

Abstract
Activation of the phosphatidylinositol-3-kinase (PI3K) pathway is one of the most frequently observed molecular alterations in many human malignancies, including head and neck squamous cell carcinoma (HNSCC). A growing body of evidence demonstrates the prime importance of the PI3K pathway at each stage of tumorigenesis, that is, tumor initiation, progression, recurrence, and metastasis. Expectedly, targeting the PI3K pathway yields some promising results in both preclinical studies and clinical trials for certain cancer patients. However, there are still many questions that need to be answered, given the complexity of this pathway and the existence of its multiple feedback loops and interactions with other signaling pathways. In this paper, we will summarize recent advances in the understanding of the PI3K pathway role in human malignancies, with an emphasis on HNSCC, and discuss the clinical applications and future direction of this field.
1. Introduction
The phosphatidylinositol-3-kinase (PI3K) signaling pathway is one of the pathways most commonly activated in human cancers [1]. It is a major downstream signaling component of receptor tyrosine kinases (RTKs) and is critical for the regulation of cell proliferation, growth, differentiation, migration, and survival [2]. Thus, it represents one of the most promising targets for cancer prevention and therapy [3].
Mounting reports of original studies and reviews have been published, highlighting the paramount importance of this pathway in human cancers. To avoid redundancy with previous publications, in this paper we will focus on summarizing recent progress in PI3K pathway research in head and neck squamous cell carcinoma (HNSCC). Specifically, this will include considerations of the molecular alterations seen in some components of the PI3K pathway, as well as functional studies of the role of the PI3K pathway in HNSCC initiation, invasion, and metastasis studied both in vitro and in vivo, with a particular focus on the use of genetically engineered mouse models (GEMMs). Finally, we will explore the potential of the PI3K pathway as a target for chemoprevention and cancer therapy.
2. Common Molecular Alterations of HNSCCs
HNSCC refers to squamous cell carcinomas (SCCs) arising from the oral cavity, tongue, pharyngeal, and laryngeal regions. As the 6th most common human cancer worldwide, they generate about 600,000 new cases and 350,000 cancer deaths each year [4, 5]. HNSCCs usually occur at a relatively late age and at a higher frequency in males possessing the well-known etiological factors of tobacco and/or alcohol usage [4, 5]. Recently, however, the incidence of HNSCC is increasing in women of a relatively young age, correlating with human papilloma virus (HPV) infection [4, 5].
Historically, the best known molecular alterations in HNSCC were the inactivation of tumor suppressors, such as p16 and p53, and activation of oncogenes, such as EGFR and Stat3 [5, 6]. We have studied the role of transforming growth factor beta (TGFβ) pathway in HNSCC using both human HNSCC samples and GEMM approaches [7–10]. Our studies indicate that inactivation of the type II receptor of TGFβ (TGFβRII) and the downstream signal mediator of TGFβ, Smad4, plays a crucial role in HNSCC development and progression [8, 10]. Perhaps the most comprehensive studies on molecular alterations of HNSCC came from two recent publications describing whole exome sequencing on human HNSCC samples [11, 12]. Two major results were generated from these papers: (1) the discovery of novel molecular alterations of the Notch signaling pathway in human HNSCC samples and (2) the validation of the PI3K pathway as one of the major targets for molecular alterations in human HNSCC samples, including alterations of the oncogene PIK3CA and the tumor suppressor gene PTEN. These two papers, together with many previous reports, clearly demonstrate the importance of the PI3K pathway in HNSCC. Furthermore, they suggest its possible involvement in every aspect of HNSCC development and progression, including tumor initiation, invasion, recurrence, resistance to therapeutics, and metastasis.
3. PI3K Signaling Pathway
PI3Ks are a family of enzymes that phosphorylate the 3-OH group on phosphatidylinositols. There are three classes of PI3Ks, with IA PI3K being the type most widely implicated in human cancers [13]. Class IA PI3K primarily phosphorylate phosphatidylinositol-4,5,bisphosphate [PI(4,5)P2] in the plasma membrane to generate the second messenger phosphatidylinositol-3,4,5,trisphosphate [PI(3,4,5)P3]. These enzymes are heterodimers, consisting of a p85 regulatory and p110 catalytic subunit [13]. Class IA PI3K are most often activated by RTK signaling and indirectly by Ras. Upon RTK signaling, p85 binds to either phosphotyrosine residues or adaptor molecules. This binding serves both to recruit the p85-p110 heterodimer to the plasma membrane and to relieve the basal inhibition of p110 by p85. p110 then phosphorylates PI(4,5)P2 to generate PI(3,4,5)P3. The 3-phosphatase PTEN (phosphatase and tensin homologue) dephosphorylates PI(3,4,5)P3 and catalyses the reverse reaction. PI(3,4,5)P3 binds a subset of pleckstrin homology domain-containing proteins, including 3-phosphoinositide-dependent protein kinase (PDK1) and protein kinase B (also called AKT) to the plasma membrane. Once there, AKT is phosphorylated at Thr308 by PDK1 and Ser473 by the mammalian target of rapamycin (mTOR) complex 2 (mTORC2) [14]. It is believed that AKT is the central signal mediator of the canonical PI3K signaling pathway. However, recent studies also suggest that the link between PI3K and AKT can be uncoupled [15–18] and that oncogenic PI3K signaling can be transmitted through an AKT-independent pathway, further adding to the complexity of PI3K signal transduction. AKT phosphorylates numerous downstream targets that regulate a wide array of cellular processes important in tumor development and progression [19]. One of its major effectors is mTOR complex 1 (mTORC1), which is activated in multiple human cancers and is one of the major targets in the PI3K pathway for chemoprevention and therapy [14] (Figure 1).
Figure 1.
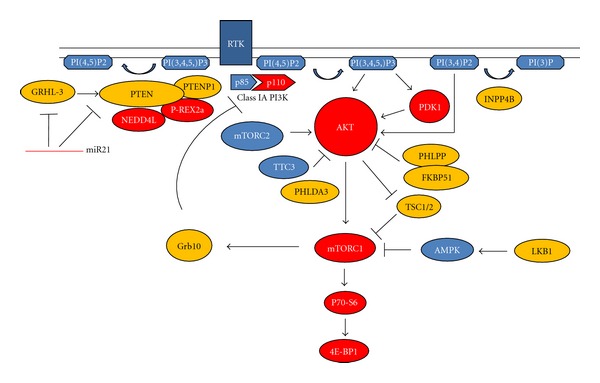
Schematic of the PI3K/AKT/mTOR pathway and its interacting molecules. Red: molecules have oncogenic property. Yellow: molecules have tumor suppression property.
In human HNSCC, molecular alterations at the levels of both expression and function have been identified. These include gain-of-function mutations and amplifications in PIK3CA (the gene coding p110α, the catalytic subunit of PI3K), loss of heterozygosity and inactivating mutations in PTEN, and overexpression/activation of AKT and mTOR signaling [5, 6]. Several reports utilizing the GEMM approach, including our own studies, have confirmed the functional importance of these molecular alterations in HNSCC development and progression. In the following sub-sections, we will summarize several molecular alterations and functional studies, particularly those using GEMMs, involving molecular components of the PI3K pathway in HNSCC tumorigenesis.
3.1. PIK3CA
PIK3CA, the gene coding for the catalytic subunit p110α of PI3K, is one of the most commonly mutated oncogenes in multiple human malignancies (3441 mutated samples among a total of 27725 samples, about 12%, according to the Catalogue of Somatic Mutations in Cancer (COSMIC) Database (http://www.sanger.ac.uk/genetics/CGP/cosmic/)). Most of these mutations are clustered in exon 9 and exon 20, which corresponds to the helical domain mutant E545K, and the kinase domain mutant H1047R, respectively. Almost all PIK3CA mutations are gain-of-function mutants, further supporting its oncogenic role in human malignancies [1]. In human HNSCC samples, the PIK3CA mutation rate is about 10% [20] but is relatively higher (20%) in HNSCC arising from a pharyngeal site [21]. In addition to somatic mutations, genomic amplification of PIK3CA has also been reported in several human cancers [1]. Interestingly, a significantly higher percentage of PIK3CA gene amplification was noted in squamous cell carcinoma, compared to adenocarcinoma in lung [22]. In human HNSCC tissue samples, over 30% of cases involving PIK3CA amplification involve the candidate gene residing in the common amplification region of 3q26.3 in human HNSCC samples [23, 24].
PIK3CA alterations have been associated with cancer recurrence [25], metastasis [26, 27], and poor prognosis [28, 29] in a variety of human cancers. In HNSCC, PIK3CA alterations correlate with an advanced stage [30, 31], vascular invasion [24], and lymph node metastasis [32]. Interestingly, in breast cancer cell motility and metastatic potential are differentially enhanced depending on whether their mutations are localized at the helical or kinase domain. An overexpression of the helical domain through mutation E545K of PIK3CA produces a more severe metastatic phenotype compared to that of the kinase domain mutation H1047R [33].
Although the activated forms of PIK3CA, generated through either mutation or amplification, are transforming in vitro [1, 13], their oncogenic potential in vivo has only recently been assessed through the GEMM approach. While deletion or inactivation of PIK3CA significantly impairs oncogenic transformation [34] and produces a significant resistance to Ras-oncogene-induced tumorigenesis [35], overexpression of PIK3CA results in hyperplasia in ovarian surface epithelium [36] and can predispose mammary glands to neoplastic transformation [37]. Moreover, a knock-in PIK3CA H1047R mutant is sufficient to induce lung and breast cancer development [38–40]. Work being done in our own lab suggests that these trends apply to HNSCC as well. Using a HNSCC inducible transgenic mouse line that we had previously developed [9], we observe a strong oncogenic role of PIK3CA in HNSCC at both initiation and progression when it is overexpressed in head and neck epithelium (L. Du et al.'s manuscript in preparation).
The underlying molecular mechanisms of PI3K-driven oncogenesis are still unclear. Although AKT is largely regarded as the dominant mediator of oncogenic PI3K signaling [2], recent studies suggest that the link between PI3K and AKT can be uncoupled [15–18]. For example, PDK1, but not AKT, is activated in some breast cancers with PIK3CA mutations [15] (Figure 1). Using a mouse model of breast cancer conditionally expressing the PIK3CA H1047R mutant, Liu et al. have shown that PIK3CA-driven mammary tumors occur via both PI3K-pathway-dependent and PI3K-pathway-independent mechanisms, suggesting the complexity of the PI3K-driven oncogenic mechanisms [40]. To this point, sophisticated PIK3CA-GEMMs for a variety of cancer types may prove to be powerful tools in revealing the role of PIK3CA in a context- and stage-specific manner.
3.2. Other PI3K Molecules
Besides the common alterations of the PIK3CA gene encoding the catalytic p110α subunit of class IA PI3K, somatic mutations in the PIK3R1 gene encoding the regulatory subunit p85α have been detected in multiple human cancers [41], including endometrium (26%), colon (5%), central nervous system (4%), breast (2%), pancreatic (2%), and skin (1%) (adapted from the COSMIC Database). Interestingly, somatic mutations of PIK3R1 are fairly common (7%, 3/41) in human HNSCC samples, with two missense mutations and one in-frame insertion [12]. The functional consequence of these mutants seems oncogenic as the mutants weaken an inhibitory interaction while retaining a stabilizing interaction between p85α and p110α, resulting in an activation of PI3K signaling [41]. However, p85α has also been shown to positively regulate PTEN [42], and reduced expression of p85α correlates with decreased PTEN expression [43]. Furthermore, deletion of PIK3R1 in mouse liver resulted in aggressive hepatocellular carcinomas with pulmonary metastasis, suggesting a tumor suppressor role [43]. Thus, further characterization of PIK3R1 mutants using both in vitro and in vivo approaches is warranted to reveal its role in HNSCC tumorigenesis.
Similar to the oncogenic role of the p110α catalytic subunit, the other isoforms of the catalytic subunit, p110β, p110γ, and p110δ, have been shown to be oncogenic in experimental settings although there are no reports of molecular alterations of these isoform subunits in human cancer samples [44]. p110β has been studied the most among these isoform subunits, and most of the results came from the p110β-GEMM approaches. Using a PTEN-GEMM for prostate cancer, Jia et al. showed that ablation of p110β, but not p110α, impeded prostate tumorigenesis [45]. On the other hand, overexpression in a constitutively activated form of the p110β isoform induced prostate intraepithelial neoplasia in mice [46]. Furthermore, knock-in of a catalytically inactive form of p110β blocked tumor development in an ERBB2-GEMM for breast cancer [47]. Future research, using both human samples and GEMM approaches and aiming to assess the roles of these isoform subunits in HNSCC, will surely produce interesting results.
3.3. PTEN
PTEN acts as a negative regulator for the PI3K signaling by dephosphorylating PI(3,4,5)P3 and is the second most commonly mutated tumor suppressor in human cancers [48]. It is estimated that the overall frequency of PTEN mutations in sporadic human cancers is about 12% (2044 mutated samples among a total of 17452 samples according to the COSMIC Database), with endometrium cancer displaying the highest frequency among those considered (38%, 690 mutated samples among a total of 1837 samples in the COSMIC database). Somatic mutation of PTEN in SCCs of the head and neck is about 3% (22 mutated samples among a total of 745 samples in the COSMIC database) but is higher in SCCs of skin (14%, 92 mutated samples among a total of 658 samples in the COSMIC database). Compared to the relatively less common somatic mutation rate, loss of PTEN expression was more common (~30%) in human HNSCCs [49]. Loss of heterozygosity at chromosome 10q near PTEN was detected in over 70% of the PTEN-mutated HNSCCs [50], suggesting an inactivation of a typical tumor suppressor. Promoter hypermethylation of PTEN has also been detected in multiple PTEN expression-lacking human cancers [51, 52]. This is, however, infrequent (~5%) in human HNSCC samples [53]. Nonetheless, loss of PTEN expression has been correlated with tumor prognosis and incorporated into the grading system used for human HNSCC patients [49, 54].
The mechanisms driving the loss of PTEN expression in human cancers are still unclear. Mutations of PIK3R1 and PIK3R2 have been shown to affect PTEN stability [55]. Posttranscriptional regulation of PTEN by the developmental transcription factor GRHL-3 has been shown to correlate with PTEN loss in SCCs in both the skin and the head and neck [56]. Another potential mechanism for PTEN loss is through posttranscriptional regulation by recently discovered mRNAs, namely miR-21, miR-26a, and miR-106b-25, all of which have been identified as PTEN-targeting mRNAs [56–58]. We have shown recently that miR-9 level is positively correlated with PTEN level in human HNSCC cell lines [59]. PTEN expression level is also regulated posttranslationally. For example, the ubiquitin ligase NEDD4-1 has been shown to negatively correlate with PTEN level [60]. Whether NEDD4-1 overexpression accounts for a subset of PTEN loss in human HNSCC samples requires further investigation (Figure 1).
Other molecules closely related to PTEN have also been found to be altered in multiple human cancers. PIP3 RAC exchanger 2a (P-REX2a) has been implicated as a PTEN-interacting protein and antagonizes PTEN in human cancers [61]. Similar to the phosphatase activity of PTEN in the PI3K/AKT signaling pathway, inositol polyphosphate 4-phosphatase type II (INPP4B) is able to suppress the PI3K/AKT signaling pathway and behaves as a tumor suppressor in at least breast and ovarian cancers [62]. Lastly, the PTEN pseudogene PTENP1 has been shown to regulate PTEN level and acts as a tumor suppressor in human cancers [63] (Figure 1).
One of the major consequences of PTEN alteration is the activation of its main downstream targets AKT and mTOR, which are oncogenic in HNSCC tumorigenesis and are attractive targets for cancer therapies [48]. However, recent studies have identified several novel pathways downstream of PTEN. For example, the JNK signaling pathway has been found to be a functional target of PTEN and is significantly associated with PTEN loss [64]. Protein synthesis by the RNA-dependent protein kinase (PKR) and the subunit of eukaryotic translation initiation factor 2 (eIF2) phosphorylation pathway is also required for tumor suppression by PTEN [65]. These results generate potential therapeutic targets to act alongside targeting of the canonical PI3K signaling pathway.
Given the potent tumor suppressor role of PTEN in multiple human cancers, GEMMs possessing tissue-specific deletion of PTEN have been created to better understand PTEN in tumorigenesis [66]. The most striking PTEN-GEMM for human cancer is the prostate PTEN deletion model, in which mice with a single deletion of PTEN in prostate cells developed metastatic prostate cancer [66]. PTEN deletion also resulted in spontaneous tumor development in other organs, such as breast, lung, bladder, and skin, with a wide range of tumor onset pattern and penetrance [66]. Furthermore, PTEN deletion increases susceptibility of mice to the induction of lung cancer by the tobacco carcinogen NNK [4-(methylnitrosamino)-1-(3-pyridyl)-1-1-butanone] suggesting a role for PTEN in tobacco-induced tumor initiation [67]. Additionally, PTEN deletion enhances tumor development and progression in the presence of additional molecular changes, such as Ras and p53 [68, 69]. Using head-and-neck-specific GEMMs, we deleted PTEN specifically in the head and neck region and observed both premalignant lesions and tumor development. This head-and-neck-specific PTEN-GEMM can be utilized as a model for testing chemoprevention and therapeutic approaches targeting PI3K pathway (J. P. Shen et al.'s manuscript in preparation).
3.4. AKT
The serine/threonine kinase AKT is the central mediator of the canonical PI3K pathway and mediates multiple cellular processes, including cell survival, proliferation, angiogenesis, metabolism, and protein translation through numerous downstream signaling proteins [19]. There are many publications covering almost every aspect of AKT relation to human cancers. For this particular paper, we will focus on the following topics: (1) molecular alterations of AKT in human cancers, (2) isoform-specific role of AKT in human cancers, (3) tobacco carcinogen-induced AKT activation, (4) in vivo role of AKT in human cancers, and (5) regulation of AKT by interacting proteins.
3.4.1. Molecular Alterations of AKT in Human Cancers
Given its central node position in the canonical PI3K pathway, AKT can be activated by either upstream PIK3CA activation or PTEN inactivation. This subsequent AKT activation, together with molecular alterations of AKT itself, represents one of the most frequent molecular changes in human cancers and provides rationale for targeting AKT as a therapeutic approach.
All three isoforms of AKT, that is AKT1, AKT2, and AKT3, have been reported to be altered in various human cancers [70, 71]. Somatic mutation occurs most frequently in AKT1 and almost exclusively manifests as the E17K mutation [72, 73]. This AKT1 E17K somatic mutation was detected in about 5% of breast cancers and 3% of both thyroid and urinary cancers (adapted from the COSMIC database). Although the AKT1 E17K mutation has not been identified so far in human SCCs of head and neck according to a single report [74], this mutation has been found in human SCCs of lung [75]. Mutations of AKT2 have been sporadically reported in various human cancers, but none of these mutations occur at the corresponding position of the E17K in AKT1 [71]. Of particular note, somatic mutation of AKT2 is relatively common in endometrial carcinoma [71]. Mutations of AKT3 at both E17K and other sites were reported in melanoma [76] and endometrial carcinoma [71]. Unfortunately, as of yet there are no studies examining AKT2 and AKT3 mutations in human HNSCC samples. It is also worth noting that, although several studies have shown the oncogenic properties of these AKT mutations in vitro, a confirmation of their functional significance requires further investigation and demonstration of validity in vivo.
In addition to somatic mutation, overexpression of AKT isoforms, particularly AKT2, has been reported in multiple human cancers [77]. Gene amplification of AKT2 was reported in human ovarian and pancreatic cancers [77]. Also, overexpression of AKT2 at the messenger level has been shown in breast and colon cancers and seems to correlate with cancer migration, invasion, and metastasis [78, 79]. Interestingly, AKT2 has been shown to be transcriptionally regulated by the master regulator of epithelial-mesenchymal transition (EMT), Twist, and is associated with tumor progression and metastasis [78, 79]. Overexpression of AKT1 and AKT3 has only been shown in human gastric cancer [77] and melanoma [80], respectively. In human HNSCC samples the overexpression of AKT2, but not AKT1 or AKT3, has been reported in one study [81].
Pan-AKT activation through phosphorylation of Ser437 and Thr308 is fairly common in multiple human cancers [19, 70]. Evaluation of these phosphorylation sites yields prognostic value in human lung cancer [82] and predicts chemotherapeutic benefit in breast cancer [83]. Persistent AKT activation is also common in human HNSCC samples, and occurs as early as the premalignancy stage, including dysplasia and carcinoma in situ, suggesting that AKT activation is an early event in human HNSCC tumorigenesis [6, 84, 85]. However, reports have also shown AKT activation to correlate with a poor clinical outcome in human HNSCC patients [86, 87]. The role of AKT activation in human HNSCC development and progression is still in need of further investigation.
3.4.2. Isoform-Specific Role of AKT in Human Cancer
Although AKT1, 2, and 3 share high sequence homology, clinical studies suggest the existence of isoform-specific roles of AKT in multiple human cancers [19, 70]. This is further validated by experimental studies using both in vitro and in vivo approaches. While AKT1 and AKT2 play a similar role in regulating cell survival and proliferation, they behave distinctly in their regulation of cell migration and EMT [88, 89]. For example, AKT1 knockdown induces cell migration and EMT in breast cancer cell lines, while AKT2 knockdown suppresses these behaviors [90]. This is further exemplified in breast cancer mouse models: while overexpression of AKT1 accelerates ErbB-2 mediated mammary tumorigenesis and suppresses tumor invasion [91], overexpression of AKT2 markedly increases the incidence of pulmonary metastases in breast cancer [92]. These data suggest that AKT1 acts as a metastasis suppressor, while AKT2 as a metastasis promoter, further warranting the need to use isoform-specific AKT inhibitors in clinical management of cancer patients.
The underlying mechanisms regulating the isoform-specific roles of AKT are still unclear. Distinct downstream targets of each AKT isoform might mediate this separate signaling transduction and be responsible for the distinct behavior of AKT isoforms in human breast cancer progression and metastasis. A recent report showing regulation of mRNA-200, which plays a critical role in cell migration and EMT, by the ratio of AKT1 to AKT2 [93] suggests another potential mechanism for the isoform-specific roles of AKT. Whether these trends are context or stage specific is still unclear. As of yet, there are no human HNSCC studies that address these questions though they are needed to guide future clinical trials on HNSCC patients using AKT isoform-specific inhibitors.
3.4.3. Tobacco Exposure and AKT Activation
Though tobacco exposure is one of most important etiological factors in HNSCC tumorigenesis, its underlying molecular mechanisms remain poorly understood [4]. In addition to the formation of DNA adducts, tobacco carcinogens, such as NNK, activate several signal transduction pathways, including AKT, in both normal and cancer cells in the lung [94]. We have shown that, in both HNSCC tumors and the adjacent mucosa, AKT is activated at a higher frequency in HNSCC patients who are smokers compared to those who are nonsmokers [95]. Also, adding physiologically relevant concentrations of NNK to normal head and neck epithelial cells and HNSCC cell lines will rapidly and constitutively activate AKT in a dose-dependent and time-dependent manner. Finally, we demonstrated that NNK exposure to mouse head and neck epithelium results in epithelial hyperproliferation and reduced apoptosis, which is correlated with AKT activation [95]. These studies suggest that AKT activation plays a pivotal role in mediating tobacco-induced HNSCC carcinogenesis and that it may be an effective target for chemoprevention.
3.4.4. In Vivo Role of AKT in Human Cancers
The precise functional consequence of AKT activation cannot be assessed without in vivo studies. This is particularly critical for the evaluation of the isoform-specific role of AKT in context- and stage-specific manners. Current in vivo models of AKT activation overwhelmingly confirm its oncogenic role at differing levels of potency in various cancer types. However, at least in breast cancer models, the distinct roles of different AKT isoforms in cancer progression and metastasis have been observed as described previously. Mice constitutively overexpressing an activated AKT driven by a keratin 5 promoter developed both skin and head and neck tumors and an increased sensitivity to skin carcinogenesis [96, 97]. The oral lesions seen in these specimens were mostly epithelial dysplasia, their malignant transition hampered by the induction of premature senescence. This suggests that AKT activation is an early event but not sufficient by itself to HNSCC tumorigenesis [97]. This is further supported by the introduction of p53 loss, which synergizes with AKT activation to develop metastatic HNSCC [97].
3.4.5. Regulation of AKT by Interacting Proteins
Numerous proteins similar to PTEN have been identified as regulators of AKT activation and stability. PH domain leucine-rich repeat protein phosphatase (PHLPP) has been shown to attenuate AKT signaling through the regulation of distinct AKT isoforms [98]. Deletion or loss of expression of PHLPP has been reported in a significant fraction of colon and prostate cancers [99, 100]. FK506-binding protein 51 (FKBP51) has been shown to act as a scaffolding protein for AKT and PHLPP, as well as promote the dephosphorylation of AKT. Furthermore, FKBP51 is downregulated in pancreatic cancer samples and cell lines [101]. Posttranslational modifications of AKT through ubiquitinating proteins such as TTC3 have also been reported [102]; however, this protein role in oncogenic AKT activation has not been studied. Lastly, the p53 target gene PH domain-only protein (PHLDA3) has been found to compete with the PH domain of AKT for binding to membrane lipids, thereby inhibiting AKT translocation to the cellular membrane and, thus, its activation. Consistent with its function, loss of the PHLDA3 genomic locus is frequently observed in primary human lung cancer samples [103]. However, such studies have not yet been reported in HNSCC cases (Figure 1).
3.5. PDK1
Although it is widely accepted that AKT activation acts as the primary oncogenic mediator in canonical PI3K signaling [2], as has been stated previously in this paper, recent studies suggest that the link between PI3K and AKT can be uncoupled [15–18]. For example, AKT signaling is diminished in human breast cancer cell lines and clinical samples harboring PIK3CA mutation. In lieu of AKT, these cells make use of a signaling pathway involving the PI3K effector PDK1 and its downstream substrate SGK3 [15]. Compared to AKT, there are few studies of PDK1 in human cancers. Increased gene copy numbers of PDK1 have been found in 21% of breast cancer samples, and total PDK1 mRNA and protein have been observed to be overexpressed in the majority of human breast cancer [104]. Overexpression of PDK1 promotes invasion and activation of matrix metalloproteinase [105], while downregulation of PDK1 inhibits migration and experimental metastases of human breast cancer cells [106]. Introduction of a hypomorphic mutation of PDK1 in a PTEN cancer mouse model suppresses tumorigenesis [107], confirming the oncogenic role of PDK1 as one of the downstream effectors of the PI3K/PTEN signaling, and suggesting PDK1 as a promising anticancer target. However, the role of PDK1 and its correlation with the canonical PI3K/PTEN/AKT pathway in HNSCC development and progression has not been assessed yet.
3.6. mTOR and Its Related Molecules
Among the numerous molecules that could act as downstream effectors of the PI3K/AKT pathway, mTOR is of particular interest. mTOR assembles into at least two distinct complexes, that is, mTORC1 and mTORC2. mTORC2 contains Rictor, SIN1 and mLST8/GbL and acts upstream of AKT to phosphorylate the Ser473 of AKT. In contrast, mTORC1 contains Raptor, PRAS40, and mLST8/GbL and acts as a major downstream target of AKT. mTORC1 regulates cell growth by controlling key eukaryotic translational regulators, including p70-S6 kinase, and the eukaryotic translational initiation factor, 4E binding protein 1 (4E-BP1) [14]. In addition, the growth factor receptor-bound protein 10 (Grb10) has been recently identified as an mTORC1 substrate. The mTORC1-mediated phosphorylation stabilizes Grb10, leading to the inhibition of the PI3K and ERK-MAPK pathways. Interestingly, Grb10 is frequently downregulated in various human cancers, with the loss of Grb10 and PTEN being mutually exclusive. This indicates Grb1 as being a tumor suppressor, potentially regulated by mTORC1 [108] (Figure 1).
In addition to directly activating mTORC1, AKT also phosphorylates tuberous sclerosis complex (TSC) 1 and TSC2, releasing their inhibition of the Ras-like small G protein, Rheb, which in turn activates mTORC1. During conditions of low nutrient availability, mTOR signaling is normally inhibited by AMP activated protein kinase (AMPK), which is activated by its upstream serine/threonine kinase LKB1 (Figure 1). Interestingly, these negative regulators of mTOR, that is, TSC1, TSC2, and LKB1, are tumor suppressors, and germline mutations of TSC1/2 or LKB1 cause hamartomas and predisposition to multiple malignancies in humans [14]. The critical connection of mTOR to the PI3K/AKT pathway has led to the prediction that the targeting of mTOR may be useful in cancer therapy. Indeed, using the mTOR inhibitor, rapamycin, yields promising results for multiple human cancers [109].
In human HNSCC, activation of mTOR/p70-S6/4E-BP1 pathway is a frequent event in clinical specimens and cell lines [110]. Amplification of Rictor has been reported in one study [111]. LOH of TSC1/2 and DNA methylation of TSC2 have been reported in human HNSCC samples [112], and overexpression of TSC2 inhibits cell growth both in vitro and in vivo [113]. A somatic mutation of LKB1, which leads to the loss of growth inhibition, has been found in a human HNSCC patient [114]. Decreased nuclear LKB1 levels have been shown to correlate with HNSCC metastasis [115]. Consistent with the effects of anti-mTOR therapy in other cancers, inhibition of mTOR by rapamycin displays a potent antitumor effect in HNSCC in vitro [110], in oral carcinogenesis model [116], and in a HNSCC-GEMM [117]. Targeting mTOR has also been shown to be a possible adjuvant therapy for microscopic residual disease in human HNSCC patients [118]. However, studies exploring the frequencies of these molecular alterations and their mechanisms in HNSCC tumorigenesis are still lacking.
4. PI3K Pathway as Target for Chemoprevention and Therapy
4.1. Chemoprevention
Activation of PI3K/AKT/mTOR pathway has been illustrated as an early event in multiple human cancers, suggesting that targeting the PI3K/AKT/mTOR pathway may have chemopreventive value. This is further exemplified by tobacco exposure activation of AKT/mTOR in multiple tobacco-related malignancies, including HNSCC [95, 119]. The tobacco-associated NNK and the DNA adduct-forming agent 4-nitroquinoline-1-oxide (4NQO) activate AKT/mTOR as early as premalignancy stage [95, 119, 120]. Inhibition of this pathway by either Deguelin, a natural compound belonging to the rotenoid family [121], or Metformin, an antidiabetes medicine, has been shown to possess chemopreventive effects on a tobacco carcinogen-induced lung tumorigenesis model [122]. In addition, mTOR inhibition by rapamycin has been shown to prevent early onset of HNSCC tumorigenesis in both the 4NQO-induced HNSCC mouse model [116] and a HNSCC-GEMM [117]. Additionally, resveratrol, a phytoalexin enriched in red grapes, strawberries, and peanuts, has been found to be a potent chemoprevention agent for many cancers [123]. One of the major mechanisms of its chemopreventive effect is through inhibition of the PI3K/AKT/mTOR pathway [124]. It will be interesting to study its chemopreventive effect on both the 4-NQO-induced HNSCC mouse model and the HNSCC-GEMMs.
4.2. Targeted Therapy
Personalized cancer therapies with selective molecular targets have emerged as a novel class of anticancer agents, with demonstrated clinical efficacy and less toxicity than conventional therapies [125]. In this situation, the PI3K/AKT/mTOR pathway has been extensively studied in almost all human malignancies including HNSCC and in both experimental and clinical settings [126]. Multiple drugs have been designed to target this pathway, making it the most “druggable” pathway for targeted therapies of human cancers. This has been summarized and reviewed in many articles. Given the scope of this paper, we will only comment on a few aspects. For a more detailed explanation of progress in PI3K targeted therapy on HNSCC, please refer to any of the several excellent reviews available on the topic [3, 5, 126, 127].
(1) Identification of Biomarkers for the Personalized Cancer Therapy Targeting the PI3K Pathway —
Since the concept of personalized cancer therapy is based on the identification of a subset of patients whose tumors carry specific molecular alterations, biomarker identification is critical for predicting the effectiveness of targeted therapy [125]. For example, both PIK3CA mutations and nuclear phosphorylation of AKT are shown as biomarkers for the effectiveness of PI3K inhibitors for human cancer patients [128, 129]. In addition, human cancer patients harboring PIK3CA mutations are sensitive to targeted therapy using the mTOR inhibitor everolimus, while human cancer patients carrying Kras mutations are resistant to the treatment [130]. This is further confirmed in a PIK3CA-GEMM and Kras-GEMM for lung cancer. While NVP-BEZ235, a dual pan-PI3K and mTOR inhibitor, led to marked tumor regression in the PIK3CA-GEMM, it did not affect tumor growth significantly when treating the Kras-GEMM unless it was used in combination with a MEK inhibitor [38]. Finally, a recent report of the screening of over three hundred nonredundant PI3K-pathway-relevant phosphopeptides identified PRAS40, a molecule involved in protein phosphorylation, as a biomarker correlated with PI3K pathway activation and AKT inhibitor sensitivity [131].
(2) Activation of PI3K/AKT/mTOR Pathway as a Resistance Mechanism to Targeted Therapy or Radiotherapy —
EGFR targeted therapy is the first FDA-approved protocol for treating human HNSCC patients [132]. However, resistance to EGFR therapy remains a major obstacle to positive clinical outcomes [127]. Activation of the canonical PI3K/AKT/mTOR pathway seems to be associated with resistance to EGFR inhibitor in multiple human cancers [133]. However, a recent study showed that an EGFR-activating mutation resistant to targeted therapy activates the mTORC2-NF-κB signaling pathway in an AKT-independent manner in glioblastoma patients [134]. Further studies are necessary to investigate the role of both canonical and noncanonical PI3K pathways in resistance to EGFR therapy in human HNSCC patients. HNSCC is relatively sensitive to radiotherapy [135]. However, activation of the PI3K/AKT/mTOR pathway is implicated in all major mechanisms of radioresistance, including intrinsic radioresistance, tumor cell proliferation, and hypoxia [135]. Thus, blocking the PI3K/AKT/mTOR pathway has great potential to enhance the effectiveness of radiotherapy for HNSCC patients.
(3) Synergistic Effect of Combination with Other Receptor Tyrosine Kinase Targeted Therapies —
Recent evidence of multiple feedback loops and interactions with other signaling pathways highlights the complexity of PI3K signaling. Using an inducible PIK3CA-GEMM for breast cancer, Liu et al. identified c-Myc elevation as a potential mechanism by which tumors develop resistance to PI3K-targeted therapies [40]. Moreover, inhibition of AKT induces activation of upstream RTK signaling pathways, such as HER3 [136], and mTOR inhibition causes activation of AKT signaling [137] or MAPK pathway [138]. These studies suggest that combination therapies of PI3K-targeted therapy together with targeting c-Myc, Her3, or MAPK pathway, may be more effective for the treatment of certain human cancer patients.
5. Prospectus
Mounting evidence clearly shows both the paramount importance of the PI3K pathway in the tumorigenesis of many human malignancies including HNSCC, and the promising results of targeting this pathway for treatment of human cancer patients. However, there are still many questions that need to be answered. Compared to the extensive studies on PIK3CA, there are few studies on the other subunits of class IA PI3Ks and their interactions with PIK3CA and PTEN. Studies of classes II and III of PI3Ks in human cancers are generally lacking. Although several proteins interacting with PTEN or AKT have been shown to play a role in tumorigenesis, more studies must be undertaken to discover novel molecules modulating the PI3K pathway, and assess their roles in tumorigenesis. In addition to the relatively linear canonical PI3K/AKT pathway, more and more noncanonical pathways are expected to be identified. Furthermore, the newly discovered mRNAs described in this paper add yet another layer of complexity to our understanding of the molecular regulation of the PI3K signaling pathway. Integrative mapping of molecular alterations in human cancers, particularly in HNSCC samples, is highly demanding. Utilization of multiple molecular approaches, especially GEMMs of the PI3K signaling pathway, will help us to better understand the complexity of this pathway in human cancers, as well as in context and stage-specific manners. Ultimately, these studies will yield identifiable biomarkers for improved clinical diagnosis and prognosis, contributing to strategies of therapy and prevention that will allow for the better management of human cancers and better outcomes for human patients.
Acknowledgments
The authors would like to thank Drs. Steve Weber, Jacob Minor, Fang Zhang, Yu Cao, Matthew Whinery, and Francis Hall of Dr. Lu's lab for their excellent work contributing in this paper. The authors would also like to thank Drs. Molly Kulesz-Martin of the Oregon Health and Science University and Antonio Jimeno of the University of Colorado Anschutz Medical Campus for providing human HNSCC samples from the two institutional IRB-approved HNSCC tumor resources. This work is supported by grant no. R01DE021788 from the National Institute of Dental and Craniofacial Research and grants from the University of Colorado Cancer Center, the Colorado Cancer League, the American Cancer Society, the Dermatology Foundation, and the THANC foundation to Dr. S.-L. Lu.
References
- 1.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27(41):5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nature Reviews Genetics. 2006;7(8):606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 3.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nature Reviews Cancer. 2009;9(8):550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 4.Argiris A, Karamouzis MV, Raben D, Ferris RL. Head and neck cancer. The Lancet. 2008;371(9625):1695–1709. doi: 10.1016/S0140-6736(08)60728-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nature Reviews Cancer. 2010;11:9–22. doi: 10.1038/nrc2982. [DOI] [PubMed] [Google Scholar]
- 6.Molinolo AA, Amornphimoltham P, Squarize CH, Castilho RM, Patel V, Gutkind JS. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncology. 2009;45(4-5):324–334. doi: 10.1016/j.oraloncology.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu SL, Reh D, Li AG, et al. Overexpression of transforming growth factor beta1 in head and neck epithelia results in inflammation, angiogenesis, and epithelial hyperproliferation. Cancer Research. 2004;64(13):4405–4410. doi: 10.1158/0008-5472.CAN-04-1032. [DOI] [PubMed] [Google Scholar]
- 8.Lu SL, Herrington H, Reh D, et al. Loss of transforming growth factor-beta type II receptor promotes metastatic head-and-neck squamous cell carcinoma. Genes and Development. 2006;20(10):1331–1342. doi: 10.1101/gad.1413306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu SL, Herrington H, Wang XJ. Mouse models for human head and neck squamous cell carcinomas. Head and Neck. 2006;28(10):945–954. doi: 10.1002/hed.20397. [DOI] [PubMed] [Google Scholar]
- 10.Bornstein S, White R, Malkoski S, et al. Smad4 loss in mice causes spontaneous head and neck cancer with increased genomic instability and inflammation. Journal of Clinical Investigation. 2009;119(11):3408–3419. doi: 10.1172/JCI38854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agrawal N, Frederick MJ, Pickering CR, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1 . Science. 2011;333(6046):1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stransky N, Egloff AM, Tward AD, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333(6046):1154–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao L, Vogt PK. Class I PI3K in oncogenic cellular transformation. Oncogene. 2008;27(41):5486–5496. doi: 10.1038/onc.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12(1):9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 15.Vasudevan KM, Barbie DA, Davies MA, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009;16(1):21–32. doi: 10.1016/j.ccr.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vogt PK, Gymnopoulos M, Hart JR. PI 3-kinase and cancer: changing accents. Current Opinion in Genetics and Development. 2009;19(1):12–17. doi: 10.1016/j.gde.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fan QW, Cheng C, Knight ZA, et al. EGFR signals to mTOR through PKC and independently of Akt in glioma. Science Signaling. 2009;2(55, article ra4) doi: 10.1126/scisignal.2000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lauring J, Cosgrove DP, Fontana S, et al. Knock in of the AKT1 E17K mutation in human breast epithelial cells does not recapitulate oncogenic PIK3CA mutations. Oncogene. 2010;29(16):2337–2345. doi: 10.1038/onc.2009.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiu W, Schonleben F, Li X, et al. PIK3CA mutations in head and neck squamous cell carcinoma. Clinical Cancer Research. 2006;12(5):1441–1446. doi: 10.1158/1078-0432.CCR-05-2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qiu W, Tong GX, Manolidis S, Close LG, Assaad AM, Su GH. Novel mutant-enriched sequencing identified high frequency of PIK3CA mutations in pharyngeal cancer. International Journal of Cancer. 2008;122(5):1189–1194. doi: 10.1002/ijc.23217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Massion PP, Taflan PM, Shyr Y, et al. Early involvement of the phosphatidylinositol 3-kinase/Akt pathway in lung cancer progression. American Journal of Respiratory and Critical Care Medicine. 2004;170(10):1088–1094. doi: 10.1164/rccm.200404-487OC. [DOI] [PubMed] [Google Scholar]
- 23.Pedrero JM, Carracedo D, Pinto CM, Jr., et al. Frequent genetic and biochemical alterations of the PI 3-K/AKT/PTEN pathway in head and neck squamous cell carcinoma. International Journal of Cancer. 2005;114(2):242–248. doi: 10.1002/ijc.20711. [DOI] [PubMed] [Google Scholar]
- 24.Estilo CL, O-charoenrat P, Ngai I, et al. The role of novel oncogenes squamous cell carcinoma-related oncogene and phosphatidylinositol 3-kinase p110alpha in squamous cell carcinoma of the oral tongue. Clinical Cancer Research. 2003;9(6):2300–2306. [PubMed] [Google Scholar]
- 25.He Y, Van’t Veer LJ, Mikolajewska-Hanclich I, et al. PIK3CA mutations predict local recurrences in rectal cancer patients. Clinical Cancer Research. 2009;15(22):6956–6962. doi: 10.1158/1078-0432.CCR-09-1165. [DOI] [PubMed] [Google Scholar]
- 26.Saal LH, Holm K, Maurer M, et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Research. 2005;65(7):2554–2559. doi: 10.1158/0008-5472-CAN-04-3913. [DOI] [PubMed] [Google Scholar]
- 27.Akagi I, Miyashita M, Makino H, et al. Overexpression of PIK3CA is associated with lymph node metastasis in esophageal squamous cell carcinoma. International Journal of Oncology. 2009;34(3):767–775. doi: 10.3892/ijo_00000202. [DOI] [PubMed] [Google Scholar]
- 28.Ogino S, Nosho K, Kirkner GJ, et al. PIK3CA mutation is associated with poor prognosis among patients with curatively resected colon cancer. Journal of Clinical Oncology. 2009;27(9):1477–1484. doi: 10.1200/JCO.2008.18.6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aleskandarany MA, Rakha EA, Ahmed MA, et al. PIK3CA expression in invasive breast cancer: a biomarker of poor prognosis. Breast Cancer Research and Treatment. 2010;122(1):45–53. doi: 10.1007/s10549-009-0508-9. [DOI] [PubMed] [Google Scholar]
- 30.Kozaki KI, Imoto I, Pimkhaokham A, et al. PIK3CA mutation is an oncogenic aberration at advanced stages of oral squamous cell carcinoma. Cancer Science. 2006;97(12):1351–1358. doi: 10.1111/j.1349-7006.2006.00343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Woenckhaus J, Steger K, Werner E, et al. Genomic gain of PIK3CA and increased expression of p110alpha are associated with progression of dysplasia into invasive squamous cell carcinoma. Journal of Pathology. 2002;198(3):335–342. doi: 10.1002/path.1207. [DOI] [PubMed] [Google Scholar]
- 32.Fenic I, Steger K, Gruber C, Arens C, Woenckhaus J. Analysis of PIK3CA and Akt/protein kinase B in head and neck squamous cell carcinoma. Oncology Reports. 2007;18(1):253–259. [PubMed] [Google Scholar]
- 33.Pang H, Flinn R, Patsialou A, et al. Differential enhancement of breast cancer cell motility and metastasis by helical and kinase domain mutations of class IA phosphoinositide 3-kinase. Cancer Research. 2009;69(23):8868–8876. doi: 10.1158/0008-5472.CAN-09-1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao JJ, Cheng H, Jia S, et al. The p110alpha isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(44):16296–16300. doi: 10.1073/pnas.0607899103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gupta S, Ramjaun AR, Haiko P, et al. Binding of Ras to phosphoinositide 3-kinase p110alpha is required for Ras- driven tumorigenesis in mice. Cell. 2007;129(5):957–968. doi: 10.1016/j.cell.2007.03.051. [DOI] [PubMed] [Google Scholar]
- 36.Liang S, Yang N, Pan Y, et al. Expression of activated PIK3CA in ovarian surface epithelium results in hyperplasia but not tumor formation. PLoS ONE. 2009;4(1) doi: 10.1371/journal.pone.0004295. Article ID e4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Renner O, Blanco-Aparicio C, Grassow M, Canamero M, Leal JF, Carnero A. Activation of phosphatidylinositol 3-kinase by membrane localization of p110alpha predisposes mammary glands to neoplastic transformation. Cancer Research. 2008;68(23):9643–9653. doi: 10.1158/0008-5472.CAN-08-1539. [DOI] [PubMed] [Google Scholar]
- 38.Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nature Medicine. 2008;14(12):1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meyer DS, Brinkhaus H, Muller U, Muller M, Cardiff RD, Bentires-Alj M. Luminal expression of PIK3CA mutant H1047R in the mammary gland induces heterogeneous tumors. Cancer Research. 2011;71(13):4344–4351. doi: 10.1158/0008-5472.CAN-10-3827. [DOI] [PubMed] [Google Scholar]
- 40.Liu P, Cheng H, Santiago S, et al. Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K pathway-dependent and PI3K pathway-independent mechanisms. Nature Medicine. 2011;17:1116–1120. doi: 10.1038/nm.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jaiswal BS, Janakiraman V, Kljavin NM, et al. Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell. 2009;16(6):463–474. doi: 10.1016/j.ccr.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chagpar RB, Links PH, Pastor MC, et al. Direct positive regulation of PTEN by the p85 subunit of phosphatidylinositol 3-kinase. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(12):5471–5476. doi: 10.1073/pnas.0908899107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taniguchi CM, Winnay J, Kondo T, et al. The phosphoinositide 3-kinase regulatory subunit p85alpha can exert tumor suppressor properties through negative regulation of growth factor signaling. Cancer Research. 2010;70(13):5305–5315. doi: 10.1158/0008-5472.CAN-09-3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nature Reviews Molecular Cell Biology. 2010;11(5):329–341. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 45.Jia S, Liu Z, Zhang S, et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008;454(7205):776–779. doi: 10.1038/nature07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee SH, Poulogiannis G, Pyne S, et al. A constitutively activated form of the p110beta isoform of PI3-kinase induces prostatic intraepithelial neoplasia in mice. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(24):11002–11007. doi: 10.1073/pnas.1005642107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ciraolo E, Iezzi M, Marone R, et al. Phosphoinositide 3-kinase p110beta activity: key role in metabolism and mammary gland cancer but not development. Science Signaling. 2008;1(36, article ra3) doi: 10.1126/scisignal.1161577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Keniry M, Parsons R. The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene. 2008;27(41):5477–5485. doi: 10.1038/onc.2008.248. [DOI] [PubMed] [Google Scholar]
- 49.Lee JI, Soria JC, Hassan KA, et al. Loss of PTEN expression as a prognostic marker for tongue cancer. Archives of Otolaryngology. 2001;127(12):1441–1445. doi: 10.1001/archotol.127.12.1441. [DOI] [PubMed] [Google Scholar]
- 50.Poetsch M, Lorenz G, Kleist B. Detection of new PTEN/MMAC1 mutations in head and neck squamous cell carcinomas with loss of chromosome 10. Cancer Genetics and Cytogenetics. 2002;132(1):20–24. doi: 10.1016/s0165-4608(01)00509-x. [DOI] [PubMed] [Google Scholar]
- 51.Mirmohammadsadegh A, Marini A, Nambiar S, et al. Epigenetic silencing of the PTEN gene in melanoma. Cancer Research. 2006;66(13):6546–6552. doi: 10.1158/0008-5472.CAN-06-0384. [DOI] [PubMed] [Google Scholar]
- 52.Soria JC, Lee HY, Lee JI, et al. Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clinical Cancer Research. 2002;8(5):1178–1184. [PubMed] [Google Scholar]
- 53.Huang KH, Huang SF, Chen IH, Liao CT, Wang HM, Hsieh LL. Methylation of RASSF1A, RASSF2A, and HIN-1 is associated with poor outcome after radiotherapy, but not surgery, in oral squamous cell carcinoma. Clinical Cancer Research. 2009;15(12):4174–4180. doi: 10.1158/1078-0432.CCR-08-2929. [DOI] [PubMed] [Google Scholar]
- 54.Squarize CH, Castilho RM, Pinto DS., Jr. Immunohistochemical evidence of PTEN in oral squamous cell carcinoma and its correlation with the histological malignancy grading system. Journal of Oral Pathology and Medicine. 2002;31(7):379–384. doi: 10.1034/j.1600-0714.2002.00142.x. [DOI] [PubMed] [Google Scholar]
- 55.Cheung LW, Hennessy BT, Li J, et al. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discovery. 2011;1:170–185. doi: 10.1158/2159-8290.CD-11-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Darido C, Georgy SR, Wilanowski T, et al. Targeting of the tumor suppressor GRHL3 by a miR-21-dependent proto-oncogenic network results in PTEN loss and tumorigenesis. Cancer Cell. 2011;20:635–648. doi: 10.1016/j.ccr.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 57.Kim H, Huang W, Jiang X, Pennicooke B, Park PJ, Johnson MD. Integrative genome analysis reveals an oncomir/ oncogene cluster regulating glioblastoma survivorship. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(5):2183–2188. doi: 10.1073/pnas.0909896107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Poliseno L, Salmena L, Riccardi L, et al. Identification of the miR-106b∼25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Science Signaling. 2010;3(117, article ra29) doi: 10.1126/scisignal.2000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Minor J, Wang X, Zhang F, et al. Methylation of microRNA-9 is a specific and sensitive biomarker for oral and oropharyngeal squamous cell carcinomas. Oral Oncology. 2012;48:73–78. doi: 10.1016/j.oraloncology.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang X, Trotman LC, Koppie T, et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell. 2007;128(1):129–139. doi: 10.1016/j.cell.2006.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fine B, Hodakoski C, Koujak S, et al. Activation of the PI3K pathway in cancer through inhibition of PTEN by exchange factor P-REX2a. Science. 2009;325(5945):1261–1265. doi: 10.1126/science.1173569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gewinner C, Wang ZC, Richardson A, et al. Evidence that inositol polyphosphate 4-phosphatase Type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell. 2009;16(2):115–125. doi: 10.1016/j.ccr.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465(7301):1033–1038. doi: 10.1038/nature09144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vivanco I, Palaskas N, Tran C, et al. Identification of the JNK signaling pathway as a functional target of the tumor suppressor PTEN. Cancer Cell. 2007;11(6):555–569. doi: 10.1016/j.ccr.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 65.Mounir Z, Krishnamoorthy JL, Robertson GP, et al. Tumor Suppression by PTENR equires the activation of the PKR-eIF2alpha phosphorylation pathway. Science Signaling. 2009;2(102, article ra85) doi: 10.1126/scisignal.2000389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nature Reviews Cancer. 2011;11(4):289–301. doi: 10.1038/nrc3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hollander MC, Balogh AR, Liwanag J, et al. Strain-specific spontaneous and NNK-mediated tumorigenesis in Pten+/− mice. Neoplasia. 2008;10(8):866–872. doi: 10.1593/neo.08406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hill R, Calvopina JH, Kim C, et al. PTEN loss accelerates KrasG12D-induced pancreatic cancer development. Cancer Research. 2010;70(18):7114–7124. doi: 10.1158/0008-5472.CAN-10-1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Puzio-Kuter AM, Castillo-Martin M, Kinkade CW, et al. Inactivation of p53 and Pten promotes invasive bladder cancer. Genes and Development. 2009;23(6):675–680. doi: 10.1101/gad.1772909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005;24(50):7455–7464. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- 71.Dutt A, Salvesen HB, Greulich H, Sellers WR, Beroukhim R, Meyerson M. Somatic mutations are present in all members of the AKT family in endometrial carcinoma. British Journal of Cancer. 2009;101:1218–1219. doi: 10.1038/sj.bjc.6605301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carpten JD, Faber AL, Horn C, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448(7152):439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 73.Bleeker FE, Felicioni L, Buttitta F, et al. AKT1E17K in human solid tumours. Oncogene. 2008;27(42):5648–5650. doi: 10.1038/onc.2008.170. [DOI] [PubMed] [Google Scholar]
- 74.Eom HS, Kim MS, Hur SY, Yoo NJ, Lee SH. Absence of oncogenic AKT1E17K mutation in prostate, esophageal, laryngeal and urothelial carcinomas, hepatoblastomas, gastrointestinal stromal tumors and malignant meningiomas. Acta Oncologica. 2009;48(7):1084–1085. doi: 10.1080/02841860902878152. [DOI] [PubMed] [Google Scholar]
- 75.Malanga D, Scrima M, De Marco C, et al. Activating E17K mutation in the gene encoding the protein kinase AKT1 in a subset of squamous cell carcinoma of the lung. Cell Cycle. 2008;7(5):665–669. doi: 10.4161/cc.7.5.5485. [DOI] [PubMed] [Google Scholar]
- 76.Davies MA, Stemke-Hale K, Tellez C, et al. A novel AKT3 mutation in melanoma tumours and cell lines. British Journal of Cancer. 2008;99(8):1265–1268. doi: 10.1038/sj.bjc.6604637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bellacosa A, Testa JR, Moore R, Larue L. A portrait of AKT kinases: human cancer and animal models depict a family with strong individualities. Cancer Biology and Therapy. 2004;3(3):268–275. doi: 10.4161/cbt.3.3.703. [DOI] [PubMed] [Google Scholar]
- 78.Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD, Wang LH. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Research. 2007;67(5):1979–1987. doi: 10.1158/0008-5472.CAN-06-1479. [DOI] [PubMed] [Google Scholar]
- 79.Rychahou PG, Kang J, Gulhati P, et al. Akt2 overexpression plays a critical role in the establishment of colorectal cancer metastasis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(51):20315–20320. doi: 10.1073/pnas.0810715105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stahl JM, Sharma A, Cheung M, et al. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Research. 2004;64(19):7002–7010. doi: 10.1158/0008-5472.CAN-04-1399. [DOI] [PubMed] [Google Scholar]
- 81.Iamaroon A, Krisanaprakornkit S. Overexpression and activation of Akt2 protein in oral squamous cell carcinoma. Oral Oncology. 2009;45(10):e175–e179. doi: 10.1016/j.oraloncology.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 82.Tsurutani J, Fukuoka J, Tsurutani H, et al. Evaluation of two phosphorylation sites improves the prognostic significance of Akt activation in non-small-cell lung cancer tumors. Journal of Clinical Oncology. 2006;24(2):306–314. doi: 10.1200/JCO.2005.02.4133. [DOI] [PubMed] [Google Scholar]
- 83.Yang SX, Costantino JP, Kim C, et al. Akt phosphorylation at Ser473 predicts benefit of paclitaxel chemotherapy in node-positive breast cancer. Journal of Clinical Oncology. 2010;28(18):2974–2981. doi: 10.1200/JCO.2009.26.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Amornphimoltham P, Sriuranpong V, Patel V, et al. Persistent activation of the Akt pathway in head and neck squamous cell carcinoma: a potential target for UCN-01. Clinical Cancer Research. 2004;10(12):4029–4037. doi: 10.1158/1078-0432.CCR-03-0249. [DOI] [PubMed] [Google Scholar]
- 85.Molinolo AA, Hewitt SM, Amornphimoltham P, et al. Dissecting the Akt/mammalian target of rapamycin signaling network: emerging results from the head and neck cancer tissue array initiative. Clinical Cancer Research. 2007;13(17):4964–4973. doi: 10.1158/1078-0432.CCR-07-1041. [DOI] [PubMed] [Google Scholar]
- 86.Massarelli E, Liu DD, Lee JJ, et al. Akt activation correlates with adverse outcome in tongue cancer. Cancer. 2005;104(11):2430–2436. doi: 10.1002/cncr.21476. [DOI] [PubMed] [Google Scholar]
- 87.Yu Z, Weinberger PM, Sasaki C, et al. Phosphorylation of Akt (Ser473) predicts poor clinical outcome in oropharyngeal squamous cell cancer. Cancer Epidemiology Biomarkers and Prevention. 2007;16(3):553–558. doi: 10.1158/1055-9965.EPI-06-0121. [DOI] [PubMed] [Google Scholar]
- 88.Irie HY, Pearline RV, Grueneberg D, et al. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. Journal of Cell Biology. 2005;171(6):1023–1034. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yoeli-Lerner M, Yiu GK, Rabinovitz I, Erhardt P, Jauliac S, Toker A. Akt blocks breast cancer cell motility and invasion through the transcription factor NFAT. Molecular Cell. 2005;20(4):539–550. doi: 10.1016/j.molcel.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 90.Dillon RL, Muller WJ. Distinct biological roles for the akt family in mammary tumor progression. Cancer Research. 2010;70(11):4260–4264. doi: 10.1158/0008-5472.CAN-10-0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hutchinson JN, Jin J, Cardiff RD, Woodgett JR, Muller WJ. Activation of Akt-1 (PKB-alpha) can accelerate ErbB-2-mediated mammary tumorigenesis but suppresses tumor invasion. Cancer Research. 2004;64(9):3171–3178. doi: 10.1158/0008-5472.can-03-3465. [DOI] [PubMed] [Google Scholar]
- 92.Dillon RL, Marcotte R, Hennessy BT, Woodgett JR, Mills GB, Muller WJ. Akt1 and Akt2 play distinct roles in the initiation and metastatic phases of mammary tumor progression. Cancer Research. 2009;69(12):5057–5064. doi: 10.1158/0008-5472.CAN-08-4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Iliopoulos D, Polytarchou C, Hatziapostolou M, et al. MicroRNAs differentially regulated by Akt isoforms control EMT and stem cell renewal in cancer cells. Science Signaling. 2009;2(92, article ra62) doi: 10.1126/scisignal.2000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.West KA, Brognard J, Clark AS, et al. Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. Journal of Clinical Investigation. 2003;111(1):81–90. doi: 10.1172/JCI16147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weber SM, Bornstein S, Li Y, et al. Tobacco-specific carcinogen nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone induces AKT activation in head and neck epithelia. International Journal of Oncology. 2011;39:1193–1198. doi: 10.3892/ijo.2011.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Segrelles C, Lu J, Hammann B, et al. Deregulated activity of Akt in epithelial basal cells induces spontaneous tumors and heightened sensitivity to skin carcinogenesis. Cancer Research. 2007;67(22):10879–10888. doi: 10.1158/0008-5472.CAN-07-2564. [DOI] [PubMed] [Google Scholar]
- 97.Moral M, Segrelles C, Lara MF, et al. Akt activation synergizes with Trp53 loss in oral epithelium to produce a novel mouse model for head and neck squamous cell carcinoma. Cancer Research. 2009;69(3):1099–1108. doi: 10.1158/0008-5472.CAN-08-3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brognard J, Sierecki E, Gao T, Newton AC. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Molecular Cell. 2007;25(6):917–931. doi: 10.1016/j.molcel.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 99.Liu J, Weiss HL, Rychahou P, Jackson LN, Evers BM, Gao T. Loss of PHLPP expression in colon cancer: role in proliferation and tumorigenesis. Oncogene. 2009;28(7):994–1004. doi: 10.1038/onc.2008.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen M, Pratt CP, Zeeman ME, et al. Identification of PHLPP1 as a tumor suppressor reveals the role of feedback activation in PTEN-mutant prostate cancer progression. Cancer Cell. 2011;20:173–186. doi: 10.1016/j.ccr.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pei H, Li L, Fridley BL, et al. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell. 2009;16(3):259–266. doi: 10.1016/j.ccr.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Suizu F, Hiramuki Y, Okumura F, et al. The E3 ligase TTC3 facilitates ubiquitination and degradation of phosphorylated Akt. Developmental Cell. 2009;17(6):800–810. doi: 10.1016/j.devcel.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 103.Kawase T, Ohki R, Shibata T, et al. PH domain-only protein PHLDA3 is a p53-regulated repressor of Akt. Cell. 2009;136(3):535–550. doi: 10.1016/j.cell.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 104.Maurer M, Su T, Saal LH, et al. 3-Phosphoinositide-dependent kinase 1 potentiates upstream lesions on the phosphatidylinositol 3-kinase pathway in breast carcinoma. Cancer Research. 2009;69(15):6299–6306. doi: 10.1158/0008-5472.CAN-09-0820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Xie Z, Yuan H, Yin Y, Zeng X, Bai R, Glazer RI. 3-Phosphoinositide-dependent Protein Kinase-1 (PDK1) promotes invasion and activation of matrix metalloproteinases. BMC Cancer. 2006;6, article 77 doi: 10.1186/1471-2407-6-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu Y, Wang J, Wu M, et al. Down-regulation of 3-phosphoinositide-dependent protein kinase-1 levels inhibits migration and experimental metastasis of human breast cancer cells. Molecular Cancer Research. 2009;7(6):944–954. doi: 10.1158/1541-7786.MCR-08-0368. [DOI] [PubMed] [Google Scholar]
- 107.Bayascas JR, Leslie NR, Parsons R, Fleming S, Alessi DR. Hypomorphic mutation of PDK1 suppresses tumorigenesis in PTEN+/− mice. Current Biology. 2005;15(20):1839–1846. doi: 10.1016/j.cub.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 108.Yu Y, Yoon SO, Poulogiannis G, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011;332(6035):1322–1326. doi: 10.1126/science.1199484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zoncu R, Efeyan A, Sabatini DM. MTOR: from growth signal integration to cancer, diabetes and ageing. Nature Reviews Molecular Cell Biology. 2011;12(1):21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Amornphimoltham P, Patel V, Sodhi A, et al. Mammalian target of rapamycin, a molecular target in squamous cell carcinomas of the head and neck. Cancer Research. 2005;65(21):9953–9961. doi: 10.1158/0008-5472.CAN-05-0921. [DOI] [PubMed] [Google Scholar]
- 111.Morris LG, Taylor BS, Bivona TG, et al. Genomic dissection of the epidermal growth factor receptor (EGFR)/PI3K pathway reveals frequent deletion of the EGFR phosphatase PTPRS in head and neck cancers. Proceedings of the National Academy of Sciences of the United States. 2011;108:19024–19029. doi: 10.1073/pnas.1111963108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chakraborty S, Mohiyuddin SM, Gopinath KS, Kumar A. Involvement of TSC genes and differential expression of other members of the mTOR signaling pathway in oral squamous cell carcinoma. BMC Cancer. 2008;8, article 163 doi: 10.1186/1471-2407-8-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kawaguchi S, Harada K, Supriatno, Yoshida H, Sato M. Overexpression of tuberous sclerosis complex 2 exerts antitumor effect on oral cancer cell lines. Oral Oncology. 2003;39(8):836–841. doi: 10.1016/s1368-8375(03)00106-4. [DOI] [PubMed] [Google Scholar]
- 114.Qiu W, Schonleben F, Thaker HM, Goggins M, Su GH. A novel mutation of STK11/LKB1 gene leads to the loss of cell growth inhibition in head and neck squamous cell carcinoma. Oncogene. 2006;25(20):2937–2942. doi: 10.1038/sj.onc.1209325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kline ER, Muller S, Pan L, Tighiouart M, Chen ZG, Marcus AI. Localization-specific LKB1 loss in head and neck squamous cell carcinoma metastasis. Head Neck. 2011;33:1501–1512. doi: 10.1002/hed.21638. [DOI] [PubMed] [Google Scholar]
- 116.Czerninski R, Amornphimoltham P, Patel V, Molinolo AA, Gutkind JS. Targeting mammalian target of rapamycin by rapamycin prevents tumor progression in an oral-specific chemical carcinogenesis model. Cancer Prevention Research. 2009;2(1):27–36. doi: 10.1158/1940-6207.CAPR-08-0147. [DOI] [PubMed] [Google Scholar]
- 117.Raimondi AR, Molinolo A, Gutkind JS. Rapamycin prevents early onset of tumorigenesis in an oral-specific K-ras and p53 two-hit carcinogenesis model. Cancer Research. 2009;69(10):4159–4166. doi: 10.1158/0008-5472.CAN-08-4645. [DOI] [PubMed] [Google Scholar]
- 118.Nathan CO, Amirghahari N, Rong X, et al. Mammalian target of rapamycin inhibitors as possible adjuvant therapy for microscopic residual disease in head and neck squamous cell cancer. Cancer Research. 2007;67(5):2160–2168. doi: 10.1158/0008-5472.CAN-06-2449. [DOI] [PubMed] [Google Scholar]
- 119.West KA, Linnoila IR, Belinsky SA, Harris CC, Dennis PA. Tobacco carcinogen-induced cellular transformation increases activation of the phosphatidylinositol 3′-Kinase/Akt pathway in vitro and in vivo . Cancer Research. 2004;64(2):446–451. doi: 10.1158/0008-5472.can-03-3241. [DOI] [PubMed] [Google Scholar]
- 120.Vitale-Cross L, Czerninski R, Amornphimoltham P, Patel V, Molinolo AA, Gutkind JS. Chemical carcinogenesis models for evaluating molecular-targeted prevention and treatment of oral cancer. Cancer Prevention Research. 2009;2(5):419–422. doi: 10.1158/1940-6207.CAPR-09-0058. [DOI] [PubMed] [Google Scholar]
- 121.Lee HY, Oh SH, Woo JK, et al. Chemopreventive effects of deguelin, a novel Akt inhibitor, on tobacco-induced lung tumorigenesis. Journal of the National Cancer Institute. 2005;97(22):1695–1699. doi: 10.1093/jnci/dji377. [DOI] [PubMed] [Google Scholar]
- 122.Memmott RM, Mercado JR, Maier CR, Kawabata S, Fox SD, Dennis PA. Metformin prevents tobacco carcinogen—induced lung tumorigenesis. Cancer Prevention Research. 2009;3:1066–1076. doi: 10.1158/1940-6207.CAPR-10-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Athar M, Back JH, Kopelovich L, Bickers DR, Kim AL. Multiple molecular targets of resveratrol: anti-carcinogenic mechanisms. Archives of Biochemistry and Biophysics. 2009;486:95–102. doi: 10.1016/j.abb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Frojdo S, Cozzone D, Vidal H, Pirola L. Resveratrol is a class IA phosphoinositide 3-kinase inhibitor. Biochemical Journal. 2007;406(3):511–518. doi: 10.1042/BJ20070236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.McDermott U, Settleman J. Personalized cancer therapy with selective kinase inhibitors: an emerging paradigm in medical oncology. Journal of Clinical Oncology. 2009;27(33):5650–5659. doi: 10.1200/JCO.2009.22.9054. [DOI] [PubMed] [Google Scholar]
- 126.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nature Reviews Drug Discovery. 2009;8(8):627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Freudlsperger C, Burnett JR, Friedman JA, Kannabiran VR, Chen Z, Van Waes C. EGFR-PI3K-AKT-mTOR signaling in head and neck squamous cell carcinomas: attractive targets for molecular-oriented therapy. Expert Opinion on Therapeutic Targets. 2011;15(1):63–74. doi: 10.1517/14728222.2011.541440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.O’Brien C, Wallin JJ, Sampath D, et al. Predictive biomarkers of sensitivity to the phosphatidylinositol 3′ kinase inhibitor GDC-0941 in breast cancer preclinical models. Clinical Cancer Research. 2010;16(14):3670–3683. doi: 10.1158/1078-0432.CCR-09-2828. [DOI] [PubMed] [Google Scholar]
- 129.Wallin JJ, Guan J, Prior WW, et al. Nuclear phospho-Akt increase predicts synergy of PI3K inhibition and doxorubicin in breast and ovarian cancer. Science Translational Medicine. 2010;2(48) doi: 10.1126/scitranslmed.3000630. Article ID 48ra66. [DOI] [PubMed] [Google Scholar]
- 130.Di Nicolantonio F, Arena S, Tabernero J, et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. Journal of Clinical Investigation. 2010;120(8):2858–2866. doi: 10.1172/JCI37539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Andersen JN, Sathyanarayanan S, Di Bacco A, et al. Pathway-based identification of biomarkers for targeted therapeutics: personalized oncology with PI3K pathway inhibitors. Science Translational Medicine. 2010;2(43) doi: 10.1126/scitranslmed.3001065. Article ID 43ra55. [DOI] [PubMed] [Google Scholar]
- 132.Bernier J, Bentzen SM, Vermorken JB. Molecular therapy in head and neck oncology. Nature Reviews Clinical Oncology. 2009;6(5):266–277. doi: 10.1038/nrclinonc.2009.40. [DOI] [PubMed] [Google Scholar]
- 133.Brand TM, Iida M, Wheeler DL. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biology and Therapy. 2011;11(9):777–792. doi: 10.4161/cbt.11.9.15050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tanaka K, Babic I, Nathanson D, et al. Oncogenic EGFR signaling activates an mTORC2-NF-kappaB pathway that promotes chemotherapy resistance. Cancer Discovery. 2011;1:524–538. doi: 10.1158/2159-8290.CD-11-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bussink J, van der Kogel AJ, Kaanders JH. Activation of the PI3-K/AKT pathway and implications for radioresistance mechanisms in head and neck cancer. The Lancet Oncology. 2008;9(3):288–296. doi: 10.1016/S1470-2045(08)70073-1. [DOI] [PubMed] [Google Scholar]
- 136.Chandarlapaty S, Sawai A, Scaltriti M, et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19(1):58–71. doi: 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rodrik-Outmezguine VS, Chandarlapaty S, Pagano NC, et al. MTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. CancerDiscovery. 2011;1:248–259. doi: 10.1158/2159-8290.CD-11-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Carracedo A, Ma L, Teruya-Feldstein J, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. Journal of Clinical Investigation. 2008;118(9):3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]