

Abstract
The burgeoning interest in the field of epigenetics has precipitated the need to develop approaches to strengthen causal inference when considering the role of epigenetic mediators of environmental exposures on disease risk. Epigenetic markers, like any other molecular biomarker, are vulnerable to confounding and reverse causation. Here, we present a strategy, based on the well-established framework of Mendelian randomization, to interrogate the causal relationships between exposure, DNA methylation and outcome. The two-step approach first uses a genetic proxy for the exposure of interest to assess the causal relationship between exposure and methylation. A second step then utilizes a genetic proxy for DNA methylation to interrogate the causal relationship between DNA methylation and outcome. The rationale, origins, methodology, advantages and limitations of this novel strategy are presented.
Keywords: DNA methylation, Mendelian randomization, confounding, reverse causation, mediation
Introduction
There is considerable anticipation of future improvements in disease prevention and treatment following on from advances in genomics and epigenomics,1,2 although also some scepticism in this regard.3,4 A particular quality of some genomic methodologies is their ability to separate causal from non-causal associations,5–7 with consequent identification of where interventions will change the course of disease development or progression. In this article, we will outline how methods that increase the strength of causal inference with respect to environmentally modifiable risk factors (Mendelian randomization) can be adapted to include epigenetic markers,8 an approach we term ‘two-step epigenetic Mendelian randomization’.
Epigenetics, once the domain of developmental biologists and then cancer researchers, has now permeated many areas of clinical medical research, following the trend set by genomics over the past decade. Although there still remains a large element of the unknown surrounding the characteristics and functional relevance of epigenetic variation, it has stimulated considerable interest. Much of the interest in epigenetics focuses on the environmentally responsive, mitotically heritable elements that have the ability to regulate gene expression (Box 1). However, the plastic nature of epigenetic patterns means that although epigenetic variation may be associated with phenotypic traits, it can be difficult to disentangle cause from consequence.
Box 1 Epigenetic inheritance.
The term epigenetics, popularized by Conrad Waddington in the early 1940s [reprinted in this issue of the IJE],17 has acquired a range of definitions in a variety of contexts but commonly alludes to the study of ‘heritable’ changes in gene function that do not entail changes to the DNA sequence itself.18 As Ho and Burggren point out, different disciplines interpret ‘inheritance’ in varying ways from the colloquial to the scientific.18 Thus inheritance can be considered at the cellular,19 population20 or cultural level.21
From the cellular perspective—and on the assumption that mechanistically we are interested in the propagation of epigenetic marks themselves and not solely the inheritance of associated phenotypic traits—use of the term ‘heritable’ is ambiguous. It refers to a mitotically heritable state that allows the perpetuation of epigenetic patterns through a particular cell lineage post-differentiation, as opposed to the meiotic transmission of epigenetic patterns. There are many illustrations of the ambiguity surrounding the notion of ‘epigenetic inheritance’, a recent paper cautioning that the assumptions of the Mendelian randomization approach may be violated by epigenetic inheritance being one such example.22 In humans, there are to date no robust examples of environmentally induced epigenetic changes that are transgenerationally inherited, although there is evidence that this might occur in other species.23–26 To complicate matters further, some epigenetic modifications are perpetuated across cell divisions but are not directly concerned with the regulation of gene expression.27
Somatic mitotic stability
The replication and transmission of epigenetic patterns during cellular proliferation should be considered as ‘mitotic stability’ as opposed to ‘inheritance’.28 Skinner proposes that this nomenclature be adopted to remove the current confusion around germ-line-mediated epigenetic inheritance. Mitotic stability embraces the concept that an environmental exposure might modify the epigenome and this alteration would then be stably perpetuated down a cell lineage, with the potential to permanently influence somatic cell function. This provides a plausible mechanism for the developmental origins of disease in later life. Epigenetic stability, however, is not an absolute prerequisite, as a transient epigenetic change could set in motion a persistent physiological effect. Rather little is known about the mechanisms by which epigenetic patterns are inherited, with the exception of the mitotic transmission of DNA methylation which is relatively well understood.29–31
Germ-line epigenetic inheritance
The erasure of epigenetic patterns during primordial germ cell development and early embryogenesis is evidence against the postulate that environmentally acquired epigenetic changes, even if acquired in the germ cell, might persist across multiple generations. However, environmental factors, notably the fungicide vinclozilin,32 stress responses33 and nutritional challenges,34 have been associated with transgenerational epigenetic inheritance in animal models, although it is often difficult to dissect evidence of transmission of epigenetic marks per se from transmission of the exposure itself.35 Evidence for epigenetic inheritance that is genetically driven exists in humans; MLH1 being an example of a gene harbouring an epigenetic variant which has been shown to be transmitted transgenerationally.36 Furthermore, RNA molecules are likely to play a role in the transmission of epigenetic information across generations. RNA-mediated effects upon phenotype have been observed in invertebrate species37 and may be pertinent to humans.
Transgenerational effects on phenotypic variation
Observations that exposures in the F0 generation can induce phenotypic changes in subsequent generations have turned to the field of epigenetics to explain such non-Mendelian phenomena.26,38–40 There are a number of models that could account for the transgenerational transmission of phenotypic variation in the absence of an inherited genetic factor including the influence of the parents’ genetically determined phenotypic traits on offspring, cultural transmission or semi-stable epigenetic mechanisms.41 Epigenetic mechanisms are not predicated on these non-genetic transgenerational phenomena, although epigenetic mechanisms might play a contributory role. However, as has been pointed out,42,43,44 phenotypic differences in isogenic organisms (such as drosophila and mice) are not in any meaningful way transgenerationally transmissible.45 The current flurry of excitement in relation to transgenerational epigenetics influences—indexed by coverage in popular science books46,47—is not driven by substantive evidence of the quantitative importance of such inheritance mechanisms in relation to the clearly established Mendelian forms of inheritance.42
There are few known robust epigenetic marker–phenotype associations outside of developmental syndromes or cancer.9 Recent literature points to a role for epigenetic variation in a range of phenotypes including neurological diseases (Parkinson's disease, Alzheimer's disease, bipolar disorder),10,11 obesity,12,13 diabetic nephropathy,14 osteoarthritis15 and ageing,16 but in many instances these are correlations without robust evidence of causality.
Epigenetic biomarkers of disease prediction and prognosis have also been identified, notably in the oncology field where peripheral blood cell DNA has been found to be a sensitive biomarker of disease risk in ovarian and bladder cancers,55,56 although these require replication. Epigenetic signatures in DNA derived from circulating tumour cells in plasma are also emerging as useful diagnostic and prognostic tools.57 In these instances, a causal relationship between epigenetic marker and phenotype is not a prerequisite for an informative predictive biomarker. Indeed, the integration of genomic and epigenomic information into disease risk prediction models alongside conventional clinical information is becoming a realistic possibility, though examples of the application of such integrated genomic and epigenomic approaches are only just beginning to emerge.58
The considerable but so far unrealized hopes for the utility of epigenetic data will become testable as new technologies that allow accrual of high volumes of epigenetic data are implemented. Indeed, the ability to generate such data at feasible cost is somewhat preceding capacity to control and harness its potential for real benefit (see Box 2 for an overview of methods of epigenetic profiling). It is therefore imperative to develop strategies to facilitate the optimal interpretation of these data. The various strategies that have been employed to disentangle observations inevitably complicated by confounding, measurement error and/or reverse causation can be applied to epigenetic associations.59
Box 2 Measuring DNA methylation.
An important first step in examining the extent to which DNA methylation may mediate causal effects of modifiable exposures on disease is to understand how DNA methylation is measured. To reduce DNA methylation to its basic unit, this can be defined as 5′methyl cytosine or the addition of a methyl group to a cytosine base. However, this process does not occur randomly across the genome, it occurs preferentially at cytosine–guanine dinucleotides (CpG sites), the distribution of which varies hugely across the genome.48 Of particular relevance to the current paper are the observations that CpG sites cluster in CpG islands, commonly in gene regulatory regions, and secondly they often display a correlation structure similar to SNP linkage disequilibrium (LD) structure. Furthermore, although a binary phenomenon at each individual CpG site (each cytosine base can only be methylated or un-methylated), at the level of a DNA sample, even taken from a single cell type, the level of methylation quantified will reflect the proportions of DNA template strands with methylation marks, often represented as a percentage of methylated DNA compared with total ‘input’ DNA. Variation in DNA methylation may arise due to differences to one or both alleles (known as allele specific methylation), although most methods to quantify DNA methylation do not detect allelic imbalance.49,50 An understanding of what is actually being measured is essential for the correct interpretation of DNA methylation data.
The development of genomic technologies for the analysis of genetic variation has been exploited in epigenomics and has resulted in rapid advances in the methods available to assay DNA methylation. Measurement can be at the global level (where an ‘overview’ of the methylation status of the genome is provided by measuring a representative sub-set of sites or regions), at the genome-wide level (site-specific or region-specific analysis depending on the technology used but with much higher resolution than global approaches) or in a targeted gene-specific manner (paralleling a SNP candidate gene approach where genes are defined through a prior discovery phase or biological prioritization). These methods tend to produce a ratio of methylated : unmethylated DNA which is commonly interpreted as a percentage. Details of specific methods can be found elsewhere.50–52 The choice of mode of measurement of DNA methylation is relevant to a two-step epigenetic Mendelian randomization approach. If considering DNA methylation as an outcome, then many alternatives exist to measure global methylation, e.g. LINE-1, Alu, Sat2 or LUMA assays,52,53 gene-specific or genome-wide methylation levels. The recent epigenome-wide association study (EWAS) reporting a profound (and robust) influence of smoking on a single CpG site of 27 000 sites analysed,54 suggests that localised effects of specific exposures might be expected rather than generic influences across the genome. However, when considering DNA methylation as an exposure (in Step 2 of the two-step epigenetic Mendelian randomization approach) a site-specific measure of DNA methylation is imperative. This is due to the identification of and reliance upon a cis-SNP at the same locus that can proxy for DNA methylation at that specific site (see Figures 4–6 for illustrative examples).
Here, we propose a two-step epigenetic Mendelian randomization strategy that draws together the principles of two established analysis strategies, namely Mendelian randomization60 and genetical genomics61 (also termed integrative genomics62). This is a two-step approach: In Step 1, the causal impact of modifiable risk factors on epigenetic signatures is established. In the second step, the causal nature of these epigenetic markers on a health-related outcome is interrogated. We previously referred to this approach as ‘genetical epigenomics’,8 by analogy with genetical genomics, but now consider the more descriptive label we apply to it preferable.
This method can be applied to studies of exposures in postnatal life—from infancy to adulthood—that potentially influence later disease risk, and also to intra-uterine factors that modify later health, within the developmental origins of health and disease (DOHaD) framework.
Mendelian randomization
The Mendelian randomization approach is predicated on the principle that if a genetic variant (e.g. Fat mass and obesity associated gene, FTO) either alters the level of, or mirrors the biological effects of, an environmentally modifiable exposure (e.g. obesity) that itself alters disease risk (e.g. blood pressure), then this genetic variant should also be related to disease risk to the degree predicted by the joint effects of the genetic variant on the modifiable exposure and of the modifiable exposure on the outcome. Instrumental variable methods of analysis63 can be applied in the Mendelian randomization setting64,65 to produce quantitative estimates of the magnitude (with confidence intervals) of the causal influence of the modifiable exposure on health outcome. Common genetic polymorphisms that have a well-characterized biological function (or are proxies for such variants) can therefore be utilized to estimate the causal effect of a suspected environmentally modifiable exposure on disease risk.5,60,65–68 The variants should not have an association with the disease outcome except through their link with the modifiable risk process of interest.
It may seem counterintuitive to study genetic variants as proxies for environmentally modifiable exposures rather than measure the exposures themselves. However, there are several crucial advantages of utilizing genetic variants in this manner, which are detailed below.
Confounding
Unlike most environmentally modifiable exposures, genetic variants are not generally associated with the wide range of behavioural, social and physiological factors that can confound epidemiological associations. Thus, when a genetic variant is used as a proxy for an environmentally modifiable exposure, it is unlikely to be confounded in the way that direct measures of the exposure will be, and this lack of confounding has been empirically demonstrated in several data sets.5,69 Furthermore, aside from the effects of population structure,70 such variants will not be associated with other genetic variants, except through LD. Thus, if population stratification is addressed (through restriction to an ethnically homogeneous sample and/or genomic control) only the minute proportion of the genome in LD with the genetic variant under study will be associated with the variant under investigation.
Reverse causation
Inferences drawn from observational studies may be subject to bias due to reverse causation. Disease processes could influence levels of exposures such as alcohol intake (either through symptoms of disease influencing desire to drink alcohol or through medical advice consequent on diagnosis), or intermediate phenotypes, such as body mass index (BMI), cholesterol levels and C-reactive protein (CRP). This is of particular relevance to epigenetic studies where reverse causation has the potential to be a major issue (that is, the trait or disease state itself alters the epigenome and not vice versa). However, germ-line genetic variants associated with average alcohol intake or levels of intermediate phenotypes will not be influenced by the onset of disease.
Temporal variation and measurement error
A genetic variant will reflect long-term levels of exposure, and, if the variant is considered to be a proxy for such exposure, it will not suffer from the measurement error inherent in phenotypes that have high levels of variability.66 For example, groups defined by cholesterol level-related genotype will, over a long period, experience cumulative differences in cholesterol levels. For individuals, blood cholesterol is variable over time, and the use of single measures of cholesterol will underestimate the true strength of association between cholesterol and, for instance, coronary heart disease (CHD). Indeed, use of the Mendelian randomization approach predicts the strength of association that is in line with randomized controlled trial findings of effects of cholesterol lowering, when the increasing benefits seen over the relatively short trial period are projected to the expectation for a lifetime.71
In the Mendelian randomization framework, the associations of genotype with outcomes are of interest because they offer strengthened inference about the action of the environmentally modifiable risk factors that the genotypes proxy for, rather than what they say about genetic mechanisms per se. Mendelian randomization studies are aimed at informing strategies to reduce disease risk through influencing the non-genetic component of modifiable risk processes. The Mendelian randomization strategy is being increasingly applied in cardiovascular disease, diabetes, cancer and infectious disease epidemiology, investigating such issues as the causal influence of alcohol intake, BMI, CRP, lipid levels and sex hormone binding globulin on disease risk.72–84 Figure 1 illustrates this approach using the example of BMI and blood pressure.84
Figure 1.
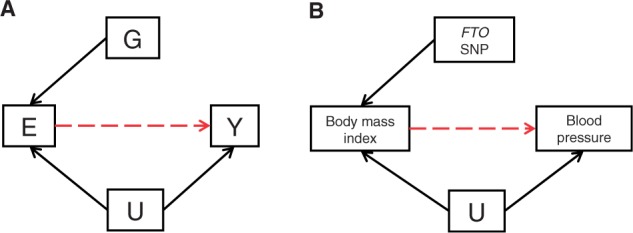
Mendelian randomization: using genetic variants as instrumental variables to establish whether an exposure is causally related to a disease or trait. (A) Instrumental variable (genetic variation) [G] acts as a proxy/instrumental variable for environmental exposure [E], postulated to influence disease [Y]. G is independent of unmeasured confounders or [U]. G only influences Y if E →Y is causal (red dashed line). (B) The influence of BMI on blood pressure using the FTO variant as an instrumental variable is shown as an example. A robust association between FTO and blood pressure is indicative of a causal association between BMI and blood pressure.81
Genetical genomics
Analogous to Mendelian randomization, approaches that have been termed ‘genetical genomics’ have utilized cis (local to methylation site) genetic variation related to transcription abundance85 to identify which RNAs are causally related to disease.62,86–88 RNAs, i.e. nucleic acids that translate genomic sequence information into proteins (Box 3), are here treated as the intermediate phenotypes that lie between genetic variation and disease-related phenotype. The implication is that modifying levels of these components of the transcriptome by, for example, pharmacotherapeutic methods would reduce the risk of disease (Figure 2). The terminology sometimes used when this method is applied is that it helps separate ‘causal’ from ‘reactive’ RNAs—the latter being related to disease risk because disease phenotypes influence their levels, i.e. through reverse causation.6 This is clearly illustrated in influential work of Schadt et al.62,89 As with intermediate phenotypes in the Mendelian randomization framework, RNA levels can also be associated with confounding factors and suffer from reverse causation, being phenotypic rather than genotypic. In the integrative genetics models, there is often assessment of a very large number of transcript abundances, with the ultimate aim of identifying causal networks from a morass of interrelated causal and non-causal elements,6 whereas Mendelian randomization studies have tended to consider a single modifiable putative risk factor at a time, although expansion to multiple phenotypic domains has been proposed.90
Box 3 RNA.
Non-coding RNAs
ncRNA—non protein coding RNAs. These are abundant in the genome, with over 3000 recognized human ncRNA genes. These RNA species are involved in diverse functions including protein biosynthesis and the splicing of messenger RNAs and have been implicated in disease.91 A nomenclature has been established around the evolving field of RNA research, the main categories pertinent to epigenetics are outlined briefly below.
miRNA—microRNA genes encode primary transcripts (pri-miRNAs) that are processed into pre-miRNAs which ultimately form mature miRNAs. These are single-stranded molecules of 19–25 nucleotides in length that bind to the 3′ untranslated region (3′UTR) of genes to inhibit transcription. With around 1000 miRNAs (http://www.mirbase.org) and more than 20 000 genes miRNAs tend to be pleiotropic. miRNAs are themselves transmitted between generations and it is postulated that they may play a role in the transgenerational transmission of epigenetic patterns.92
lncRNA—RNA transcripts of more than 200 nucleotides that do not encode proteins, known as long non-coding RNAs. Similar to miRNA, this form of ncRNA is involved in regulation of gene expression, possibly through interaction with enhancer regions as opposed to 3′UTRs.93
piRNA—Piwi-interacting RNAs are small ncRNAs of 25–33 nucleotides in length. They function in the defence of germ-line cells against transposons and are a feature of mammalian genomes.
Other RNA species
mRNA—messenger RNA is transcribed from the DNA sequence and mediates the transfer of genetic information from the nucleus to the ribosome. The mRNA fraction of any cell or tissue represents those genes actively being transcribed at the time of RNA extraction.
tRNA—transfer RNAs are small RNA molecules that are required for translation of mRNA into proteins, through physically transporting amino acids to the ribosome for assembly into a polypeptide chain.
rRNA–ribosomal RNAs form complexes with ribosomal proteins to form the large structures required for the physical translation of mRNA into proteins.
Figure 2.

Integrative genomics: using genetic variants as a causal anchor to demonstrate that modulation of gene expression levels at a specific locus can influence the incidence of a disease or trait and to discount the possibility that reverse causation is at play, i.e. that the disease state is not acting to alter gene expression levels. (A) Instrumental variable (genetic variation) [G] acts as a proxy for gene expression level [R], postulated to influence disease [Y]. G is independent of unmeasured confounder or [U]. G only influences Y if R →Y is causal (red dashed line). (B) The relationship between SNPs at the 17q21 locus (ORMDL3 gene), transcript levels of genes in Epstein–Barr virus-transformed lymphoblastoid cell lines and childhood asthma is shown as an example.86 The SNPs associated with childhood asthma were consistently and strongly associated (P < 10−22) in cis with transcript levels of ORMDL3, making these SNPs useful instrumental variables in this setting
Epigenetic markers as intermediate/mediating phenotypes
Pathways between modifiable exposures and disease generally involve mitotically stable changes in regulation of gene expression, which is the focus of epigenetic investigations, since modifications in cellular function are often necessary for the development of persistently pathological tissue. Although there is no agreed definition of epigenetics (see, for example, the seven definitions summarised by Ho and Burggren 201018), a conventional viewpoint considers it to be potentially measurable, mitotically stable modifications of DNA other than DNA sequence variation.94–96 Some popular expositions have focused on meiotically stable DNA modifications,97,98 perhaps capitalizing on interest generated by the notion of inheritance of acquired characteristics and the echoes of Lamarckianism this entails (Box 1).99 While there is evidence that such inter-generationally transmissible epigenetic processes exist,100–102 their importance is unclear and is certainly of less relevance to disease prevention or treatment than within-generation epigenetic processes. Within generation here includes in utero influences on the offspring epigenome, as the epigenetic marks are not transmitted, merely the exposure is acting upon two generations (mother and child) simultaneously and might be described as an early life exposure with respect to the offspring.102 The focus of most molecular epigenomic studies to date has, for pragmatic reasons, been on DNA methylation—the binding of methyl groups to cytosine bases,12–16,55–58,94–96 though other processes, in particular histone modifications and non-coding RNAs (see Box 3 for an overview of non-coding RNA species), are also involved.103 In this article, we focus on DNA methylation in relation to common complex disease and illustrate the potential to extend Mendelian randomization approaches to examine the role of DNA methylation as a mediator on the causal pathway between modifiable exposures and disease risk.
In molecular epidemiological terms, DNA methylation can be considered as an intermediate phenotype, but one that is closely proximal to the germ-line genome. Epigenetic biomarkers, like many other molecular biomarkers, are vulnerable to confounding by the ‘usual’ factors; age, sex, socio-economic position, diet, smoking, alcohol intake, etc. However, unlike other biomarkers, epigenetic patterns (explicitly DNA methylation) have a very close relationship with underlying genetic architecture. DNA methylation patterns can correlate closely with local genetic variants.48,104 Mendelian randomization approaches then allow these genetic variants to be used to circumvent the issue of potential confounding of the methylation patterns. Indeed, methylation changes can be directly introduced through polymorphic variation, i.e. the ablation or addition of a CpG (methylation) site—this is discussed in greater detail later.
An important role for DNA methylation in common complex disease is assumed in many of the reviews of epigenetic processes in different clinical domains published in recent times.10,105 The primary hypothesis is that environmental factors influence the epigenome which alters the regulation of gene expression and thus modulates disease risk. This framework is based upon the assumption that changes to the epigenome are an intermediate step on the causal pathway to disease, however, reverse causation and confounding are often difficult to discount. Indeed, within this field, it is probably fair to say that currently there is more speculation than robust and replicable data.
The relationship between genotype and epigenotype
Recent studies of human brain tissue have highlighted that a large proportion of inter-individual variation in DNA methylation is associated with common cis-acting genetic variation, i.e. genetic variation that is local to the DNA methylation site.106,107 This was recently corroborated in an extensive genomic, epigenomic and transcriptomic analysis of HapMap cell line DNA that reported a predominance of cis-acting SNPs with respect to DNA methylation levels, as opposed to more distal trans effects.48,108 Furthermore, where such genetic determinants of DNA methylation exist in a heterozygous state, they may result in allele-specific methylation (ASM),49 i.e. differences in methylation between two paired alleles. Estimates suggest that ~20% of heterozygous SNPs (mainly those located at CpG sites) are associated with ASM.104,109 DNA methylation shows regional correlation, although this requires formally establishing in each experimental- or tissue-specific setting and should not be an a priori assumption. It should therefore be feasible to identify cis-SNPs that correlate with methylation across a specific region, allowing methylation patterns to be considered at the haplotype level; a concept promoted by Bell and colleagues.110 In this way, the use of genetic variants as a proxy for DNA methylation levels, in particular the abundance of cis-acting elements, adds substantially to the feasibility of the two-step epigenetic Mendelian randomization approach.
Mediation of effects of exposures on disease outcomes
In epidemiological studies, the identification and analysis of mediation is often a key focus. For example, higher BMI is associated with elevated risk of CHD, and some of this association may reflect a causal influence of BMI on blood pressure, which in turn influences CHD risk. In this situation, blood pressure would be a partial mediator of the influence of BMI on CHD, with the important implication that therapeutically modifying blood pressure could break this link. Figure 3 illustrates these processes, which are sometimes referred to as direct and indirect effects. The figure also highlights how particular sources of bias and confounding can occur in such mediation analyses111–114 and the potential for residual confounding (caused by what has been referred to as collider bias) and measurement error in the mediator113 to distort interpretations of such data. Below we describe how a two-step epigenetic Mendelian randomization approach can be used to examine the role of DNA methylation as a mediator of the causal pathway between modifiable environmental exposures and disease outcomes.
Figure 3.

Mediation: a modifiable causal risk factor [E] for disease [Y] exerts its causal effect (at least in part) via the effect of E on X (the mediator) and through the causal effect of X on Y. U1 and U2 represent all confounders for the association of E with Y and X with Y, respectively. U1 and U2 can include different characteristics. In simple multivariable analyses to test this hypothesis it is tempting to adjust the association of E with Y for U1 and declare that this is the total causal effect of E on Y and then to adjust further for X; any resulting attenuation of the U1 adjusted association of E with Y following further adjustment for X is considered to represent the amount of the causal effect of E on Y that is mediated by X. However, by conditioning on X a pathway between U2 and E is produced and hence this association (E with Y) is now confounded by U2. In this situation X is said to be a collider between and E and U2.115 Furthermore, measurement error in X will bias the assessment of its mediation. Thus, both U1 and U2 require separate consideration and this can be achieved in the two-step epigenetic Mendelian randomization framework
A two-step epigenetic Mendelian randomization approach
A two-step epigenetic Mendelian randomization approach first requires a genetic proxy of the modifiable exposure which is related to DNA methylation (the mediator), and secondly, a genetic proxy of methylation is used to evaluate the relationship between this methylation mediator and the disease outcome or trait (Figure 4). Either step could, however, be used in isolation to interrogate the causal relationship between exposure and DNA methylation or the relationship between methylation and outcome.
Figure 4.
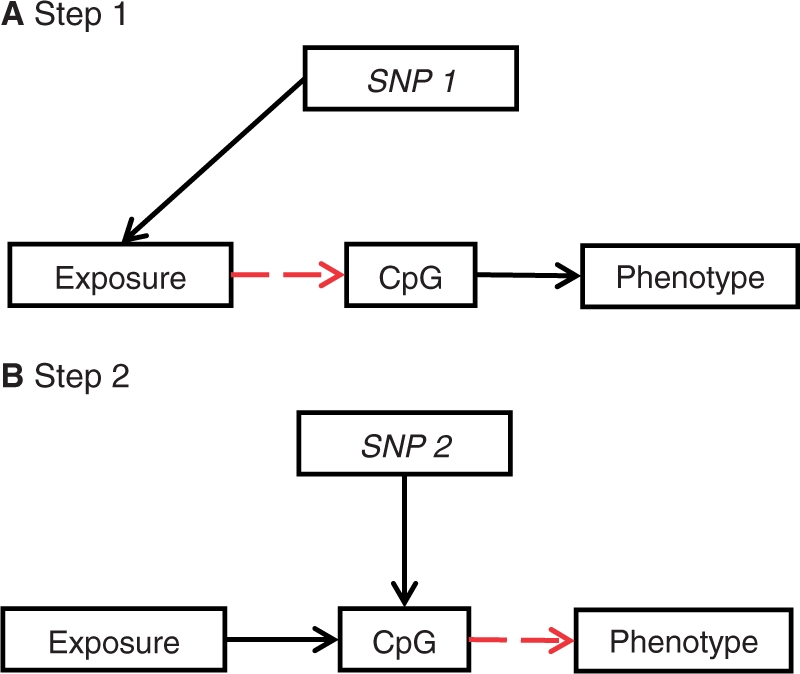
Two-step epigenetic Mendelian randomization: applying the principle of Mendelian randomization to DNA methylation as an intermediate phenotype. Genetic variants can be used as instrumental variables in a two-step framework to establish whether DNA methylation is on the causal pathway between exposure and disease. An overview of the two-step framework of this approach is shown. (A) First, an SNP is used to proxy for the environmentally modifiable exposure of interest and (B) secondly, a different SNP is used to proxy for DNA methylation levels
Genetic proxies for environmentally modifiable exposures
Many (but not all) of the genetic proxies, or instrumental variables, validated to date in a Mendelian randomization framework could be applied in a two-step epigenetic Mendelian randomization approach. For example, genetic proxies for smoking behaviour,115 alcohol consumption,116,118 BMI,81 lipid profiles119 or inflammation markers66 have been used in Mendelian randomization studies and could be applied to assess whether these environmental factors impact upon the epigenome. Care is required to ensure that the instrument chosen does not directly influence DNA methylation itself, an example being the use of FTO as an instrument for BMI as the gene product has been shown to have DNA demethylase activity.120 The main challenge arises when considering exactly what measure to use as the outcome, i.e. ‘the epigenome’. As alluded to earlier, we limit our discussion here to DNA methylation. It has been postulated that some regions of the epigenome are more environmentally responsive than others.49 However, most appraisals of environmentally induced epigenetic perturbation to date have either involved the assessment of global DNA methylation using assays such as LINE-1, Alu or Sat2, or have adopted an agnostic genome-wide approach, with large numbers of markers across the genome. An EWAS approach can be applied to interrogate potential associations between a given environmental exposure and DNA methylation as a phenotypic outcome.50 Few examples of this exist, two notable ones being the influence of smoking54 and age16 on DNA methylation (recognizing that age is not an environmental exposure per se). Future EWASs in relation to other exposures will provide additional robust signals (Box 2).
In the absence of an EWAS, when considering methylation as an outcome where does one look with respect to a specific exposure? Much focus has been placed upon CpG-rich regions (or islands) in the regulatory regions of genes, primarily based on the rationale that this is the mechanism whereby DNA methylation regulates gene expression, and partly driven by the technology and platforms available. More recently, both promoter and intragenic methylation have been shown to have different relationships with transcriptional regulation.121 There is also a growing recognition that CpG sites more distal to the promoter might show more variation.122–124 However, there is no consensus as yet on which variably methylated regions are most vulnerable to environmental influences, if indeed there is a difference.
As an alternative to a genome-wide appraisal, a candidate gene approach can provide a suitable means of identifying environmentally responsive epigenetic loci. An example of such is the analysis of the epigenetic regulation of the glucocorticoid receptor (NR3C1) gene in human brain tissue from suicide victims who were exposed to child abuse.125 This human study followed extensive analysis of this locus in a rodent model of maternal postnatal care by the same research group.126,35 In both contexts, the methylation levels of specific CpG sites within the Nc3r1/NR3C1 gene promoter region were analysed. Using this approach, one cannot conclude that this is the only stress responsive region of the methylome; however, the locus would still be appropriate for use in a two-step epigenetic Mendelian randomization approach. Indeed, a comprehensive analysis of chromosome 18, which harbours Nc3r1 in this rodent model, has been undertaken. This showed that co-ordinated regional epigenetic changes spanning over 100 kb on this chromosome are evident in response to maternal care.127
Parallels with candidate genetic association studies and the contemporary, robust genome-wide association study are relevant, where initial candidate gene studies, although fruitful on occasion, generally lacked the statistical power required to identify replicable associations in a situation where there was a high level of multiple statistical testing.128 Similarly, it is likely that many candidate DNA methylation studies will not survive wider replication. Whichever approach is selected, where to search for epigenetic variation is clearly a fundamental prerequisite to identifying environmentally induced variation.
Genetic proxies for DNA methylation
In the second step of a two step epigenetic Mendelian randomization approach a genetic proxy for methylation levels is required. This may take the form of a cis-SNP, i.e. a SNP in the vicinity of the CpG site that correlates with methylation levels. Various terminologies have been proposed to describe polymorphisms that influence local methylation propensity including ‘sequence-influenced methylation polymorphism’ (SIMP),104 CpG-SNP,109,130,131 cis-SNP131 and epiallele.132 It is possible to locate such potential SNPs or proxies by interrogating the SNP architecture flanking the CpG site or differentially methylated region of interest and assessing the correlation between methylation levels at the site of interest and genotype. This approach has recently been used to investigate the observed association between methylation of the TACSTD2 gene promoter and childhood adiposity, where a cis-SNP 162bp from the CpG sites assayed correlated highly with methylation levels and could thus be used as a proxy12. There will also be instances where a CpG site is ablated or lost due to the presence of a polymorphism. Such a methylation-removing or methylation-introducing SNP might be termed an mSNP.107,129 However, mSNPs are not ideal instruments for a two-step epigenetic Mendelian randomization approach as there is a one-to-one relationship between the SNP and methylation, meaning that it cannot be discerned whether the SNP is acting through epigenetic mechanisms or other functional pathways. mSNPs can be used to proxy for average methylation over a region, however.
It is also useful at this juncture to identify whether the SNP selected to act as a proxy for methylation maps to a transcription factor binding site, enhancer or other motif that may have the potential to introduce pleiotropic effects. It is plausible that a SNP which correlates highly with a methylation site may also have functional consequences independent of its association with local methylation marks.
Instrumental variables analysis applied to two-step epigenetic Mendelian randomization studies
We propose the application of an instrumental variables (IV) analysis to the two-step epigenetic Mendelian randomization approach outlined in Figure 4. Thus, taking the scenario shown in Figure 5A, the application of IV analysis to the postulated link between smoking, altered methylation and cardiovascular disease is provided as an example. This example is based upon the recently reported association between DNA methylation at a single CpG site at the F2RL3 locus and smoking54, together with the use of the CHRN3/5 variant, which is an established instrumental variant for smoking intensity134. IV analysis allows the generation of an unbiased estimate of the modifiable exposure (here smoking) by the genotype (the instrumental variable, here the CHRNA3/5 variant134) and then, to use those predicted values to estimate the association between the exposure (smoking) and the outcome (here methylation of the F2RL3 locus). What is called a test of endogeneity in the econometrics literature is then conducted to evaluate the level of agreement between the regression slope from the conventional analysis and the predicted causal association from the IV analysis.65 Agreement between the two estimates suggests that the observational association is not a seriously biased or confounded estimate of the causal effect. If the two estimates are clearly distinct this suggests either the conventional analysis is producing a confounded or biased estimate of the causal effect or there is violation of the assumptions of the IV analysis, as discussed later. In Step 2 of the epigenetic Mendelian randomization approach, when considering the position of DNA methylation on the causal pathway between an environmental exposure and disease (e.g. smoking, methylation, cardiovascular disease), IV analysis must then be repeated with methylation as the predictor rather than the outcome (Figure 5B). For this second step, an instrumental variable is required that correlates with methylation levels in the smoking responsive CpG locus (here F2RL3). This should be a cis variant, i.e. an SNP in the vicinity of the F2RL3 CpG site. As outlined in the previous section, many SNPs that correlate with methylation levels are found to be cis variants.48,107 Collectively, this two-step process can provide a more reliable assessment of the causal pathway between the environmental exposure (smoking), the mediator (DNA methylation) and the outcome (cardiovascular disease).
Figure 5.

Two-step epigenetic Mendelian randomization applied to smoking and cardiovascular disease: (A) instrumental variable [G] acts as a proxy for environmental exposure [E], postulated to influence disease [Y] via altered DNA methylation [X]. G is independent of unmeasured confounder [U]. G only influences X if E →X is causal (red dashed line). This is shown in the left hand side of Panel A. This is illustrated by considering the influence of smoking on cardiovascular disease risk using the CHRN3/5 variant as an instrumental variable for smoking intensity, as shown on the right hand side of Panel A. This variant has been used previously as an instrument to assess the causal relationship between smoking and BMI.133 The CpG site of interest could be the site in F2RL3 recently reported by Breitling et al.54 (B) In a second step, an alternative proxy, here an SNP in the same gene in which methylation is measured (F2RL3), is used, as shown in the two diagrams in Panel B to assess the causal relationship between X and Y or F2RL3 methylation and CVD
The approach can be applied to assess the effect of in utero exposures, using maternal genotype as a proxy for maternal exposure in the first step and offspring genotype as a proxy for methylation in the second step. An example in this regard with respect to maternal alcohol exposure and offspring cognition is outlined in Figure 6, where altered DNA methylation in the infant might plausibly mediate some of the effects of this exposure.
Figure 6.

Applying a two-step epigenetic Mendelian randomization approach to in utero influences on offspring outcomes: (A) maternal instrumental variable [Gm] acts as a proxy for environmental influences [Em] on fetal DNA methylation during pregnancy [Xo] and subsequent offspring outcome [Yo]. Gm only influences Xo if Em→Xo is causal (red dashed line). The postulated influence of maternal alcohol intake during pregnancy on offspring cognition using the maternal ADH1B variant as an instrumental variable is shown as an example.116 (B) An additional proxy, which correlates with methylation at the alcohol responsive CpG site(s), is then applied in a second step. In this instance, the offspring's genotype [Go] is used as a proxy for methylation
Strengths and weaknesses
Although predicated on the now well-established Mendelian randomization framework, concrete examples of the application of the two-step epigenetic Mendelian randomization approach are required. A recent report by Terry et al. details support for gene-specific DNA methylation being associated with exposures including benzene, air pollution, arsenic, cigarette smoking and alcohol drinking.135 These exposure–methylation associations could be interrogated utilizing the first step of a two-step epigenetic Mendelian randomization approach. It must be recognized, however, that reported associations between environmental factors and both global and gene-specific DNA methylation are often modest in size and lack the robustness of equivalent contemporary genetic association studies. For the second step, robust instruments for DNA methylation, such as the F2RL3 CpG site associated with smoking exposure, will be required.54 The relationship of variably methylated regions with underlying DNA sequence requires more detailed interrogation to elicit a greater number of cis-acting variants robustly associated DNA methylation levels.
Tissue specificity is clearly an important feature of epigenetic patterns, explaining how over 200 tissue types can arise from the same genotype. Inherent in this observation is the assumption that genotype must only be partially correlated with DNA methylation patterns—to allow tissue-specific methylation signatures to exist on a background of uniform genotype. Therefore, genetic proxies for methylation levels may be tissue specific and will require tissue-specific validation if being used in DNA samples from tissues other than peripheral blood. Detailed information of DNA methylation patterns across multiple tissues is gradually becoming available as initiatives to sequence reference methylomes gain momentum. It will be possible within the foreseeable future to assess the relationship between genetic variation and DNA methylation in a tissue-specific manner using openly accessible data sources. Limitations include that these data are often generated on diseased tissue and that they usually have very little information on environmental exposures or other relevant covariates.
It is recognised that Mendelian randomization has certain limitations,60 and these apply equally to the two-step epigenetic Mendelian randomization approach. These include the need for larger sample sizes than have generally been used to date in epigenetic epidemiology studies to ensure robust findings; problems introduced by population stratification that could generate spurious genetic variant—epigenotype and genetic variant—outcome associations; reintroduced confounding through genetic pleiotropy and linkage disequilibrium and complexities introduced by developmental compensation to genetic perturbations. Potential limitations are elaborated on in Table 1, and approaches to evaluating the contribution or avoidance of these problems. A strategy to overcome the important issue of pleiotropy—where a genetic variant has more than one direct correlate that would invalidate conclusions based on the assumption of a single pathway—is the use of multiple genetic instruments (including potentially many combinations of independent instruments). In the Mendelian randomization context, in some cases, it may be possible to identify two separate genetic variants, which are not in linkage disequilibrium with each other, but which both serve as proxies for the environmentally modifiable risk factor of interest. If both variants are related to the outcome of interest and point to the same underlying association, then it becomes much less plausible that reintroduced confounding explains the association, since it would have to be acting in the same way for these two unlinked variants. This can be likened to randomized controlled trials of different blood pressure-lowering agents, which work through different mechanisms and have different potential side effects. If the different agents produce the same reductions in cardiovascular disease risk, then it is unlikely that this is through agent-specific (pleiotropic) effects of the drugs; rather, it points to blood pressure lowering as being key. The latter is indeed what is in general observed.137 In the Mendelian randomization setting, two distinct genetic variants acting as instruments for higher body fat content have been used to demonstrate that greater adiposity is related to higher bone mineral density.138 With the large number of genetic variants that are being identified in genome-wide association studies in relation to particular phenotypes it is possible to generate many independent combinations of such variants, and from these combinations many independent instrumental variable estimates of the causal associations between an environmentally modifiable risk factor and a disease outcome can be obtained. The independent estimates will not be plausibly influenced by any common pleiotropy or LD-induced confounding, and therefore if they display consistency this provides strong evidence against the notion that reintroduced confounding is generating the associations. The same principles can be applied in two-step epigenetic Mendelian randomization studies (Figure 7).
Table 1.
Limitations of Mendelian randomization and two-step epigenetic Mendelian randomization studies
| Limitation | Role in Mendelian randomization (MR) studies | Role in two-step epigenetic Mendelian randomization (TSMR) studies | Approaches to evaluating or avoiding the limitation |
|---|---|---|---|
| Low statistical power | MR studies are often of low power and effect estimates are imprecise because of this | Currently, poorly defined as expected strength of environment—epigenome and epigenome—disease associations not well established | Increase sample size and/or combine genetic variants so they explain more of the variance of the intermediate phenotype (e.g. methylation in TSMR studies) |
| Population stratification generating spurious genetic variant—intermediate phenotype or genetic variant—disease associations | A potential problem but can be avoided by applying standard genetic epidemiology methodologies | A potential problem and currently it is unknown whether genetic variant—methylation associations will be more or less subject to population stratification | Restrict analyses to ethnically homogeneous groups and/or apply correction methods using ancestrally informative markers or principal components from genome-wide data |
| Re-introduced confounding though pleiotropy | A genetic variant may directly influence more than one post-transcriptional process. Known to be the case for some genetic variants | As in MR studies | When possible utilize cis variants with respect to the intermediate phenotype under study, as these may be less likely to have pleiotropic effects. Apply multiple instrument approaches with more than one independent genetic variant as unlikely pleiotropy will generate the same associations for different instruments |
| LD-induced confounding | LD is useful in genetic association studies as it allows marker SNPs to proxy for un-genotyped causal SNPs. However, this can re-introduce confounding if LD leads to the association of SNPs related to more than one post-transcriptional process. This case will be similar to the pleiotropy situation | As in MR studies | Studies can be carried out in populations with different LD structures. Approaches to avoiding distortion by pleiotropy will also counter problems due to LD |
| Canalization/developmental compensation | During development compensatory processes may be generated that counter the phenotypic perturbation consequent on the genetic variant utilized as an instrument | As in MR studies. As epigenetic processes are particularly important during development it could be anticipated that such developmental compensation could theoretically be occurring | Requires context-specific knowledge. One generic approach to which the context-specific information can be added is to examine the genetic variant—intermediate phenotype association at different stages of life. If not evident during early life (e.g. not detectable at birth or in infancy, but emerge later) then canalization is less likely to be occurring. Such a situation is seen with respect to the association of variation in FTO and adiposity136 |
| Lack of genetic variants to proxy for modifiable exposure of interest | No reliable genetic variant associations for many intermediate phenotypes of interest, although an increasing number of these now identified | For Step 1, as for MR studies. Identification of cis variants to proxy for methylation differences is in its infancy | Continued genome-wide and sequencing-based studies |
| Complexity of associations | Without adequate biological knowledge misleading inferences regarding intermediate phenotypes and disease may be drawn | As in MR studies | Increased biological understanding of genotype—phenotype links |
Figure 7.
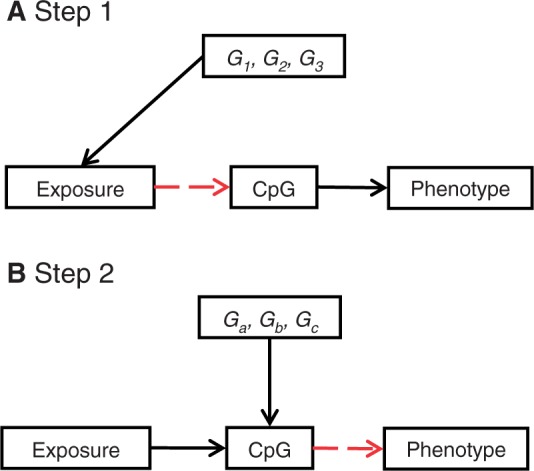
Multiple instruments: a potential advantage of a two-step epigenetic Mendelian randomization approach is the exploitation of genetic heterogeneity where multiple instrumental variables can be combined to strengthen causal inference for a role of DNA methylation in the causal pathway between exposure and disease. (A) Genetic heterogeneity might occur when several genes [G1, G2, G3] are robustly associated with the exposure of interest and can be used as separate instruments to interrogate the association of the exposure and DNA methylation. The influence of multiple lipid-altering SNPs might be one example.139 Collectively, the combination of lipid-altering variants can be used to repeatedly assess the causal relationship between lipid levels and DNA methylation. (B) A multiple instrument approach is also possible in step 2 where several uncorrelated cis-SNPs [Ga, Gb, Gc] might be identified that impact upon DNA methylation at a particular site or across a particular genomic region. These could then be used as separate instruments to interrogate the relationship between methylation and phenotype
Summary and potential applications
Two-step epigenetic Mendelian randomization has the potential to contribute to furthering understanding of the causal role of DNA methylation in mediating environmental influences on common complex disease, overcoming the potential for confounding and reverse causation. In translational terms, two-step epigenetic Mendelian randomization has the potential to help distinguish between intervention targets (truly causal) and epiphenomena (non-causal) which may, nevertheless, be informative diagnostic or prognostic biomarkers.
Funding
C.L.R. and G.D.S. received funding from a number of sources including the Medical Research Council, Biotechnology and Biological Sciences Research Council, Wellcome Trust, European Union FP7 (IRSES GEoCoDE) and various medical charities. This work does not represent that funded from any single source.
Acknowledgements
Thanks to Ezra Susser, John Lynch, Nic Timpson, Luisa Zuccolo, Catherine Potter, Karin Michels and Alexandra Binder for comments on an earlier draft of this article and in particular to Debbie Lawlor for her helpful comments on mediation.
Conflict of interest: None declared
KEY MESSAGES.
Establishing the causal role of epigenetic variation on the pathway linking exposure to disease is crucial to the development of epigenetic-based interventions. Approaches are required to strengthen causal inference.
Two-step epigenetic Mendelian randomization provides a framework for strengthening causal inference with regard to epigenetic factors, namely DNA methylation.
The principles of Mendelian randomization and genetical genomics have been merged to form a two-step analysis framework, termed two-step epigenetic Mendelian randomization, which first assesses the causal relationship between exposure and DNA methylation and secondly, between DNA methylation and outcome.
Incorporating epigenetic markers into studies of modifiable risk factors and disease outcomes in this way will help in further understanding of causal pathways to disease.
References
- 1.Hamm CA, Costa FF. The impact of epigenomics on future drug design and new therapies. Drug Discov Today. 2011;16:626–35. doi: 10.1016/j.drudis.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 2.Feero WG, Guttmacher AE, Collins FS. Genomic medicine–an updated primer. N Engl J Med. 2010;362:2001–11. doi: 10.1056/NEJMra0907175. [DOI] [PubMed] [Google Scholar]
- 3.Vineis P, Pearce NE. Genome-wide association studies may be misinterpreted: genes versus heritability. Carcinogenesis. 2011;32:1295–98. doi: 10.1093/carcin/bgr087. [DOI] [PubMed] [Google Scholar]
- 4.Le Fanu J. The disappointments of the double helix: a master theory. J R Soc Med. 2010;103:43–45. doi: 10.1258/jrsm.2009.09k077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ebrahim S, Davey Smith G. Mendelian randomization: can genetic epidemiology help redress the failures of observational epidemiology? Hum Genet. 2008;123:15–33. doi: 10.1007/s00439-007-0448-6. [DOI] [PubMed] [Google Scholar]
- 6.Schadt EE. Molecular networks as sensors and drivers of common human diseases. Nature. 2009;461:218–223. doi: 10.1038/nature08454. [DOI] [PubMed] [Google Scholar]
- 7.Rockman MV. Reverse engineering the genotype-phenotype map with natural genetic variation. Nature. 2008;456:738–44. doi: 10.1038/nature07633. [DOI] [PubMed] [Google Scholar]
- 8.Relton CL, Davey Smith G. Epigenetic epidemiology of common complex disease: prospects for prediction, prevention, and treatment. PLoS Med. 2010;7:e1000356. doi: 10.1371/journal.pmed.1000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lim DH, Maher ER. Genomic imprinting syndromes and cancer. Adv Genet. 2010;70:145–75. doi: 10.1016/B978-0-12-380866-0.60006-X. [DOI] [PubMed] [Google Scholar]
- 10.Urdinguio RG, Sanchez-Mut JV, Esteller M. Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol. 2009;8:1056–72. doi: 10.1016/S1474-4422(09)70262-5. [DOI] [PubMed] [Google Scholar]
- 11.Kaminsky Z, Tochigi M, Jia P, et al. A multi-tissue analysis identifies HLA complex group 9 gene methylation differences in bipolar disorder. Mol Psychiatry. doi: 10.1038/mp.2011.64. 2011; doi:10.1038/mp.2011.64 [Epub] [DOI] [PubMed] [Google Scholar]
- 12.Groom A, Potter C, Swan DC, et al. Postnatal Growth and DNA Methylation Are Associated With Differential Gene Expression of the TACSTD2 Gene and Childhood Fat Mass. Diabetes. 2012;61:391–400. doi: 10.2337/db11-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Godfrey KM, Sheppard A, Gluckman PD, et al. Epigenetic gene promoter methylation at birth is associated with child's later adiposity. Diabetes. 2011;60:1528–34. doi: 10.2337/db10-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bell CG, Teschendorff AE, Rakyan VK, Maxwell AP, Beck S, Savage DA. Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Med Genomics. 2010;3:33. doi: 10.1186/1755-8794-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reynard LN, Bui C, Canty-Laird EG, Young DA, Loughlin J. Expression of the osteoarthritis-associated gene GDF5 is modulated epigenetically by DNA methylation. Hum Mol Genet. 2011;20:3450–60. doi: 10.1093/hmg/ddr253. [DOI] [PubMed] [Google Scholar]
- 16.Hernandez DG, Nalls MA, Gibbs JR, et al. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum Mol Genet. 2011;20:1164–72. doi: 10.1093/hmg/ddq561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waddington CH. The epigenotype. Endeavour. 1942;1:18–20. Reprinted in Int J Epidemiol 2012;41:10–13. [Google Scholar]
- 18.Ho DH, Burggren WW. Epigenetics and transgenerational transfer: a physiological perspective. J Exp Biol. 2010;213:3–16. doi: 10.1242/jeb.019752. [DOI] [PubMed] [Google Scholar]
- 19.Bird A. Perceptions of epigenetics. Nature. 2007;447:396–98. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 20.Jablonka E, Raz G. Transgenerational epigenetic inheritance: prevalence, mechanisms, and implications for the study of heredity and evolution. Q Rev Biol. 2009;84:131–76. doi: 10.1086/598822. [DOI] [PubMed] [Google Scholar]
- 21.Jablonka E, Lamb MJ. Evolution in Four Dimensions: Genetic, Epigenetic, Behavioral, and Symbolic Variation in the History of Life. Cambridge: MIT Press; 2005. [Google Scholar]
- 22.Ogbuanu IU, Zhang H, Karmaus W. Can we apply the Mendelian randomization methodology without considering epigenetic effects? Emerg Themes Epidemiol. 2009;6:3 doi: 10.1186/1742-7622-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seong KH, Li D, Shimizu H, Nakamura R, Ishii S. Inheritance of stress-induced, ATF-2-dependent epigenetic change. Cell. 2011;145:1049–61. doi: 10.1016/j.cell.2011.05.029. [DOI] [PubMed] [Google Scholar]
- 24.Morgan DK, Whitelaw E. The case for transgenerational epigenetic inheritance in humans. Mamm Genome. 2008;19:394–97. doi: 10.1007/s00335-008-9124-y. [DOI] [PubMed] [Google Scholar]
- 25.Rakyan VK, Chong S, Champ ME, et al. Transgenerational inheritance of epigenetic states at the murine Axin(Fu) allele occurs after maternal and paternal transmission. Proc Natl Acad Sci USA. 2003;100:2538–43. doi: 10.1073/pnas.0436776100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ng SF, Lin RC, Laybutt DR, Barres R, Owens JA, Morris MJ. Chronic high-fat diet in fathers programs beta-cell dysfunction in female rat offspring. Nature. 2010;467:963–66. doi: 10.1038/nature09491. [DOI] [PubMed] [Google Scholar]
- 27.Dupont C, Armant DR, Brenner CA. Epigenetics: definition, mechanisms and clinical perspective. Semin Reprod Med. 2009;27:351–57. doi: 10.1055/s-0029-1237423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skinner MK. Environmental epigenetic transgenerational inheritance and somatic epigenetic mitotic stability. Epigenetics. 2011;6:838–42. doi: 10.4161/epi.6.7.16537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daxinger L, Whitelaw E. Transgenerational epigenetic inheritance: more questions than answers. Genome Res. 2010;20:1623–28. doi: 10.1101/gr.106138.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin C, Zhang Y. Mechanisms of epigenetic inheritance. Curr Opin Cell Biol. 2007;19:266–72. doi: 10.1016/j.ceb.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 31.Zaidi SK, Young DW, Montecino M, et al. Bookmarking the genome: maintenance of epigenetic information. J Biol Chem. 2011;286:18355–61. doi: 10.1074/jbc.R110.197061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anway MD, Cupp AS, Uzumcu M, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science. 2005;308:1466–69. doi: 10.1126/science.1108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matthews SG, Phillips DI. Transgenerational inheritance of stress pathology. Exp Neurol. 2011 doi: 10.1016/j.expneurol.2011.01.009. doi:10.1016/j.expneurol.2011.01.009 [Epub] [DOI] [PubMed] [Google Scholar]
- 34.Sinclair KD, Karamitri A, Gardner DS. Dietary regulation of developmental programming in ruminants: epigenetic modifications in the germline. Soc Reprod Fertil Suppl. 2010;67:59–72. [PubMed] [Google Scholar]
- 35.Kappeler L, Meaney MJ. Epigenetics and parental effects. Bioessays. 2010;32:818–27. doi: 10.1002/bies.201000015. [DOI] [PubMed] [Google Scholar]
- 36.Crepin M, Dieu MC, Lejeune S, et al. Evidence of constitutional MLH1 epimutation associated to transgenerational inheritance of cancer susceptibility. Hum Mutat. 2011;33:180–188. doi: 10.1002/humu.21617. [DOI] [PubMed] [Google Scholar]
- 37.Vastenhouw NL, Brunschwig K, Okihara KL, Muller F, Tijsterman M, Plasterk RH. Gene expression: long-term gene silencing by RNAi. Nature. 2006;442:882. doi: 10.1038/442882a. [DOI] [PubMed] [Google Scholar]
- 38.Carone BR, Fauquier L, Habib N, et al. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell. 2010;143:1084–96. doi: 10.1016/j.cell.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waterland RA, Travisano M, Tahiliani KG, Rached MT, Mirza S. Methyl donor supplementation prevents transgenerational amplification of obesity. Int J Obes. 2008;32:1373–79. doi: 10.1038/ijo.2008.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li CC, Cropley JE, Cowley MJ, Preiss T, Martin DI, Suter CM. A sustained dietary change increases epigenetic variation in isogenic mice. PLoS Genet. 2011;7:e1001380. doi: 10.1371/journal.pgen.1001380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Day T, Bonduriansky R. A unified approach to the evolutionary consequences of genetic and nongenetic inheritance. Am Nat. 2011;178:E18–36. doi: 10.1086/660911. [DOI] [PubMed] [Google Scholar]
- 42.Davey Smith G. Epigenesis for Epidemiologists: does evo-devo have implications for population health research and practice? Int J Epidemiol. 2012;41:236–47. doi: 10.1093/ije/dys016. [DOI] [PubMed] [Google Scholar]
- 43.Charlesworth D, Charlesworth B. Not quite a revolution. The Guardian Review. 3 September 2011, p. 15. [Google Scholar]
- 44.Haldane J. Some principles of causal analysis in genetics. Erkenntnis. 1936;6:346–57. [Google Scholar]
- 45.Lynch M. The rate of polygenic mutation. Genet Res. 1988;51:137–48. doi: 10.1017/s0016672300024150. [DOI] [PubMed] [Google Scholar]
- 46.Francis R. Epigenetics: The Ultimate Mystery of Inheritance. New York: W.W. Norton & Company, Incorp; 2011. [Google Scholar]
- 47.Carey N. The Epigenetics Revolution: How Modern Biology is Rewriting Our Understanding of Genetics, Disease and Inheritance. London: Icon Books Ltd;; 2011. [Google Scholar]
- 48.Bell JT, Pai AA, Pickrell JK, et al. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 2011;12:R10. doi: 10.1186/gb-2011-12-1-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meaburn EL, Schalkwyk LC, Mill J. Allele-specific methylation in the human genome: implications for genetic studies of complex disease. Epigenetics. 2010;5:578–82. doi: 10.4161/epi.5.7.12960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet. 2011;12:529–41. doi: 10.1038/nrg3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet. 2010;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- 52.Nelson HH, Marsit CJ, Kelsey KT. “Global methylation” in exposure biology and translational medical science. Environ Health Perspect. 2011;119:1528–33. doi: 10.1289/ehp.1103423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karimi M, Luttropp K, Ekstrom TJ. Global DNA methylation analysis using the luminometric methylation assay. Methods Mol Biol. 2011;791:135–44. doi: 10.1007/978-1-61779-316-5_11. [DOI] [PubMed] [Google Scholar]
- 54.Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet. 2011;88:450–57. doi: 10.1016/j.ajhg.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marsit CJ, Koestler DC, Christensen BC, Karagas MR, Houseman EA, Kelsey KT. DNA methylation array analysis identifies profiles of blood-derived DNA methylation associated with bladder cancer. J Clin Oncol. 2011;29:1133–39. doi: 10.1200/JCO.2010.31.3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Teschendorff AE, Menon U, Gentry-Maharaj A, et al. An epigenetic signature in peripheral blood predicts active ovarian cancer. PLoS One. 2009;4:e8274. doi: 10.1371/journal.pone.0008274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kneip C, Schmidt B, Seegebarth A, et al. SHOX2 DNA methylation is a biomarker for the diagnosis of lung cancer in plasma. J Thorac Oncol. 2011 doi: 10.1097/JTO.0b013e318220ef9a. 6:1632–38. [DOI] [PubMed] [Google Scholar]
- 58.Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Davey Smith G. Assessing intrauterine influences on offspring health outcomes: can epidemiological studies yield robust findings? Basic Clin Pharmacol Toxicol. 2008;102:245–56. doi: 10.1111/j.1742-7843.2007.00191.x. [DOI] [PubMed] [Google Scholar]
- 60.Davey Smith G, Ebrahim S. 'Mendelian randomization': can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32:1–22. doi: 10.1093/ije/dyg070. [DOI] [PubMed] [Google Scholar]
- 61.Li J, Burmeister M. Genetical genomics: combining genetics with gene expression analysis. Hum Mol Genet. 2005;14(Spec No. 2):R163–69. doi: 10.1093/hmg/ddi267. [DOI] [PubMed] [Google Scholar]
- 62.Schadt EE, Lamb J, Yang X, et al. An integrative genomics approach to infer causal associations between gene expression and disease. Nat Genet. 2005;37:710–17. doi: 10.1038/ng1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Greenland S. An introduction to instrumental variables for epidemiologists. Int J Epidemiol. 2000;29:722–29. doi: 10.1093/ije/29.4.722. [DOI] [PubMed] [Google Scholar]
- 64.Thomas DC, Conti DV. Commentary: the concept of 'Mendelian Randomization'. Int J Epidemiol. 2004;33:21–25. doi: 10.1093/ije/dyh048. [DOI] [PubMed] [Google Scholar]
- 65.Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27:1133–63. doi: 10.1002/sim.3034. [DOI] [PubMed] [Google Scholar]
- 66.Davey Smith G, Harbord R, Ebrahim S. Fibrinogen, C-reactive protein and coronary heart disease: does Mendelian randomization suggest the associations are non-causal? QJM. 2004;97:163–66. doi: 10.1093/qjmed/hch025. [DOI] [PubMed] [Google Scholar]
- 67.Davey Smith G, Ebrahim S. What can mendelian randomisation tell us about modifiable behavioural and environmental exposures? BMJ. 2005;330:1076–79. doi: 10.1136/bmj.330.7499.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sheehan NA, Didelez V, Burton PR, Tobin MD. Mendelian randomisation and causal inference in observational epidemiology. PLoS Med. 2008;5:e177. doi: 10.1371/journal.pmed.0050177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Davey Smith G, Lawlor DA, Harbord R, Timpson N, Day I, Ebrahim S. Clustered environments and randomized genes: a fundamental distinction between conventional and genetic epidemiology. PLoS Med. 2007;4:e352. doi: 10.1371/journal.pmed.0040352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Palmer LJ, Cardon LR. Shaking the tree: mapping complex disease genes with linkage disequilibrium. Lancet. 2005;366:1223–34. doi: 10.1016/S0140-6736(05)67485-5. [DOI] [PubMed] [Google Scholar]
- 71.Ebrahim S, Lawlor DA, Shlomo YB, et al. Alcohol dehydrogenase type 1C (ADH1C) variants, alcohol consumption traits, HDL-cholesterol and risk of coronary heart disease in women and men: British Women's Heart and Health Study and Caerphilly cohorts. Atherosclerosis. 2008;196:871–78. doi: 10.1016/j.atherosclerosis.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 72.Timpson NJ, Lawlor DA, Harbord RM, et al. C-reactive protein and its role in metabolic syndrome: mendelian randomisation study. Lancet. 2005;366:1954–59. doi: 10.1016/S0140-6736(05)67786-0. [DOI] [PubMed] [Google Scholar]
- 73.Davey Smith G, Lawlor DA, Harbord R, et al. Association of C-reactive protein with blood pressure and hypertension: life course confounding and mendelian randomization tests of causality. Arterioscler Thromb Vasc Biol. 2005;25:1051–56. doi: 10.1161/01.ATV.0000160351.95181.d0. [DOI] [PubMed] [Google Scholar]
- 74.Casas JP, Shah T, Cooper J, et al. Insight into the nature of the CRP-coronary event association using Mendelian randomization. Int J Epidemiol. 2006;35:922–31. doi: 10.1093/ije/dyl041. [DOI] [PubMed] [Google Scholar]
- 75.Keavney B, Danesh J, Parish S, et al. Fibrinogen and coronary heart disease: test of causality by ‘Mendelian randomization’. Int J Epidemiol. 2006;35:935–43. doi: 10.1093/ije/dyl114. [DOI] [PubMed] [Google Scholar]
- 76.Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331–39. doi: 10.1001/jama.2009.801. [DOI] [PubMed] [Google Scholar]
- 77.Thanassoulis G, O'Donnell CJ. Mendelian randomization: nature's randomized trial in the post-genome era. JAMA. 2009;301:2386–88. doi: 10.1001/jama.2009.812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zacho J, Tybjaerg-Hansen A, Jensen JS, Grande P, Sillesen H, Nordestgaard BG. Genetically elevated C-reactive protein and ischemic vascular disease. N Engl J Med. 2008;359:1897–908. doi: 10.1056/NEJMoa0707402. [DOI] [PubMed] [Google Scholar]
- 79.Timpson NJ, Wade KH, Davey Smith G. Mendelian Randomization: Application to Cardiovascular Disease. Current Hypertension Reports. 2012;14:29–37. doi: 10.1007/s11906-011-0242-7. [DOI] [PubMed] [Google Scholar]
- 80.Shah SH, de Lemos JA. Biomarkers and cardiovascular disease: determining causality and quantifying contribution to risk assessment. JAMA. 2009;302:92–93. doi: 10.1001/jama.2009.949. [DOI] [PubMed] [Google Scholar]
- 81.Timpson NJ, Harbord R, Davey Smith G, Zacho J, Tybjaerg-Hansen A, Nordestgaard BG. Does greater adiposity increase blood pressure and hypertension risk?: Mendelian randomization using the FTO/MC4R genotype. Hypertension. 2009;54:84–90. doi: 10.1161/HYPERTENSIONAHA.109.130005. [DOI] [PubMed] [Google Scholar]
- 82.Brennan P, McKay J, Moore L, et al. Obesity and cancer: Mendelian randomization approach utilizing the FTO genotype. Int J Epidemiol. 2009;38:971–75. doi: 10.1093/ije/dyp162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ding EL, Song Y, Manson JE, et al. Sex hormone-binding globulin and risk of type 2 diabetes in women and men. N Engl J Med. 2009;361:1152–63. doi: 10.1056/NEJMoa0804381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Scott JA, Berkley JA, Mwangi I, et al. Relation between falciparum malaria and bacteraemia in Kenyan children: a population-based, case-control study and a longitudinal study. Lancet. 2011;378:1316–23. doi: 10.1016/S0140-6736(11)60888-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jansen RC, Nap JP. Genetical genomics: the added value from segregation. Trends Genet. 2001;17:388–91. doi: 10.1016/s0168-9525(01)02310-1. [DOI] [PubMed] [Google Scholar]
- 86.Moffatt MF, Kabesch M, Liang L, et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448:470–73. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- 87.Chen Y, Zhu J, Lum PY, et al. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;52:429–35. doi: 10.1038/nature06757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Emilsson V, Thorleifsson G, Zhang B, et al. Genetics of gene expression and its effect on disease. Nature. 2008;452:423–28. doi: 10.1038/nature06758. [DOI] [PubMed] [Google Scholar]
- 89.Drake TA, Schadt EE, Lusis AJ. Integrating genetic and gene expression data: application to cardiovascular and metabolic traits in mice. Mamm Genome. 2006;17:466–79. doi: 10.1007/s00335-005-0175-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Davey Smith G. Random allocation in observational data: how small but robust effects could facilitate hypothesis-free causal inference. Epidemiology. 2011;22:460–63. doi: 10.1097/EDE.0b013e31821d0426. [DOI] [PubMed] [Google Scholar]
- 91.Wright MW, Bruford EA. Naming 'junk': human non-protein coding RNA (ncRNA) gene nomenclature. Hum Genomics. 2011;5:90–98. doi: 10.1186/1479-7364-5-2-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rassoulzadegan M, Grandjean V, Gounon P, Vincent S, Gillot I, Cuzin F. RNA-mediated non-mendelian inheritance of an epigenetic change in the mouse. Nature. 2006;441:469–74. doi: 10.1038/nature04674. [DOI] [PubMed] [Google Scholar]
- 93.Orom UA, Shiekhattar R. Long non-coding RNAs and enhancers. Curr Opin Genet Dev. 2011;21:194–98. doi: 10.1016/j.gde.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Feinberg AP, Irizarry RA, Fradin D, et al. Personalized epigenomic signatures that are stable over time and covary with body mass index. Sci Transl Med. 2010;2:49ra67. doi: 10.1126/scitranslmed.3001262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Feinberg AP. Genome-scale approaches to the epigenetics of common human disease. Virchows Arch. 2010;456:13–21. doi: 10.1007/s00428-009-0847-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Feinberg AP, Irizarry RA. Evolution in health and medicine Sackler colloquium: Stochastic epigenetic variation as a driving force of development, evolutionary adaptation, and disease. Proc Natl Acad Sci USA. 2010;107(Suppl. 1):1757–64. doi: 10.1073/pnas.0906183107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cloud J. Why your DNA isn't your destiny. Time Magazine. 6 January 2010. [Google Scholar]
- 98.Harrell E. The Human Epigenome. Time Magazine. 8 October 2009. [Google Scholar]
- 99.Haig D. Weismann Rules! OK? Epigenetics and the Lamarckian temptation. Biol Phil. 2007;22:415–28. [Google Scholar]
- 100.Waterland RA, Travisano M, Tahiliani KG. Diet-induced hypermethylation at agouti viable yellow is not inherited transgenerationally through the female. FASEB J. 2007;21:3380–85. doi: 10.1096/fj.07-8229com. [DOI] [PubMed] [Google Scholar]
- 101.Jablonka E, Lamb MJ. The inheritance of acquired epigenetic variations. J Theor Biol. 1989;139:69–83. doi: 10.1016/s0022-5193(89)80058-x. [DOI] [PubMed] [Google Scholar]
- 102.Waterland RA, Kellermayer R, Laritsky E, et al. Season of conception in rural gambia affects DNA methylation at putative human metastable epialleles. PLoS Genet. 2010;6:e1001252. doi: 10.1371/journal.pgen.1001252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kendrick SF, O'Boyle G, Mann J, et al. Acetate, the key modulator of inflammatory responses in acute alcoholic hepatitis. Hepatology. 2010;51:1988–97. doi: 10.1002/hep.23572. [DOI] [PubMed] [Google Scholar]
- 104.Hellman A, Chess A. Extensive sequence-influenced DNA methylation polymorphism in the human genome. Epigenetics Chromatin. 2010;3:11. doi: 10.1186/1756-8935-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Baccarelli A, Rienstra M, Benjamin EJ. Cardiovascular epigenetics: basic concepts and results from animal and human studies. Circ Cardiovasc Genet. 2010;3:567–73. doi: 10.1161/CIRCGENETICS.110.958744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gibbs JR, van der Brug MP, Hernandez DG, et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010;6:e1000952. doi: 10.1371/journal.pgen.1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang D, Cheng L, Badner JA, et al. Genetic control of individual differences in gene-specific methylation in human brain. Am J Hum Genet. 2010;86:411–19. doi: 10.1016/j.ajhg.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pai AA, Bell JT, Marioni JC, Pritchard JK, Gilad Y. A genome-wide study of DNA methylation patterns and gene expression levels in multiple human and chimpanzee tissues. PLoS Genet. 2011;7:e1001316. doi: 10.1371/journal.pgen.1001316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shoemaker R, Deng J, Wang W, Zhang K. Allele-specific methylation is prevalent and is contributed by CpG-SNPs in the human genome. Genome Res. 2010;20:883–89. doi: 10.1101/gr.104695.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bell CG, Finer S, Lindgren CM, et al. Integrated genetic and epigenetic analysis identifies haplotype-specific methylation in the FTO type 2 diabetes and obesity susceptibility locus. PLoS One. 2010;5:e14040. doi: 10.1371/journal.pone.0014040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Robins JM, Greenland S. Identifiability and exchangeability for direct and indirect effects. Epidemiology. 1992;3:143–55. doi: 10.1097/00001648-199203000-00013. [DOI] [PubMed] [Google Scholar]
- 112.Cole SR, Hernan MA. Fallibility in estimating direct effects. Int J Epidemiol. 2002;31:163–65. doi: 10.1093/ije/31.1.163. [DOI] [PubMed] [Google Scholar]
- 113.Blakely T. Commentary: estimating direct and indirect effects-fallible in theory, but in the real world? Int J Epidemiol. 2002;31:166–67. doi: 10.1093/ije/31.1.166. [DOI] [PubMed] [Google Scholar]
- 114.Hafeman DM. Confounding of indirect effects: a sensitivity analysis exploring the range of bias due to a cause common to both the mediator and the outcome. Am J Epidemiol. 2011;174:710–17. doi: 10.1093/aje/kwr173. [DOI] [PubMed] [Google Scholar]
- 115.Cole SR. Illustrating bias due to conditioning on a collider. Int J Epidemiol. 2010;39:417–20. doi: 10.1093/ije/dyp334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Timofeeva MN, McKay JD, Davey Smith G, et al. Genetic polymorphisms in 15q25 and 19q13 loci, cotinine levels, and risk of lung cancer in EPIC. Cancer Epidemiol Biomarkers Prev. 2011;20:2250–61. doi: 10.1158/1055-9965.EPI-11-0496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zuccolo L, Fitz-Simon N, Gray R, et al. A non-synonymous variant in ADH1B is strongly associated with prenatal alcohol use in a European sample of pregnant women. Hum Mol Genet. 2009;18:4457–66. doi: 10.1093/hmg/ddp388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chen L, Davey Smith G, Harbord RM, Lewis SJ. Alcohol intake and blood pressure: a systematic review implementing a Mendelian randomization approach. PLoS Med. 2008;5:e52. doi: 10.1371/journal.pmed.0050052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Benn M, Tybjaerg-Hansen A, Stender S, Frikke-Schmidt R, Nordestgaard BG. Low-density lipoprotein cholesterol and the risk of cancer: a mendelian randomization study. J Natl Cancer Inst. 2011;103:508–19. doi: 10.1093/jnci/djr008. [DOI] [PubMed] [Google Scholar]
- 120.Gerken T, Girard CA, Tung YC, et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science. 2007;318:1469–72. doi: 10.1126/science.1151710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Brenet F, Moh M, Funk P, et al. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS One. 2011;6:e14524. doi: 10.1371/journal.pone.0014524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hansen KD, Timp W, Bravo HC, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43:768–75. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ji H, Ehrlich LI, Seita J, et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature. 2010;467:338–42. doi: 10.1038/nature09367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Irizarry RA, Ladd-Acosta C, Wen B, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–86. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.McGowan PO, Sasaki A, D'Alessio AC, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12:342–48. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cameron NM, Shahrokh D, Del Corpo A, et al. Epigenetic programming of phenotypic variations in reproductive strategies in the rat through maternal care. J Neuroendocrinol. 2008;20:795–801. doi: 10.1111/j.1365-2826.2008.01725.x. [DOI] [PubMed] [Google Scholar]
- 127.McGowan PO, Suderman M, Sasaki A, et al. Broad epigenetic signature of maternal care in the brain of adult rats. PLoS One. 2011;6:e14739. doi: 10.1371/journal.pone.0014739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Colhoun HM, McKeigue PM, Davey Smith G. Problems of reporting genetic associations with complex outcomes. Lancet. 2003;361:865–72. doi: 10.1016/s0140-6736(03)12715-8. [DOI] [PubMed] [Google Scholar]
- 129.Sigurdsson MI, Smith AV, Bjornsson HT, Jonsson JJ. HapMap methylation-associated SNPs, markers of germline DNA methylation, positively correlate with regional levels of human meiotic recombination. Genome Res. 2009;19:581–89. doi: 10.1101/gr.086181.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tycko B. Allele-specific DNA methylation: beyond imprinting. Hum Mol Genet. 2010;19:R210–20. doi: 10.1093/hmg/ddq376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bell CG. Integration of genomic and epigenomic DNA methylation data in common complex diseases by haplotype-specific methylation analysis. Personalized Medicine. 2011;8:243–51. doi: 10.2217/pme.11.14. [DOI] [PubMed] [Google Scholar]
- 132.Finer S, Holland ML, Nanty L, Rakyan VK. The hunt for the epiallele. Environ Mol Mutagen. 2011;52:1–11. doi: 10.1002/em.20590. [DOI] [PubMed] [Google Scholar]
- 133.Freathy RM, Kazeem GR, Morris RW, et al. Genetic variation at CHRNA5-CHRNA3-CHRNB4 interacts with smoking status to influence body mass index. Int J Epidemiol. 2011;40:1617–28. doi: 10.1093/ije/dyr077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tobacco and Genetics Consortium. Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet. 2010;42:441–47. doi: 10.1038/ng.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Terry MB, Delgado-Cruzata L, Vin-Raviv N, Wu HC, Santella RM. DNA methylation in white blood cells: association with risk factors in epidemiologic studies. Epigenetics. 2011;6:828–37. doi: 10.4161/epi.6.7.16500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sovio U, Mook-Kanamori DO, Warrington NM, et al. Association between common variation at the FTO locus and changes in body mass index from infancy to late childhood: the complex nature of genetic association through growth and development. PLoS Genet. 2011;7:e1001307. doi: 10.1371/journal.pgen.1001307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Law MR, Morris JK, Wald NJ. Use of blood pressure lowering drugs in the prevention of cardiovascular disease: meta-analysis of 147 randomised trials in the context of expectations from prospective epidemiological studies. BMJ. 2009;338:b1665. doi: 10.1136/bmj.b1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Timpson NJ, Sayers A, Davey-Smith G, Tobias JH. How does body fat influence bone mass in childhood? A Mendelian randomization approach. J Bone Miner Res. 2009;24:522–33. doi: 10.1359/jbmr.081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Teslovich TM, Musunuru K, Smith AV, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–13. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]