: a kinetic and thermodynamic analysis. Biochemistry. 2003;42:9028–9040. doi: 10.1021/bi0346493. [DOI] [PubMed] [Google Scholar]
- 9.Russell R, Jordan R, McMacken R. Kinetic characterization of the ATPase cycle of the DnaK molecular chaperone. Biochemistry. 1998;37:596–607. doi: 10.1021/bi972025p. [DOI] [PubMed] [Google Scholar]
- 10.Mayer MP, Schröder H, Rüdiger S, Paal K, Laufen T, Bukau B. Multistep mechanism of substrate binding determines chaperone activity of Hsp70. Nat Struct Mol Biol. 2000;7:586–593. doi: 10.1038/76819. [DOI] [PubMed] [Google Scholar]
- 11.Pierpaoli EV, Gisler SM, Christen P. Sequence-specific rates of interaction of target peptides with the molecular chaperones DnaK and DnaJ. Biochemistry. 1998;37:16741–16748. doi: 10.1021/bi981762y. [DOI] [PubMed] [Google Scholar]
- 12.Szabo A, Langer T, Schröder H, Flanagan J, Bukau B, Hartl FU. The ATP hydrolysis-dependent reaction cycle of the Escherichia coli Hsp70 system DnaK, DnaJ, and GrpE. Proc Natl Acad Sci USA. 1994;91:10345–10349. doi: 10.1073/pnas.91.22.10345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han W, Christen P. Mechanism of the targeting action of DnaJ in the DnaK molecular chaperone system. J Biol Chem. 2003;278:19038–19043. doi: 10.1074/jbc.M300756200. [DOI] [PubMed] [Google Scholar]
- 14.Laufen T, Mayer MP, Beisel C, Klostermeier D, Mogk A, Reinstein J, Bukau B. Mechanism of regulation of Hsp70 chaperones by DnaJ cochaperones. Proc Natl Acad Sci USA. 1999;96:5452–5457. doi: 10.1073/pnas.96.10.5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Packschies L, Theyssen H, Buchberger A, Bukau B, Goody RS, Reinstein J. GrpE accelerates nucleotide exchange of the molecular chaperone DnaK with an associative displacement mechanism. Biochemistry. 1997;36:3417–3422. doi: 10.1021/bi962835l. [DOI] [PubMed] [Google Scholar]
- 16.Siegenthaler RK, Christen P. Tuning of DnaK chaperone action by nonnative protein sensor DnaJ and thermosensor GrpE. J Biol Chem. 2006;281:34448–34456. doi: 10.1074/jbc.M606382200. [DOI] [PubMed] [Google Scholar]
- 17.Shi L, Kataoka M, Fink AL. Conformational characterization of DnaK and its complexes by small-angle X-ray scattering. Biochemistry. 1996;35:3297–3308. doi: 10.1021/bi951984l. [DOI] [PubMed] [Google Scholar]
- 18.Chen Z, Kurt N, Rajagopalan S, Cavagnero S. Secondary structure mapping of DnaK-bound protein fragments: chain helicity and local helix unwinding at the binding site. Biochemistry. 2006;45:12325–12333. doi: 10.1021/bi0612263. [DOI] [PubMed] [Google Scholar]
- 19.Kurt N, Cavagnero S. Nonnative helical motif in a chaperone-bound protein fragment. Biophys J. 2008;94:L48–L50. doi: 10.1529/biophysj.107.127647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schröder H, Langer T, Hartl FU, Bukau B. DnaK, DnaJ and GrpE form a cellular chaperone machinery capable of repairing heat-induced protein damage. EMBO J. 1993;12:4137–4144. doi: 10.1002/j.1460-2075.1993.tb06097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herbst R, Schäfer U, Seckler R. Equilibrium intermediates in the reversible unfolding of firefly (Photinus pyralis) luciferase. J Biol Chem. 1997;272:7099–7105. doi: 10.1074/jbc.272.11.7099. [DOI] [PubMed] [Google Scholar]
- 22.Herbst R, Gast K, Seckler R. Folding of firefly (Photinus pyralis) luciferase: aggregation and reactivation of unfolding intermediates. Biochemistry. 1998;37:6586–6597. doi: 10.1021/bi972928i. [DOI] [PubMed] [Google Scholar]
- 23.Spudich GM, Miller EJ, Marqusee S. Destabilization of the Escherichia coli RNase H kinetic intermediate: switching between a two-state and three-state folding mechanism. J Mol Biol. 2004;335:609–618. doi: 10.1016/j.jmb.2003.10.052. [DOI] [PubMed] [Google Scholar]
- 24.Dabora JM, Marqusee S. Equilibrium unfolding of Escherichia coli ribonuclease H: characterization of a partially folded state. Protein Sci. 1994;3:1401–1408. doi: 10.1002/pro.5560030906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bogatyreva NS, Osypov AA, Ivankov DN. KineticDB: a database of protein folding kinetics. Nucleic Acids Res. 2009;37:D342–D346. doi: 10.1093/nar/gkn696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Linke K, Wolfram T, Bussemer J, Jakob U. The roles of the two zinc binding sites in DnaJ. J Biol Chem. 2003;278:44457–44466. doi: 10.1074/jbc.M307491200. [DOI] [PubMed] [Google Scholar]
- 27.Gamer J, Multhaup G, Tomoyasu T, McCarty JS, Rüdiger S, Schönfeld HJ, Schirra C, Bujard H, Bukau B. A cycle of binding and release of the DnaK, DnaJ and GrpE chaperones regulates activity of the Escherichia coli heat shock transcription factor sigma32. EMBO J. 1996;15:607–617. [PMC free article] [PubMed] [Google Scholar]
- 28.Palleros DR, Shi L, Reid KL, Fink AL. Hsp70-protein complexes. Complex stability and conformation of bound substrate protein. J Biol Chem. 1994;269:13107–13114. [PubMed] [Google Scholar]
- 29.Swain JF, Schulz EG, Gierasch LM. Direct comparison of a stable isolated Hsp70 substrate-binding domain in the empty and substrate-bound states. J Biol Chem. 2006;281:1605–1611. doi: 10.1074/jbc.M509356200. [DOI] [PubMed] [Google Scholar]
- 30.Swain JF, Dinler G, Sivendran R, Montgomery DL, Stotz M, Gierasch LM. Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol Cell. 2007;26:27–39. doi: 10.1016/j.molcel.2007.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rüdiger S, Schneider-Mergener J, Bukau B. Its substrate specificity characterizes the DnaJ co-chaperone as a scanning factor for the DnaK chaperone. EMBO J. 2001;20:1042–1050. doi: 10.1093/emboj/20.5.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schönfeld HJ, Schmidt D, Zulauf M. Investigation of the molecular chaperone DnaJ by analytical ultracentrifugation. Anal Ultracentrifugation. 1995;99:7–10. [Google Scholar]
- 33.Pierpaoli EV, Sandmeier E, Schönfeld HJ, Christen P. Control of the DnaK chaperone cycle by substoichiometric concentrations of the co-chaperones DnaJ and GrpE. J Biol Chem. 1998;273:6643. doi: 10.1074/jbc.273.12.6643. [DOI] [PubMed] [Google Scholar]
- 34.Tang YC, Chang HC, Roeben A, Wischnewski D, Wischnewski N, Kerner MJ, Hartl FU, Hayer-Hartl M. Structural features of the GroEL-GroES nano-cage required for rapid folding of encapsulated protein. Cell. 2006;125:903–914. doi: 10.1016/j.cell.2006.04.027. [DOI] [PubMed] [Google Scholar]
- 35.Ekman D, Björklund Å, Frey-Skött J, Elofsson A. Multi-domain proteins in the three kingdoms of life: orphan domains and other unassigned regions. J Mol Biol. 2005;348:231–243. doi: 10.1016/j.jmb.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 36.Kurt N, Rajagopalan S, Cavagnero S. Effect of hsp70 chaperone on the folding and misfolding of polypeptides modeling an elongating protein chain. J Mol Biol. 2006;355:809–820. doi: 10.1016/j.jmb.2005.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grimshaw J, Jelesarov I, Schonfeld HJ, Christen P. Reversible thermal transition in GrpE, the nucleotide exchange factor of the DnaK heat-shock system. J Biol Chem. 2001;276:6098–6104. doi: 10.1074/jbc.M009290200. [DOI] [PubMed] [Google Scholar]
- 38.Flynn GC, Pohl J, Flocco MT, Rothman JE. Peptide-binding specificity of the molecular chaperone BiP. Nature. 1991;353:726–730. doi: 10.1038/353726a0. [DOI] [PubMed] [Google Scholar]
- 39.Randall LL, Hardy SJS. SecB, one small chaperone in the complex milieu of the cell. Cell Mol Life Sci. 2002;59:1617–1623. doi: 10.1007/PL00012488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Popp S, Packschies L, Radzwill N, Vogel KP, Steinhoff HJ, Reinstein J. Structural dynamics of the DnaK-peptide complex. J Mol Biol. 2005;347:1039–1052. doi: 10.1016/j.jmb.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 41.Slepenkov SV, Witt S. The unfolding story of the Escherichia coli Hsp70 DnaK: is DnaK a holdase or an unfoldase? Mol Microbiol. 2002;45:1197–1206. doi: 10.1046/j.1365-2958.2002.03093.x. [DOI] [PubMed] [Google Scholar]
- 42.Sharma SK, De Los Rios P, Christen P, Lustig A, Goloubinoff P. The kinetic parameters and energy cost of the Hsp70 chaperone as a polypeptide unfoldase. Nat Chem Biol. 2010;6:914–920. doi: 10.1038/nchembio.455. [DOI] [PubMed] [Google Scholar]
- 43.Buchberger A, Valencia A, McMacken R, Sander C, Bukau B. The chaperone function of DnaK requires the coupling of ATPase activity with substrate binding through residue E171. EMBO J. 1994;13:1687–1695. doi: 10.1002/j.1460-2075.1994.tb06433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Theyssen H, Schuster HP, Packschies L, Bukau B, Reinstein J. The second step of ATP binding to DnaK induces peptide release. J Mol Biol. 1996;263:657–670. doi: 10.1006/jmbi.1996.0606. [DOI] [PubMed] [Google Scholar]
- 45.Klostermeier D, Seidel R, Reinstein J. Functional properties of the molecular chaperone DnaK from Thermus thermophilus. J Mol Biol. 1998;279:841–853. doi: 10.1006/jmbi.1998.1816. [DOI] [PubMed] [Google Scholar]
- 46.Zylicz M, Yamamoto T, McKittrick N, Sell S, Georgopoulos C. Purification and properties of the dnaJ replication protein of Escherichia coli. J Biol Chem. 1985;260:7591–7598. [PubMed] [Google Scholar]
- 47.Buchberger A, Theyssen H, Schröder H, McCarty JS, Virgallita G, Milkereit P, Reinstein J, Bukau B. Nucleotide-induced conformational changes in the ATPase and substrate binding domains of the DnaK chaperone provide evidence for interdomain communication. J Biol Chem. 1995;270:16903–16910. doi: 10.1074/jbc.270.28.16903. [DOI] [PubMed] [Google Scholar]
- 48.Rodriguez F, Arsène-Ploetze F, Rist W, Rüdiger S, Schneider-Mergener J, Mayer MP, Bukau B. Molecular basis for regulation of the heat shock transcription factor by the DnaK and DnaJ Chaperones. Mol Cell. 2008;32:347–358. doi: 10.1016/j.molcel.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 49.Schönfeld HJ, Schmidt D, Schröder H, Bukau B. The DnaK chaperone system of Escherichia coli: quaternary structures and interactions of the DnaK and GrpE components. J Biol Chem. 1995;270:2183–2189. doi: 10.1074/jbc.270.5.2183. [DOI] [PubMed] [Google Scholar]
- 50.Raschke TM, Kho J, Marqusee S. Confirmation of the hierarchical folding of RNase H: a protein engineering study. Nat Struct Biol. 1999;6:825–830. doi: 10.1038/12277. [DOI] [PubMed] [Google Scholar]
- 51.Russell R, Karzai AW, Mehl AF, McMacken R. DnaJ dramatically stimulates ATP hydrolysis by DnaK: insight into targeting of Hsp70 proteins to polypeptide substrates. Biochemistry. 1999;38:4165–4176. doi: 10.1021/bi9824036. [DOI] [PubMed] [Google Scholar]
- 52.Silberg JJ, Hoff KG, Vickery LE. The Hsc66-Hsc20 chaperone system in Escherichia coli: chaperone activity and interactions with the DnaK-DnaJ-GrpE system. J Bacteriol. 1998;180:6617–6624. doi: 10.1128/jb.180.24.6617-6624.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Levenberg K. A method for the solution of certain nonlinear problems in least squares. Quart Appl Math. 1944;2:164–168. [Google Scholar]
- 54.Marquardt DW. An algorithm for least-squares estimation of nonlinear parameters. J Soc Ind Appl Math. 1963;11:431–441. [Google Scholar]
- 55.More J. The Levenberg-Marquardt algorithm: implementation and theory. Lect Notes Math. 1978;630:105–116. [Google Scholar]
- 56.Wojdyr M. Fityk: a general-purpose peak fitting program. J Appl Crystallogr. 2010;43:1126–1128. [Google Scholar]
- 57.Keck JL, Marqusee S. Substitution of a highly basic helix/loop sequence into the RNase H domain of human immunodeficiency virus reverse transcriptase restores its Mn2+-dependent RNase H activity. Proc Natl Acad Sci USA. 1995;92:2740–2744. doi: 10.1073/pnas.92.7.2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Walpole RE, Myers RH, Myers SL, Ye K. Probability and statistics for engineers and scientists. New Jersey: Pearson Prentice Hall; 2007. [Google Scholar]