

Abstract
An exome-sequencing study of families with multiple breast-cancer-affected individuals identified two families with XRCC2 mutations, one with a protein-truncating mutation and one with a probably deleterious missense mutation. We performed a population-based case-control mutation-screening study that identified six probably pathogenic coding variants in 1,308 cases with early-onset breast cancer and no variants in 1,120 controls (the severity grading was p < 0.02). We also performed additional mutation screening in 689 multiple-case families. We identified ten breast-cancer-affected families with protein-truncating or probably deleterious rare missense variants in XRCC2. Our identification of XRCC2 as a breast cancer susceptibility gene thus increases the proportion of breast cancers that are associated with homologous recombination-DNA-repair dysfunction and Fanconi anemia and could therefore benefit from specific targeted treatments such as PARP (poly ADP ribose polymerase) inhibitors. This study demonstrates the power of massively parallel sequencing for discovering susceptibility genes for common, complex diseases.
Main Text
Currently, only approximately 30% of the familial risk for breast cancer has been explained, leaving the substantial majority unaccounted for.1 Recently, exome sequencing has been demonstrated to be a powerful tool for identifying the underlying cause of rare Mendelian disorders. However, diseases such as breast cancer present substantially increased complexity in terms of locus, allelic and phenotypic heterogeneity, and relationships between genotype and phenotype.
As part of a collaborative (Leiden University Medical Centre, the Spanish National Cancer Center, and The University of Melbourne) project involving the exome capture and massively parallel sequencing of multiple-case breast-cancer-affected families, we applied whole-exome sequencing to DNA from multiple affected relatives from 13 families (family structure and sample availability were considered before the affected relatives were chosen). Bioinformatic analysis of the resulting exome sequences identified a protein-truncating mutation, c.651_652del (p.Cys217∗), in X-ray repair cross complementing gene-2 (XRCC2 [MIM 600375; NM_005431.1]) in the peripheral-blood DNA of a man participating in the Australian Breast Cancer Family Registry2 (ABCFR; Figure 1A); this man (III-4 in Figure 1A) had been diagnosed with breast cancer at 29 years of age, and his mother (II-3), sister (III-5), and cousin (III-1) had been diagnosed with breast cancer at 37, 41, and 34 years of age, respectively. The cousin (III-1), who had also been selected for exome sequencing, did not carry this mutation, the sister's DNA was Sanger sequenced and was found to carry the mutation, and there was no DNA available for testing of the mother. Exome sequencing of three individuals from a family participating in a Dutch research study of multiple-case breast-cancer-affected families identified a probably deleterious missense mutation (c.271C>T [p.Arg91Trp] in XRCC2) (Figure 2) in two sisters (II-6 and II-8 in Figure 1B) diagnosed with breast cancer at 40 and 48 years of age, respectively, but not in their cousin (II-1), who was diagnosed at 47 years of age.
Figure 1.
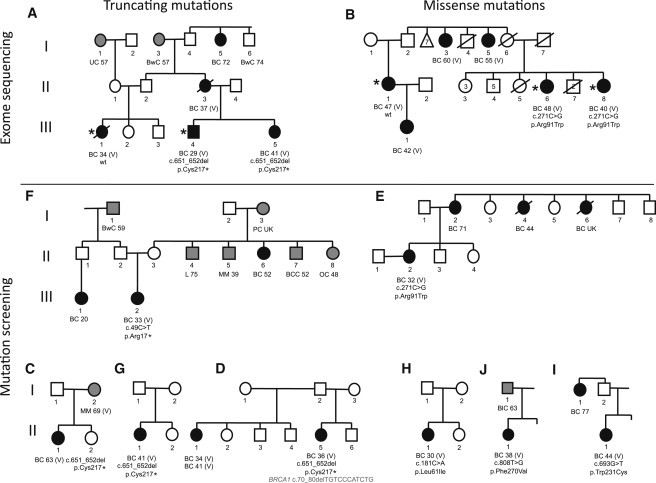
Pedigrees of Families Found to Carry XRCC2 Mutations
Mutation status is indicated for all family members for whom a DNA sample was available. Cancer diagnosis and age of onset are indicated for affected members. Asterisks indicate that DNA underwent exome sequencing (libraries for 50 bp fragment reads were prepared according to the SOLiD Baylor protocol 2.1 and the Nimblegen exome-capture protocol v.1.2 with some variations). The following abbreviations are used: BC, breast cancer (black filled symbols); PC, pancreatic cancer; BwC, bowel cancer; UC, uterine cancer; MM, malignant melanoma; UK, unknown age; BlC, bladder cancer; OC, ovarian cancer; BCC, basal cell carcinoma; L, lung cancer; (all gray-filled symbols); V, verified cancer (via cancer registry or pathology report); and wt, wild-type. Some symbols represent more than one person as indicated by a numeral.
Figure 2.
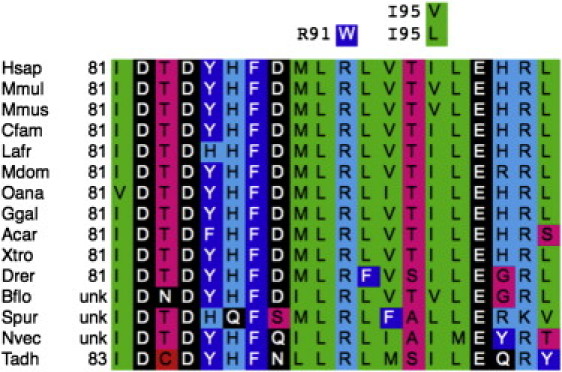
XRCC2 Multiple-Sequence Alignment Centered on Position Arg91
Missense substitutions observed in this interval are given with the missense residue directly above the corresponding human reference sequence residue. The following abbreviations are used: Hsap, Homo sapiens; Mmul, Macaca mulatta; Mmus, Mus musculus; Cfam, Canis familiaris; Lafr, Loxodonta africana; Mdom, Monodelphis domestica; Oana, Ornithorhynchus anatinus; Ggal, Gallus gallus; Acar, Anolis coralinensis; Xtro, Xenopus tropicalis; Drer, Danio rerio; Bflo, Branchiostoma floridae; Spur, Strongylocentrotus purpuratus; Nvec, Nematostella vectensis; and Tadh, Trichoplax adhaerans. The alignment, or updated versions thereof, is available at the Align-GVGD website (see Web Resources).
Genotyping of XRCC2 mutations c.651_652del (p.Cys217∗) and c.271C>T (p.Arg91Trp) in 1,344 cases and 1,436 controls from the Melbourne Collaborative Cohort Study3 (MCCS) and the ABCFR revealed one control (II-2, Figure 1C) who carried c.651_652del (p.Cys217∗). Intriguingly, this control individual's sister (II-1) was diagnosed with breast cancer at 63 years of age, and her mother (I-2) was diagnosed with melanoma at 69 years of age (Figure 1C, Tables 1 and 2).
Table 1.
Mutation Screening in Multiple-Case Breast Cancer Families
| Rare XRCC2 Variants | Effect on Protein | Align-GVGDa | SIFTb | PolyPhen-2.1 (HumDiv) | Case or Control | Pedigree (Study Source) | Age and Origin of Carrier |
|---|---|---|---|---|---|---|---|
| Truncating variants | |||||||
| c.651_652del | p.Cys217∗ | − | − | − | case | Figure 1A (ABCFR)e | 29, white |
| c.651_652del | p.Cys217∗ | − | − | − | casec | Figure 1C (kConFab) | 36, white |
| c.651_652del | p.Cys217∗ | − | − | − | control | Figure 1D (MCCS) | 72, white |
| Missense substitutions | |||||||
| c.271C>T | p.Arg91Trp | C65 | 0.00 | probably damaging | case | Figure 1B (Dutch)e | 40, white |
| c.271C>T | p.Arg91Trp | C65 | 0.00 | probably damaging | cased | Figure 1E (ABCFR) | 32, white |
| c.283A>C | p.Ile95Val | C0 | 0.34 | benign | case | − (kConFab) | 59, white |
| c.283A>G | p.Ile95Leu | C0 | 0.41 | benign | case | − (kConFab) | 70, white |
| c.283A>C | p.Ile95Val | C0 | 0.34 | benign | case | − (BRICOH) | 68, white |
| Silent substitution | |||||||
| c.582G>T | p.Thr194Thr | − | − | − | case | − (kConFab) | 60, white |
The following abbreviations are used: ABCFR; Australian Breast Cancer Family Registry; kConFab, Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer; MCCS, Melbourne Collaborative Cohort Study; and BRICOH, Beckman Research Institute of City of Hope.
Protein multiple sequence alignment (PMSA) used for obtaining scores for Align-GVGD: from Human to Branchiostoma floridae (Bflo).
PMSA used for obtaining scores for SIFT: from Human to Trichoplax (Tadh).
This woman also carries BRCA1 c.70_80del (p.Cys24Serfs∗13).
This carrier of p.Arg91Trp was identified through both the ABFCR multiple-case family screening and the BCFR-IARC (Breast Cancer Family Registry-International Agency for Research on Cancer) case-control screening.
Family included in the exome-sequencing phase.
Table 2.
Case-Control Mutation Screening Applied to the BCFR Population-Based Study
| Rare XRCC2 Variants | Effect on Protein | Align-GVGDa | SIFTb | PolyPhen-2.1 (HumDiv) | Case (n = 1,308) or Control (n = 1,120) | Pedigree (BCFR) | Age and Origin of Carrier |
|---|---|---|---|---|---|---|---|
| Truncating variants | |||||||
| c.49C>T | p.Arg17∗ | − | − | − | case | Figure 1F | 33, white |
| c.46G>T | p.Ala16Ser | C0 | 0.24 | benign | case | − | 44, East Asian |
| c.181C>A | p.Leu61Ile | C0 | 0.00 | possibly damaging | case | Figure 1H | 30, East Asian |
| c.271C>T | p.Arg91Trp | C65 | 0.00 | probably damaging | casec | Figure 1E | 32, white |
| c.283A>G | p.Ile95Val | C0 | 0.34 | benign | control | − | 44, white |
| c.693G>T | p.Trp231Cys | C65 | 0.00 | probably damaging | cased | Figure 1I | 44, East Asian |
| c.808T>G | p.Phe270Val | C45 | 0.00 | probably damaging | case | Figure 1J | 38, African |
| Silent substitution | |||||||
| c.354G>A | p.Val118Val | − | − | − | cased | − | 44, East Asian |
| 5′ UTR variants | |||||||
| c.-1G>A | ? | − | − | − | casee | − | 32, white |
The following abbreviation is used: BCFR, Breast Cancer Family Registry.
Protein multiple sequence alignment (PMSA) used for obtaining scores for Align-GVGD: from Human to Branchiostoma floridae (Bflo).
PMSA used for obtaining scores for SIFT: from Human to Trichoplax (Tadh).
This carrier of p.Arg91Trp was identified through both the ABFCR multiple-case family screening and the BCFR-IARC (Breast Cancer Family Registry-International Agency for Research on Cancer) case-control screening.
This 44-year-old East Asian case carries p.Trp231Cys and p.Val118Val.
This case is considered a “noncarrier” in the analysis.
XRCC2, a RAD51 paralog, was cloned because of its ability to complement the DNA-damage sensitivity of the irs1 hamster cell line.4 Cells derived from Xrcc2-knockout mice exhibit profound genetic instability as a result of homologous recombination (HR) deficiency.5 XRCC2 is highly conserved, and most truncations of the protein destroy its ability to protect cells from the effects of the DNA cross-linking agent mitomycin C.6 The involvement of the HR DNA repair genes BRCA1 (MIM 113705), BRCA2 (MIM 600185), ATM (MIM 607585), CHEK2 (MIM 604373), BRIP1 (MIM 605882), PALB2 (MIM 610355), and RAD51C (MIM 602774) in breast cancer risk emphasizes the importance of this mechanism in the etiology of breast cancer.7–9 Biallelic mutations in three of these genes are associated with Fanconi anemia (FA), and, most interestingly, Shamseldin et al.10 have recently reported a homozygous frameshift mutation in XRCC2 as being associated with a previously unrecognized form of FA. XRCC2 binds directly to the C-terminal portion of the product of the breast cancer susceptibility pathway gene RAD51 (MIM 179617), which is central to HR.6,11 XRCC2 also complexes in vivo with RAD51B (RAD51L1 [MIM 602948]), the product of the breast and ovarian cancer susceptibility gene RAD51C9 and the product of the ovarian cancer risk gene RAD51D (MIM 602954),12,13 and localizes to sites of DNA damage.6 Cells deficient in XRCC2 also show centrosome disruption, a key component of mitotic-apparatus dysfunction, which is often linked to the onset of mitotic catastrophe. XRCC2 is important in preventing chromosome missegregation leading to aneuploidy.14 Studies of common genetic variation in XRCC2 have reported some evidence of association with breast cancer risk (e.g., rs3218408),15 subtle effects on DNA-repair capacity,16 and poor survival associated with rs3218536 (XRCC2, Arg188His).15
On the basis of the exome-sequencing results, the subsequent genotyping of the two probably pathogenic variants in the MCCS and ABCFR, the rarity of these variants, and the biochemical plausibility of XRCC2, we conducted two further studies in parallel. The first study was case-control mutation screening of XRCC2 (with high-resolution melt [HRM] curve analysis followed by Sanger-sequencing confirmation) in an additional series of 1,308 cases with early-onset breast cancer and 1,120 frequency-matched controls recruited through population-based sampling by the Breast Cancer Family Registry2 (BCFR; Supplemental Data, available online); the BCFR sampling was recently carried out for the characterization of the breast cancer risk associated with variants in ATM and CHEK2.17,18 The second study was mutation screening of XRCC2 in a series of index cases from multiple-case breast-cancer-affected families and a series of male breast cancer cases.
The case-control mutation screening identified two cases that carried protein-truncating variants in XRCC2: individual III-2 had c.49C>T (p.Arg17∗) (Figure 1F), and individual II-1 had c.651_652del (p.Cys217∗) (Figure 1G). Five cases carried singleton missense substitutions ranging from probably deleterious to relatively innocuous (according to in silico prediction). One control carried a relatively innocuous missense substitution (Table 2). In addition, a case diagnosed with breast cancer at 32 years of age carried a G>A substitution located one nucleotide prior to the start codon.
We graded the rare missense variants by using three computational tools: SIFT, Polyphen2.1, and Align-GVGD. Differences in grading between these tools were minor. Depending on which of the three computational tools we used to grade the missense substitutions, the statistical significances of the differences in the frequency and severity distributions of protein-truncating variants and rare missense substitutions between cases and controls from the case-control mutation-screening study fell in the range of p = 0.01–0.02 (adjusted for race, study center, and age). There were six probably deleterious variants (predicted deleterious by at least two prediction algorithms) in the cases and none in the controls, corresponding to a p value by Fisher's exact test of 0.02. All together, the case-control mutation-screening data provide statistical support for the hypothesis that rare, evolutionarily unlikely sequence variation in XRCC2 is associated with increased risk of breast cancer.
Mutation screening (by Sanger sequencing) of XRCC2 in the index cases of 689 multiple-case breast-cancer-affected families participating in the BCFR and the Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer19 (kConFab) plus 150 male breast cancer cases participating in a US-based study of male breast cancer (Beckman Research Institute of the City of Hope20) and kConFab revealed three rare coding-sequence alterations. We identified a second family (from the kConFab resource) with an index case who carried XRCC2 c.651_652del (p.Cys217∗); this individual (II-5, Figure 1D) also carried a truncating mutation in BRCA1 (c.70_80del [p.Cys24Serfs∗13]). We identified an ABCFR index case (II-2, Figure 1E and Figure 2) who carried the previously identified missense substitution, XRCC2 c.271C>T (p.Arg91Trp). We also identified a male breast cancer case who carried a relatively innocuous missense substitution, c.283A>C (p.Ile95Leu).
In addition to the protein-truncating mutations and the above-described missense variants, a number of missense, silent, and intronic variants were also observed in XRCC2, and common SNPs that were reported in public databases such as dbSNP, HapMap, or the 1,000 Genomes Project were also identified. These included the common coding SNP c.563G>A (p.Arg188His) (rs3218536), one silent substitution, three 5′UTR variants, five 3′UTR variants, and six intronic variants in the vicinity of exon-intron boundaries. All these variants were predicted to be neutral according to various in silico predictions tools (Supplemental Data, Tables 1 and 2). For common SNPs (>1% in controls), no difference in allele frequency was observed between cases and controls in the BCFR series.
The genetic studies included in this report received approval from The University of Melbourne Human Research Ethics Committee, the International Agency for Research on Cancer institutional review board (IRB), and the local IRBs of every center from which we report findings.
Of the six distinct rare variants predicted to severely affect protein function and identified in our work, two were truncating mutations, and four were missense changes. Although most recognized pathogenic mutations in the major breast cancer susceptibility genes are protein truncating, there is evidence that missense mutations might be the more prominent of some more recently-identified breast cancer susceptibility genes. For example, in comprehensive studies of ATM and CHEK2, the proportion of probably deleterious or pathogenic rare sequence variants that are missense changes is often over 50%. More relevantly, estimates of breast cancer risk are higher for missense variants than they are for protein-truncating variants. This has been observed through case-control mutation-screening analyses of ATM and CHEK217,18 and through a pedigree analysis21 of ATM; in these analyses, the breast cancer risk associated with one specific missense mutation approaches the average risk associated with pathogenic BRCA2 mutations. A very recent analysis of PALB2 mutations found no difference in the frequency of missense mutations between two case groups (contralateral and unilateral breast cancer cases),22 suggesting that the contribution of missense mutations to breast cancer risk might vary between susceptibility genes.
Our finding of XRCC2 as a breast cancer susceptibility gene expands the proportion of breast cancer that is associated with rare mutations in the HR-DNA-repair pathways and the number of breast cancer susceptibility genes in which biallelic mutations are associated with FA; the precise contribution of mutation in these genes will become clearer as more whole-exome-sequencing (or whole-genome-sequencing) and targeted-pathway-sequencing studies are performed. XRCC2 mutations appear to be very rare, even in the context of multiple-case families; they appear in 1 of 66 (1.5%) early-onset female breast cancer cases with a strong family history of the disease present in the ABCFR, compared to 9 (14%) BRCA1 mutations, 6 (9%) BRCA2 mutations, 3 (5%) TP53 (MIM 191170) mutations, and 2 (3%) PALB2 mutations.
These frequencies are consistent with data from both breast cancer linkage studies that have suggested that no single gene is likely to account for a large fraction of the remaining familial aggregation of breast cancer5 and reports from recent candidate-gene sequencing studies that have associated other members of the HR pathway with breast cancer susceptibility.23,24 Although mutations in HR-DNA-repair genes are rare, it is important to identify people whose breast cancer is associated with HR-DNA-repair dysfunction because they could benefit from specific targeted treatments such as PARP inhibitors. Unaffected relatives of people with a mutation in a HR-DNA-repair gene could also be offered predictive testing and subsequent clinical management and genetic counseling on the basis of their mutation status. The identification of a family with rare mutations in both XRCC2 and BRCA1 illustrates the complexity of the underlying genetic architecture of breast cancer susceptibility for some families and the challenges for personalized risk-prediction models that are incorporating an increasing array of risk factors, which include rare mutations in breast cancer susceptibility genes and more common genetic variation. Currently, estimating the relative importance of the XRCC2 mutation to the breast cancer risk for members of this family is difficult because of the presence of a BRCA1 protein-truncating mutation in the proband in addition to the XRCC2 mutation. Many examples have been described of individuals and families carrying deleterious mutations in more than one proven breast cancer susceptibility gene; one such example is the co-observation of BRCA1, BRCA2, ATM, and CHEK2 mutations.21,25
This study demonstrates the power of massively parallel sequencing in the discovery of additional breast cancer susceptibility genes when used with an appropriate study design. Our approach could be applied to other common, complex diseases with components of unexplained heritability.
Acknowledgments
This work was supported by Cancer Council Victoria (grant 628774), the National Institutes of Health (R01CA155767 and R01CA121245), the Australian National Health and Medical Research Council (grant 466668), The University of Melbourne (infrastructure award to J.L.H.), a Victorian Life Sciences Computation Initiative grant (VR00353) on its Peak Computing Facility at the University of Melbourne, and an initiative of the Victorian Government and Dutch Cancer Society (grant UL 2009-4388). The research resources, including the Melbourne Collaborative Cohort Study, the Australian Breast Cancer Family Study, the Breast Cancer Family Registry, and the Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer, are further acknowledged in the supplementary information. We wish to thank Nivonirina Robinot and Geoffroy Durand for their technical help during the case-control mutation screening at the International Agency for Research on Cancer, Georgia Chenevix-Trench for her support of and contribution to the establishment of the case-control mutation-screening study, and Greg Wilhoite for sequencing the male breast cancer cases at the Beckman Research Institute of City of Hope. This work and partial support for S.L.N. was provided by the Morris and Horowitz Families Endowment. Work at the Spanish National Cancer Center was partially funded by the Spanish Association Against Cancer and Health Ministry (FIS08/1120). M.C.S. is a National Health and Medical Research Council (NHMRC) Senior Research Fellow and a Victorian Breast Cancer Research Consortium (VBCRC) Group Leader. J.L.H. is a NHMRC Australia Fellow and a VBCRC Group Leader. T.N.-D. is a Susan G. Komen for the Cure Postdoctoral Fellow.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Align-GVGD, http://agvgd.iarc.fr/alignments
GATK v.1.0.4418, http://gatk.sourceforge.net/
Genome Viewer (IGV v.1.5.48), http://www.broadinstitute.org/software/igv/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Picard v.1.29, http://sourceforge.net/projects/picard/
PolyPhen2.1, http://genetics.bwh.harvard.edu./pph2/
SIFT, http://sift.jcvi.org/
SOLiD Baylor protocol 2.1, http://www.hgsc.bcm.tmc.edu/documents/Preparation_of_SOLiD_Capture_Libraries.pdf
UCSC Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway
References
- 1.Turnbull C., Rahman N. Genetic predisposition to breast cancer: Past, present, and future. Annu. Rev. Genomics Hum. Genet. 2008;9:321–345. doi: 10.1146/annurev.genom.9.081307.164339. [DOI] [PubMed] [Google Scholar]
- 2.John E.M., Hopper J.L., Beck J.C., Knight J.A., Neuhausen S.L., Senie R.T., Ziogas A., Andrulis I.L., Anton-Culver H., Boyd N., Breast Cancer Family Registry The Breast Cancer Family Registry: An infrastructure for cooperative multinational, interdisciplinary and translational studies of the genetic epidemiology of breast cancer. Breast Cancer Res. 2004;6:R375–R389. doi: 10.1186/bcr801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giles, G.G., and R, E.D. (2002). The Melbourne Collaborative Cohort Study. IARC Sci Publ 156, 2. [PubMed]
- 4.Cartwright R., Tambini C.E., Simpson P.J., Thacker J. The XRCC2 DNA repair gene from human and mouse encodes a novel member of the recA/RAD51 family. Nucleic Acids Res. 1998;26:3084–3089. doi: 10.1093/nar/26.13.3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deans B., Griffin C.S., O'Regan P., Jasin M., Thacker J. Homologous recombination deficiency leads to profound genetic instability in cells derived from Xrcc2-knockout mice. Cancer Res. 2003;63:8181–8187. [PubMed] [Google Scholar]
- 6.Tambini C.E., Spink K.G., Ross C.J., Hill M.A., Thacker J. The importance of XRCC2 in RAD51-related DNA damage repair. DNA Repair (Amst.) 2010;9:517–525. doi: 10.1016/j.dnarep.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 7.Moynahan M.E., Chiu J.W., Koller B.H., Jasin M. Brca1 controls homology-directed DNA repair. Mol. Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- 8.Moynahan M.E., Pierce A.J., Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol. Cell. 2001;7:263–272. doi: 10.1016/s1097-2765(01)00174-5. [DOI] [PubMed] [Google Scholar]
- 9.Meindl A., Hellebrand H., Wiek C., Erven V., Wappenschmidt B., Niederacher D., Freund M., Lichtner P., Hartmann L., Schaal H. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat. Genet. 2010;42:410–414. doi: 10.1038/ng.569. [DOI] [PubMed] [Google Scholar]
- 10.Shamseldin H.E., Elfaki M., Alkuraya F.S. Exome sequencing reveals a novel Fanconi group defined by XRCC2 mutation. J. Med. Genet. 2012;49:184–186. doi: 10.1136/jmedgenet-2011-100585. [DOI] [PubMed] [Google Scholar]
- 11.Gao L.-B., Pan X.-M., Li L.-J., Liang W.-B., Zhu Y., Zhang L.-S., Wei Y.-G., Tang M., Zhang L. RAD51 135G/C polymorphism and breast cancer risk: A meta-analysis from 21 studies. Breast Cancer Res. Treat. 2011;125:827–835. doi: 10.1007/s10549-010-0995-8. [DOI] [PubMed] [Google Scholar]
- 12.Loveday C., Turnbull C., Ramsay E., Hughes D., Ruark E., Frankum J.R., Bowden G., Kalmyrzaev B., Warren-Perry M., Snape K., Breast Cancer Susceptibility Collaboration (UK) Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat. Genet. 2011;43:879–882. doi: 10.1038/ng.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu N., Schild D., Thelen M.P., Thompson L.H. Involvement of Rad51C in two distinct protein complexes of Rad51 paralogs in human cells. Nucleic Acids Res. 2002;30:1009–1015. doi: 10.1093/nar/30.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griffin C.S., Simpson P.J., Wilson C.R., Thacker J. Mammalian recombination-repair genes XRCC2 and XRCC3 promote correct chromosome segregation. Nat. Cell Biol. 2000;2:757–761. doi: 10.1038/35036399. [DOI] [PubMed] [Google Scholar]
- 15.Lin W.-Y., Camp N.J., Cannon-Albright L.A., Allen-Brady K., Balasubramanian S., Reed M.W.R., Hopper J.L., Apicella C., Giles G.G., Southey M.C. A role for XRCC2 gene polymorphisms in breast cancer risk and survival. J. Med. Genet. 2011;48:477–484. doi: 10.1136/jmedgenet-2011-100018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rafii S., O'Regan P., Xinarianos G., Azmy I., Stephenson T., Reed M., Meuth M., Thacker J., Cox A. A potential role for the XRCC2 R188H polymorphic site in DNA-damage repair and breast cancer. Hum. Mol. Genet. 2002;11:1433–1438. doi: 10.1093/hmg/11.12.1433. [DOI] [PubMed] [Google Scholar]
- 17.Le Calvez-Kelm F., Lesueur F., Damiola F., Vallée M., Voegele C., Babikyan D., Durand G., Forey N., McKay-Chopin S., Robinot N., Breast Cancer Family Registry Rare, evolutionarily unlikely missense substitutions in CHEK2 contribute to breast cancer susceptibility: results from a breast cancer family registry case-control mutation-screening study. Breast Cancer Res. 2011;13:R6. doi: 10.1186/bcr2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tavtigian S.V., Oefner P.J., Babikyan D., Hartmann A., Healey S., Le Calvez-Kelm F., Lesueur F., Byrnes G.B., Chuang S.-C., Forey N., Australian Cancer Study. Breast Cancer Family Registries (BCFR) Kathleen Cuningham Foundation Consortium for Research into Familial Aspects of Breast Cancer (kConFab) Rare, evolutionarily unlikely missense substitutions in ATM confer increased risk of breast cancer. Am. J. Hum. Genet. 2009;85:427–446. doi: 10.1016/j.ajhg.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mann G.J., Thorne H., Balleine R.L., Butow P.N., Clarke C.L., Edkins E., Evans G.M., Fereday S., Haan E., Gattas M., Kathleen Cuningham Consortium for Research in Familial Breast Cancer Analysis of cancer risk and BRCA1 and BRCA2 mutation prevalence in the kConFab familial breast cancer resource. Breast Cancer Res. 2006;8:R12. doi: 10.1186/bcr1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding Y.C., Steele L., Chu L.-H., Kelley K., Davis H., John E.M., Tomlinson G.E., Neuhausen S.L. Germline mutations in PALB2 in African-American breast cancer cases. Breast Cancer Res. Treat. 2011;126:227–230. doi: 10.1007/s10549-010-1271-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldgar D.E., Healey S., Dowty J.G., Da Silva L., Chen X., Spurdle A.B., Terry M.B., Daly M.J., Buys S.M., Southey M.C., BCFR. kConFab Rare variants in the ATM gene and risk of breast cancer. Breast Cancer Res. 2011;13:R73. doi: 10.1186/bcr2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tischkowitz M., Capanu M., Sabbaghian N., Li L., Liang X., Vallée M.P., Tavtigian S.V., Concannon P., Foulkes W.D., Bernstein L., The WECARE Study Collaborative Group Rare germline mutations in PALB2 and breast cancer risk: A population-based study. Hum Mutat. 2012;33:674–680. doi: 10.1002/humu.22022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rahman N., Seal S., Thompson D., Kelly P., Renwick A., Elliott A., Reid S., Spanova K., Barfoot R., Chagtai T., Breast Cancer Susceptibility Collaboration (UK) PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat. Genet. 2007;39:165–167. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seal S., Thompson D., Renwick A., Elliott A., Kelly P., Barfoot R., Chagtai T., Jayatilake H., Ahmed M., Spanova K., Breast Cancer Susceptibility Collaboration (UK) Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat. Genet. 2006;38:1239–1241. doi: 10.1038/ng1902. [DOI] [PubMed] [Google Scholar]
- 25.Turnbull C., Seal S., Renwick A., Warren-Perry M., Hughes D., Elliott A., Pernet D., Peock S., Adlard J.W., Barwell J., Breast Cancer Susceptibility Collaboration (UK), EMBRACE Gene-gene interactions in breast cancer susceptibility. Hum. Mol. Genet. 2012;21:958–962. doi: 10.1093/hmg/ddr525. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.