

1. INTRODUCTION
1.1 Allosteric Modulation – A Historical Perspective
Early ideas regarding allosterism emerged over 50 years ago, but gained little traction in the receptor field due to limitations in molecular pharmacology and screening technology.1,2 Allosterism is a critical biochemical mechanism, as it enables proteins to sense changes in their environment and respond to them; Fenton has recently referred to this as ‘second secret of life’, preceded only by the genome.1–3 The term allostery comes from the Greek allos (
 λλoς), “other”, and stereos (στερεÒς), “solid (object)”, meaning that an allosteric site of a regulatory protein is physically distinct from the classic, active, site.1–4–8 In terms of receptor-based small molecule drug discovery, the binding site for the endogenous ligand is referred to as the orthosteric site.1,4–8 In this setting, an allosteric modulator is a small molecule that binds at a topographically distinct allosteric site, and either potentiates or inhibits the binding and/or signaling of an orthosteric ligand.1,4–8 Fueled by the clinical success of the first allosteric modulator drugs 1–4 (benzodiazepines, referred to as ‘benzo’ or BZD), which potentiate the effect of the neurotransmitter γ-aminobutyric acid (GABA) at the ionotropic GABAA receptor, the concept of allosteric modulation for a wide range of molecular targets has gained momentum in modern drug discovery (Figure 1).4,9 Benzodiazepines, for example, possess a number of modes of pharmacology and include positive allosteric modulators (PAMs), which potentiate GABAA receptor response, negative allosteric modulators (NAMs), which decrease channel activity and modulate the ability of these GABAergic receptors to elicit sedative, hypnotic, and anxiolytic effects. In addition to PAMs and NAMs, silent allosteric modulators (SAMs, or neutral allosteric ligands) bind at allosteric sites and can block the activity of PAMs and NAMs but, importantly, have no effect on orthosteric ligand responses. In contrast to the potentially deadly effects of direct acting GABAA agonists, allosteric modulation of GABAA by the benzodiazepine class has proven clinically safe and effective.4,9 With advances in molecular pharmacology and screening technology, allosteric modulators have now been developed for other ion channels, kinases, phospholipases and 7 Transmembrane Spanning Receptors (7TMRs, also known as G protein-coupled receptors (GPCRs)).1,4–8,10–15
λλoς), “other”, and stereos (στερεÒς), “solid (object)”, meaning that an allosteric site of a regulatory protein is physically distinct from the classic, active, site.1–4–8 In terms of receptor-based small molecule drug discovery, the binding site for the endogenous ligand is referred to as the orthosteric site.1,4–8 In this setting, an allosteric modulator is a small molecule that binds at a topographically distinct allosteric site, and either potentiates or inhibits the binding and/or signaling of an orthosteric ligand.1,4–8 Fueled by the clinical success of the first allosteric modulator drugs 1–4 (benzodiazepines, referred to as ‘benzo’ or BZD), which potentiate the effect of the neurotransmitter γ-aminobutyric acid (GABA) at the ionotropic GABAA receptor, the concept of allosteric modulation for a wide range of molecular targets has gained momentum in modern drug discovery (Figure 1).4,9 Benzodiazepines, for example, possess a number of modes of pharmacology and include positive allosteric modulators (PAMs), which potentiate GABAA receptor response, negative allosteric modulators (NAMs), which decrease channel activity and modulate the ability of these GABAergic receptors to elicit sedative, hypnotic, and anxiolytic effects. In addition to PAMs and NAMs, silent allosteric modulators (SAMs, or neutral allosteric ligands) bind at allosteric sites and can block the activity of PAMs and NAMs but, importantly, have no effect on orthosteric ligand responses. In contrast to the potentially deadly effects of direct acting GABAA agonists, allosteric modulation of GABAA by the benzodiazepine class has proven clinically safe and effective.4,9 With advances in molecular pharmacology and screening technology, allosteric modulators have now been developed for other ion channels, kinases, phospholipases and 7 Transmembrane Spanning Receptors (7TMRs, also known as G protein-coupled receptors (GPCRs)).1,4–8,10–15
Figure 1.

Benzodiazepines, the first allosteric modulators with clinical success, and marketed as GABAA allosteric modualtors. A generic benzodiazepine scaffold 1 highlighting the classical substitution patterns. 2 (Librium™) was the first benzodiazepine launched by Hoffmann-La Roche in 1960, and many other congeners followed such as 3 (Valium™) and the tricylic analog 4 (Xanax™).
1.2 7TMRs Structure and Ligands
7TMRs are the largest class of cell surface receptors, accounting for over 30% of currently marketed drugs and over 50% of all known drugs.4–7 7TMRs are plasma membrane proteins that receive stimuli (in the form of hormones, neurotransmitters, light, ions or odorants) on the extracellular surface to alter receptor conformation, which in turn activates signaling cascades and effector systems located within the intracellular cytosol via coupling to G proteins and other accessory proteins.4–7 Much of our understanding of the basic structure and function of 7TMRs is based on biochemical, genetic, imaging, and molecular pharmacological research, as crystal structures of 7TMRs (Rhodopsin, opsin, beta2 and beta 1 (agonist and antagonist bound), dopamine D3, Adenosine 2A (agonist and antagonist bound), chemokine CXCR4, histamine H1) have only recently been solved definitively.4–7,16–32 However, these crystal structures have powered the development of homology models for multiple 7TMRs, and afforded avenues for ligand design efforts. Structurally, all 7TMRs possess seven transmembrane helices, three extracellular and three intracellular loops, with an extracellular N-terminal tail and an intracellular C-terminal tail (Figure 2).4–7,16–32 The heptahelical transmembrane domain is largely hydrophobic whereas the extracellular (e1–e3) and intracellular (i1–i3) segments, or loops, are generally hydrophilic as would be anticipated for amino acids exposed to the phospholipid-rich membrane and the water-rich environments, respectively. The seven transmembrane helices are each approximately two-dozen amino acids long, while the C- and N-terminal tails as well as the loops can vary widely in length with up to hundreds of amino acids.4–7,16–32 Based on sequence homology and functional roles, 7TMRs commonly divided into three main Families (or classes): A (e.g., M1 mAChR), B (e.g., CRF1) and C (e.g., mGlu5) (Figure 2). The families are readily distinguished by comparing their amino acid sequences; Family B are distinguished from Family A by the presence of a larger extracellular loop and Family C have a large, bi-lobed N-terminal Venus Fly Trap (VFT) domain. A second major difference between the families concerns the location of the orthosteric binding site and the nature of the orthosteric ligand. As shown in Figure 2, the orthosteric binding site of many Family A 7TMRs is located with the 7TM domain whereas the orthosteric binding site is located in the large extracellular loop within Family B and within the VFT domain in Family C. The orthosteric ligands for Family A and C are neurotransmitters, for example, 5 (acetylcholine, for the mAChRs) and 9 (glutamate, for the mGluRs), respectively.4–7 The orthosteric ligands for Family B 7TMRs are large peptide ligands with usually >30 amino acids, such as the 41 amino acid peptide 7 (hCRF) for corticotrophin releasing factor 1 (CRF1). In contrast, allosteric ligands are structurally distinct from orthosteric ligands and bind at distinct sights, often, but not always, topologically distant from the orthosteric site.4–7 For example, the Family A M1 mAChR PAM 6 (BQCA),33 is believed to bind in a region above the TMs among the extracellular loops, whereas the Family B PAM, 8 (DMP696),34 and the Family C NAM, 10 (MPEP),35,36 bind within the TM domains.
Figure 2.
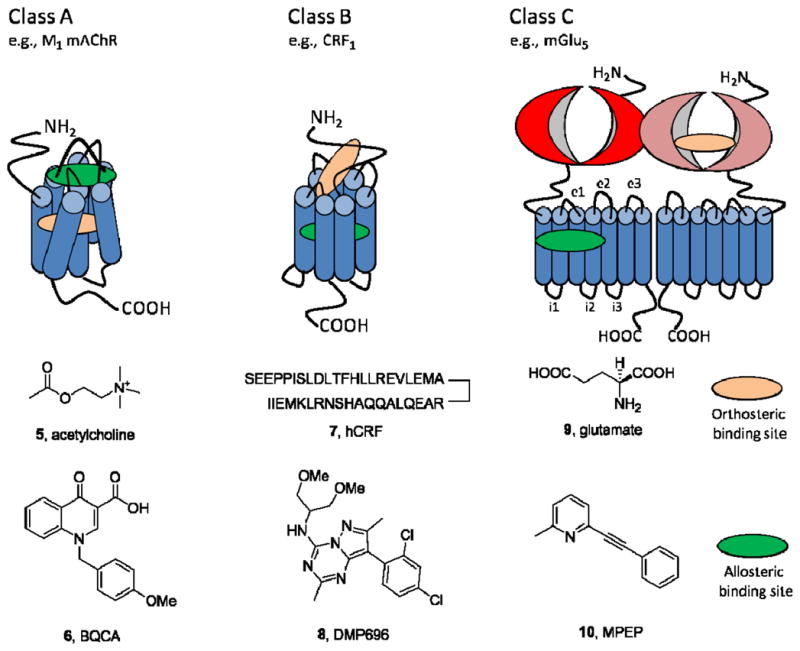
Structural topology of typical orthosteric and allosteric sites of Family A, B and C 7TMRs, highlighting representative orthosteric and allosteric ligands for each Family.
Are there naturally occurring allosteric modulators? This question is invariably posed during any discussion of allosteric modulators, and one must understand the complexity of identifying such ligands within the chemical diversity of ligands within the human body.1,2,37 However, a few natural allosteric modulators have been described, including the unnatural amino acid D-serine (an allosteric modulator of the NMDA receptor),38 L-phenylalanine and L-tryptophan (allosteric modulators of the calcium receptor)39 and the tetrepeptide Leu-Ser-Ala-Leu, also known as 5-HT moduline (an allosteric modulator of the 5-HT1B receptor).40,41
1.3 Orthosteric and Allosteric 7TMR Pharmacology
Historically, almost all of the FDA-approved drugs that act at 7TMRs bind at the orthosteric site and regulate receptor function by either classical agonism (directly stimulating a receptor response), inverse agonism (blocking constitutive receptor activity) or competitive antagonism (blocking the binding of the native agonist).4–8 This is somewhat expected as the many of these ligands were discovered by employing assays that biased targeting of the orthosteric binding site. Despite this success, synthetic ligands exist for only a fraction of the known 7TMRs, and many efforts have failed to produce highly selective compounds suitable as drug leads due to the highly conserved orthosteric binding site across a family of 7TMRs and/or due to unfavorable physicochemical and drug metabolism/pharmacokinetic (DMPK) properties of synthetic orthosteric ligands. In many cases, direct acting agonists are either toxic, or lead to receptor desensitization, internalization or down-regulation due to being ‘turned-on’ for prolonged periods.4–8
In recent years, extraordinary progress has been made in the discovery, chemical optimization, pharmacological understanding and, in some cases, clinical development of allosteric modulators for multiple 7TMRs to treat a wide range of peripheral and CNS pathologies (Tables 1–3).4–8, 10,11, 42–180 This is due in large part to the development of functional assays that allow discovery of ligands that modulate a receptor without regard to the binding site, and in effect, identify ligands that do not bind at the orthosteric site. These new allosteric ligands include PAMs, NAMs, SAMs, as well as allosteric agonists (allosteric compounds that activate the receptor in the absence of the orthosteric ligand), partial antagonists (ligands that fully occupy the NAM site, but only partially block receptor signaling) and ago-PAMs (PAMs that have inherent allosteric agonist activity).4–8,10,11,42–44 It should be noted, however, that many reported “allosteric” agonists may actually be “bitopic” ligands, that is, hybrid orthosteric/allosteric ligands that bind to both the orthosteric and allosteric sites within a given 7TMR.181–183 Allosteric ligands offer numerous advantages: 1) allosteric binding sites may be under less evolutionary pressure for their conservation, thus enabling high subtype selectivity to be achieved, 2) the effects of an allosteric modulator are saturable; once allosteric sites are occupied, no additional effects are observed, ie., a ‘ceiling effect’ (this is in contrast to “ceiling” of a partial agonist that will vary with receptor density and stimulus response coupling; this is thus far more variable than a ceiling level driven by cooperativity (at the level of binding, that is; i.e., an alpha of 10 sets a limit of 10, whereas a partial agonist can scale up to a full agonist or down to an antagonist depending on the tissue and the disease)), 3) a modulator that lacks agonistic activity will only exert its effects when the endogenous agonist is present, resulting in temporal and spatial activity (also referred to as state dependence) of the endogenous ligand and 4) improved chemical tractability.4–8 While the majority of these advantages over orthosteric ligands have been realized, allosteric modulation is far from a panacea for drug discovery, and there are many caveats to consider. First, the lack of evolutionary pressure on allosteric sites can, and has, led to significant species differences, which complicates preclinical pharmacodynamic and safety studies in mice/rats/dogs if a primary assay employs recombinant human receptor, or vice versa.4–8,10,11 Second, the state-dependence of allosteric modulators could be a liability in degenerative pathologies due to the progressive loss of endogenous orthosteric tone. For example, Alzheimer’s disease is characterized by a decrease in cholinergic tone with disease progression, rendering an mAChR PAM ineffective over time as there is no acetycholine to potentiate; in these situations, an allosteric agonist might prove more optimal for disorders in which the orthosteric ligand is lost as the disease progresses.4–8,10,11,42–44,181–183
Table 1.
Reported Allosteric Modulators of Family A G Protein-Coupled Receptors
| Receptor | Modulator example(s) |
|---|---|
| Adenosine A1 | (PD 81723; PD 117975; PD 78416; PD 71605; LUF 5484; T-62);45 (VCP 520; VCP 333)46 |
| Adenosine A2 | Amiliorides |
| Adenosine A3 | VU5455Z; VU8504Z; DU124183; [LUF6000 (compound 3 in paper)]47 (AM 251; 2-arachidonylglycerol or 2-AG)48,49 |
| Adrenoceptor α1 | Amilorides; benzodiazepines; conopeptide; ρ-TIA |
| Adrenoceptor α2A, α2B | Amilorides; sodium ions50 |
| Adrenoceptor β2 | Zinc |
| Cannabinoid CB1 | Org27569;51 Org27759;52 PSNCBAM-1;53 (JHW 007, RTI-371)54 |
| Chemokine CXCR1 | Reparixin;55 SCH527123 (Compound 2–27 inhibit CXCL8 which activates CXCR1, overall inhibition of CXCR1);56,57 SCH- 47983358 |
| Chemokine CXCR2 | Reparixin;55 SCH527123;56 SB656933;59 DF2162;59 SCH-47983358 |
| Chemokine CXCR3 | IP-10, I-TAC |
| Chemokine CXCR4 | RSVM;60 ASLW;60 trichosanthin;61 plerixafor62 |
| Chemokine CCR1 | BX-471;63 CP-481–715;64 UCB3562565 |
| Chemokine CCR3 | UCB3562565 |
| Chemokine CCR5 | Trichosanthin;61 TAK779;66 Aplaviroc; AK602; 873140;66,67 AK530;68 TAKK 220;69 SCH351125; ancriviroc;70 vicriviroc;71 Maraviroc72 |
| Dopamine D1 | Zinc73 |
| Dopamine D2 | Amiloride,74 zinc |
| Endothelin ETA | aspirin, sodium salicylate75 |
| Gonadotropin-releasing hormone receptor (GnRH) | Furan Derivitive -1 (bitopic); TAK-01376 |
| GH secretagogue | L-629,429, GHRP-6, MK-67777,78 |
| Luteinizing hormone | Org 41841, [3H]Org 4355379 |
| mAChR M1 | Brucine; BQCA; TBPB; AC-42; 77-LH-28-1; N-DMC;3,43,80,81 VU0119498;82 staurosporine; ML169;83 ML137;84 ML07185 |
| mAChR M2 | McN-A-343; BR384; gallamine;86 W84;87 AC-42; 77-LH-28-188 |
| mAChR M3 | VU0119498;82 amiodarone; N-ethylamiodarone |
| mAChR M4 | LY2033298;89,90 VU0010010;91 VU0152099;92 VU015210092,93 ML108; 92, Thiochrome; 3,43,80,81 WIN 62577; alcuronium; ML17393 |
| mAChR M5 | ML129;94 VU0119498;82 VU0365114;95 VU0400265;96 ML17295 |
| Neurokinin NK1 | Heparin |
| Opioid μ, δ | Cannabidiol97 |
| Purine P2Y1 | 2,2-o-pyridylisatogen tosylate |
| Serotonin 5HT1B/1D | 5HT-modulin |
| Serotonin 5HT2A, 5HT2 | Oleamine98 |
| Serotonin 5HT2C | Oleamine;98 PNU-69176E99 |
GH, growth hormone; mAChR, muscarininc acetylcholine receptor
Table 3.
Reported Allosteric Modulators of Family C G Protein-Coupled Receptors
| Receptor | Modulator Example(s) |
|---|---|
| Calcium sensing receptor | NPS 467; NPS 568; L-amino acids;112–114 cinacalcet;115 NPS 2143;116 calhex 231;117 SB-423557;118 SB-423562;119 calindol; ronacalceret37 |
| GABAB | BHF177;120 rac-BHFF; BHFI;121 CGP7930; GS39783; CGP13501122–125 |
| mGluR1 | (−)-CPCCOEt;126 BAY36-7620;127 R214127;128 EM-TBPC;129 JNJ16259685;130 YM-298198;131 A841720;132 FTIDC;133 YM- 230888;134 CFMMC;135 VU-71;136 Ro 01-6128; Ro 67-4853; Ro 67-7476;137 Ro 07-11401138,139 |
| mGluR2 | LY181837; LY487379; 3-MPPTS; cyPPTS; 2,2,2-TEMPS; CBiPES; 140–143 BINA;144 GSK1331258;145 MNI-136; MNI-137146–148 |
| mGluR3 | MNI-136; MNI-137146,147 |
| mGluR4 | (−)-PHCCC;149,150 ML128;151 VU0001171; VU0080241; VU0092145; VU0155041;152–154 VU0359516;155 SIB-1893156,157 |
| mGluR5 | MPEP;35 MTEP;158 Fenobam;159 VU0285683; VU0360172;160 DMeOB; DFB; DCB;161 VU0365396; VU0357121;162 CPPHA;163,164 CDPPB;165,166 VU-29;167 ADX-47273168–171 |
| mGluR7 | AMN082;172 MDIP; MMPIP173 |
| T1R1 | S807; IMP37,174–176 |
| T1R2 | S819; SE-2; SE-337,177,178 |
| T1R3 | Cyclamate; lactisole37, 179,180 |
mGluR, metabotropic glutamate receptor; T1R, taste receptors (sweet and umami)
By their very nature, allosteric ligands promote distinct conformations of 7TMRs such that the interactive properties of the receptor towards orthosteric ligands, as well as intracellular cytosolic proteins, can be modified in a ligand- and signaling protein-specific manner. This phenomenon has been termed “probe dependence”184 and has substantial implications for the functional characterization and classification of allosteric modulators, as well as challenges associated with assigning quantitative parameters to facilitate allosteric ligand SAR. For example, the allosteric modulator 11 (LY2033298) positively modulates the binding affinity of the orthosteric agonist, ACh at the M4 muscarinic receptor, but is neutral when tested against the orthosteric antagonist 12, ([3H]QNB).90 Use of the endogenous agonist in a compound screen would thus reveal the allosteric activity of a ligand such as 11, whereas a radioligand-based screen using 12 as the probe would fail to identify 11 (Figure 3). This highlights the requirement for careful consideration in the choice of orthosteric ligands to assess the effects of an allosteric modulator. Although the endogenous agonist for a given 7TMR should be the orthosteric probe of choice, this may not always be possible due to issues such as compound stability or in situations such as screening for ligands for orphan 7TMRs where the endogenous agonist is not known. In these cases, the use of a surrogate orthosteric probe is common, but the ensuing pharmacology may prove to be misleading due to the potential for differential probe dependence between the modulator and the surrogate agonist relative to the therapeutically relevant endogenous agonist. These considerations also extend to the potential for off-target activities of allosteric modulators. Although the aforementioned mAChR allosteric ligand 11 is a selective PAM for the M4 mAChR when tested against ACh, it displays remarkable positive and negative allosteric effects at the M2 mAChR when tested against other orthosteric agonists, such as oxotremorine and xanomeline; if the latter agents were used as surrogates to characterize mAChR activity in modulator screens, then the resultant pharmacology would reflect activity at an undesired target (e.g. M2 mAChR) in addition to the desired target (e.g. M4 mAChR).90 Finally, there are many 7TMRs that have more than one endogenous orthosteric agonist, but which may not all respond the same way to allosteric ligands. A striking example of this phenomenon was recently observed at the glucagon-like peptide 1 (GLP1) receptor, where the small molecule allosteric agonist 13 (Novo Nordisk’s Compound 2) had no effect on the signaling of the endogenous orthosteric peptide agonist GLP1(7–36) but significantly potentiated the signaling of another endogenous GLP1 receptor peptide, oxyntomodulin (Figure 3).110
Figure 3.

Structures of GPCR allosteric ligands 11, 13, 14 and 15 that demonstrate the concept of ‘probe dependence’, with 12, an mAChR orthosteric radioligand discussed in the text.
As a final point, when an allosteric ligand binds to a 7TMR, the receptor adopts a unique, novel conformation (vide infra), enabling it to activate any number of downstream signaling cascades to the exclusion of other possible receptor states.4–8,10,11 Here, probe-dependence manifested at the level of the cytosolic interacting protein (e.g., G protein; β-arrestin etc.) would result in signal pathway-dependent allosteric modulation. This has been coined ‘stimulus-bias’, stimulus-trafficking’, ‘differential receptor trafficking’ or ‘functional selectivity’.185–187 In short, an allosteric ligand may activate all downstream signaling cascades or a ‘surgical’ selection of cascades with clear ramifications in pharmacodynamic models; therefore, it is critical to possess the requisite assays to understand how an allosteric ligand will modulate multiple signaling events. For example, the mGlu5 PAM 14 (CPPHA, Family C 7TMR) was shown to have differential effects on DHPG-mediated calcium signaling and ERK1/2 phosphorylation in a native astrocyte system.188 In addition, an allosteric modulator of the M1 muscarinic receptor 15 (VU0029767) potentiates ACh-mediated intracellular calcium mobilization, but not phospholipase D activation; therefore, depending on the pathway/assay assessed 15 would alternatively be classed as a PAM or a SAM, respectively (Figure 3).82
2. MODE OF ACTION OF ALLOSTERIC MODUALTORS
7TMRs are highly flexible proteins capable of assuming multiple conformations, of which some are active, some are inactive (pharmacologically silent), and some are partially active. In fact, 7TMRs should be thought of as ensembles of tertiary conformations, randomly sampled by the receptor, and very subtle changes (as small as 1Å) can engender profound effects on receptor activity.1,4–8,10,11 Thus, when an allosteric modulator binds to a site topographically distinct from the orthosteric site, a change in receptor conformation occurs that can modify receptor activity in either a positive, negative or neutral direction. As mentioned before, the allosterically bound receptor is a ‘new’ receptor type, with novel behavior and activity potential. Oligomerization of GPCRs, as either hetero- or homodimers adds additional opportunities for modulation and probe dependence.189 Operationally, 7TMR allosteric modulators exhibit affinity modulation, efficacy modulation or varying degrees of both modes of modulation (Figure 4A). With affinity modulation, the conformational change in the 7TMR upon allosteric ligand binding can affect either the association or dissociation rate (or both) of the orthosteric ligand. 1,4–8,10,11 For example, a PAM that displays affinity modulation will result in a more potent orthosteric ligand (agonist). For efficacy modulation, the conformational change in the 7TMR upon allosteric ligand binding leads to a change in signaling capacity (also termed intrinsic efficacy) and thereby either facilitates or inhibits receptor coupling to downstream effectors. There are two models proposed to account for the interactions between the allosteric and orthosteric ligand (Figures 4B and C).1,4,7 In one model, termed the ‘allosteric hot wire’, the allosteric modulation site is directly linked to the orthosteric site through specific pathways.1,190,191 In a more recent model, termed ‘global allosteric modulation’, allosteric communication to the orthosteric site relies on long range interaction through order/disorder transitions from multiple receptor conformations (i.e., population dynamics).192,193 A number of mass-action schemes, based on variants of the ternary complex model, have been presented to describe the molecular effects of allosteric ligands on orthosteric pharmacology in terms of one or more “cooperativity factors”, which indicate the magnitude and direction of an allosteric modulator-mediated stabilization of different 7TMR states.4 From these models, it can be appreciated that the functional potency of an allosteric modulator will depend not only on its affinity for the allosteric site, but also on the degree of cooperativity with the orthosteric ligand. Thus, a PAM (e.g., 11) may bind to the receptor with weak affinity but possess potent functional activity due to a high cooperativity factor.4,10,90 In contrast, another PAM (e.g, benzodiazepines) may bind with very high affinity but low positive cooperativity, thus also displaying potent functional activity. The low affinity observed with some PAMs can preclude them from serving as radioligands and PET tracers, and the affinity/functional activity balance must be carefully assessed when considering receptor occupancy, for example, as a potential biomarker strategy.
Figure 4.
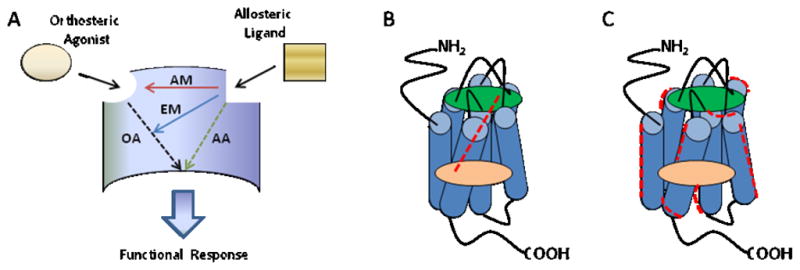
Mode of action of 7TMR allosteric modulators. A) Allosteric ligands bind to a site topographically distinct from the orthosteric site on the 7TMR to modulate either the affinity (AM, affinity modulation) or efficacy (EM, efficacy modulation). This is in contrast to direct orthosteric agonism (OA) by the native ligand or allosteric agonism (AA) by the allosteric ligand alone. B) The ‘hot wire’ mode of allostery, suggesting a direct energy link between the allosteric binding site (green) and the orthosteric binding site (peach). C) The ‘global allosteric modulation’ mode, suggesting changes at the orthosteric site are derived from global conformational variants within an ensemble of conformations.
Unfortunately, most mass-action based molecular models of allosteric modulation contain too many parameters to be fitted to real experimental data, and thus cannot be used to rationalize structure-activity studies in a manner that can inform drug candidate selection matrices. A useful means for placing these issues on a more practical quantitative level is through the use of an “operational” model of allosterism and agonism, which has been developed to describe allosteric effects in terms of a minimum number of experimentally accessible parameters.194 This model is illustrated conceptually in (Figure 5), and the equation describing the signaling of an orthosteric agonist in the presence of an allosteric modulator according to the model is as follows:
where E is the effect, [A] and [B] are the concentrations and KA, and KB are the equilibrium dissociation constants of the orthosteric and allosteric ligand, respectively, α is the cooperativity factor describing the allosteric effect of each ligand on the other’s binding affinity, β is a scaling factor (from zero to infinity) that quantifies the magnitude by which the allosteric modulator modifies the efficacy of the orthosteric agonist at a given signal pathway, and the parameters τA and τB relate to the ability of the orthosteric and allosteric ligands, respectively, to promote receptor activation (direct agonism); these latter parameters incorporate the intrinsic efficacy of each ligand, the total density of receptors, and the efficiency of stimulus-response coupling. The parameters Em and n denote the maximal possible system response and the slope factor of the transducer function that links occupancy to response, respectively.4,10,194
Figure 5.

Schematic representation of the parameters underlying the operational model of allosterism and agonism. Parameters are defined in the main text.
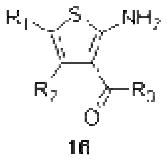
Importantly, the operational model can be fitted to experimentally derived data to provide estimates of some, or all, of its parameters.47,52,90,195–197 At a minimum, there are three key parameters that can be routinely derived from application of this model to most functional screening data, as long as full concentration-response and curve-shift relationships are determined. These three parameters are: the allosteric modulator KB, which provides information on the interaction of the allosteric ligand with the allosteric binding pocket on the free receptor, the composite cooperativity parameter, αβ, which provides information on the overall allosteric effect on the orthosteric agonist in the chosen functional assay, and the modulator efficacy parameter, τB, which provides information on the ability of the allosteric ligand to promote agonism in its own right in the absence of orthosteric ligand. Table 4 illustrates an example of such allosteric modulator SAR determined through analysis of the functional effects of a series of 2-amino-3-benzoylthiophenes (2A3BT) on A1 adenosine receptor-mediated ERK1/2 phosphorylation.46 From this analysis, it can be seen that, for instance, the increase in trifluoromethylphenyl substitutions to the R2 group of the 2A3BT scaffold can increase positive cooperativity while having a detrimental effect on modulator affinity (compare 16b to 16d, and 16g to 16h), whereas conformational constraint in the R2/R3 regions tends to convert positive modulators into highly negative modulators (e.g., 16n, 16r). It should also be noted that the reference compound, 16a, progressed into Phase IIB clinical trials for the treatment of neuropathic pain (King Pharmaceuticals) prior to failing due to lack of efficacy; it is possible that this failure may be attributed to the rather low degree of positive cooperativity, as revealed by the application of the operational model to the in vitro data. Although speculative, this finding highlights some of the advantages of operational modeling when applied to allosteric SAR, namely, the ability to link the chemistry to the key, measured, biological parameters, and the ability to facilitate hypothesis generation to understand biological mechanisms and their relevance to the desired therapeutic profile. For instance, one can ask questions such as: how much cooperativity (αβ) or allosteric agonism (τB) is required to achieve in vivo efficacy? How do structural modifications affect compound affinity (KB) versus cooperativity (αβ)? The latter is important as these properties are not correlated and thus different structural manipulations can change them in different directions. Probe dependence will manifest as different αβ values depending on the orthosteric agonist used and/or the signal pathway being assessed as a readout of receptor activation. In terms of drug discovery programs, these insights can be used to more rationally inform the design of candidate selection matrices for drugs acting allosterically. 46,47,52,90,195–197
Table 4.
Allosteric Operational Model Parameters Describing the Functional Effect of Various 2-Amino-3-Benzoylthiophenes on ERK1/2 Phosphorylation mediated by the Orthosteric Agonist, R-PIA, at Adenosine A1 Receptors.
 | ||||
|---|---|---|---|---|
| Compound | Structure | pK3 | logαβ(αβ) | logτB(τB) |
| 16a(T62)a | 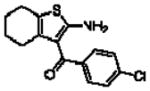 |
5.49 ± 0.09 | 0.58 ± 0.08 (3.8) | −0.32 ± 0.05(0.5) |
|
16b 9aa |
 |
6.37 ± 0.16 | 0.38 ±0.07 (2.4) | 0.33 ± 0.04 (0.5) |
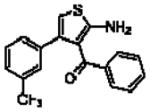
| 16c(VCP333)a | 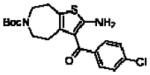 |
5.23 ± 0.25 | 0.64 ± 0.12(4.4) | −1.34 ± 0.75 (0.05) |
| 16d(9o)a | 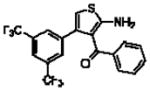 |
5.22 ± 0.07 | 1.06 ± 0.08 (11) | 0.62 ± 0.24 (4.1) |
| 16e(13c)a | 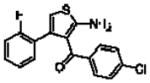 |
5.10 ± 0.18 | 1.04 ± 0.20(11) | 0.13 ± 0.21(2.7) |
| 16f(13d)a | 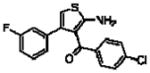 |
5.88 ± 0.16 | 0.67 ± 0.10(4.7) | −0.30 ± 0.08 (0.5) |
| 16g(13a)a | 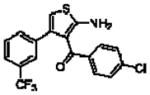 |
5.74 ± 0.12 | 0.55 ± 0.06(3.5) | −0.48 ± 0.08 (0.3) |
| 16h(13o)a | 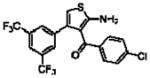 |
5.46 ± 0.32 | 1.31 ± 0.40(20) | 0.50 ± 0.20(3.2) |
| 16i(12j)a | 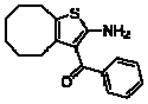 |
5.08 ± 0.17 | 0.46 ± 0.07(2.9) | −0.41 ± 0.09(0.4) |
| 16j(25b)a | 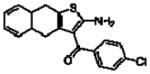 |
6.01 ± 0.16 | 0.44 ± 0.07(2.7) | −0.48 ± 0.08 (0.3) |
| 16k(25f)a | 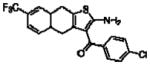 |
5.11 ± 0.41 | 0.58 ± 0.07(3.8) | −1000(ĩ0) |
| 16l(25d)a | 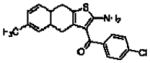 |
5.12 ± 0.32 | 0.82 ± 0.16 (6.6) | −1000(ĩ0) |
| 16m(12o)a | 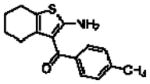 |
5.10 ± 0.14 | 0.98 ± 0.14 (10) | 0.31 ± 0.08 (2.0) |
| 16n(17d)a | 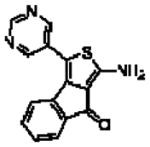 |
6.67 ± 0.13 | −1000(~0) | −1000(~0) |
| 16o(22d)a | 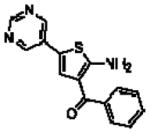 |
5.82 ± 0.10 | −1000(~0) | −1000(~0) |
| 16p(22a)a | 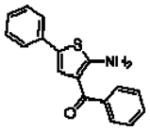 |
6.36 ± 0.13 | −1000(~0) | −1000(~0) |
| 16q(2o)a |  |
6.07 ± 0.05 | −1000(~0) | −1000(~0) |
| 16r(13b)a | 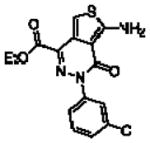 |
8.30 ± 0.24 | −1000(~0) | −1000(~0) |
Compound nomendature refers to compound identifier in the manuscript in which it originally appeared
3. IN VITRO PHARMACOLOGY OF ALLOSTERIC MODULATORS
The development of high-throughput functional (kinetic) assays, have enabled scientists to perform screens of large compound collections and identify small molecules capable of modulating the activity of a receptor through novel, allosteric mechanisms.4,160,198–201 While there are multiple approaches and technologies to accomplish this for 7TMRs, one of the most common approaches measures receptor-induced mobilization of intracellular calcium using an imaging-based plate reader that makes simultaneous measurements of calcium levels in each well of a multi-well-well microplate containing cells transfected with the receptor of interest and loaded with calcium-sensitive fluorescent dye. In the early stages of drug discovery for allosteric modulators of 7TMRs, HTS campaigns targeted the identification of either PAMs or NAMs, running single-point screens with either an EC20 or EC80 concentration of the orthosteric agonist, respectively. More recently, ‘triple add’ protocols have supplanted ‘single add’ screens to allow). for, in a single assay, the identification of PAMs, NAMs, agonists and antagonists (Figure 6A).4,160,198–201 Due to the emerging concept of ‘molecular switches’ (see below),202 this screening paradigm is also optimal as for use as a program’s primary assay, to identify subtle structural changes leading to opposing modes of pharmacology.
Figure 6.

Functional assays, measuring calcium fluorescence as a surrogate for 7TMR receptor activation, employed to identify and profile 7TMR allosteric modulators. A). The ‘triple add’ paradigm for both HTS campaigns and primary assay for lead optimization. Vehicle (black) trace where an EC20 of orthosteric agonist is added 120 seconds into kinetic run, followed by an EC80 of orthosteric agonist. Compounds are added at T=0, and an agonist (green), elicits calcium fluorescence immediately upon addition. Secondary assays with orthosteric radioligands and/or mutant receptors will determine if the compound is an orthosteric or allosteric agonist. An antagonist (red) will block both the EC20 and the EC80; once again, secondary assays will distinguish competitive from noncompetitive (NAM) antagonists. A pure PAM (blue) will not elicit receptor activation alone, but will potentiate the EC20 to varying degrees of efficacy, while an ago-PAM (orange) will activate the receptor alone, plus potentiate the EC20. Hence, a single assay protocol will identify agonists, allosteric agonists, PAMs, ago-PAMs, antagonists and NAMs. B) In vitro pharmacology of an mAChR PAM, once again with a calcium fluorescence readout. The PAM has no effect alone on receptor activation, but in the presence of an EC20 (or sub-threshold concentration of orthosteric agonist, ACh in this case), a classical concentration-response-curve (CRC) results, from which an EC50 for potentiation can be calculated. Also, the %Max, the degree of potentiation above the EC20, can be measured, and both the EC50 and %Max must be optimized. C) “Fold-Shift” assay. Here, the concentration of the orthosteric agonist (ACh) is held constant, and increasing concentrations of the PAM cause a parallel leftward shift of the ACh CRC, in effect making ACh a more potent agonist.
As shown in Figure 6B, a prototypical PAM has no effect on the transfected cells in the absence of the orthosteric agonist, but in the presence of a sub-threshold concentration of the orthosteric agonist, increasing PAM concentrations provide a classical concentration-response-curve (CRC) from which an EC50 for potentiation can be quantified as an empirical measure of modulator potency under a defined set of assay conditions.4,5 This also affords a %Max value, the maximum increase in activity of the orthosteric agonist above the EC20, i.e., from an EC20 to an EC90. In this type of experimental paradigm, the observed EC50 reflects both the affinity of the modulator for the allosteric site, as well as the degree of cooperativity that it exerts on the orthosteric ligand. In contrast, the %Max value only reflects the cooperativity of the interaction. Each of these parameters must be optimized, as it is possible to have allosteric modulators with high potency and low %Max (reflecting high affinity and low cooperativity), weak potency and high %Max (low affinity and high cooperativity) and various combinations thereof. If the assay is run in an alternative mode, whereby the complete orthosteric agonist CRC is determined in the absence or presence of a fixed concentration of allosteric modulator, then another property that can be evaluated and optimized is the degree of the agonist curve ‘fold-shift’, either to the left (potentiation) or right/down (antagonism) of an agonist CRC (Figure 6C).4,5 This is typically reported as a fold-shift at a single concentration, such as a 30-fold shift at 10 μM. Optimally, if multiple modulator concentrations are utilized in this latter type of experiment, then the data can be fitted to the operational model (above) and direct estimates of affinity and cooperativity can be obtained. Irrespective, each of the properties described in the preceding section (% Max, potency, fold-shift or operational model affinity, cooperativity and efficacy) must be simultaneously optimized for optimal in vivo efficacy.4,5 A common question posed in small molecule allosteric modulators programs is: which of these parameters is most important for in vivo efficacy? Unfortunately, the answer is not clear and may vary by 7TMR, allosteric binding site, allosteric ligand chemotype and therapeutic indication. For now, a general empirical guideline followed by most researchers in the field is to optimize for potency (EC50), and % Max and aim for at least a 5-fold shift of the agonist CRC; however, it should be notes that estimates of fold-shift for some Family A and C 7TMR allosteric ligands may approach >70-fold, depending on the assay.4,5 It should be noted that other properties of a CNS compound, for example, the ability of a compound to penetrate the blood brain barrier, the clearance of a compound from the brain and systemic compartments, the amount of compound that is non-protein bound and able to interact with the target, and the in vivo situation being assessed (for example, how long does a receptor need to be occupied with drug to maintain efficacy), certainly must be balanced with in vitro pharmacology profiles when assessing in vivo efficacy and potency of allosteric modulators.
4. STRUCTURE-ACTIVITY-RELATIONSHIPS (SAR) within mGluRs and mAChRs
Another common observation from numerous structure activity relationship (SAR) studies performed to date on 7TMR allosteric modulators is the finding of ‘flat’ or ‘shallow’ SAR for different classes, often making optimization of micromolar potency ligands either difficult or impossible.1,4–12 The literature in this arena is filled with tales of heroic optimization campaigns wherein 100s or 1000s of compounds were synthesized and evaluated, affording only 5–10% active molecules. However, there are also cases wherein SAR is robust and tractable, though rare. Thus, for the chemical optimization of allosteric ligands,4,197,202 focused, iterative library synthesis provides a distinct advantage over traditional singleton approaches; however, multiple dimensions of a scaffold must be surveyed to identify regions tolerant of modification. As SAR is often extremely ‘shallow’, the concept of walking fluorine atoms around an allosteric ligand, i.e., ‘the fluorine walk’ has achieved some success in identifying positions tolerant of change. For example, 6 displayed ‘shallow’ SAR, and multiple attempts at optimization, adding groups larger than fluorine (methyl, OR, Cl, Br, alkyl, etc…) to multiple positions (ie, R1 and/or R2) on the scaffold of 6 led to primarily inactive compounds 17.33,203 The strategic installation of fluorine atoms to both the core R1 and the benzyl side chain R2 of 6 led to compounds with significant improvements in M1 PAM activity (Figure 7), but only in the 5- (18), 8- (19) or 5,8-positions (20) – introduction of fluorine atoms in any other positions led to inactive compounds. Once identified, these new fluorinated cores opened up new avenues for diverse functionalization on the benzyl core that were not tolerated on the parent core, leading to potent M1 PAMs such as 21 (M1 EC50 = 41 nM).203 A very similar ‘fluorine walk’ strategy has proven successful in developing sub-micromolar M1 PAMs in other chemotypes.83,84 It is important to note that in these cases, and many other published studies, only fluorine substitutions were tolerated, making this approach a potential first tier strategy in allosteric ligand optimization.
Figure 7.

SAR within the M1 PAM 6 Chemotype. Groups R1 and R2 other than F, as in 17, were not tolerated and afforded inactive compounds. Walking fluorine atoms around the core identified three positions, leading to cores 18–20, that engendered M1 PAM activity. Re-optimization with the fluorinated cores led to potent M1 PAMs such as 21.
Adding to the ‘flat’ or ‘shallow’ allosteric structure activity relationships (SAR), the emerging concept of ‘molecular switches’, i.e., subtle structural changes that can change the mode of pharmacology (from PAM to NAM and/or SAM) and/or subtype selectivity within a family of receptors, threatens to diminish the application of rational drug design approaches to allosteric modulators.202 Moreover, unexpected occurrences of ‘molecular switches’ require alterations to routine screening paradigms for optimization efforts. Here, the ‘triple add’ approach is ideal, 4,160,198–201 identifying allosteric agonists, PAMs, and NAMs in a single screen. Although examples of ‘molecular switches’ developed for mGluRs and mAChRs are presented herein, this subtle effect has been reported for allosteric kinase and phospholipase ligands as well.202
In the field of mGluRs, the first allosteric modulators disclosed were (−)-ethyl-(7E)-7-hydroxyimino-1,7α-dihydrocyclopropa[b]chromene-1α-carboxylate (CPCCOEt, mGlu1 selective) and 10 (mGlu5 selective), both NAMs, followed by the earliest mGlu5 PAMs, 3,3′-difluorobenzaldazine (DFB), 14, and 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide (CDPPB).4,44, While various molecular switches on the difluorobenzaldazine scaffold had the ability to engender the full range of PAM, NAM and SAM pharmacology (structures not shown),161 an even more surprising example of pharmacological mode switching can be seen in Figure 8. A functional HTS with the mGlu5 receptor identified the “partial antagonist” 22, which maximally inhibited 71% of the glutamate response with an mGlu5 IC50 = 486 nM.202,204–206 This simple and very low molecular weight hit was rapidly explored using a parallel iterative library approach to reveal numerous molecular switches. Introduction of a 3′-methyl group transformed the partial antagonist into a very potent full NAM 23 (mGlu5 IC50 = 7.5 nM). This methyl group exquisitely illustrates the idea of a “molecular switch” as it is moved just one position over to provide compound 24, which is now a weak PAM (mGlu5 EC50 = 3.3 μM) with the ability to potentiate a sub-maximal dose of glutamate up to a full glutamate response (99% Glu Max). Further optimization within this series of PAMs provided compound 25; a very potent mGlu5 PAM which possessed robust in vivo efficacy in a rodent amphetamine-induced hyperlocomotion assay. 202,204–206
Figure 8.

Subtle ‘molecular switches’ within a series of mGlu5 allosteric ligands giving rise to partial antagonists 22, full NAMs 23, and PAMs 24 and 25.
Also within the mGluR field, molecular switches have been reported that profoundly affect the receptor subtype selectivity of a given allosteric ligand, and also the mode of pharmacology. Figure 9 shows 26 ((−)-PHCCC), which possess both mGlu4 PAM and mGlu1 NAM activity.155 Introduction of a weakly basic nitrogen to arrive at 27 (VU0359516) effectively abolished all mGlu1 NAM activity to provide a pure mGlu4 PAM (EC50 > 30 μM versus mGlu1–3,5–8) with good potency and efficacy (mGlu4 EC50 = 380 nM, Glu Max = 121%, 20-fold shift).155 Subsequent to this work, an mGlu2 FRET-based binding assay identified the fluorinated analog 28 which displayed an mGlu2 Ki = 6.6 μM, while at the same time, lacked all functional activity at mGlu2 and mGlu3.207 Importantly, 28 could also block (silence) the activities of related mGlu2 and mGlu3 allosteric modulators 29–31: the definition of an mGlu2/3 SAM. As alluded to above, the position of the fluorine in 28 represented a productive location for the introduction of alternate molecular switches (Figure 9, X = Cl, Me, OMe) which could transform an mGlu1 NAM/mGlu4 PAM 26 or an mGlu2/3 SAM 28 into a series of mGlu2 NAM/mGlu3 PAMs 29–31.207
Figure 9.

Subtle ‘molecular switches’ within a series of mGlu4 allosteric ligands providing a selective mGlu4 PAM 27, or allosteric ligands that cross over into the Group II family of mGlus and displaying mGlu2 and mGlu3 PAM, NAM and SAM activities.
These types of molecular switches that govern receptor subtype selectivity have similarly been reported in the mAChR literature. Figure 10 shows 32, which was an attractive, low-molecular weight PAM hit from a functional HTS assay with the M1 mAChR.82,84,94–96 Although originally identified as only an M1 PAM, subsequent characterization revealed 32 to be a non-selective Gq-coupled mAChR PAM displaying activity at the M1,3,5 receptor subtypes, while being devoid of activity at the Gi-coupled M2 and M4 mAChRs.82 This interesting selectivity for the different G protein-coupled signaling pathways spurred medicinal chemistry efforts to determine if subtle molecular switches could be discovered that might provide selectivity for a single mAChR subtype. The first break through came with the discovery that placing a 5-trifluoromethoxy substituent on the isatin core lead to a profound preference for M5 PAM activity. This molecular switch employed during an exploration of substituents about the benzyl group ultimately provided 33, (X = Me, ML129) and 34 (R = Ph, ML172) the first and most highly selective M5 PAMs, respectively, reported to date.94–96 Further structural modifications around the non-selective lead 32 revealed that replacing the bromine with various aryl groups identified the N-methyl pyrazole as a powerful molecular switch for establishing high levels of M1 selectivity. Fine tuning this M1 PAM activity through the strategic introduction of a fluorine atom, ie, the ‘fluorine walk’ provided the highly selective M1 PAM 35 (ML137).84 Ongoing studies exploring the utility of these and other allosteric ligands promise to reveal numerous additional molecular switches and as these compounds progress into more animal models and detailed DMPK evaluations it will only be a matter of time before metabolism-induced molecular switches are reported.201 It should be noted that such molecular switches may not be observed within all allosteric ligand scaffolds, or at all allosteric binding sites; in fact, some allosteric ligands and binding sites are considered ‘molecular locks’, with robust tractable SAR.
Figure 10.

Subtle ‘molecular switches’ within a series of mAChR allosteric ligands that afford highly selective M5 PAMs 33 and 34 or a selective M1 PAM 35.
5. Opportunities for CNS Disorders and Therapeutics
Despite these challenges, allosteric ligands have enabled researchers to develop small molecule tools (Tables 1–3) 4–8, 10,11, 42–180 with exquisite selectivity for a particular target, not possible with orthosteric ligands, and achieve proof of concept in preclinical models of various CNS disorders. For example, mGlu5 NAMs have provided preclinical target validation in models of anxiety, fragile X syndrome, chronic pain, migraine and GERD.170 In fact, mGlu5 NAMs are one of the most advanced with multiple compounds in clinical development, and displaying efficacy in Phase II (Table 5).208–230 Diverse chemotypes of both mGlu5171 and mGlu2148 PAMs have shown robust activity in preclinical models of schizophrenia and cognition, while selective mGlu4 PAMs157 have validated the target for both pain and Parkinson’s disease.4
Table 5.
mGlu5 NAM Clinical Compounds
| Drug Name | Structure | Organization | Clinical Trials |
|---|---|---|---|
| fenobam™ (NPL-2009)208–210 | 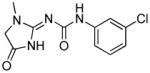 |
Neuropharm | anxiety, fragile X syndrome |
| raseglurant™ (ADX10059)211–214 | 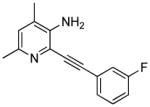 |
Addex | anxiety, GERD, migraine |
| dipraglurant™ (ADX48621)211–216 | Addex | PD-LID | |
| AFQ056217–222 |  |
Novartis | fragile X syndrome, PD-LID, GERD, nicotine addiction |
| AZD2066223,224 | Structure Unknown | AstraZeneca | GERD, chronic neuropathic pain, major depressive disorder, diabetic neuropathy |
| AZD2516223,225 | Structure Unknown | AstraZeneca | GERD, chronic neuropathic pain, major depressive disorder |
| RG7090226 | Structure Unknown | Roche | treatment resistant depression |
| RO4917523227 | Structure Unknown | Roche | fragile X syndrome, treatment resistant depression |
| STX107228,229 | Structure Unknown | Seaside Therapeutics | fragile X syndrome |
The biogenic amine receptors, the prototypes for promiscuous pharmacology, have benefited greatly from allosteric approaches.4,42–180 For example, the mAChRs have been targets of interest for multiple CNS disorders since the 1950s, but the highly conserved orthosteric (acethycholine) binding site led to unselective small molecules. Due to the therapeutic relevance of the mAChRs, multiple companies advanced orthosteric, pan-mAChR agonists into the clinic and noted efficacy in Phase II and Phase III trials in Alzheimer’s disease and schizophrenia patients; however, the adverse events from activation of peripheral M2 and M3 prevented further development. Recently, allosteric ligands, both allosteric/bitopic agonists and PAMs, have been developed for M1, M4 and M5.4,43 These tools are now dissecting the individual contributions of these three mACh receptor subtypes in the clinical efficacy of pan-mAChR agonists. Importantly, this is but one example against the backdrop of the allosteric ligands in Tables1–3, 4,42–180 that are validating discrete targets within large families of receptors, and shedding light on the therapeutic potential of GPCRs, long obscured by the lack of selective tool compounds.
Finally, it is very important to point out that allosteric ligands have been advanced into marketed therapeutics, suggesting the approach of targeting allosteric mechanisms is sound and tractable (Figure 11). 36 is a PAM of the calcium sensing receptor (a Family C GPCR) and is used to treat hyperparathyroidism.115 37 is a NAM of chemokine receptor 5 (a Family A GPCR) that inhibits HIV entry into cells and is used to treat HIV infections.72 Thus, both PAMs and NAMs have advanced to the market, with many other allosteric ligands in clinical trials and late preclinical development.4
Figure 11.

Structures of the two marketed GPCR allosteric modulators. 36, a PAM of the calcium sensing receptor and 37, a NAM of CCR5.
6. Conclusion
The broad uptake of the concept of allosteric modulation has led to a renaissance in pharmacological approaches towards 7TMR pharmacology, promising new and exciting avenues to the pursuit of receptor-subtype and pathway-selective small molecules. Although the promises of this approach are apparent, there remain significant challenges with regards to optimal means for detecting, validating and quantifying allosteric ligand effects in a manner that routinely informs SAR and candidate selection matrices, and is also ultimately predictive of therapeutic efficacy. To meet these challenges, further convergence is required between the disciplines of pharmacology, which can shed insight into the nature and mechanisms underlying phenomena such as probe dependence and pathway-biased modulation, medicinal/synthetic chemistry, which can overcome the issue of ‘flat’ allosteric SAR and explore the full potential of molecular switches in creating new allosteric behaviors, and structural biology, which can identity the underlying structural basis of allosteric ligand binding and the associated receptor conformations that mediate allosteric effects. Fortunately, recent work in the field is illustrating how truly translational approaches towards 7TMR pharmacology promise to overcome many of these challenges and realize the therapeutic potential of allosteric modulators as novel medicines.
Table 2.
Reported Allosteric Modulators of Family B G Protein-Coupled Receptors
| Receptor | Modulator Example(s) |
|---|---|
| CRF1 receptor | NBI 35965;100 NBI 27914;101 antalarmin;102 DMP696;34 SSR125543A;103,104 DMP904;34 NBI 30775/R121919105,106 |
| CGRP receptor | Compounds 1, 3 and 4107 |
| Glucagon | L-168049;108 DAB; and CP-91149109 |
| GLP-1 receptor | T-0632; NovoNordisk compound 1–6;110 compound 2111 |
CGRP, calcitonin gene related peptide; CRF1, corticotrophin releasing factor 1; GLP-1, glucagon-like peptide 1
Acknowledgments
The authors acknowledge funding from NIH for support of our research in allosteric modulators of GPCRS, and in particular NIMH (MH082867), NIDA (DA023947) and the MLPCN (U54MH084659).
Biographies
Bruce J. Melancon, Ph.D. is a medicinal chemist for the Vanderbilt Center for Neuroscience Drug Discovery (VCNDD) at Vanderbilt University. Bruce completed his doctoral work in 2008 at the University of Notre Dame under the direction of Professor Richard E. Taylor on the development of methodology for asymmetric syntheses of 1,2-disubstituted cyclopropanes via cationic pathways. Subsequently, he received an NIH postdoctoral fellowship at Vanderbilt University Medical Center under the direction of Professor Gary A. Sulikowski in chemical biology working on a variety of projects including small-molecule inhibitors of Wnt. Bruce accepted a senior staff scientist position in the VCNDD in 2010. His research focuses on the development of allosteric modulators of mAChR M1 and M4 for the treatment of schizophrenia and Alzheimer’s disease.
Corey R. Hopkins, Ph.D. is Associate Director of Medicinal Chemistry for the Vanderbilt Center for Neuroscience Drug Discovery (VCNDD) at Vanderbilt University. Corey completed his doctorate in 2002 with Professor Peter Wipf at the University of Pittsburgh on the total synthesis of tetrazomine and naphthyridinomycin/bioxalomycin class of compounds. He also developed novel ring expansion methodology to make pharmacophore analogs of Dnacin. Corey moved to Sanofi-Aventis Pharmaceuticals in 2001 and later became Senior Research Investigator in CNS Medicinal Chemistry. He worked on therapeutic targets for several diseases including multiple sclerosis, Rheumatoid arthritis, osteoporosis, respiratory diseases and autoimmune disorders. In 2008, Corey accepted a Research Assistant Professor position in the Department of Pharmacology and serves as Co-Director of the Vanderbilt Specialized Chemistry Center for Accelerated Probe Development.
Kyle A. Emmitte, Ph.D. is Associate Director of Medicinal Chemistry for the Vanderbilt Center for Neuroscience Drug Discovery (VCNDD) at Vanderbilt University. Kyle completed his doctorate in 2001 with Professor Michael T. Crimmins at the University of North Carolina at Chapel Hill on the development of asymmetric alkylation–ring-closing metathesis strategies for the synthesis of medium-ring ether natural products. Kyle joined GlaxoSmithKline in Research Triangle Park, NC, and made significant contributions to the discovery of GSK461364, an inhibitor of polo-like kinase 1. Kyle also co-led the team that discovered GSK1904529, an inhibitor of insulin-like growth factor-1 receptor. In 2008, Kyle accepted a Research Assistant Professor position in the Department of Pharmacology and serves as Co-Director of the Vanderbilt Specialized Chemistry Center for Accelerated Probe Development.
Michael R. Wood, Ph.D. is Associate Director of Medicinal Chemistry for the Vanderbilt Center for Neuroscience Drug Discovery (VCNDD) at Vanderbilt University. Mike completed his doctorate in 1995 with Professor Bruce H. Lipshutz at the University of California, Santa Barbara where he developed catalytic copper conditions for the 1,4-addition of functionalized organometallic reagents. In 1996, he accepted a postdoctoral position with Professor David A. Evans at Harvard University and concluded the first total synthesis of the Vancomycin aglycon. In 1998, he moved to Merck and Co., Inc. to develop antagonists of Bradykinin B1 and CGRP receptors. In 2009, Mike accepted a Research Assistant Professor position in the Department of Pharmacology and serves as the Co-Director of the Vanderbilt Specialized Chemistry Center for Accelerated Probe Development.
Colleen Niswender, Ph.D. is the Director of Molecular Pharmacology for the Vanderbilt Center for Neuroscience Drug Discovery (VCNDD) at Vanderbilt University. Colleen obtained her doctorate in pharmacology in 1996 under the direction of Professor Ronald Emeson at Vanderbilt University studying regulation of RNA editing in the mammalian central nervous system and characterized molecular determinants regulating RNA editing events within the AMPA subtype glutamate receptor, GluR2, and the G protein-coupled 5-HT2C serotonin receptor. She pursued postdoctoral studies with Professor G. Stanley McKnight at the University of Washington. She accepted an Associate Professor position in Pharmacology in 2004 serving as Director of Molecular Pharmacology. She has extensive experience with GPCRs, cell signaling, assay development, HTS and lead optimization, and concepts of allosteric modulation.
Arthur Christopolous, Ph.D. is a Professor and National Health and Medical Research Council (NHMRC) Senior Research Fellow at Monash University (Melbourne, Australia). Arthur received his doctorate in Pharmacology from the Victorian College of Pharmacy at Monash University in 1997 and then pursued postdoctoral studies at the University of Minnesota. In 1999, Arthur returned to the University of Melbourne as a Senior Research Fellow. He currently serves as the co-Director of the Drug Discovery Biology Laboratory at the Monash Institute of Pharmaceutical Sciences & Department of Pharmacology. His research interests include understanding novel modes of regulation of GPCRs to identify novel targets or approaches for drug discovery, investigating virtually all levels of GPCR structure/function, including analysis of the functional significance of single nucleotide polymorphisms and RNA-editing.
P. Jeffrey Conn, Ph.D. is the Director of the Vanderbilt Center for Neuroscience Drug Discovery (VCNDD) at Vanderbilt University. Conn received his doctorate in Pharmacology from Vanderbilt in 1986 and pursued postdoctoral studies at Yale University. He accepted a faculty position in Pharmacology at Emory University in 1988 establishing himself as a leader in studies of neurotransmitter receptor function. In 2000, he moved to Merck and Co., Inc. as director of the Department of Neuroscience. In 2003, Conn moved to Vanderbilt University and founded the VCNDD. He serves as Editor-in-Chief of Molecular Pharmacology. Dr. Conn is the Lee E. Limbard Professor of Pharmacology at Vanderbilt University. Dr. Conn’s current research is focused on development of novel treatment strategies for schizophrenia, Parkinson’s disease, and other disorders.
Craig W. Lindsley, Ph.D. is Director of Medicinal Chemistry for the Vanderbilt Center for Neuroscience Drug Discovery (VCNDD) at Vanderbilt University. Craig received his doctorate in 1996 from the University of California, Santa Barbara, and pursued postdoctoral studies at Harvard University. In 2001, Craig moved to Merck & Co., Inc. and developed a streamlined approach for lead optimization, resulting in delivery of six preclinical candidates. He also provided preclinical proof-of-concept for the first isoenzyme selective, allosteric AKT kinase inhibitors, the first mGluR5 and M1 PAMs. In 2006, Craig accepted Associate Professor appointments in Pharmacology and Chemistry at Vanderbilt University. He serves as Editor-in-Chief of ACS Chemical Neuroscience. Now full professor, Craig serves as Principal Investigator of the Vanderbilt Specialized Chemistry Center for Accelerated Probe Development.
Footnotes
Central Nervous System (CNS), G Protein-Coupled Receptor (GPCR), Seven Transmembrane Receptor (7TMR), Transmembrane (TM), allosteric, positive allosteric modulator (PAM), negative allosteric modulator (NAM), silent allosteric modulator (SAM), benzodiazepines (BZD),γ-aminobutyric acid (GABAA), Venus Fly Trap (VFT), Muscarinic Acetylcholine receptor (mAChR), acetylcholine (ACh), metabotropic glutamate receptor (mGluR), N-methyl D-aspartate (NMDA), 5-hydroxytryptamine receptor 1B (5-HT1B), drug metabolism/pharmacokinetic (DMPK), structure-activity relationship (SAR), glucagon-like peptide 1 (GLP1), dihydroxyphenylglycine (DHPG), Positron Emission Tomography (PET), concentration-response-curve (CRC), fluorescence resonance energy transfer (FRET), high-throughput screening (HTS), Gastroesophageal reflux disease (GERD), (−)-ethyl-(7E)-7-hydroxyimino-1,7α-dihydrocyclopropa[b]chromene-1α-carboxylate (CPCCOEt), 3,3′-difluorobenzaldazine (DFB), 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide (CDPPB), 2-Methyl-6-(phenylethynyl)pyridine (MPEP), [3H]-quinuclidinyl benzylate ([3H]QNB).
References
- 1.Kenakin T, Miller LJ. Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev. 2010;62:265–304. doi: 10.1124/pr.108.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Umbarger HE. Evidence for a negative feedback mechanism in the biosynthesis of isoleucine. Science. 1956;123:848. doi: 10.1126/science.123.3202.848. [DOI] [PubMed] [Google Scholar]
- 3.Fenton AW. Allostery: an illustrated definition for the ‘second secret of life’. Trends Biochem Sci. 2008;33:420–425. doi: 10.1016/j.tibs.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conn PJ, Christopolous A, Lindsley CW. Allosteric Modulators of GPCRs as a Novel Approach to Treatment of CNS Disorders. Nat Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bridges TM, Lindsley CW. G-protein coupled receptors: From classical modes of modulation to allosteric mechanisms. ACS Chem Biol. 2008;3:530–542. doi: 10.1021/cb800116f. [DOI] [PubMed] [Google Scholar]
- 6.Christopoulos A, Kenakin T. G protein-coupled receptors allosterism and complexing. Pharmacol Rev. 2002;54:323–374. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- 7.Christopoulos A. Allosteric binding sites on cell surface receptors: novel targets for drug discovery. Nat Rev Drug Discov. 2002;1:198–210. doi: 10.1038/nrd746. [DOI] [PubMed] [Google Scholar]
- 8.Kenakin TP. 7TM receptor allostery: putting numbers to shapeshifting proteins. Trends Pharmacol Sci. 2009;30:460–469. doi: 10.1016/j.tips.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 9.Mohler HF, Fritschy JM, Rudolph U. A new benzodiazepine pharmacology. J Pharmacol Exp Ther. 2002;300:2–8. doi: 10.1124/jpet.300.1.2. [DOI] [PubMed] [Google Scholar]
- 10.Langmead CJ, Christopoulos A. Allosteric agonists of 7TM receptors: expanding the pharmacological toolbox. Trends Pharmacol Sci. 2006;27:475–481. doi: 10.1016/j.tips.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 11.May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- 12.Lewis JA, Lebois EP, Lindsley CW. Allosteric modulators of kinases and GPCRs. Design principles and structural diversity. Curr Opin Chem Biol. 2008;12:269–279. doi: 10.1016/j.cbpa.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 13.Scott SA, Selvy PE, Buck J, Cho HP, Criswell TL, Thomas AL, Armstrong MD, Arteaga C, Lindsley CW, Brown HA. Design of isoform-selective phospholipase D inhibitors that modulate cancer cell invasiveness. Nat Chem Bio. 2009;5:108–117. doi: 10.1038/nchembio.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hogg RC, Buisson B, Bertrand D. Allosteric modulation of ligand-gated ion channels. Biochem Pharmacol. 2005;70:1267–1276. doi: 10.1016/j.bcp.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 15.Bogoyevitch MA, Fairlie DP. A new paradigm for protein kinase inhibition: blocking phosphorylation without directly targeting ATP binding. Drug Discov Today. 2007;12:622–633. doi: 10.1016/j.drudis.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 16.Deupi X, Standfuss J. Strucutral insights into agonist-induced activation of G protein-coupled receptors. Curr Opin Struct Bio. doi: 10.1016/j.sbi.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 17.Li J, Edwards PC, Burghammer M, Villa C, Schertler GF. Structure of bovine rhodopsin in a trigonal crystal form. J Mol Biol. 2004;343:1409–1438. doi: 10.1016/j.jmb.2004.08.090. [DOI] [PubMed] [Google Scholar]
- 18.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. The retinal conformation and its environment in rhodopsin in light of a new 2.2 A ° crystal structure. J Mol Biol. 2004;342:571–583. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 19.Standfuss J, Xie G, Edwards PC, Burghammer M, Oprian DD, Schertler GF. Crystal structure of a thermally stable rhodopsin mutant. J Mol Biol. 2007;372:1179–1188. doi: 10.1016/j.jmb.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kobilka B, Schertler GF. New G-protein-coupled receptor crystal structures: insights and limitations. Trends Pharmacol Sci. 2008;29:79–83. doi: 10.1016/j.tips.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 21.Tate CG, Schertler GF. Engineering G protein-coupled receptors to facilitate their structure determination. Curr Opin Struct Biol. 2009;19:386–395. doi: 10.1016/j.sbi.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 22.Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. The 2. 6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature. 2008;454:183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- 28.Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 29.Standfuss J, Edwards PC, D’Antona A, Fransen M, Xie G, Oprian DD, Schertler GF. The structural basis of agonist- induced activation in constitutively active rhodopsin. Nature. 2011;471:656–660. doi: 10.1038/nature09795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choe HW, Kim YJ, Park JH, Morizumi T, Pai EF, Krauss N, Hofmann KP, Scheerer P, Ernst OP. Crystal structure of metarhodopsin II. Nature. 2011;471:651–655. doi: 10.1038/nature09789. [DOI] [PubMed] [Google Scholar]
- 31.Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma L, Seager M, Wittman M, Bickel N, Burno M, Jones K, Graufelds VK, Xu G, Pearson M, McCampbell A, Gaspar R, Shughrue P, Danzinger A, Regan C, Garson S, Doran S, Kreatsoulas C, Veng L, Lindsley CW, Shipe W, Kuduk S, Jacobson M, Sur C, Kinney G, Seabrook GR, Ray WJ. Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc Natl Acad Sci USA. 2009;106:15950–15955. doi: 10.1073/pnas.0900903106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li YW, Fitzgerald L, Wong H, Lelas S, Zhang G, Lindner MD, Wallace T, McElroy J, Lodge NJ, Gilligan P, Zaczek R. The pharmacology of DMP696 and DMP904, non-peptidergic CRF1 receptor antagonists. CNS Drug Rev. 2005;11:21–52. doi: 10.1111/j.1527-3458.2005.tb00034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gasparini F, Lingenhohl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S, Varney MA, Johnson EC, Hess SD, Rao SP, Sacaan AI, Santori EM, Velicelebi G, Kuhn R. 2-Methyl-6-(phenylethynyl)-pyrimidine (MPEP), a potent, selective and systemically active mGluR5 receptor antagonist. Neuropharmacology. 1999;38:1493–1503. doi: 10.1016/s0028-3908(99)00082-9. [DOI] [PubMed] [Google Scholar]
- 36.Lindsley JE, Rutter J. Whence cometh the allosterome? Proc Natl Acad Sci USA. 2006;103:10533–10535. doi: 10.1073/pnas.0604452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Urwyler S. Allosteric modulation of Family C G protein-coupled receptors: from molecular insights to therapeutic perspectives. Pharmacol Rev. 2011;63:59–126. doi: 10.1124/pr.109.002501. [DOI] [PubMed] [Google Scholar]
- 38.Tsai G, Coyle JT. Glutamatergic mechanisms in schizophrenia. Annu Rev Pharmacol Toxicol. 2002;42:165–179. doi: 10.1146/annurev.pharmtox.42.082701.160735. [DOI] [PubMed] [Google Scholar]
- 39.Lee HJ, Mun HC, Lewis NC, Crouch MF, Culverston EL, Mason RS, Conigrave AD. Allosteric activation of the extracellular Ca2+-sensing receptor by L-amino acids enhances ERK1/2 phosphorylation. Biochem J. 2007;404:141–149. doi: 10.1042/BJ20061826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Massot O, Rosselle JC, Fillion MP, Grimbaldi B, Cloez-Tayarani I, Fugelli A, Prudhomme N, Seguin L, Rosseau B, Plantefol M. 5-Hydroxytryptamine-moduline, a new endogenous cerebral peptide, controls the serotonergic activity via a specific interaction with 5-hydroxytrypatime 1B/1D receptors. Mol Pharmacol. 1996;50:752–762. [PubMed] [Google Scholar]
- 41.Rouselle JC, Massot O, Delepierre M, Zifa E, Rosseau B, Fillion G. Isolation and characterization of an endogenous peptide form rat brain interacting specifically with the serotonergic 1B receptor subtypes. J Biol Chem. 1996;271:726–735. doi: 10.1074/jbc.271.2.726. [DOI] [PubMed] [Google Scholar]
- 42.Bridges TM, LeBois EP, Hopkins CR, Wood MR, Jones JK, Conn PJ, Lindsley CW. Antipsychotic potential of muscarinic allosteric modulation. Drug News & Perspect. 2010;23:229–240. doi: 10.1358/dnp.2010.23.4.1416977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Conn PJ, Jones C, Lindsley CW. Subtype selective allosteric modulators of muscarinic receptors for the treatment of CNS disorders. Trends in Pharm Sci. 2009;30:148–156. doi: 10.1016/j.tips.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Conn PJ, Lindsley CW, Jones C. Allosteric modulation of metabotropic glutamate receptors as a novel approach for the treatment of schizophrenia. Trends in Pharm Sci. 2009;30:25–31. doi: 10.1016/j.tips.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aurelio L, Valant C, Figler H, Flynn BL, Linden J, Sexton PM, Christopoulos A, Scammells PJ. 3- and 6-Substituted 2-amino-4,5,6,7-tetrahydrothieno[2,3-c]pyridines as A1 adenosine receptor allosteric modulators and antagonists. Bioorg Med Chem. 2009;17:7353–7361. doi: 10.1016/j.bmc.2009.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Valant C, Aurelio L, Urmaliya VB, White P, Scammells PJ, Sexton PM, Christopoulos A. Delineating the mode of action of adenosine A1 receptor allosteric modulators. Mol Pharmacol. 2010;78:444–455. doi: 10.1124/mol.110.064568. [DOI] [PubMed] [Google Scholar]
- 47.Aurelio L, Valant C, Flynn BL, Sexton PM, Christopoulos A, Scammells PJ. Allosteric modulators of the adenosine A1 receptor: synthesis and pharmacological evaluation of 4-substituted 2-amino-3-benzoylthiophenes. J Med Chem. 2009;52:4543–4547. doi: 10.1021/jm9002582. [DOI] [PubMed] [Google Scholar]
- 48.Kim Y, de Castro S, Gao ZG, Ijzerman AP, Jacobson KA. Novel 2- and 4-substituted 1H-imidazo[4,5-c]quinolin-4-amine derivatives as allosteric modulators of the A3 adenosine receptor. J Med Chem. 2009;52:2098–2108. doi: 10.1021/jm801659w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lane JR, Beukers MW, Muilder-Krieger T, IJzerman AP. The endocannabinoid 2-arachidonylglycerol is a negative allosteric modulator of the human A3 adenosine receptor. Biochemical Pharmacology. 2010;79:48–56. doi: 10.1016/j.bcp.2009.07.024. [DOI] [PubMed] [Google Scholar]
- 50.Pihlavisto M, Sjöholm B, Scheinin M, Wurster S. Modulation of agonist binding to recombinant human a2-adrenoceptors by sodium ions. Biochim Biophys Acta. 1998;1448:135–146. doi: 10.1016/s0167-4889(98)00118-9. [DOI] [PubMed] [Google Scholar]
- 51.Swaminath G, Steenhuis J, Kobilka B, Lee TW. Allosteric modulation of β2-adrenergic receptor by Zn(2+) Mol Pharmacol. 2002;61:65–72. doi: 10.1124/mol.61.1.65. [DOI] [PubMed] [Google Scholar]
- 52.Price MR, Baillie GL, Thomas A, Stevenson LA, Easson M, Goodwin R, McLean A, McIntosh L, Goodwin G, Walker G, Westwood P, Marrs J, Thomson F, Cowley P, Christopoulos A, Pertwee RG, Ross RA. Allosteric modulation of the cannabinoid CB1 receptor. Mol Pharmacol. 2005;68:1484–1495. doi: 10.1124/mol.105.016162. [DOI] [PubMed] [Google Scholar]
- 53.Horswill JG, Bali U, Shaaban S, Keily JF, Jeevaratnam P, Babbs AJ, Reynet C, Wong Kai In P. PSNCBAM-1, a novel allosteric antagonist at cannabinoid CB1 receptors with hypophagic effects in rats. Br J Pharmacol. 2007;152:805–814. doi: 10.1038/sj.bjp.0707347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Navarro HA, Howard JL, Pollard GT, Carroll FI. Positive allosteric modulation of the human cannabinoid (CB) receptor by RTI-371, a selective inhibitor of the dopamine transporter. Br J Pharmacol. 2009;156:1178–1184. doi: 10.1111/j.1476-5381.2009.00124.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marsh DR, Flemming JM. Inhibition of CXCR1 and CXCR2 chemokine receptors attenuates acute inflammation, preserves gray matter and diminishes autonomic dysreflexia after spinal cord injury. Spinal Cord. 2011;49:337–344. doi: 10.1038/sc.2010.127. [DOI] [PubMed] [Google Scholar]
- 56.Sablone MR, Cesta MC, Moriconi A, Aramini A, Bizzarri C, Di Giacinto C, Di Bitondo R, Gloaguen I, Aschi M, Crucianelli M, Bertini R, Allegretti M. Structure-Activity Relationship of novel phenylacetic CXCR1 inhibitors. Bioorg Med Chem Lett. 2009;19:4026–4030. doi: 10.1016/j.bmcl.2009.06.027. [DOI] [PubMed] [Google Scholar]
- 57.Varney ML, Singh S, Li A, Mayer-Ezell R, Bond R, Singh RK. Small molecule antagonists for CXCR2 and CXCR1 inhibit human colon cancer liver metastases. Cancer Lett. 2011;300:180–188. doi: 10.1016/j.canlet.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chapman RW, Phillips JE, Hipkin RW, Curran AK, Lundell D, Fine JS. CXCR2 antagonists for the treatment of pulmonary disease. Pharmacol Ther. 2009;121:55–68. doi: 10.1016/j.pharmthera.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 59.Singh S, Sadanandam A, Nannuru KC, Varney ML, Mayer-Ezell R, Bond R, Singh RK. Small-molecule antagonists for CXCR2 and CXCR1 inhibit human melanoma growth by decreasing tumor cell proliferation, survival, and angiogenesis. Clin Cancer Res. 2009;15:2380–2386. doi: 10.1158/1078-0432.CCR-08-2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sachpatzidis A, Benton BK, Manfredi JP, Wang H, Hamilton A, Dohlman HG, Lolis E. Identification of allosteric peptide agonists of CXCR4. J Biol Chem. 2003;278:896–907. doi: 10.1074/jbc.M204667200. [DOI] [PubMed] [Google Scholar]
- 61.Zhao J, Ben LH, Wu YL, Hu W, Ling K, Xin SM, Nie HL, Ma L, Pei G. Anti-HIV agent trichosanthin enhances the capabilities of chemokines to stimulate chemotaxis and G protein activation, and this is mediated through interaction of trichosanthin and chemokine receptors. J Exp Med. 1999;190:101–111. doi: 10.1084/jem.190.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cashen AF, Nervi B, DiPersio J. AMD3100: CXCR4 antagonist and rapid stem cell-mobilizing agent. Future Oncol. 2007;3:19–27. doi: 10.2217/14796694.3.1.19. [DOI] [PubMed] [Google Scholar]
- 63.Vaidehi N, Schlyer S, Trabanino RJ, Floriano WB, Abrol R, Sharma S, Kochanny M, Koovakat S, Dunning L, Liang M, Fox JM, de Mendonça FL, Pease JE, Goddard WA, III, Horuk R. Predictions of CCR1 chemokine receptor structure and BX 471 antagonist binding followed by experimental validation. J Biol Chem. 2006;281:27613–27620. doi: 10.1074/jbc.M601389200. [DOI] [PubMed] [Google Scholar]
- 64.Gladue RP, Cole SH, Roach ML, Tylaska LA, Nelson RT, Shepard RM, McNeish JD, Ogborne KT, Neote KS. The human specific CCR1 antagonist CP-481,715 inhibits cell infiltration and inflammatory responses in human CCR1 transgenic mice. J Immunol. 2006;176:3141–3148. doi: 10.4049/jimmunol.176.5.3141. [DOI] [PubMed] [Google Scholar]
- 65.Sabroe I, Peck MJ, Van Keulen BJ, Jorritsma A, Simmons G, Clapham PR, Williams TJ, Pease JE. A small molecule antagonist of chemokine receptors CCR1 and CCR3. Potent inhibition of eosinophil function and CCR3-mediated HIV-1 entry. J Biol Chem. 2000;275:25985–25992. doi: 10.1074/jbc.M908864199. [DOI] [PubMed] [Google Scholar]
- 66.Watson C, Jenkinson S, Kazmierski W, Kenakin T. The CCR5 receptor-based mechanism of action of 873140, a potent allosteric noncompetitive HIV entry inhibitor. Mol Pharmacol. 2005;67:1268–1282. doi: 10.1124/mol.104.008565. [DOI] [PubMed] [Google Scholar]
- 67.Nichols WG, Steel HM, Bonny T, Adkison K, Curtis L, Millard J, Kabeya K, Clumeck N. Hepatotoxicity observed in clinical trials of aplaviroc (GW873140) Antimicrob Agents Chemother. 2008;52:858–865. doi: 10.1128/AAC.00821-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maeda K, Nakata H, Koh Y, Miyakawa T, Ogata H, Takaoka Y, Shibayama S, Sagawa K, Fukushima D, Moravek J, Koyanagi Y, Mitsuya H. Spirodiketopiperazine-based CCR5 inhibitor which preserves CC-chemokine/CCR5 interactions and exerts potent activity against R5 human immunodeficiency virus type 1 in vitro. J Virol. 2004;78:8654–8662. doi: 10.1128/JVI.78.16.8654-8662.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tremblay CL, Giguel F, Guan Y, Chou TC, Takashima K, Hirsch MS. TAK-220, a novel small-molecule CCR5 antagonist, has favorable anti-human immunodeficiency virus interactions with other antiretrovirals in vitro. Antimicrob Agents Chemother. 2005;49:3483–3485. doi: 10.1128/AAC.49.8.3483-3485.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Strizki JM, Xu S, Wagner NE, Wojcik L, Liu J, Hou Y, Endres M, Palani A, Shapiro S, Clader JW, Greenlee WJ, Tagat JR, McCombie S, Cox K, Fawzi AB, Chou CC, Pugliese-Sivo C, Davies L, Moreno ME, Ho DD, Trkola A, Stoddart CA, Moore JP, Reyes GR, Baroudy BM. SCH-C (SCH 351125), an orally bioavailable, small molecule antagonist of the chemokine receptor CCR5, is a potent inhibitor of HIV-1 infection in vitro and in vivo. Proc Natl Acad Sci USA. 2001;98:12718–12723. doi: 10.1073/pnas.221375398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Klibanov OM. Vicriviroc, a CCR5 receptor antagonist for the potential treatment of HIV infection. Curr Opin Investig Drugs. 2009;10:845–859. [PubMed] [Google Scholar]
- 72.Meanwell NA, Kadow JF. Maraviroc, a chemokine CCR5 receptor antagonist for the treatment of HIV infection and AIDS. Curr Opin Investig Drugs. 2007;8:669–681. [PubMed] [Google Scholar]
- 73.Schetz JA, Sibley DR. Zinc allosterically modulates antagonist binding to cloned D1 and D2 dopamine receptors. J Neurochem. 1997;68:1990–1997. doi: 10.1046/j.1471-4159.1997.68051990.x. [DOI] [PubMed] [Google Scholar]
- 74.Hoare SR, Coldwell MC, Armstrong D, Strange PG. Regulation of human D(1), d(2(long)), d(2(short)), D(3) and D(4) dopamine receptors by amiloride and amiloride analogues. Br J Pharmacol. 2000;130:1045–1059. doi: 10.1038/sj.bjp.0703370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.De Mey JG, Compeer MG, Lemkens P, Meens MJ. ET(A)-receptor antagonists or allosteric modulators? Trends Pharamcol Sci. 2011;32:345–351. doi: 10.1016/j.tips.2011.02.018. [DOI] [PubMed] [Google Scholar]
- 76.Heitman LH, Ye K, Oosterom J, Ijzerman AP. Amiloride derivatives and a nonpeptidic antagonist bind at two distinct allosteric sites in the human gonadotropin-releasing hormone receptor. Mol Pharmacol. 2008;73:1808–1815. doi: 10.1124/mol.107.043521. [DOI] [PubMed] [Google Scholar]
- 77.Holst B, Frimurer TM, Mokrosinski J, Halkjaer T, Cullberg KB, Underwood CR, Schwartz TW. Overlapping binding site for the endogenous agonist, small-molecule agonists, and ago-allosteric modulators on the ghrelin receptor. Mol Pharmacol. 2009;75:44–59. doi: 10.1124/mol.108.049189. [DOI] [PubMed] [Google Scholar]
- 78.Janovick JA, Maya-Núñez G, Ulloa-Aguirre A, Huhtaniemi IT, Dias JA, Verbost P, Conn PM. Increased plasma membrane expression of human follicle-stimulating hormone receptor by a small molecule thienopyr(im)idine. Mol Cell Endocrinol. 2009;298:84–88. doi: 10.1016/j.mce.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Heitman LH, Oosterom J, Bonger KM, Timmers CM, Wiegerinck PH, Ijzerman AP. [3H]Org 43553, the first low-molecular-weight agonistic and allosteric radioligand for the human luteinizing hormone receptor. Mol Pharmacol. 2008;73:518–524. doi: 10.1124/mol.107.039875. [DOI] [PubMed] [Google Scholar]
- 80.Gregory KJ, Sexton PM, Christopoulos A. Allosteric modulation of muscarinic acetylcholine receptors. Curr Neuropharmacol. 2007;5:157–167. doi: 10.2174/157015907781695946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jones CK, Brady AE, Davis AA, Xiang Z, Bubser M, Tantawy MN, Kane A, Bridges TM, Kennedy JP, Bradley SR, Peterson T, Baldwin RM, Kessler R, Deutch A, Lah JL, Levey AI, Lindsley CW, Conn PJ. Novel selective allosteric activator of the M1 muscarinic acetylcholine receptor reduces amyloid processing and produces antipsychotic-like activity in rats. J Neurosci. 2008;28(41):10422–10433. doi: 10.1523/JNEUROSCI.1850-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Marlo JE, Niswender CM, Luo Q, Brady AE, Shirey JK, Rodriguez AL, Bridges TM, Williams R, Days E, Nalywajko NT, Austin C, Williams M, Xiang Y, Orton D, Brown HA, Kim K, Lindsley CW, Weaver CD, Conn PJ. Identification and characterization of novel allosteric potentiators of M1 muscarinic receptors reveals multiple modes of activity. Mol Pharm. 2009;75:577–588. doi: 10.1124/mol.108.052886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Reid PR, Bridges TM, Sheffler DJ, Cho HP, Lewis LM, Days E, Daniels JS, Jones CK, Niswender CM, Weaver CD, Conn PJ, Lindsley CW, Wood MR. Discovery and optimization of a novel, selective and brain penetrant M1 positive allosteric modulator (PAM): the development of ML169, an MLPCN probe. Bioorg Med Chem Lett. 2011;21:2697–2701. doi: 10.1016/j.bmcl.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bridges TM, Kennedy JP, Cho HP, Conn PJ, Lindsley CW. Chemical optimization of an M1, M3, M5 positive allosteric modulator (PAM) lead. Part II. Development of a highly selective M1 PAM. Bioorg Med Chem Lett. 2010;20:1972–1975. doi: 10.1016/j.bmcl.2010.01.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lebois EP, Bridges TM, Dawson ES, Kennedy Jp, Xiang Z, Jadhav SB, Yin H, Meiler J, Jones CK, Conn PJ, Weaver CD, Lindsley CW. Discovery and development of novel subtype-selective M1 allosteric agonists for the investigation of M1 receptor function. ACS Chemical Neurosci. 2010;1:104–121. doi: 10.1021/cn900003h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Suga H, Ehlert FJ. Investigating the interaction of McN-A-343 with the M2 muscarinic receptor using its nitrogen mustard derivative. Biochem Pharmacol. 2010;79:1025–1035. doi: 10.1016/j.bcp.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Maier-Peuschel M, Frölich N, Dees C, Hommers LG, Hoffmann C, Nikolaev VO, Lohse MJ. A fluorescence resonance energy transfer-based M2 muscarinic receptor sensor reveals rapid kinetics of allosteric modulation. J Biol Chem. 2010;285:8793–8800. doi: 10.1074/jbc.M109.098517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.May LT, Avlani VA, Langmead CJ, Herdon HJ, Wood MD, Sexton PM, Christopoulos A. Structure-function studies of allosteric agonism at M2 muscarinic acetylcholine receptors. Mol Pharmacol. 2007;72:463–476. doi: 10.1124/mol.107.037630. [DOI] [PubMed] [Google Scholar]
- 89.Lanzafame AA, Sexton PM, Christopoulos A. Interaction studies of multiple binding sites on m4 muscarinic acetylcholine receptors. Mol Pharmacol. 2006;70:736–746. doi: 10.1124/mol.106.024711. [DOI] [PubMed] [Google Scholar]
- 90.Leach K, Loiacono RE, Felder CC, McKinzie DL, Mogg A, Shaw DB, Sexton PM, Christopoulos A. Molecular mechanisms of action and in vivo validation of an M4 muscarinic acetylcholine receptor allosteric modulator with potential antipsychotic properties. Neuropsychopharmacol. 2010;35:855–869. doi: 10.1038/npp.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shirey JK, Xiang Z, Orton D, Brady AE, Johnson KA, Williams R, Ayala JE, Rodriguez AL, Wess J, Weaver D, Niswender CM, Conn PJ. An allosteric potentiator of M4 mAChR modulates hippocampal synaptic transmission. Nat Chem Biol. 2008;4:42–50. doi: 10.1038/nchembio.2007.55. [DOI] [PubMed] [Google Scholar]
- 92.Brady A, Jones CK, Bridges TM, Kennedy PJ, Thompson AD, Breininger ML, Gentry PR, Yin H, Jadhav SB, Shirey J, Conn PJ, Lindsley CW. Centrally active allosteric potentiators of the M4 muscarinic acetylcholine receptor reverse amphetamine-induced hyperlocomotion behavior in rats. J Pharm & Exp Ther. 2008;327:941–953. doi: 10.1124/jpet.108.140350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kennedy JP, Bridges TM, Gentry PR, Brogan JT, Brady AE, Shirey JK, Jones CK, Conn PJ, Lindsley CW. Synthesis and structure-activity-relationships of allosteric potentiators of the M4 muscarinic acetylcholine receptor. ChemMedChem. 2009;4:1600–1607. doi: 10.1002/cmdc.200900231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bridges TM, Marlo JE, Niswender CM, Jones JK, Jadhav SB, Gentry PR, Weaver CD, Conn PJ, Lindsley CW. Discovery of the first highly M5-preferring muscarinic acetylcholine receptor ligand, an M5 positive allosteric modulator derived from a series of 5-trifluoromethoxy N-benzyl isatins. J Med Chem. 2009;52:3445–3448. doi: 10.1021/jm900286j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bridges TM, Kennedy JP, Cho HP, Conn PJ, Lindsley CW. Chemical optimization of an M1, M3, M5 positive allosteric modulator (PAM) lead. Part I. Development of a highly selective M5 PAM. Bioorg Med Chem Lett. 2010;20:558–562. doi: 10.1016/j.bmcl.2009.11.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bridges TM, Kennedy JP, Hopkins CR, Conn PJ, Lindsley CW. Heterobiaryl and heterobiaryl ether derived M5 positive allosteric modulators. Bioorg Med Chem Lett. 2010;20:5617–5622. doi: 10.1016/j.bmcl.2010.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kathmann M, Flau K, Redmer A, Tränkle C, Schlicker E. Cannabidiol is an allosteric modulator at mu- and delta-opioid receptors. Naunyn Schmiedebergs Arch Pharmacol. 2006;372:354–361. doi: 10.1007/s00210-006-0033-x. [DOI] [PubMed] [Google Scholar]
- 98.Thomas EA, Carson MJ, Neal MJ, Sutcliffe JG. Unique allosteric regulation of 5-hydroxytryptamine receptor-mediated signal transduction by oleamide. Proc Natl Acad Sci USA. 1997;94:14115–14119. doi: 10.1073/pnas.94.25.14115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Im WB, Chio CL, Alberts GL, Dinh DM. Positive allosteric modulator of the human 5-HT2C receptor. Mol Pharmacol. 2003;64:78–84. doi: 10.1124/mol.64.1.78. [DOI] [PubMed] [Google Scholar]
- 100.Hoare SR, Fleck BA, Gross RS, Crowe PD, Williams JP, Grigoriadis DE. Allosteric ligands for the corticotropin releasing factor type 1 receptor modulate conformational states involved in receptor activation. Mol Pharmacol. 2008;73:1371–1380. doi: 10.1124/mol.107.042978. [DOI] [PubMed] [Google Scholar]
- 101.Hoare SR, Brown BT, Santos MA, Malany S, Betz SF, Grigoriadis DE. Single amino acid residue determinants of non-peptide antagonist binding to the corticotropin-releasing factor1 (CRF1) receptor. Biochem Pharmacol. 2006;72:244–255. doi: 10.1016/j.bcp.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 102.Hoare SR, Sullivan SK, Ling N, Crowe PD, Grigoriadis DE. Mechanism of corticotropin-releasing factor type I receptor regulation by nonpeptide antagonists. Mol Pharmacol. 2003;63:751–765. doi: 10.1124/mol.63.3.751. [DOI] [PubMed] [Google Scholar]
- 103.Gully D, Geslin M, Serva L, Fontaine E, Roger P, Lair C, Darre V, Marcy C, Rouby PE, Simiand J, Guitard J, Gout G, Steinberg R, Rodier D, Griebel G, Soubrié P, Pascal M, Pruss R, Scatton B, Maffrand JP, Le Fur G. 4-(2-Chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]5-methyl-N-(2-propynyl)-1,3-thiazol-2-amine hydrochloride (SSR125543A): a potent and selective corticotrophin-releasing factor(1) receptor antagonist. I. Biochemical and pharmacological characterization. J Pharmacol Exp Ther. 2002;301:322–332. doi: 10.1124/jpet.301.1.322. [DOI] [PubMed] [Google Scholar]
- 104.Griebel G, Simiand J, Steinberg R, Jung M, Gully D, Roger P, Geslin M, Scatton B, Maffrand JP, Soubrié P. 4-(2-Chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4-methylphenyl)ethyl]5-methyl-N-(2-propynyl)-1, 3-thiazol-2-amine hydrochloride (SSR125543A), a potent and selective corticotrophin-releasing factor(1) receptor antagonist. II. Characterization in rodent models of stress-related disorders. J Pharmacol Exp Ther. 2002;301:333–345. doi: 10.1124/jpet.301.1.333. [DOI] [PubMed] [Google Scholar]
- 105.Hoare SR. Allosteric modulators of class B G-protein-coupled receptors. Curr Neuropharmacol. 2007;5:168–179. doi: 10.2174/157015907781695928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chen C, Wilcoxen KM, Huang CQ, Xie YF, McCarthy JR, Webb TR, Zhu YF, Saunders J, Liu XJ, Chen TK, Bozigian H, Grigoriadis DE. Design of 2,5-dimethyl-3-(6-dimethyl-4-methylpyridin-3-yl)-7-dipropylaminopyrazolo[1,5-a]pyrimidine (NBI 30775/R121919) and structure--activity relationships of a series of potent and orally active corticotropin-releasing factor receptor antagonists. J Med Chem. 2004;47:4787–4798. doi: 10.1021/jm040058e. [DOI] [PubMed] [Google Scholar]
- 107.Salvatore CA, Mallee JJ, Bell IM, Zartman CB, Williams TM, Koblan KS, Kane SA. Identification and pharmacological characterization of domains involved in binding of CGRP receptor antagonists to the calcitonin-like receptor. Biochemistry. 2006;45:1881–1887. doi: 10.1021/bi052044w. [DOI] [PubMed] [Google Scholar]
- 108.Petersen KF, Sullivan JT. Effects of a novel glucagon receptor antagonist (Bay 27-9955) on glucagon-stimulated glucose production in humans. Diabetologia. 2001;44:2018–2024. doi: 10.1007/s001250100006. [DOI] [PubMed] [Google Scholar]
- 109.Latsis T, Andersen B, Agius L. Diverse effects of two allosteric inhibitors on the phosphorylation state of glycogen phosphorylase in hepatocytes. Biochem J. 2002;368:309–316. doi: 10.1042/BJ20021070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Koole C, Wootten D, Simms J, Valant C, Sridhar R, Woodman OL, Miller LJ, Summers RJ, Christopoulos A, Sexton PM. Allosteric ligands of the glucagon-like peptide 1 receptor (GLP-1R) differentially modulate endogenous and exogenous peptide responses in a pathway-selective manner: implications for drug screening. Mol Pharmacol. 2010;78:456–465. doi: 10.1124/mol.110.065664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Koole C, Wootten D, Simms J, Valant C, Miller LJ, Christopoulos A, Sexton PM. Polymorphism and ligand dependent changes in human glucagon-like peptide-1 receptor (GLP-1R) function: allosteric rescue of loss of function mutation. Mol Pharmacol. 2011 doi: 10.1124/mol.111.072884. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nemeth EF, Steffey ME, Hammerland LG, Hung BC, Van Wagenen BC, DelMar EG, Balandrin MF. Calcimimetics with potent and selective activity on the parathyroid calcium receptor. Proc Natl Acad Sci USA. 1998;95:4040–4045. doi: 10.1073/pnas.95.7.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang Z, Qiu W, Quinn SJ, Conigrave AD, Brown EM, Bai M. Three adjacent serines in the extracellular domains of the CaR are required for L-amino acid-mediated potentiation of receptor function. J Biol Chem. 2002;277:33727–33735. doi: 10.1074/jbc.M200976200. [DOI] [PubMed] [Google Scholar]
- 114.Zhang Z, Jiang Y, Quinn SJ, Krapcho K, Nemeth EF, Bai M. L-phenylalanine and NPS R-467 synergistically potentiate the function of the extracellular calcium-sensing receptor through distinct sites. J Biol Chem. 2002;277:33736–33741. doi: 10.1074/jbc.M200978200. [DOI] [PubMed] [Google Scholar]
- 115.Nemeth EF, Heaton WH, Miller M, Fox J, Balandrin MF, Van Wagenen BC, Colloton M, Karbon W, Scherrer J, Shatzen E, Rishton G, Scully S, Qi M, Harris R, Lacey D, Martin D. Pharmacodynamics of the type II calcimimetic compound cinacalcet HCl. J Pharmacol Exp Ther. 2004;308:627–635. doi: 10.1124/jpet.103.057273. [DOI] [PubMed] [Google Scholar]
- 116.Nemeth EF, Delmar EG, Heaton WL, Miller MA, Lambert LD, Conklin RL, Gowen M, Gleason JG, Bhatnagar PK, Fox J. Calcilytic compounds: potent and selective Ca2+ receptor antagonists that stimulate secretion of parathyroid hormone. J Pharmacol Exp Ther. 2001;299:323–331. [PubMed] [Google Scholar]
- 117.Kessler A, Faure H, Petrel C, Rognan D, Césario M, Ruat M, Dauban P, Dodd RH. N1-Benzoyl-N2-[1-(1-naphthyl)ethyl]-trans-1,2-diaminocyclohexanes: Development of 4-chlorophenylcarboxamide (calhex 231) as a new calcium sensing receptor ligand demonstrating potent calcilytic activity. J Med Chem. 2006;49:5119–5128. doi: 10.1021/jm051233+. [DOI] [PubMed] [Google Scholar]
- 118.Kumar S, Matheny CJ, Hoffman SJ, Marquis RW, Schultz M, Liang X, Vasko JA, Stroup GB, Vaden VR, Haley H, Fox J, DelMar EG, Nemeth EF, Lago AM, Callahan JF, Bhatnagar P, Huffman WF, Gowen M, Yi B, Danoff TM, Fitzpatrick LA. An orally active calcium-sensing receptor antagonist that transiently increases plasma concentrations of PTH and stimulates bone formation. Bone. 2010;46:534–542. doi: 10.1016/j.bone.2009.09.028. [DOI] [PubMed] [Google Scholar]
- 119.Marquis RW, Lago AM, Callahan JF, Rahman A, Dong X, Stroup GB, Hoffman S, Gowen M, DelMar EG, Van Wagenen BC, Logan S, Shimizu S, Fox J, Nemeth EF, Roethke T, Smith BR, Ward KW, Bhatnagar P. Antagonists of the calcium receptor. 2. Amino alcohol-based parathyroid hormone secretagogues. J Med Chem. 2009;52:6599–6605. doi: 10.1021/jm900563e. [DOI] [PubMed] [Google Scholar]
- 120.Guery S, Floersheim P, Kaupmann K, Froestl W. Syntheses and optimization of new GS39783 analogues as positive allosteric modulators of GABA B receptors. Bioorg Med Chem Lett. 2007;17:6206–6211. doi: 10.1016/j.bmcl.2007.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Malherbe P, Masciadri R, Norcross RD, Knoflach F, Kratzeisen C, Zenner MT, Kolb Y, Marcuz A, Huwyler J, Nakagawa T, Porter RH, Thomas AW, Wettstein JG, Sleight AJ, Spooren W, Prinssen EP. Characterization of (R, S)-5,7-di-tert-butyl-3-hydroxy-3-trifluoromethyl-3H-benzofuran-2-one as a positive allosteric modulator of GABA B receptors. Br J Pharmacol. 2008;154:797–811. doi: 10.1038/bjp.2008.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Urwyler S, Mosbacher J, Lingenhoehl K, Heid J, Hofstetter K, Froestl W, Bettler B, Kaupmann K. Positive allosteric modulation of native and recombinant gamma-aminobutyric acid(B) receptors by 2,6-Di-tert-butyl-4-(3-hydroxy-2,2-dimethyl-propyl)-phenol (CGP7930) and its aldehyde analog CGP13501. Mol Pharmacol. 2001;60:963–971. [PubMed] [Google Scholar]
- 123.Jacobson LH, Cryan JF. Evaluation of the anxiolytic-like profile of the GABA B receptor positive modulator CGP7930 in rodents. Neuropharmacology. 2008;54:854–862. doi: 10.1016/j.neuropharm.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 124.Brusberg M, Ravnefjord A, Martinsson R, Larsson H, Martinez V, Lindström E. The GABA(B) receptor agonist, baclofen, and the positive allosteric modulator, CGP7930, inhibit visceral pain-related responses to colorectal distension in rats. Neuropharmacology. 2009;56:362–367. doi: 10.1016/j.neuropharm.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 125.Urwyler S, Pozza MF, Lingenhoehl K, Mosbacher J, Lampert C, Froestl W, Koller M, Kaupmann K. N, N′-Dicyclopentyl-2-methylsulfanyl-5-nitro-pyrimidine-4, 6-diamine (GS39783) and structurally related compounds: novel allosteric enhancers of gamma-aminobutyric acid B receptor function. J Pharmacol Exp Ther. 2003;307:322–330. doi: 10.1124/jpet.103.053074. [DOI] [PubMed] [Google Scholar]
- 126.Litschig S, Gasparini F, Rueegg D, Stoehr N, Flor PJ, Vranesic I, Prézeau L, Pin JP, Thomsen C, Kuhn R. CPCCOEt, a noncompetitive metabotropic glutamate receptor 1 antagonist, inhibits receptor signaling without affecting glutamate binding. Mol Pharmacol. 1999;55:453–461. [PubMed] [Google Scholar]
- 127.Carroll FY, Stolle A, Beart PM, Voerste A, Brabet I, Mauler F, Joly C, Antonicek H, Bockaert J, Müller T, Pin JP, Prézeau L. BAY36-7620: a potent non-competitive mGlu1 receptor antagonist with inverse agonist activity. Mol Pharmacol. 2001;59:965–973. [PMC free article] [PubMed] [Google Scholar]
- 128.Lavreysen H, Janssen C, Bischoff F, Langlois X, Leysen JE, Lesage AS. [3H]R214127: a novel high-affinity radioligand for the mGlu1 receptor reveals a common binding site shared by multiple allosteric antagonists. Mol Pharmacol. 2003;63:1082–1093. doi: 10.1124/mol.63.5.1082. [DOI] [PubMed] [Google Scholar]
- 129.Malherbe P, Kratochwil N, Knoflach F, Zenner MT, Kew JN, Kratzeisen C, Maerki HP, Adam G, Mutel V. Mutational analysis and molecular modeling of the allosteric binding site of a novel, selective, noncompetitive antagonist of the metabotropic glutamate 1 receptor. J Biol Chem. 2003;278:8340–8347. doi: 10.1074/jbc.M211759200. [DOI] [PubMed] [Google Scholar]
- 130.Lavreysen H, Wouters R, Bischoff F, Nóbrega Pereira S, Langlois X, Blokland S, Somers M, Dillen L, Lesage AS. JNJ16259685, a highly potent, selective and systemically active mGlu1 receptor antagonist. Neurpharmacology. 2004;47:961–972. doi: 10.1016/j.neuropharm.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 131.Kohara A, Toya T, Tamura S, Watabiki T, Nagakura Y, Shitaka Y, Hayashibe S, Kawabata S, Okada M. Radioligand binding properties and pharmacological characterization of 6-amino-N-cyclohexyl-N,3-dimethylthiazolo[3,2-a]benzimidazole-2-carboxamide (YM-298198), a high-affinity, selective, and noncompetitive antagonist of metabotropic glutamate receptor type 1. J Pharmacol Exp Ther. 2005;315:163–169. doi: 10.1124/jpet.105.087171. [DOI] [PubMed] [Google Scholar]
- 132.El-Kouhen O, Lehto SG, Pan JB, Chang R, Baker SJ, Zhong C, Hollingsworth PR, Mikusa JP, Cronin EA, Chu KL, McGaraughty SP, Uchic ME, Miller LN, Rodell NM, Patel M, Bhatia P, Mezler M, Kolasa T, Zheng GZ, Fox GB, Stewart AO, Decker MW, Moreland RB, Brioni JD, Honore P. Blockade of mGluR1 receptor results in analgesia and disruption of motor and cognitive performances: effects of A-841720, a novel non-competitive mGluR1 receptor antagonist. Br J Pharmacol. 2006;149:761–774. doi: 10.1038/sj.bjp.0706877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Suzuki G, Kimura T, Satow A, Kaneko N, Fukuda J, Hikichi H, Sakai N, Maehara S, Kawagoe-Takaki H, Hata M, Azuma T, Ito S, Kawamoto H, Ohta H. Pharmacological characterization of a new, orally active and potent allosteric metabotropic glutamate receptor 1 antagonist, 4-[1-(2-fluoropyridin-3-yl)-5-methyl-1H-1,2,3-triazol-4-yl]-N-isopropyl-N-methyl-3,6-dihydropyridine-1(2H)-carboxamide (FTIDC) J Pharmacol Exp Ther. 2007;321:1144–1153. doi: 10.1124/jpet.106.116574. [DOI] [PubMed] [Google Scholar]
- 134.Kohara A, Nagakura Y, Kiso T, Toya T, Watabiki T, Tamura S, Shitaka Y, Itahana H, Okada M. Antinociceptive profile of a selective metabotropic glutamate receptor 1 antagonist YM-230888 in chronic pain rodent models. Eur J Pharmacol. 2007;571:8–16. doi: 10.1016/j.ejphar.2007.05.030. [DOI] [PubMed] [Google Scholar]
- 135.Fukuda J, Suzuki G, Kimura T, Nagatomi Y, Ito S, Kawamoto H, Ozaki S, Ohta H. Identification of a novel transmembrane domain involved in the negative modulation of mGluR1 using a newly discovered allosteric mGluR1 antagonist, 3-cyclohexyl-5-fluoro-6-methyl-7-(2-morpholin-4-ylethoxy)-4H-chromen-4-one. Neuropharmacology. 2009;57:438–445. doi: 10.1016/j.neuropharm.2009.06.017. [DOI] [PubMed] [Google Scholar]
- 136.Hemstapat K, de Paulis T, Chen Y, Brady AE, Grover VK, Alagille D, Tamagnan GD, Conn PJ. A novel class of positive allosteric modulators of metabotropic glutamate receptor subtype 1 interact with a site distinct from that of negative allosteric modulators. Mol Pharmacol. 2006;70:616–626. doi: 10.1124/mol.105.021857. [DOI] [PubMed] [Google Scholar]
- 137.Knoflach F, Mutel V, Jolidon S, Kew JN, Malherbe P, Vieira E, Wichmann J, Kemp JA. Positive allosteric modulators of metabotropic glutamate 1 receptor: characterization, mechanism of action, and binding site. Proc Natl Acad Sci USA. 2001;98:13402–13407. doi: 10.1073/pnas.231358298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Vieira E, Huwyler J, Jolidon S, Knoflach F, Mutel V, Wichmann J. Fluorinated 9H-xanthene-9-carboxylic acid oxazol-2-yl-amides as potent, orally available mGlu1 receptor enhancers. Bioorg Med Chem Lett. 2009;19:1666–1669. doi: 10.1016/j.bmcl.2009.01.108. [DOI] [PubMed] [Google Scholar]
- 139.Owen DR. Recent advances in the medicinal chemistry of the metabotropic glutamate receptor 1 (mGlu1) ACS Chem Neurosci. 2011;2:394–401. doi: 10.1021/cn2000124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Johnson MP, Baez M, Jagdmann GE, Jr, Britton TC, Large TH, Callagaro DO, Tizzano JP, Monn JA, Schoepp DD. Discovery of allosteric potentiators for the metabotropic glutamate 2 receptor: synthesis and subtype selectivity of N-(4-(2-methoxyphenoxy)phenyl)-N-(2,2,2-trifluoroethylsulfonyl)pyrid-3-ylmethylamine. J Med Chem. 2003;46:3189–3192. doi: 10.1021/jm034015u. [DOI] [PubMed] [Google Scholar]
- 141.Johnson MP, Barda D, Britton TC, Emkey R, Hornback WJ, Jagdmann GE, McKinzie DL, Nisenbaum ES, Tizzano JP, Schoepp DD. Metabotropic glutamate 2 receptor potentiators: receptor modulation, frequency-dependent synaptic activity, and efficacy in preclinical anxiety and psychosis model(s) Psychopharmacology (Berl) 2005;179:271–283. doi: 10.1007/s00213-004-2099-9. [DOI] [PubMed] [Google Scholar]
- 142.Barda DA, Wang ZQ, Britton TC, Henry SS, Jagdmann GE, Coleman DS, Johnson MP, Andis SL, Schoepp DD. SAR study of a subtype selective allosteric potentiator of metabotropic glutamate 2 receptor, N-(4-phenoxyphenyl)-N-(3-pyridinylmethyl)ethanesulfonamide. Bioorg Med Chem Lett. 2004;14:3099–3102. doi: 10.1016/j.bmcl.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 143.Galici R, Echemendia NG, Rodriguez AL, Conn PJ. A selective allosteric potentiator of metabotropic glutamate (mGlu) 2 receptors has effects similar to an orthosteric mGlu2/3 receptor agonist in mouse models predictive of antipsychotic activity. J Pharmacol Exp Ther. 2005;315:1181–1187. doi: 10.1124/jpet.105.091074. [DOI] [PubMed] [Google Scholar]
- 144.Galici R, Jones CK, Hemstapat K, Nong Y, Echemendia NG, Williams LC, de Paulis T, Conn PJ. Biphenyl-indanone A, a positive allosteric modulator of the metabotropic glutamate receptor subtype 2, has antipsychotic- and anxiolytic-like effects in mice. J Pharmacol Exp Ther. 2006;318:173–185. doi: 10.1124/jpet.106.102046. [DOI] [PubMed] [Google Scholar]
- 145.D’Alessandro PL, Corti C, Roth A, Ugolini A, Sava A, Montanari D, Bianchi F, Garland SL, Powney B, Koppe EL, Rocheville M, Osborne G, Perez P, de la Fuente J, De Los Frailes M, Smith PW, Branch C, Nash D, Watson SP. The identification of structurally novel, selective, orally bioavailable positive modulators of mGluR2. Bioorg Med Chem Lett. 2010;20:759–762. doi: 10.1016/j.bmcl.2009.11.032. [DOI] [PubMed] [Google Scholar]
- 146.Hemstapat K, Da Costa H, Nong Y, Brady AE, Luo Q, Niswender CM, Tamagnan GD, Conn PJ. A novel family of potent negative allosteric modulators of group II metabotropic glutamate receptors. J Pharmacol Exp Ther. 2007;322:254–264. doi: 10.1124/jpet.106.117093. [DOI] [PubMed] [Google Scholar]
- 147.Woltering TJ, Adam G, Alanine A, Wichmann J, Knoflach F, Mutel V, Gatti S. Synthesis and characterization of 8-ethynyl-1,3-dihydro-benzo[b][1,4]diazepin-2-one derivatives: new potent non-competitive metabotropic glutamate receptor 2/3 antagonists. Part 1. Bioorg Med Chem Lett. 2007;17:6811–6815. doi: 10.1016/j.bmcl.2007.10.026. [DOI] [PubMed] [Google Scholar]
- 148.Sheffler DJ, Pinkerton AB, Dahl R, Markou A, Cosford NDP. Recent progress in the synthesis and characterization of group II metabotropic glutamate receptor allosteric modulators. ACS Chem Neurosci. 2011;2:382–393. doi: 10.1021/cn200008d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Maj M, Bruno V, Dragic Z, Yamamoto R, Battaglia G, Inderbitzin W, Stoehr N, Stein T, Gasparini F, Vranesic I, Kuhn R, Nicoletti F, Flor PJ. (−)-PHCCC, a positive allosteric modulator of mGluR4: characterization, mechanism of action, and neuroprotection. Neropharmacology. 2003;45:895–906. doi: 10.1016/s0028-3908(03)00271-5. [DOI] [PubMed] [Google Scholar]
- 150.Marino MJ, Williams DL, Jr, O’Brien JA, Valenti O, McDonald TP, Clements MK, Wang R, DiLella AG, Hess JF, Kinney GG, Conn PJ. Allosteric modulation of group III metabotropic glutamate receptor 4: a potential approach to Parkinson’s disease treatment. Proc Natl Acad Sci USA. 2003;100:13668–13673. doi: 10.1073/pnas.1835724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Engers DW, Niswender CM, Weaver CD, Jadhav S, Menon UN, Zamorano R, Conn PJ, Lindsley CW, Hopkins CR. Synthesis and evaluation of a series of heterobiarylamides that are centrally penetrant metabotropic glutamate receptor 4 (mGluR4) positive allosteric modulators (PAMs) J Med Chem. 2009;52:4115–4118. doi: 10.1021/jm9005065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Niswender CM, Johnson KA, Weaver CD, Jones CK, Xiang Z, Luo Q, Rodriguez AL, Marlo JE, de Paulis T, Thompson AD, Days EL, Nalywajko T, Austin CA, Williams MB, Ayala JE, Williams R, Lindsley CW, Conn PJ. Discovery, characterization, and antiparkinsonian effect of novel positive allosteric modulators of metabotropic glutamate receptor 4. Mol Pharmacol. 2008;74:1345–1358. doi: 10.1124/mol.108.049551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Williams R, Johnson KA, Gentry PR, Niswender CM, Weaver CD, Conn PJ, Lindsley CW, Hopkins CR. Synthesis and SAR of a novel positive allosteric modulator (PAM) of the metabotropic glutamate receptor 4 (mGluR4) Bioorg Med Chem Lett. 2009;19:4967–4970. doi: 10.1016/j.bmcl.2009.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Williams R, Niswender CM, Luo Q, Le U, Conn PJ, Lindsley CW. Positive allosteric modulators of the metabotropic glutamate receptor subtype 4 (mGluR4). Part II: Challenges in hit-to-lead. Bioorg Med Chem Lett. 2009;19:962–966. doi: 10.1016/j.bmcl.2008.11.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Williams R, Zhou Y, Niswender CM, Luo Q, Conn PJ, Lindsley CW, Hopkins CR. Re-exploration of the PHCCC Scaffold: Discovery of Improved Positive Allosteric Modulators of mGluR4. ACS Chem Neurosci. 2010;1:411–419. doi: 10.1021/cn9000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mathiesen JM, Svendsen N, Bräuner-Osborne H, Thomsen C, Ramirez MT. Positive allosteric modulation of the human metabotropic glutamate receptor 4 (hmGluR4) by SIB-1893 and MPEP. Br J Pharmacol. 2003;138:1026–1030. doi: 10.1038/sj.bjp.0705159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Robichaud AJ, Engers DW, Lindsley CW, Hopkins CR. Recent Progress in the Identification of Metabotropic Glutamate 4 Receptor Ligands and Their Potential Utility as CNS Therapeutics. ACS Chem Neurosci. 2011;2:433–449. doi: 10.1021/cn200043e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Cosford ND, Tehrani L, Roppe J, Schweiger E, Smith ND, Anderson J, Bristow L, Brodkin J, Jiang X, McDonald I, Rao S, Washburn M, Varney MA. 3-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]-pyridine: a potent and highly selective metabotropic glutamate subtype 5 receptor antagonist with anxiolytic activity. J Med Chem. 2003;46:204–206. doi: 10.1021/jm025570j. [DOI] [PubMed] [Google Scholar]
- 159.Porter RH, Jaeschke G, Spooren W, Ballard TM, Büttelmann B, Kolczewski S, Peters JU, Prinssen E, Wichmann J, Vieira E, Mühlemann A, Gatti S, Mutel V, Malherbe P. Fenobam: a clinically validated nonbenzodiazepine anxiolytic is a potent, selective, and noncompetitive mGlu5 receptor antagonist with inverse agonist activity. J Pharmacol Exp Ther. 2005;315:711–721. doi: 10.1124/jpet.105.089839. [DOI] [PubMed] [Google Scholar]
- 160.Rodriguez AL, Grier MD, Jones CK, Herman EJ, Kane AS, Smith RL, Williams R, Zhou Y, Marlo JE, Days EL, Blatt TN, Jadhav S, Menon UN, Vinson PN, Rook JM, Stauffer SR, Niswender CM, Lindsley CW, Weaver CD, Conn PJ. Discovery of novel allosteric modulators of metabotropic glutamate receptor subtype 5 reveals chemical and functional diversity and in vivo activity in rat behavioral models of anxiolytic and antipsychotic activity. Mol Pharmacol. 2010;78:1105–1123. doi: 10.1124/mol.110.067207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.O’Brien JA, Lemaire W, Chen TB, Chang RS, Jacobson MA, Ha SN, Lindsley CW, Schaffhauser HJ, Sur C, Pettibone DJ, Conn PJ, Williams DL., Jr A family of highly selective allosteric modulators of the metabotropic glutamate receptor subtype 5. Mol Pharmacol. 2003;64:731–740. doi: 10.1124/mol.64.3.731. [DOI] [PubMed] [Google Scholar]
- 162.Hammond AS, Rodriguez AL, Townsend SD, Niswender CM, Gregory KJ, Lindsley CW, Conn PJ. Discovery of a Novel Chemical Class of mGlu(5) Allosteric Ligands with Distinct Modes of Pharmacology. ACS Chem Neurosci. 2010;1:702–716. doi: 10.1021/cn100051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhao Z, Wisnoski DD, O’Brien JA, Lemaire W, Williams DL, Jr, Jacobson MA, Wittman M, Ha SN, Schaffhauser H, Sur C, Pettibone DJ, Duggan ME, Conn PJ, Hartman GD, Lindsley CW. Challenges in the development of mGluR5 positive allosteric modulators: the discovery of CPPHA. Bioorg Med Chem Lett. 2007;17:1386–1391. doi: 10.1016/j.bmcl.2006.11.081. [DOI] [PubMed] [Google Scholar]
- 164.O’Brien JA, Lemaire W, Wittmann M, Jacobson MA, Ha SN, Wisnoski DD, Lindsley CW, Schaffhauser HJ, Sur C, Duggan ME, Pettibone DJ, Conn J, Williams DL. A novel selective allosteric modulator potentiates the activity of the native metabotropic glutamate receptor subtype 5 (mGluR5) in rat forebrain. J Pharmacol Exp Therapeut. 2004;309(2):568–579. doi: 10.1124/jpet.103.061747. [DOI] [PubMed] [Google Scholar]
- 165.Kinney GG, O’Brien JA, Lemaire W, Burno M, Bickel DJ, Clements MK, Chen TB, Wisnoski DD, Lindsley CW, Tiller PR, Smith S, Jacobson MA, Sur C, Duggan ME, Pettibone DJ, Conn PJ, Williams DL., Jr A novel selective positive allosteric modulator of metabotropic glutamate receptor subtype 5 has in vivo activity and antipsychotic-like effects in rat behavioral models. J Pharmacol Exp Ther. 2005;313:199–206. doi: 10.1124/jpet.104.079244. [DOI] [PubMed] [Google Scholar]
- 166.Lindsley CW, Wisnoski DD, Leister WH, O’Brien JA, Lemaire W, Williams DL, Jr, Burno M, Sur C, Kinney GG, Pettibone DJ, Tiller PR, Smith S, Duggan ME, Hartman GD, Conn PJ, Huff JR. Discovery of positive allosteric modulators for the metabotropic glutamate receptor subtype 5 from a series of N-(1,3-diphenyl-1H- pyrazol-5-yl)benzamides that potentiate receptor function in vivo. J Med Chem. 2004;47:5825–5828. doi: 10.1021/jm049400d. [DOI] [PubMed] [Google Scholar]
- 167.de Paulis T, Hemstapat K, Chen Y, Zhang Y, Saleh S, Alagille D, Baldwin RM, Tamagnan GD, Conn PJ. Substituent effects of N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamides on positive allosteric modulation of the metabotropic glutamate-5 receptor in rat cortical astrocytes. J Med Chem. 2006;49:3332–3344. doi: 10.1021/jm051252j. [DOI] [PubMed] [Google Scholar]
- 168.Liu F, Grauer S, Kelley C, Navarra R, Graf R, Zhang G, Atkinson PJ, Popiolek M, Wantuch C, Khawaja X, Smith D, Olsen M, Kouranova E, Lai M, Pruthi F, Pulicicchio C, Day M, Gilbert A, Pausch MH, Brandon NJ, Beyer CE, Comery TA, Logue S, Rosenzweig-Lipson S, Marquis KL. ADX47273 [S-(4-fluoro-phenyl)-{3-[3-(4-fluoro-phenyl)-[1,2,4]-oxadiazol-5-yl]-piperidin-1-yl}-methanone]: a novel metabotropic glutamate receptor 5-selective positive allosteric modulator with preclinical antipsychotic-like and procognitive activities. J Pharmacol Exp Ther. 2008;327:827–839. doi: 10.1124/jpet.108.136580. [DOI] [PubMed] [Google Scholar]
- 169.Engers DW, Rodriguez AL, Oluwatola O, Hammond AS, Venable DF, Williams R, Sulikowski GA, Conn PJ, Lindsley CW. Synthesis, SAR and unanticipated pharmacological profiles of analogs of the mGluR5 ago-potentitaor ADX-47273. ChemMedChem. 2009;4:505–511. doi: 10.1002/cmdc.200800357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Emmitte KA. Recent advances in the design and development of novel negative allosteric modulators of mGlu5. ACS Chem Neurosci. 2011;2:411–432. doi: 10.1021/cn2000266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Stauffer SR. Progress towards positive allosteric modulators of the metbotropic glutamate receptor subtype 5 (mGlu5) ACS Chem Neurosci. 2011;2:450–470. doi: 10.1021/cn2000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Mitsukawa K, Yamamoto R, Ofner S, Nozulak J, Pescott O, Lukic S, Stoehr N, Mombereau C, Kuhn R, McAllister KH, van der Putten H, Cryan JF, Flor PJ. A selective metabotropic glutamate receptor 7 agonist: activation of receptor signaling via an allosteric site modulates stress parameters in vivo. Proc Natl Acad Sci USA. 2005;102:18712–18717. doi: 10.1073/pnas.0508063102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Suzuki G, Tsukamoto N, Fushiki H, Kawagishi A, Nakamura M, Kurihara H, Mitsuya M, Ohkubo M, Ohta H. In vitro pharmacological characterization of novel isoxazolopyridone derivatives as allosteric metabotropic glutamate receptor 7 antagonists. J Pharmacol Exp Ther. 2007;323:147–156. doi: 10.1124/jpet.107.124701. [DOI] [PubMed] [Google Scholar]
- 174.Li X, Staszewski L, Xu H, Durik K, Zoller M, Adler E. Human receptors for sweet and umani taste. Proc Natl Acad Sci USA. 2002;99:4692–4696. doi: 10.1073/pnas.072090199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Nelson G, Chandrashekar J, Hoon MA, Feng L, Zhao G, Ryba NJ, Zuker CS. An amino acid taste receptor. Nature. 2002;416:199–203. doi: 10.1038/nature726. [DOI] [PubMed] [Google Scholar]
- 176.Zhang F, Klebansky B, Fine RM, Liu H, Xu H, Servant G, Zoller M, Tachdijian C, Li X. Molecular mechanism for the umani taste synergism. Proc Natl Acad Sci USA. 2008;105:20930–20934. doi: 10.1073/pnas.0810174106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zhang F, Klebansky B, Fine RM, Liu H, Xu H, Servant G, Zoller M, Tachdjian C, Li X. Molecular mechanism of the sweet taste enhancers. Proc Natl Acad Sci USA. 2010;107:4752–4757. doi: 10.1073/pnas.0911660107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Servant G, Tachdjian C, Tang XQ, Werner S, Zhang F, Li X, Kamdar P, Petrovic G, Ditschun T, Java A. Positive allosteric modulators of the human sweet taste receptor enhance sweet taste. Proc Natl Acad Sci USA. 2010;107:4746–4751. doi: 10.1073/pnas.0911670107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Xu H, Staszewski L, Tang H, Adler E, Zoller M, Li X. Different functional roles of T1R subunits in the heterotrimeris taste receptors. Proc Natl Acad Sci USA. 2004;101:14258–14263. doi: 10.1073/pnas.0404384101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Jiang P, Cui M, Zhoa B, Snyder LA, Benard LM, Osman R, Margolskee RF, Max M. Identification of the cyclamate interaction site within the transmembrane domains of human sweet taste receptor subunit T1R3. J Biol Chem. 2005;280:34296–34305. doi: 10.1074/jbc.M505255200. [DOI] [PubMed] [Google Scholar]
- 181.Valant C, Gregory KJ, Hall NE, Scammells PJ, Lew MJ, Sexton PM, Christopoulos A. A novel mechanism of G protein-coupled receptor functional selectivity: muscarinic partial agonist McN-A-343 as a bitopic orhtosteric/allosteric ligand. J Biol Chem. 2008;283:29312–29321. doi: 10.1074/jbc.M803801200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Valant C, Sexton PM, Christopoulos A. Orthosteric/Allosteric Bitopic Ligands: Going Hybrid at GPCRs. Mol Int. 2009;9:125–135. doi: 10.1124/mi.9.3.6. [DOI] [PubMed] [Google Scholar]
- 183.Avlani VA, Langmead CJ, Guida E, Wood MD, Tehan BG, Hederon HJ, Watson JM, Sexton PM, Christopoulos A. Orthosteric and Allosteric Modes of Interaction of Novel Selective Agonists of the M1 Muscarinic Acetylcholine. Mol Pharm. 2010;78:94–104. doi: 10.1124/mol.110.064345. [DOI] [PubMed] [Google Scholar]
- 184.Leach K, Sexton PM, Christopoulos A. Allosteric modulators of GPCRs: Taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci. 2007;28:382–389. doi: 10.1016/j.tips.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 185.Digby GJ, Conn PJ, Lindsley CW. Orthosteric- and allosteric-induced ligand-directed trafficking at G protein-coupled receptors. Curr Opin Drug Disc & Dev. 2010;13:577–586. [PMC free article] [PubMed] [Google Scholar]
- 186.Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilla B, Weinstein H, Javitch JA, Roth BL, Christopolous A, Sexton PM, Miller KJ, Spedding M, Mailman RB. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- 187.Mathiesen JM, Ulven T, Martini L, Gerlach LO, Heinemann A, Kostenis E. Identification of indole derivatives exclusively interfering with a G proteinindependent signaling pathway of the prostaglandin D2 receptor CRTH2. Mol Pharmacol. 2005;68:393–402. doi: 10.1124/mol.104.010520. [DOI] [PubMed] [Google Scholar]
- 188.Zhang Y, Rodriguez AL, Conn PJ. Allosteric potentiators of metabotropic glutamate receptor subtype 5 have differential effects on different signaling pathways in cortical astrocytes. J Pharmacol Exp Ther. 2005;315:1212–1219. doi: 10.1124/jpet.105.090308. [DOI] [PubMed] [Google Scholar]
- 189.Smith NJ, Milligan G. Allostery at G protein-coupled receptor homo- and heteromers: uncharted pharmacological landscapes. Pharmacol Rev. 2010;62:701–725. doi: 10.1124/pr.110.002667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lockless SW, Ranganathan R. Evolutionary conserved pathways of energetic connectivity in protein families. Science. 1999;286:295–299. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- 191.Datta D, Scheer JM, Romanowski MJ, Wells JA. An allosteric circuit in caspase-1. J Mol Biol. 2008;381:1157–1167. doi: 10.1016/j.jmb.2008.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Liu J, Perumal NB, Oldfield CJ, Su EW, Uversky VN, Dunker AK. Intrinsic disorder in transcription factors. Biochemistry. 2006;45:6873–6888. doi: 10.1021/bi0602718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hilser VJ, Thompson EB. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc Natl Acad Sci USA. 2007;104:8311–8315. doi: 10.1073/pnas.0700329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Leach K, Sexton PM, Christopoulos A. Allosteric modulators of GPCRs: Taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci. 2007;28:382–389. doi: 10.1016/j.tips.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 195.Leach K, Davey AE, Felder CC, Sexton PM, Christopoulos A. The role of transmembrane domain 3 (TMIII) in the actions of orthosteric, allosteric and atypical agonists of the M4 muscarinic acetylcholine receptor. Mol Pharmacol. 2011;79:855–865. doi: 10.1124/mol.111.070938. [DOI] [PubMed] [Google Scholar]
- 196.Lu JYL, Yang Y, Gnacadja G, Christopoulos A, Reagan JD. Effect of the calcimimetic R-568 on correcting inactivating mutations in the human calcium-sensing receptor. J Pharmacol Exp Ther. 2009;331:775–786. doi: 10.1124/jpet.109.159228. [DOI] [PubMed] [Google Scholar]
- 197.Nawaratne V, Leach K, Felder CC, Sexton PM, Christopoulos A. Structural determinants of allosteric agonism and modulation at the M4 muscarinic acetylcholine receptor: Identification of ligand-specific and global activation mechanisms. J Biol Chem. 2010;285:19012–19021. doi: 10.1074/jbc.M110.125096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kennedy JP, Williams L, Bridges TM, Daniels RN, Weaver D, Lindsley CW. Application of combinatorial chemistry science on modern drug discovery. J Comb Chem. 2008;10:345–354. doi: 10.1021/cc700187t. [DOI] [PubMed] [Google Scholar]
- 199.Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 200.Maudsley S, Martin B, Luttrell LM. The origins of diversity and specificity in G protein-coupled receptor signaling. J Pharmacol Exp Ther. 2005;314:485–494. doi: 10.1124/jpet.105.083121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.McLoughlin DJ, Bertelli F, Williams C. The A, B, Cs of G protein-coupled receptor pharmacologyin assay development for HTS. Expert Opin Drug Discov. 2007;2:603–619. doi: 10.1517/17460441.2.5.603. [DOI] [PubMed] [Google Scholar]
- 202.Wood MR, Hopkins CR, Brogan JT, Conn PJ, Lindsley CW. ‘Molecular switches’ on allosteric ligands that modulate modes of pharmacology. Biochemistry. 2011;50:2403–2140. doi: 10.1021/bi200129s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Yang FV, Shipe WD, Bunda JL, Nolt MB, Wisnoski DD, Zhao Z, Barrow JC, Ray WJ, Ma L, Wittman M, Seager M, Koeplinger K, Hartman GD, Lindsley CW. Parallel synthesis of N-Birayl Quinolone Carboxylic Acids as selective M1 positive allosteric modulators. Bioorg Med Chem Lett. 2010;20:531–536. doi: 10.1016/j.bmcl.2009.11.100. [DOI] [PubMed] [Google Scholar]
- 204.Sharma S, Rodriguez A, Conn PJ, Lindsley CW. Synthesis and SAR of a mGluR5 allosteric partial antagonist lead: Unexpected modulation of pharmacology with slight structural modifications to a 5-(phenylethynyl)pyrimidine scaffold. Bioorg Med Chem Lett. 2008;18:4098–4101. doi: 10.1016/j.bmcl.2008.05.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Sharma S, Kedrowski J, Rook JM, Smith JM, Jones CK, Rodriguez AL, Conn PJ, Lindsley CW. Discovery of Molecular Switches that Modulate Modes of mGluR5 Pharmacology In Vitro and In Vivo Within a Series of Functionalized 5-(phenylethynyl)pyrimidines. J Med Chem. 2009;52:4103–4106. doi: 10.1021/jm900654c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Lamb JA, Engers DW, Niswender CM, Venable D, Conn PJ, Lindsley CW. Discovery of molecular switches within the ADX-47273 mGlu5 PAM scaffold that modulate modes of pharmacology to afford potent mGlu5 NAMs, PAMs, and partial antagonists. Bioorg Med Chem Lett. 2011;21:2711–2714. doi: 10.1016/j.bmcl.2010.11.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Schann S, Mayer S, Franchet C, Frauli M, Steinberg E, Thomas M, Baron L, Neuville P. Chemical switch of a metabotropic glutamate receptor 2 silent allosteric modulator into dual metabotropic glutamate receptor 2/3 negative/positive allosteric modulators. J Med Chem. 2010;53:8775–8779. doi: 10.1021/jm101069m. [DOI] [PubMed] [Google Scholar]
- 208.Pecknold JC, McClure DJ, Appeltauer L, Wrzesinski L, Allan T. Treatment of anxiety using fenobam (a nonbenzodiazepine) in a double-blind standard (diazepam) placebo-controlled study. J Clin Psychopharmacology. 1982;2:129–132. [PubMed] [Google Scholar]
- 209.Berry-Kravis EM, Hessl D, Coffey S, Hervey C, Schneider A, Yuhas J, Hutchinson J, Snape M, Tranfaglia M, Nguyen DV, Hagerman R. A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J Med Genet. 2009;46:266–271. doi: 10.1136/jmg.2008.063701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.http://www.neuropharm.co.uk/aboutus/portfolio_pipeline/fragile_x_programme
- 211.International Nonproprietary Names for Pharmaceutical Substances (INN) WHO Drug Information. 2009;23:1–50. [Google Scholar]
- 212.Keywood C, Wakefield M, Tack J. A proof-of-concept study evaluating the effect of ADX10059, a metabotropic glutamate receptor-5 negative allosteric modulator, on acid exposure and symptoms in gastroesophageal reflux disease. Gut. 2009;58:1192–1199. doi: 10.1136/gut.2008.162040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Marin JCA, Goadsby PJ. Glutamatergic fine tuning with ADX-10059: A novel therapeutic approach for migraine? Exp Opin Invest Drugs. 2010;19:555–561. doi: 10.1517/13543781003691832. [DOI] [PubMed] [Google Scholar]
- 214.Goadsby PJ, Keywood CG. Investigation of the role of mGluR5 inhibition in migraine: A proof of concept study of ADX10059 in acute treatment of migraine. Abstracts of Papers, 61st annual meeting of the American Academy of Neurology; Seattle, WA. Saint Paul, MN: American Academy of Neurology; 2009. p. Abstract P06.006. [Google Scholar]
- 215.Press Release. 2008. Jan 3, Addex Pharmaceuticals: Addex Announces ADX10059 Phase IIa Acute Anxiety Data. [Google Scholar]
- 216.Bessis A-S, Bolea C, Bonnet B, Epping-Jordan M, Poirier N, Poli S-M, Rocher J-P, Thollon Y. Novel Alkynyl Derivatives as Modulators of Metabotropic Glutamate Receptors. WO 2005/123703 Int Patent Appl.
- 217.Press Release. 2011. Mar 31, Addex Pharmaceuticals: Addex Initiates Phase IIa Clinical Trial of Dipraglurant-IR in Parkinson’s Disease Levodopa-Induced Dyskinesia (PD-LID) [Google Scholar]
- 218.Rouzade-Dominguez M-L, Pfannkuche H-J, Gasparini F. Use of mGluR5 (ESP. AFQ056) in GI (ESP. GERD) WO 2007/006530 Int Patent Appl.
- 219.Gasparini F. mGluR5 antagonists as potential symptomatic therapy for Fragile X Disorder. Abstracts of Papers, 239th ACS national meeting; San Francisco, CA. Washingtion, DC: American Chemical Society; p. Abstract MEDI 12031. [Google Scholar]
- 220.Berg D, Godau J, Trenkwalder C, Eggert K, Csoti I, Storch A, Gasparini F, Hariry S, Johns D, Gomez-Mancilla B. A randomized, controlled trial evaluating AFQ056 in reducing levodopa-induced dyskinesia in patients with Parkinson’s disease. Abstracts of Papers, 13th International Congress of Parkinson’s Disease and Movement Disorders; Paris, France. Milwaukee, WI: The Movement Disorder Society; 2009. p. Abstract LB-05. [Google Scholar]
- 221.Novartis: Planned filings 2011 to = ≥2015. Novartis Second Quarter 2011 Results, Novartis Investor Presentation; 2011. Jul 19, [Google Scholar]
- 222.http://clinicaltrials.gov/ct2/results?term=AFQ056
- 223.Harris G. New York Times. 2010. Apr 30, Promise seen in drug for retardation syndrome; p. A1. [Google Scholar]
- 224.AstraZeneca: NCEs Phases I and II. and Development Pipeline – Discontinued Projects vs 29 July 2010. AstraZeneca Development Pipeline. 2011 Jan 27; [Google Scholar]
- 225.http://www.clinicaltrials.gov/ct2/results?term=AZD2066
- 226.http://www.clinicaltrials.gov/ct2/results?term=AZD2516
- 227.F. Hoffman-LaRoche, Ltd. Pipeline Summary. 2010. Apr 15, Roche Pipeline 2010. [Google Scholar]
- 228.http://clinicaltrials.gov/ct2/results?term=RO4917523
- 229.Seaside Therapeutics: Novel therapies for patients with autism and Fragile X Syndrome. Corporate Fact Sheet. 2010 Quarter;:1. [Google Scholar]
- 230.http://www.seasidetherapeutics.com/compounds