

Abstract
Kaposi's sarcoma herpesvirus (KSHV) belongs to the gamma-2 Herpesviridae and is associated with three neoplastic disorders: Kaposi's sarcoma (KS), primary effusion lymphoma (PEL), and multicentric Castleman's disease (MCD). The viral latency-associated nuclear antigen 1 (LANA) is expressed in all latently KSHV-infected cells and is involved in viral latent replication and maintenance of the viral genome. We show that LANA interacts with the ubiquitin-specific protease USP7 through its N-terminal TRAF (tumor necrosis factor [TNF] receptor-associated factor) domain. This interaction involves a short sequence (amino acids [aa] 971 to 986) within the C-terminal domain of LANA with strong similarities to the USP7 binding site of the Epstein-Barr virus (EBV) EBNA-1 protein. A LANA mutant with a deletion of the identified USP7 binding site showed an enhanced ability to replicate a plasmid containing the KSHV latent origin of replication but was comparable to the wild-type LANA (LANA WT) with regard to the regulation of viral and cellular promoters. Furthermore, the LANA homologues of two other gamma-2 herpesviruses, MHV68 and RRV, also recruit USP7. Our findings suggest that recruitment of USP7 to LANA could play a role in the regulation of viral latent replication. The recruitment of USP7, and its role in herpesvirus latent replication, previously described for the latent EBNA-1 protein of the gamma-1 herpesvirus (lymphocryptovirus) EBV (M. N. Holowaty et al., J. Biol. Chem. 278:29987–29994, 2003), may thereby be a conserved feature among gammaherpesvirus latent origin binding proteins.
INTRODUCTION
Kaposi's sarcoma herpesvirus (KSHV), or human herpesvirus 8, is a gamma-2 herpesvirus (5, 33, 38). KSHV is the causative agent of Kaposi's sarcoma and two lymphoproliferative disorders—primary effusion lymphoma (PEL) and multicentric Castleman's disease (MCD) (4, 44). KSHV persists in infected cells predominantly in a latent state, during which only a small subset of genes is expressed. Among them, the latency-associated nuclear antigen 1 (LANA), encoded by open reading frame 73, is constitutively expressed in all latently KSHV-infected cells and KSHV-associated malignancies (10, 22, 37).
LANA is a multifunctional protein that plays important roles in the maintenance of the viral genome, latent genome replication, and correct episome distribution in dividing cells. It tethers the viral genome to the host cell DNA by interacting with human chromatin by means of its N- and C-terminal domains and with the terminal repeat (TR) region of the viral DNA via its C-terminal domain. Viral genome maintenance involves interaction with cellular histones and chromatin-associated proteins like MeCP2, UNG2, BRD2/4, and DEK (24, 34, 35, 48, 51). Like the Epstein-Barr virus nuclear antigen 1 (EBNA-1) and other viral DNA binding proteins, LANA recruits additional proteins to allow latent genome replication, such as members of the origin recognition complex (ORC) (9, 29, 45).
Additionally, LANA can act as a transcriptional repressor or activator of both viral and cellular promoters. It interacts with proteins or protein complexes such as CREB2/ATF4, CBP, mSIN3, or Sp1 (25, 28, 49). LANA also interacts with p53, retinoblastoma protein (pRb), and glycogen synthase kinase 3β (GSK-3β), thereby inhibiting the activation of p53-dependent promoters, inducing the activation of E2F target genes, or promoting entry into the S phase of the cell cycle (13, 14, 36).
The ubiquitin-specific protease 7 (USP7), also called HAUSP (herpesvirus-associated USP), is a deubiquitinating enzyme that regulates numerous proteins, including tumor suppressors, DNA repair proteins, proteins involved in immune responses, viral proteins, and epigenetic modulators. It was identified to be a key player in the p53-Mdm2 pathway as it can deubiquitinate both p53 and Mdm2, with higher affinity for Mdm2 leading to Mdm2 stabilization and thereby Mdm2-catalyzed degradation of p53 (7, 8, 21, 26, 27, 43). It was observed that Mdm2 and p53 bind in a mutually exclusive manner within the N-terminal tumor necrosis factor (TNF) receptor-associated factor (TRAF)-like domain of USP7, recognizing the same shallow groove on the USP7 surface (21, 43). Mdm2 makes more extensive contacts to USP7 than p53, which accounts for a higher binding affinity (21, 43). This is supported by competition assays where an Mdm2 peptide efficiently displaced a p53 peptide (21). Furthermore, consensus peptide sequences for recognition by USP7 could be described. Sheng and colleagues (43) identified P/A-X-X-S (with X as any residue) as the consensus sequence, with the serine residue being an important residue for mediating contact to substrates, as recently also confirmed for another USP7 substrate (40).
Moreover, USP7 is involved in the regulation of two further proteins with a regulatory role in the p53-Mdm2 pathway: death-domain-associated protein DAXX and MdmX (also known as Mdm4), a structural homologue of Mdm2 (30, 46). The interplay between Mdm2 and USP7 seems to be fine-tuned by DAXX, underlining the importance of a tight regulation of USP7 in cell fate decisions (46).
USP7 was originally identified as an interaction partner of ICP0 (also named Vmw110), an immediate-early gene of herpes simplex virus 1 (HSV-1) with a role in the initiation of the viral lytic life cycle (11). ICP0, a ubiquitin E3 ligase, induces auto-ubiquitination, which is counteracted by USP7, leading to stabilization of the ICP0 protein (3). ICP0 can also ubiquitinate USP7 and reduce cellular USP7 levels (2). However, it was shown that the stabilizing effect of ICP0 deubiquitination by USP7 is dominant over ICP0-induced degradation of USP7, as no reduction in USP7 levels can be observed when ICP0 is expressed in limiting amounts (2). In contrast to the cellular USP7 target proteins described so far (see above), ICP0 binds within the C-terminal domain of USP7 (18).
USP7 was further found to interact with Epstein-Barr virus EBNA-1, which is a functional homologue of LANA (19). An EBNA-1 mutant unable to interact with USP7 shows increased replication of the latent EBV origin and decreased transcriptional activation but has no effect on the segregation function of EBNA-1 (19). Additionally, it was observed that USP7 forms a complex with DNA-bound EBNA-1 and that the DNA binding activity of EBNA-1 is increased by USP7 (41). USP7 further shows augmented recruitment to the FR (family of repeats) element of oriP, the origin of latent EBV replication (41), and can decrease H2B ubiquitination together with GMPS (guanosine 5′ monophosphate synthetase), thereby influencing gene expression (41, 47). Thus, there is some evidence that the interaction of USP7 and EBNA-1 regulates plasmid maintenance and/or latent replication during EBV infection (41). It was further shown that EBNA-1 binds with higher affinity to the N-terminal domain of USP7 than p53, indicating that EBNA-1 competes with p53 for USP7 binding during infection. This is supported also by structural studies (39, 43) and may have an effect on p53 function (18).
Here, we show that LANA, as well as the LANA homologues of two other gamma-2 herpesviruses, also interact with USP7. The residues critical for USP7 binding are predominantly located in the C-terminal domain of LANA, in a short region displaying high sequence homology to EBNA-1. Similar to EBNA-1, LANA interacts with the N-terminal part of USP7, which is also involved in the interaction with the USP7 targets p53, MdmX, and Mdm2. Similar to what was previously shown for EBV, latent viral replication of the KSHV genome depends on USP7, while LANA transcriptional activity is not affected by USP7 binding.
MATERIALS AND METHODS
Expression plasmids.
All full-length LANA constructs were in a pcDNA 3.1 background. The numbering of amino acids of LANA corresponds to the KSHV BC-1 sequences (38). An expression vector for full-length LANA was described previously (50). The sequences of PCR primers used in this study, and their numbered designations, are listed in Table 1. The mutant with deletion of amino acids (aa) 971 to 986 was cloned by overlapping PCR with primers 1, 2, 3, and 4. For the cLANA Δ 971-986 construct, primer 5 was used for overlapping PCR instead of primer 1. LANA GST fusion proteins used in glutathione S-transferase (GST) pulldown experiments represented different segments of the C-terminal domain (aa 951 to 1162) of LANA and were described previously (35). Additionally, the construct C6a was generated by PCR using primers 6 and 7. To obtain the cLANA Δ 971-986 mutant in the GST vector backbone, another overlapping PCR was performed with primers 5, 3, 4, and 8.
Table 1.
Sequences of primers used for cloning different expression vectors
| Primer no. | Primer name | Sequence |
|---|---|---|
| 1 | LANA start for | 5′- ATA GAA TTC ATG GCG CCC CCG GGA ATG-3′ |
| 2 | LANA end XhoI rev | 5′- AGA CTC GAG TTA TGT CAT TTC CTG TGG AGA G-3′ |
| 3 | LANA aa 970 rev delta | 5′- TCT GTG GGG TGG TGA TGT TCT GAG TGT ATT ATC TCC TGG TGG-3′ |
| 4 | LANA aa 987 for delta | 5′- AGT AGC CCA CCA GGA GAT AAT ACA CTC AGA ACA TCA CCA CCC CAC AG-3′ |
| 5 | LANA aa 934 EcoRI for | 5′- AGA GAA TTC GAA GAG CCC ATA ATC TTG-3′ |
| 6 | LANA GST C6a for | 5′- AGA GAA TTC CCA GAC GAT GAC CCA CAA C-3′ |
| 7 | LANA GST C6a rev | 5′- AGA GCG GCC GCT CAT ACA TAG CGG-3′ |
| 8 | LANA end NotI rev | 5′- AGA GCG GCC GCT TAT GTC ATT TCC TGT GGA GAG-3′ |
| 9 | LANA aa 1 for | 5′- AGA GTC GAC ACC ATG GCG CCC CCG GGA ATG-3′ |
| 10 | LANA aa 340 rev | 5′- AGA GCG GCC GCG CTC CTC ATC TGT CTC CTG-3′ |
| 11 | LANA aa 934 SalI for | 5′- AGA GTC GAC ACC ATG GAA GAG CCC ATA ATC TTG-3′ |
| 12 | LANA aa 1162 rev | 5′- AGA GCG GCC GCG TGT CAT TTC CTG TGG AGA G-3′ |
| 13 | LANA aa 934 BamHI for | 5′- AGA GGA TCC GAA GAG CCC ATA ATC TTG-3′ |
| 14 | LANA 4A aa 977-980 for | 5′- C GAT GAC CCA CAA GCT GCC GCA GCT CGC GAA TAC CGC-3′ |
| 15 | LANA 4A aa 977-980 rev | 5′- GCG GTA TTC GCG AGC TGC GGC AGC TTG TGG GTC ATC G-3′ |
| 16 | LANA end EcoRI rev | 5′- ATA GAA TTC TTA TGT CAT TTC CTG TGG AGA G-3′ |
| 17 | LANA aa 1065-67 for | 5′- GAT TTT TTG GGG CAA TGA CGC AGC CGC ACT TAA AAA ATT ATC TCA GG-3′ |
| 18 | LANA aa 1065-67 rev | 5′- CCT GAG ATA ATT TTT TAA GTG CGG CTG CGT CAT TGC CCC AAA AAA TC-3′ |
| 19 | USP7 aa 56 for | 5′- AGA GGA TCC GAG GAG GAC ATG GAG GAT G-3′ |
| 20 | USP7 aa 205 rev | 5′- AGA CTT AAG TTA CCA CGC AAC TCC ATG GGG AGC-3′ |
Mammalian LANA constructs harboring only the N- or C-terminal domain of LANA were cloned with the following primers (in myc epitope-containing pcDNA3myc provided by S. Mittnacht, London, United Kingdom, or HA-containing pVR1255 provided by J. Stewart, Liverpool, United Kingdom): primers 9, 10, 11, 12, 2, and 13. For the cLANA 977-980/AAAA mutant, a potential USP7 binding motif in the C-terminal domain of LANA (aa 977 to 980) was changed to four alanines by site-directed mutagenesis using primers 14 and 15.
For electrophoretic mobility shift assays (EMSAs), cLANA was cloned in pGEX-6p-1 by PCR with primers 13 and 16, with full-length LANA as the template. Mutation of the PYG motif (aa 1065 to 1067) to alanine was introduced by site-directed mutagenesis using primers 17 and 18.
Full-length RRV Orf73 (1,347 bp) of RRV isolate 17577 tagged N terminally with a myc epitope was provided by R. Searles (Portland, OR). The MHV68 Orf73 construct cloned from murine herpesvirus 4 strain 68-infected cells in the HA-tag-containing pVR1255 vector was provided by J. Stewart.
The USP7 expression plasmid expressing GFP-tagged USP7 was kindly provided by Roger Everett, Glasgow, United Kingdom. An N-terminal USP7 construct was cloned based on the full-length construct by PCR with primers 19 and 20 into a GST fusion vector (pGEX-6p-1).
The plasmid containing the TR region of the KSHV genome was described previously (50). This plasmid was used to generate a 1× TR luciferase reporter vector (51).
The cyclin E luciferase reporter vector consisting of the human cyclin E promoter region cloned in front of a luciferase gene (in pGL2Basic) was from R. Weinberg's laboratory (Cambridge, MA) (16). The p53 luciferase reporter construct containing the p53-responsive promoter region of the human mdm2 gene (in pGL2Basic) was provided by M. Dobbelstein (Göttingen, Germany), as was an expression construct for HA-tagged human p53 in a pcDNA backbone.
Cells and cell culture.
HEK 293, HEK 293-T, and p53-deficient H1299 cells were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS), 50 IU/ml penicillin, and 50 μg/ml streptomycin at 37°C in a 5% CO2 incubator.
Suspension cells such as BJAB and BJAB stably infected with KSHV (S. Kati, S. Hübner, T. Rothämel, and T.F. Schulz, unpublished data), as well as the PEL cell lines BC1 and BC3, were grown in RPMI 1640 (containing l-glutamine) supplemented with 20% fetal calf serum (FCS), 50 IU/ml penicillin, and 50 μg/ml streptomycin at 37°C in a 5% CO2 incubator.
For transfection, adherent cells were grown to subconfluence in six-well plates and transfected with the indicated expression constructs with FuGENE transfection reagent (Roche) according to the manufacturer's instructions.
SDS-PAGE and immunoblotting.
Forty-eight hours after transfection with the indicated expression plasmids, cells were lysed in 300 μl TBST lysis buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100) per well. Cleared lysates were subjected to SDS-PAGE, and proteins were detected by Ponceau S staining or immunoblotting.
The following antibodies were used: mouse or rabbit α-GFP (Clontech), mouse α-actin (Millipore), rat α-LANA (ABI), mouse or rat α-HA (Roche), human α-KS serum from KSHV-seropositive patients, and polyclonal rabbit serum detecting GST fusion proteins.
GST pulldown assay.
For GST pulldown experiments, Escherichia coli Rosetta cultures transformed with GST expression plasmids or GST only were grown at 37°C in LB medium plus ampicillin and chloramphenicol. Cultures were induced at an optical density at 600 nm of 0.4 to 0.6 with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and harvested by centrifugation 5 h after induction. Pelleted cells were resuspended in 500 μl phosphate-buffered saline (PBS) with protease inhibitors, sonicated for 1 min on ice, supplemented with 1% Triton X-100, and incubated for 10 min at 4°C. Following centrifugation, the supernatant was incubated with 80 μl glutathione-Sepharose beads (Amersham Biosciences) overnight at 4°C. Beads were washed three times with PBS plus protease inhibitors and analyzed on a Coomassie-stained gel to calculate equal amounts of GST fusion proteins. HEK 293-T cells were transfected and lysed as mentioned above. Cleared lysate (300 μl) was incubated with either GST fusion proteins or GST control protein for 1 h at room temperature (RT). Beads were washed six times with PBS and analyzed by SDS-PAGE and Western blotting.
Coimmunoprecipitation.
HEK 293-T cells were cotransfected with plasmids encoding the interaction partners of interest; the amounts used are indicated in the figure legends. Forty-eight hours after transfection, cells were washed once in cold PBS and lysed in 300 μl of TBST buffer containing protease inhibitors per well of a six-well plate. For precipitation of endogenous proteins, suspension cell lines were also lysed in 300 μl TBST buffer containing protease inhibitors.
The monoclonal antibodies α-GFP (Clontech), α-HA (Roche), and α-LANA (ABI) were immobilized on protein A or G Sepharose beads (Amersham). For precipitation of myc-tagged proteins, c-myc monoclonal antibody-agarose beads (Clontech) were used.
An aliquot of 40 μl cleared cell lysates was taken (input control), and the remaining 260 μl were incubated overnight on a roller at 4°C with 12 μl of beads coupled with antibody to precipitate the protein of interest. Afterwards, samples were centrifuged (1,100 × g, 30 s) and washed extensively with 700 μl TBST (supplemented with 1% sodium deoxycholate, if higher stringency was necessary). Finally, beads were boiled with 30 μl of 5× loading buffer (62.5 mM Tris-HCl, pH 6.8, 2% [wt/vol] SDS, 10% glycerol, 50 mM dithiothreitol [DTT], 0.01% [wt/vol] bromophenol blue) for 3 min and 20 μl of the samples was analyzed by SDS-PAGE. As input control, 1/10 of the input lysate was loaded.
Transient replication assay.
Short-term replication assays were performed with HEK 293 cells cotransfected with 500 ng expression vector containing 1× TR and 250 ng pBluescript (input control) plus 500 ng of LANA expression constructs or the empty vector pcDNA3. Thirty hours after transfection, the cells were split 1:2 and allowed to grow for a further 48 h. Cells were then harvested in 150 μl of lysis buffer per well of a six-well plate (10 mM Tris-HCl, 10 mM EDTA, 0.6% SDS), and duplicate wells, obtained by splitting, were pooled. Chromosomal DNA was precipitated overnight with 0.85 M NaCl. An aliquot was collected from the supernatant to determine the expression levels of the different LANA constructs by immunoblotting. Plasmid DNA was purified from the supernatant using phenol-chloroform extraction and precipitated afterwards with ethanol. Finally, plasmid DNA was resuspended in H2O. A total of 90% of the DNA was digested with 40 U KpnI and 40 U of DpnI for 48 h, while the remaining 10% was digested with 10 U KpnI to measure input DNA. The digested DNA was separated on a 0.8% agarose gel and analyzed by Southern blotting with a 962-bp fragment of pBluescript used as an alkaline phosphatase-labeled probe (AlkPhos Direct Labeling and Detection System with CDP-Star; GE Healthcare).
EMSA.
IRD700-labeled oligonucleotides (IBA GmbH, Göttingen, Germany) were prepared as follows: 5 μl of each oligonucleotide (LANA BS top, 5′-GAG GCG GCG CGC GGC CCC ATG CCC GGG CGG GAG GCG CCG CAG GCC CCG GCG GCG TCC CCG GC-3′; LANA BS bottom, 5′-GCC GGG GAC GCC GCC GGG GCC TGC GGC GCC TCC CGC CCG GGC ATG GGG CCG CGC GCC GCC TC-3′) was mixed with 2 μl of 10× annealing buffer (200 mM Tris-HCl, pH 7.6, 500 mM NaCl, 100 mM MgCl2), and the volume was adjusted to 20 μl with H2O. This annealing reaction mixture was incubated for 5 min at 95°C, 10 min at 65°C, 10 min at 37°C, and 10 min at 20°C and cooled to 4°C. The annealed oligonucleotides were diluted 1:5 for the binding reaction.
For binding, 0.5 to 2 μl of LANA GST fusion proteins was added to 1.5 μl binding buffer (300 mM Tris-HCl, pH 7.9, 500 mM KCl, 100 mM MgCl2, 10 mM DTT, 10 mM EDTA) as well as 1.5 μl 100% glycerol, 1.5 μl of a 2.5% Tween 20 solution, 0.75 μl poly(dI-dC) (1 mg/ml), 0.75 μl of a 10-mg/ml solution of bovine serum albumin (BSA), and 1 μl of the annealed oligonucleotides. Reactions were made up with H2O to a 15-μl final volume and incubated for 30 min at RT in the dark. For competition, a 100× to 200× excess of unlabeled probe was added to the band shift mixture. Supershifts were carried out with 4 μl of a polyclonal LANA rabbit serum or a rabbit antibody not reacting with LANA for a control. For supershift, binding reaction mixtures were incubated for another 30 min after addition of the antibodies.
A 4% native acrylamide gel was prerun for 1 h at 160 V in 1× TBE. Afterwards, the band shift reaction mixtures were loaded onto the gel and separated for 2 h at 160 V. After the run, gels were immediately analyzed with the Odyssey infrared imaging system from Licor Biosciences.
Luciferase reporter assay.
For luciferase reporter assays, HEK 293, HEK 293-T, or H1299 cells were transiently cotransfected in duplicates with 50 ng of reporter plasmid and expression constructs as indicated in the figure legends. Forty-eight hours after transfection, cells were washed once with PBS and lysed in 300 μl reporter lysis buffer (Promega) per well of a six-well plate. Luciferase activity was measured in cleared lysates with a luciferase system in accordance with the manufacturer's instructions (Promega).
RESULTS
LANA interacts with USP7.
USP7 was previously reported to interact with two herpesviral proteins, HSV-1 ICP0 and EBV EBNA-1, and to be important for the life cycle of both viruses. Since LANA is a functional homologue of EBNA-1, we investigated whether LANA could also interact with USP7. We transfected HEK 293-T cells with a GFP-tagged expression construct for USP7 as well as an expression vector for LANA and precipitated protein complexes with α-LANA antibody coupled to protein G-Sepharose beads. Following washing of the immunocomplexes, the interaction was analyzed via immunoblotting using an α-GFP antibody. Transfection of empty vector served as a negative control. We found that LANA is capable of interacting with USP7 (Fig. 1A). This observation could be confirmed in a reciprocal coimmunoprecipitation experiment, in which LANA was found in a complex with precipitated USP7 (Fig. 1B). Additionally, the interaction of LANA and USP7 could be verified with coimmunoprecipitation experiments using naturally KSHV-infected PEL cell lines, as well as BJAB cells stably infected with recombinant KSHV virus (Fig. 1C and D). Endogenously expressed (untransfected) USP7 interacted with LANA in KSHV-infected cells, but not in the uninfected control BJAB cells (Fig. 1C), and in all PEL cell lines tested (BC1, BC3) (Fig. 1D).
Fig 1.

LANA interacts with USP7. (A) The LANA expression construct and a plasmid encoding GFP-tagged USP7, or the appropriate empty vectors, were cotransfected in equal amounts (1 μg each) in 293-T cells. Cells were lysed after 48 h, and protein complexes were immunoprecipitated as described in Materials and Methods with α-LANA antibody coupled to protein G-Sepharose beads. After extensive washing, beads were analyzed by SDS-PAGE and immunoblotting using α-LANA and α-GFP antibodies. (B) In a reciprocal coimmunoprecipitation experiment, protein complexes were precipitated with α-GFP antibody coupled to protein A-Sepharose beads and the interaction was visualized by staining the immunoblot with an antibody to LANA or GFP as a control. (C) Coimmunoprecipitation (Co-IP) of endogenous (i.e., not transfected) LANA with USP7 in BJAB cells stably infected with recombinant KSHV virus (BJAB rK). Parental BJAB and duplicate samples immunoprecipitated with a control IgG served as control. Co-IP and IB were performed as described for panel A. (D) Coimmunoprecipitation of endogenous LANA with endogenously expressed USP7 in different PEL cell lines. As a control, duplicate samples were immunoprecipitated with a control IgG. Co-IP and IB were performed as described for panel A.
Mapping of the USP7 binding site of LANA.
Following the observation that LANA interacts with USP7, we investigated which domains of both proteins are involved in the interaction. We started by mapping the USP7 binding site in LANA to the N-terminal or C-terminal domain. USP7 coimmunoprecipitates with the C-terminal domain of LANA but not with the N-terminal domain (Fig. 2A). To further determine the residues of LANA critical for USP7 binding, GST pulldown experiments were performed. Truncated versions of the C-terminal domain of LANA fused to GST (Fig. 2B) were expressed in bacteria, isolated, and purified by binding to GST beads, and their interaction with eukaryotically expressed plasmids of GFP-tagged USP7 or the appropriate empty vector was investigated. In the GST pulldown experiments, USP7 interacted with the constructs C4, C5, and C6 (Fig. 2B and C). In addition, the small LANA fragment C6a, comprising a region of 16 aa, interacted with USP7 (Fig. 2B and D). This region, which is located between aa 971 and 986, contains a PGPS motif (Fig. 3) that corresponds to the consensus sequence P/A-X-X-S for USP7 binding found by Sheng and coworkers (43).
Fig 2.

The region aa 971 to 986 of LANA is sufficient for USP7 binding. (A) Immunoprecipitation experiments with the N-terminal (aa 1 to 340) or C-terminal (aa 934 to 1162) domain of LANA. The different LANA expression constructs containing an HA-tag and a plasmid encoding GFP-tagged USP7 were cotransfected in equal amounts (1 μg each) in 293-T cells. As a control, the appropriate empty vectors were transfected. Immunoprecipitations were carried out as described in the legend to Fig. 1 and Materials and Methods. (B) Schematic representation of the GST-tagged truncated versions of the C-terminal domain of LANA (aa 951 to 1162) used in this study. + and (+), interaction and weak interaction, respectively, with USP7; −, lack of interaction. (C and D) GST pulldown experiments performed with LANA-GST fusion proteins (aa 951 to 1162) expressed in bacteria. Purified and adjusted amounts of each protein were incubated with eukaryotic cell lysate of 293-T cells previously transfected with an expression construct encoding GFP-tagged USP7 (top) or GFP alone (middle); input for both experiments is shown in the left of panel C. After extensive washing, protein interaction was analyzed by SDS-PAGE and immunoblotting using an α-GFP antibody. Expression of GST alone and GST fusion proteins was verified by Ponceau S staining.
Fig 3.
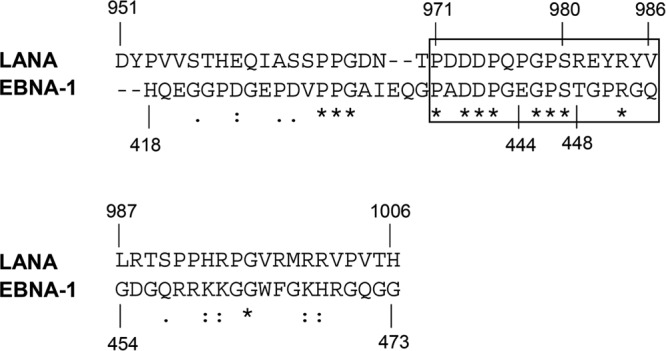
Alignment of the USP7 binding regions of LANA and EBNA-1. Alignment (originally generated with ClustalW) shows the described USP7 binding site of EBNA-1 (19) in comparison to the identified USP7 binding site in the C-terminal domain of LANA. *, Conserved amino acids; :, conserved substitutions; ., semiconserved substitutions. The rectangle marks the region aa 971 to 986.
An alignment of the USP7 binding site of LANA with the previously described USP7 binding site in EBNA-1 (19) showed that the consensus motif, as well as further residues of the identified LANA region involved in USP7 binding, are conserved in EBNA-1. Within the fragment C6 (aa 951 to 1006), the sequence of aa 971 to 986 (C6a) is the only one to show significant sequence homology between LANA and EBNA-1 (Fig. 3). These sequence similarities emphasize the importance of these residues for the interaction with USP7.
As depicted in Fig. 4, removal of aa 971 to 986 from the C-terminal LANA domain abolished binding of LANA to USP7 in GST pulldown experiments (Fig. 4A) as well as in coimmunoprecipitation experiments (Fig. 4B). In addition, the substitution of the PGPS motif in LANA (aa 977 to 980) with four alanine residues eliminated the binding of USP7 to the LANA C-terminal domain in coimmunoprecipitation assays (Fig. 4B). This finding confirms the importance of LANA aa 971 to 986, and in particular aa 977 to 980, for binding to USP7.
Fig 4.

Deletion of aa 971 to 986 from the C-terminal domain of LANA abolishes or reduces USP7 binding. (A) LANA constructs harboring a GST tag were expressed in bacterial cells, bound to glutathione-Sepharose 4B beads overnight, and washed, and adjusted amounts were incubated for 1 h with lysates of 293-T cells transiently expressing either GFP-tagged USP7 or the empty GFP vector following transfection with the respective expression construct. After extensive washing steps, beads were analyzed by SDS-PAGE and immunoblotting using an α-GFP antibody (right panel, input: left panel). Expression of GST alone and GST fusion proteins was verified by Ponceau S staining. (B) Western blots showing the results of immunoprecipitation experiments. Different LANA expression constructs containing an HA tag with either the deletion of aa 971 to 986 or an alanine substitution of the potential USP7 binding motif (aa 977 to 980 in cLANA 977–980/AAAA) and a plasmid encoding GFP-tagged USP7 were cotransfected in equal amounts (1 μg each) in 293-T cells. As a control, the appropriate empty vectors were transfected. Cells were lysed 48 h posttransfection, and protein complexes were immunoprecipitated with α-HA antibody coupled to protein-A-Sepharose beads as described in Materials and Methods and analyzed by immunoblotting using α-HA (cLANA) and α-GFP (USP7) antibodies. (C) The LANA expression constructs and a plasmid encoding GFP-tagged USP7 were cotransfected in equal amounts (1 μg each) in 293-T cells. As a control, the corresponding empty vectors were transfected. Cells were lysed after 48 h, and protein complexes were precipitated with α-LANA antibody coupled to protein-A-Sepharose beads. After extensive washing, beads were analyzed by SDS-PAGE and immunoblotting using an α-GFP antibody. (D) LANA binds to the N-terminal domain of USP7. The GST fusion construct of the N-terminal domain of USP7 was expressed in bacteria, purified, and incubated with eukaryotic cell lysate of 293-T cells previously transfected with expression constructs encoding LANA or LANA Δ 971–986. After extensive washing, protein interaction was analyzed by SDS-PAGE and immunoblotting using an α-LANA antibody (top right). Equal amounts of GST and GST-USP7 were verified by probing the membrane with an antibody to GST (bottom right).
In the next step, the deletion of the region aa 971 to 986 was introduced into a full-length LANA construct by overlapping PCR. The full-length LANA construct harboring the deletion of the potential USP7 binding site (LANA Δ 971–986) showed a strongly reduced binding to USP7 (Fig. 4C). The signal intensities of the immunoprecipitated USP7 bands in Fig. 4C (second panel from bottom) were quantified with ImageJ and normalized to the upper LANA band in the bottom panel of Fig. 4C, indicating a residual interaction of 29% (data not shown). This residual binding cannot be attributed to an additional USP7 binding site in the N-terminal domain of LANA, as the N-terminal domain of LANA was not found to interact with USP7 (Fig. 2A).
LANA binds to the N-terminal domain of USP7.
Most USP7 substrates and interaction partners like Mdm2, Mdmx, p53, and EBNA-1 were shown to bind to the N-terminal TRAF domain of USP7 (18, 21, 39, 40, 43). In contrast, the HSV-1 protein ICP0 interacts with the C-terminal part of USP7, a domain of unknown function (18). We investigated whether LANA is able to interact with the N-terminal domain of USP7 in GST pulldown experiments. The N-terminal domain of USP7 was expressed as a GST fusion protein in bacteria and purified via the GST tag. Lysates of cells transiently transfected with expression constructs of LANA or LANA Δ 971–986 were incubated with adjusted amounts of GST-USP7 protein coupled to beads. After washing, protein interaction was analyzed by immunoblotting. These experiments revealed that LANA can interact with the N-terminal TRAF domain whereas the LANA mutant lacking the USP7 binding site (aa 971 to 986) is not able to bind to the N-terminal domain of USP7 (Fig. 4D). This result suggests that LANA resembles EBNA-1, p53, Mdm2, and MdmX in targeting the N-terminal TRAF domain of USP7 and underlines the importance for the LANA region (aa 971 to 986) for interaction with the TRAF domain of USP7.
Deletion of aa 971 to 986 increases the ability of LANA to replicate latent viral DNA.
As binding of USP7 to a LANA mutant lacking aa 971 to 986 is strongly reduced (in coimmunoprecipitation assays) or absent (in GST pulldown assays), this LANA mutant was used to study the functional importance of the USP7-LANA interaction. LANA mediates episome persistence by tethering the viral episome to human chromatin and thus is important for latent replication. LANA binds via its C-terminal domain to two regions in the terminal repeats (TRs) of the KSHV genome, which are named LANA binding sites 1 and 2 (LBS1 and LBS2) (20). LANA is also sufficient to replicate a TR-containing plasmid in cells in a short-term replication assay (17, 20, 29, 51). Therefore, the mutant lacking the USP7 binding site (aa 971 to 986) was tested in a transient replication assay to analyze its replication ability.
The LANA mutant lacking aa 971 to 986 replicated a plasmid containing the KSHV latent origin of replication more efficiently than the wild-type LANA (LANA WT) (Fig. 5A). Quantification of the relative band intensity observed in three independent experiments suggests an approximately four times stronger replication by LANA Δ 971–986 in comparison to LANA WT (Fig. 5B).
Fig 5.

The LANA mutant lacking aa 971 to 986 shows enhanced replication of a plasmid containing the KSHV origin of latent replication. (A) Transient replication assay. 293 cells were cotransfected in duplicates with expression plasmids of LANA or the LANA mutant lacking the USP7 binding site (aa 971 to 986), a plasmid containing one TR subunit, and the empty vector pBluescript as a transfection control. Cells were split 1:2 around 30 h posttransfection and harvested 72 h posttransfection. Plasmid DNA was precipitated and isolated by phenol-chloroform extraction. Replicated plasmid DNA was distinguished from transfected plasmid DNA by digestion with the methylation-sensitive enzyme DpnI. DNA bands were separated in an agarose gel and visualized by Southern blotting (top two panels). Detection of expression of LANA proteins used in the experiment with an α-LANA antibody (bottom panel). (B) Quantification of the relative replication mediated by LANA or LANA Δ 971–986 calculated with ImageJ based on relative band intensity. The data shown represent an average value from three independent experiments. The difference in replication was calculated to be significant using a one-sample t test (two sided, P < 0.01). (C) The GST fusion proteins cLANA, cLANA PYG/AAA, and cLANA Δ 971–986 as well as GST alone as control were expressed in bacterial cells, precipitated by glutathione-Sepharose 4B beads, and eluted with glutathione after several washing steps. The purified GST proteins were subjected to SDS-PAGE and visualized by Coomassie staining to adjust protein amounts for the binding reaction (left panels). Electrophoretic mobility shift assays were carried out with IRD700-labeled double-stranded oligonucleotides containing LBS-1 and LBS-2. Reactions were performed with the purified and adjusted GST proteins. Unlabeled oligonucleotides were used for competition and applied at a 200× excess. For supershift (last two lanes) a rabbit serum specific for LANA was added to the binding reaction and an unrelated rabbit antibody was used for control. The arrows mark specific shifts (right panel).
Increased replication ability of LANA Δ 971–986 is not due to altered DNA binding capacity.
To investigate if the increased replication activity of the LANA mutant lacking the USP7 binding site (aa 971 to 986) could be explained by differences in the ability of both LANA proteins to bind DNA, EMSAs (electrophoretic mobility shift assays) were performed. Therefore, a fluorescently labeled (IRD700) oligonucleotide harboring both LBS-1 and LBS-2 was used. The oligonucleotide was incubated with previously expressed and purified GST-tagged cLANA proteins in the EMSA binding reaction. Protein complexes were analyzed on a native polyacrylamide gel, and bands were detected via fluorescence.
The C-terminal LANA domain lacking aa 971 to 986 bound to DNA to the same extent as cLANA WT (Fig. 5C). For both LANA proteins, a pattern of shifted bands was observed, of which those indicated by arrows in Fig. 5C are considered to represent specific shifts. Application of unlabeled oligonucleotide in great excess efficiently competed with binding of cLANA to DNA, and the bands seen in the presence of WT cLANA could be “supershifted” using an antibody to LANA. These results indicate the specific nature of the observed bands. As an additional control, a LANA mutant with a PYG motif mutated to three alanine residues was included; this mutant had previously been reported to abolish binding to the LBS regions (23). As expected, this mutant did not interact with DNA (Fig. 5C).
USP7 does not influence the transactivation function of LANA.
LANA acts as a transcriptional repressor or activator of several cellular and viral genes. Therefore, we analyzed the possible influence of USP7 binding on the role of LANA as a transactivator or repressor. Since LANA plays a role in E2F-dependent transcription, we examined the effect of USP7 binding on the activation of the cyclin E promoter, as previously shown (36). The mutant LANA Δ 971–986 was able to activate the cyclin E promoter to levels comparable to those achieved by LANA WT (Fig. 6A). Second, the full-length LANA mutant lacking the identified USP7 binding site (LANA Δ 971–986) (Fig. 6B) and the corresponding C-terminal mutant (cLANA Δ 971–986) (data not shown) were analyzed for their ability to modulate the p53-dependent activation of a p53-responsive promoter, known to be repressed by LANA (13), in p53-deficient H1299 cells. Neither of the LANA mutants was compromised in its ability to repress p53-dependent transcription (Fig. 6B and data not shown) in comparison to LANA WT.
Fig 6.

Transcriptional activation or repression by LANA is not influenced by USP7 recruitment. (A to C) Representation of luciferase reporter experiments to address the role of USP7 on LANA-mediated transcriptional activity. All results were based on duplicate samples. (A) 293-T cells were transfected with 50 ng reporter plasmid harboring the cyclin E promoter and the indicated amounts of the LANA plasmids. Transfection of empty vectors instead of protein-encoding plasmids served as controls. Cells were lysed 48 h posttransfection, and luciferase activity was measured. Expression of LANA constructs was verified by immunoblotting using a human serum recognizing LANA. (B) Reporter plasmid (50 ng) harboring a p53-responsive element from the mdm2 promoter region, 200 ng p53, and the indicated amounts of full-length LANA plasmids were cotransfected in p53-negative H1299 cells. The assay was carried out as described for panel A. (C) 293 cells were transfected with 50 ng reporter plasmid harboring the KSHV TR region in front of a luciferase gene and the indicated amounts of the LANA plasmids. The assays were carried out as described for panel A. Expression of LANA was verified by immunoblotting using a human serum recognizing LANA. Reporter assays shown in this figure (A to C) are representative of results of multiple experiments. Similar results were obtained when luciferase values were normalized to those obtained with a luciferase control vector (not shown).
LANA also represses the promoter/enhancer activity of the TR promoter region in the KSHV genome (51). We analyzed the impact of the LANA Δ 971–986 mutant on a TR reporter vector in HEK 293 cells. Both the LANA WT protein and the mutant lacking the USP7 binding site (aa 971 to 986) inhibited the TR reporter vector (Fig. 6C). Taken together, these findings indicate that recruitment of USP7 to LANA does not influence LANA transcriptional activation or repression and does not impact on the regulation of p53-dependent transcription by LANA.
USP7 interacts with the Orf73 proteins of MHV68 and RRV.
We next explored whether homologous Orf73 proteins from KSHV-related gammaherpesviruses could also interact with USP7. We focused on the Orf73 proteins from murine herpesvirus 68 (MHV68) as well as rhesus rhadinovirus (RRV). Expression constructs for both proteins were cotransfected with a USP7 expression construct or the corresponding empty vector. Protein complexes were precipitated via tags fused to the Orf73 proteins and analyzed on Western blots. In each experiment, a C-terminal LANA construct harboring the same tag served as a positive control. Both proteins, MHV68 Orf73 (Fig. 7A) and RRV Orf73 (Fig. 7B), interacted reproducibly with USP7 in coimmunoprecipitation experiments. Together with the previously investigated role of USP7 in the function of EBV EBNA-1, these findings suggest that the interaction of USP7 with Orf73 proteins is conserved among different gammaherpesviruses and underlines the possible importance of this interaction for the viral latent life cycle.
Fig 7.

LANA homologues of other gamma-2 herpesviruses also recruit USP7. (A) HA-tagged expression constructs for the C-terminal domain of LANA (aa 934 to 1162) or the latent Orf73 protein of MHV68 and a plasmid encoding GFP-tagged USP7 were cotransfected in equal amounts (1 μg each) in 293-T cells. As a control, the corresponding empty vectors were transfected. Cells were lysed after 48 h, and protein complexes were precipitated with α-HA antibody coupled to protein G-Sepharose beads and analyzed by immunoblotting using α-HA and α-GFP antibodies. (B) Myc-tagged expression constructs for the C-terminal domain of LANA (aa 934 to 1162) or the latent Orf73 protein of RRV and a plasmid encoding GFP-tagged USP7, or the appropriate control vectors, were cotransfected in equal amounts (1 μg each) in 293-T cells. Cells were lysed after 48 h, and protein complexes were precipitated with α-myc antibody coupled to agarose beads and analyzed by immunoblotting using α-myc and α-GFP antibodies.
DISCUSSION
KSHV can establish a lifelong persistent infection in humans. LANA is involved in virus persistence, as it tethers the viral episome to human chromatin. LANA binds the TR region of the KSHV genome and different histones of the human chromatin (1, 6, 15), and it is also involved in latent replication of the viral genome. To ensure viral persistence, LANA interacts with different cellular proteins. We observed that LANA, as well as the LANA homologues of two other gamma-2 herpesviruses, MHV68 and RRV, recruits the deubiquitinating enzyme USP7 (Fig. 1 and 7).
Together with previous studies on the recruitment of USP7 by EBNA-1, our findings suggest that USP7 is a cellular protein which plays an important role during gammaherpesvirus latency. The importance of USP7 is illustrated by the multitude of USP7 targets identified so far, which include several transcription factors (e.g., FOXO4), tumor suppressor proteins (e.g., PTEN, p53), and epigenetic modifiers (e.g., DNMT1). In short, USP7 is involved in cellular responses to various stress stimuli and in maintaining cellular homeostasis. Disregulation of USP7 is associated with diseases such as cancer and neurodegenerative disorders.
In the case of HSV-1 and EBV, the function of USP7 is thought to include the stabilization of HSV-1 ICP0 or to ensure p53 degradation in EBV-infected cells, respectively (2, 3, 18, 39, 43). We wanted to elucidate the functional importance of the LANA-USP7 interaction and mapped the binding site of USP7 on LANA in order to create a LANA mutant deficient for USP7 binding. This LANA mutant was used to study the role of USP7 recruitment to LANA. The C-terminal domain of LANA alone is able to mediate the interaction with USP7 (Fig. 2), and mapping of the USP7 binding site was achieved with a set of truncated C-terminal LANA constructs (Fig. 2B to D). This led to the identification of a small region of 16 aa localized between aa 971 and 986, which still interacts with USP7 (Fig. 3). In turn, the deletion of this region from the C-terminal domain of LANA abolishes interaction with USP7 (Fig. 4). Additionally, we assessed whether deletion of the identified USP7 binding region (aa 971 to 986) abrogated binding to USP7 in the context of full-length LANA. This LANA mutant retained some residual interaction with USP7 in coimmunoprecipitation assays, but it failed to interact with the USP7 TRAF domain in GST pulldown assays (Fig. 4C and D). The residual binding could not be attributed to an additional USP7 binding site in the N-terminal domain of LANA (Fig. 2A), although possible USP7 binding sites, according to the USP7 consensus binding motif (43), are present in the N-terminal part of LANA (not shown). It is possible that residual binding of USP7 to the LANA mutant occurs indirectly via another protein binding both LANA and USP7. A potential candidate could be p53, which was shown to interact directly with LANA (13) but is also a substrate of USP7.
The identified USP7 binding region was found to be conserved in EBNA-1, which is a functional homologue of, but displays only limited sequential homology to, LANA (Fig. 3). Within the USP7 binding region of LANA and EBNA-1 several amino acids are identical, highlighting the conservation of the USP7 binding site in LANA and EBNA-1.
This USP7 binding region in LANA centers on a PGPS motif that corresponds to published consensus sequences (21, 43). Substitution of this PGPS motif with alanines eliminated the binding of USP7 to the C-terminal domain of LANA (Fig. 4B). Taken together, these interaction studies revealed a potential USP7 binding site in the C-terminal domain of LANA between aa 971 and 986, including a PGPS motif that mediates contact to USP7.
With the exception of ICP0, USP7 interacts with its partners through the N-terminal TRAF domain (19). We observed that LANA also binds to the N-terminal TRAF domain of USP7 (Fig. 4D), indicating an interacting pattern similar to those of EBNA-1, p53, MdmX, and Mdm2. Nevertheless, we cannot exclude that further USP7 regions are involved in the interaction with LANA.
To investigate the functional importance of the interaction between LANA and USP7, we compared LANA WT with the deletion mutant lacking aa 971 to 986 in functional assays. A clear effect of USP7 recruitment to LANA was observed when using an assay that measures the replication of a KSHV episomal genome. In a transient replication assay, the LANA mutant lacking the USP7 binding site (aa 971 to 986) consistently showed increased replication activity (Fig. 5A and B). This is in line with observations for EBNA-1, as an EBNA-1 mutant that does not bind to USP7 was also shown to replicate the viral DNA more efficiently (19). These findings indicate that recruitment of USP7 to LANA can modulate latent viral DNA replication.
The increased replication activity of LANA upon deletion of the USP7 binding site cannot be attributed to an enhanced DNA binding ability of the mutant, as shown by EMSAs (Fig. 5C). This is in line with the currently held notion that LANA aa 1025 to 1162 form a domain required for DNA binding and dimerization (23, 42), and the USP7 binding site identified here would therefore lie outside this domain. The effect of USP7 on LANA-mediated viral genome replication could have different explanations. USP7 was shown to deubiquitinate histone H2B (41). Mono-ubiquitination of H2B was reported to influence transcription, both positively and negatively (12, 31, 32, 53), but effects on other processes, such as DNA replication, cannot be excluded. A complete lack or reduced recruitment of USP7 to LANA could therefore influence H2B mono-ubiquitination by either enhancing or inhibiting deubiquitination of H2B, which, in turn, could have an effect on viral DNA accessibility for transcription or replication. In our hands, the LANA Δ 971–986 mutant showed the same ability as LANA to activate heterologous reporters (Fig. 6A), to repress p53-mediated transcription of the mdm2 promoter (Fig. 6B), and to act as a transcriptional repressor of the LANA TR promoter (Fig. 6C). It is, therefore, in our view less likely that the recruitment of USP7 to LANA plays a role during LANA-mediated transcriptional activation or repression. Since the KSHV episome is also complexed with histones (45, 52), USP7 could influence accessibility of the viral DNA for latent replication or influence viral persistence mediated by LANA, as LANA is able to bind to histone H2B directly.
Likewise, the LANA Δ 971–986 mutant can repress p53-dependent transcription from a p53-dependent promoter to the same extent as LANA WT. This observation suggests that the recruitment of USP7 to LANA is unlikely to silence USP7, thereby modifying p53 function by increasing the amount of either ubiquitinated p53 or Mdm2, the p53 E3 ligase. In conclusion, taken together with previously published findings on the role of USP7 in EBNA-1-dependent latent replication of EBV, our findings suggest that a role of USP7 in the replication of latent viral DNA appears to be a conserved feature among gamma-1 and gamma-2 herpesviridae.
ACKNOWLEDGMENTS
We thank Melanie Scholz for cloning several LANA constructs used in this study. We are very grateful to the following people for providing us with plasmids and expression constructs: S. Mittnacht (London) for the empty vector pcDNA3myc, J. Stewart (Liverpool) for the HA-containing empty vector pVR1255 and MHV68 Orf73 in pVR1255, R. Searles (Portland, OR) for myc-tagged full-length RRV Orf73, R. Everett (Glasgow), for the expression vector of GFP-tagged USP7, R. Weinberg (Cambridge, MA), for the cyclin E luciferase reporter vector, and M. Dobbelstein (Göttingen) for the expression construct for HA-tagged p53 as well as the p53 luciferase reporter vector.
This work was supported by the EU Integrated Project INCA (LSHC-CT-18730), the DFG Priority program SPP1130, and the Collaborative Research Centre DFG SFB900 (Chronic infections, Mechanisms of microbial persistence and its control).
Footnotes
Published ahead of print 18 April 2012
REFERENCES
- 1. Barbera AJ, et al. 2006. The nucleosomal surface as a docking station for Kaposi's sarcoma herpesvirus LANA. Science 311:856–861 [DOI] [PubMed] [Google Scholar]
- 2. Boutell C, Canning M, Orr A, Everett RD. 2005. Reciprocal activities between herpes simplex virus type 1 regulatory protein ICP0, a ubiquitin E3 ligase, and ubiquitin-specific protease USP7. J. Virol. 79:12342–12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Canning M, Boutell C, Parkinson J, Everett RD. 2004. A RING finger ubiquitin ligase is protected from autocatalyzed ubiquitination and degradation by binding to ubiquitin-specific protease USP7. J. Biol. Chem. 279:38160–38168 [DOI] [PubMed] [Google Scholar]
- 4. Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186–1191 [DOI] [PubMed] [Google Scholar]
- 5. Chang Y, et al. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869 [DOI] [PubMed] [Google Scholar]
- 6. Cotter MA, II, Robertson ES. 1999. The latency-associated nuclear antigen tethers the Kaposi's sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells. Virology 264:254–264 [DOI] [PubMed] [Google Scholar]
- 7. Cummins JM, et al. 2004. Tumour suppression: disruption of HAUSP gene stabilizes p53. Nature 428:1 p following 486 [DOI] [PubMed] [Google Scholar]
- 8. Cummins JM, Vogelstein B. 2004. HAUSP is required for p53 destabilization. Cell Cycle 3:689–692 [PubMed] [Google Scholar]
- 9. Dhar SK, et al. 2001. Replication from oriP of Epstein-Barr virus requires human ORC and is inhibited by geminin. Cell 106:287–296 [DOI] [PubMed] [Google Scholar]
- 10. Dupin N, et al. 1999. Distribution of human herpesvirus-8 latently infected cells in Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma. Proc. Natl. Acad. Sci. U. S. A. 96:4546–4551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Everett RD, et al. 1997. A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J. 16:1519–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fleming AB, Kao CF, Hillyer C, Pikaart M, Osley MA. 2008. H2B ubiquitylation plays a role in nucleosome dynamics during transcription elongation. Mol. Cell 31:57–66 [DOI] [PubMed] [Google Scholar]
- 13. Friborg J, Jr, Kong W, Hottiger MO, Nabel GJ. 1999. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 402:889–894 [DOI] [PubMed] [Google Scholar]
- 14. Fujimuro M, et al. 2003. A novel viral mechanism for dysregulation of beta-catenin in Kaposi's sarcoma-associated herpesvirus latency. Nat. Med. 9:300–306 [DOI] [PubMed] [Google Scholar]
- 15. Garber AC, Shu MA, Hu J, Renne R. 2001. DNA binding and modulation of gene expression by the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Virol. 75:7882–7892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Geng Y, et al. 1996. Regulation of cyclin E transcription by E2Fs and retinoblastoma protein. Oncogene 12:1173–1180 [PubMed] [Google Scholar]
- 17. Grundhoff A, Ganem D. 2003. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus permits replication of terminal repeat-containing plasmids. J. Virol. 77:2779–2783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Holowaty MN, Sheng Y, Nguyen T, Arrowsmith C, Frappier L. 2003. Protein interaction domains of the ubiquitin-specific protease, USP7/HAUSP. J. Biol. Chem. 278:47753–47761 [DOI] [PubMed] [Google Scholar]
- 19. Holowaty MN, et al. 2003. Protein profiling with Epstein-Barr nuclear antigen-1 reveals an interaction with the herpesvirus-associated ubiquitin-specific protease HAUSP/USP7. J. Biol. Chem. 278:29987–29994 [DOI] [PubMed] [Google Scholar]
- 20. Hu J, Garber AC, Renne R. 2002. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus supports latent DNA replication in dividing cells. J. Virol. 76:11677–11687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hu M, et al. 2006. Structural basis of competitive recognition of p53 and MDM2 by HAUSP/USP7: implications for the regulation of the p53-MDM2 pathway. PLoS Biol. 4:e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kellam P, et al. 1999. Characterization of monoclonal antibodies raised against the latent nuclear antigen of human herpesvirus 8. J. Virol. 73:5149–5155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kelley-Clarke B, et al. 2007. Determination of Kaposi's sarcoma-associated herpesvirus C-terminal latency-associated nuclear antigen residues mediating chromosome association and DNA binding. J. Virol. 81:4348–4356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krithivas A, Fujimuro M, Weidner M, Young DB, Hayward SD. 2002. Protein interactions targeting the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus to cell chromosomes. J. Virol. 76:11596–11604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Krithivas A, Young DB, Liao G, Greene D, Hayward SD. 2000. Human herpesvirus 8 LANA interacts with proteins of the mSin3 corepressor complex and negatively regulates Epstein-Barr virus gene expression in dually infected PEL cells. J. Virol. 74:9637–9645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li M, Brooks CL, Kon N, Gu W. 2004. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol. Cell 13:879–886 [DOI] [PubMed] [Google Scholar]
- 27. Li M, et al. 2002. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 416:648–653 [DOI] [PubMed] [Google Scholar]
- 28. Lim C, Gwack Y, Hwang S, Kim S, Choe J. 2001. The transcriptional activity of cAMP response element-binding protein-binding protein is modulated by the latency associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Biol. Chem. 276:31016–31022 [DOI] [PubMed] [Google Scholar]
- 29. Lim C, Sohn H, Lee D, Gwack Y, Choe J. 2002. Functional dissection of latency-associated nuclear antigen 1 of Kaposi's sarcoma-associated herpesvirus involved in latent DNA replication and transcription of terminal repeats of the viral genome. J. Virol. 76:10320–10331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Meulmeester E, et al. 2005. Loss of HAUSP-mediated deubiquitination contributes to DNA damage-induced destabilization of Hdmx and Hdm2. Mol. Cell 18:565–576 [DOI] [PubMed] [Google Scholar]
- 31. Minsky N, Oren M. 2004. The RING domain of Mdm2 mediates histone ubiquitylation and transcriptional repression. Mol. Cell 16:631–639 [DOI] [PubMed] [Google Scholar]
- 32. Minsky N, et al. 2008. Monoubiquitinated H2B is associated with the transcribed region of highly expressed genes in human cells. Nat. Cell Biol. 10:483–488 [DOI] [PubMed] [Google Scholar]
- 33. Neipel F, Albrecht JC, Fleckenstein B. 1997. Cell-homologous genes in the Kaposi's sarcoma-associated rhadinovirus human herpesvirus 8: determinants of its pathogenicity? J. Virol. 71:4187–4192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ottinger M, et al. 2006. Kaposi's sarcoma-associated herpesvirus LANA-1 interacts with the short variant of BRD4 and releases cells from a BRD4- and BRD2/RING3-induced G1 cell cycle arrest. J. Virol. 80:10772–10786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Platt GM, Simpson GR, Mittnacht S, Schulz TF. 1999. Latent nuclear antigen of Kaposi's sarcoma-associated herpesvirus interacts with RING3, a homolog of the Drosophila female sterile homeotic (fsh) gene. J. Virol. 73:9789–9795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Radkov SA, Kellam P, Boshoff C. 2000. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 6:1121–1127 [DOI] [PubMed] [Google Scholar]
- 37. Rainbow L, et al. 1997. The 222- to 234-kilodalton latent nuclear protein (LNA) of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) is encoded by orf73 and is a component of the latency-associated nuclear antigen. J. Virol. 71:5915–5921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Russo JJ, et al. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. U. S. A. 93:14862–14867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Saridakis V, et al. 2005. Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1 implications for EBV-mediated immortalization. Mol. Cell 18:25–36 [DOI] [PubMed] [Google Scholar]
- 40. Sarkari F, et al. 2010. Further insight into substrate recognition by USP7: structural and biochemical analysis of the HdmX and Hdm2 interactions with USP7. J. Mol. Biol. 402:825–837 [DOI] [PubMed] [Google Scholar]
- 41. Sarkari F, et al. 2009. EBNA1-mediated recruitment of a histone H2B deubiquitylating complex to the Epstein-Barr virus latent origin of DNA replication. PLoS Pathog. 5:e1000624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schwam DR, Luciano RL, Mahajan SS, Wong L, Wilson AC. 2000. Carboxy terminus of human herpesvirus 8 latency-associated nuclear antigen mediates dimerization, transcriptional repression, and targeting to nuclear bodies. J. Virol. 74:8532–8540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sheng Y, et al. 2006. Molecular recognition of p53 and MDM2 by USP7/HAUSP. Nat. Struct. Mol. Biol. 13:285–291 [DOI] [PubMed] [Google Scholar]
- 44. Soulier J, et al. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276–1280 [PubMed] [Google Scholar]
- 45. Stedman W, Deng Z, Lu F, Lieberman PM. 2004. ORC, MCM, and histone hyperacetylation at the Kaposi's sarcoma-associated herpesvirus latent replication origin. J. Virol. 78:12566–12575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tang J, et al. 2006. Critical role for Daxx in regulating Mdm2. Nat. Cell Biol. 8:855–862 [DOI] [PubMed] [Google Scholar]
- 47. van der Knaap JA, et al. 2005. GMP synthetase stimulates histone H2B deubiquitylation by the epigenetic silencer USP7. Mol. Cell 17:695–707 [DOI] [PubMed] [Google Scholar]
- 48. Verma SC, et al. 2006. Latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus recruits uracil DNA glycosylase 2 at the terminal repeats and is important for latent persistence of the virus. J. Virol. 80:11178–11190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Verma SC, Borah S, Robertson ES. 2004. Latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus up-regulates transcription of human telomerase reverse transcriptase promoter through interaction with transcription factor Sp1. J. Virol. 78:10348–10359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Viejo-Borbolla A, et al. 2003. A domain in the C-terminal region of latency-associated nuclear antigen 1 of Kaposi's sarcoma-associated herpesvirus affects transcriptional activation and binding to nuclear heterochromatin. J. Virol. 77:7093–7100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Viejo-Borbolla A, et al. 2005. Brd2/RING3 interacts with a chromatin-binding domain in the Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 (LANA-1) that is required for multiple functions of LANA-1. J. Virol. 79:13618–13629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wong LY, Wilson AC. 2005. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen induces a strong bend on binding to terminal repeat DNA. J. Virol. 79:13829–13836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhao Y, et al. 2008. A TFTC/STAGA module mediates histone H2A and H2B deubiquitination, coactivates nuclear receptors, and counteracts heterochromatin silencing. Mol. Cell 29:92–101 [DOI] [PubMed] [Google Scholar]