

Abstract
G protein-coupled receptors (GPCRs) are the largest class of cell-surface receptors, and these membrane proteins exist in equilibrium between inactive and active states.1-13 Conformational changes induced by extracellular ligands binding to GPCRs result in a cellular response through the activation of G-proteins. The A2A adenosine receptor (A2AAR) is responsible for regulating blood flow to the cardiac muscle and is important in the regulation of glutamate and dopamine release in the brain.14 In this study, we have successfully raised a mouse monoclonal antibody against human A2AAR that prevents agonist but not antagonist binding to the extracellular ligand-binding pocket. The structure of the A2AAR-antibody Fab fragment (Fab2838) complex reveals that the fragment, unexpectedly, recognises the intracellular surface of A2AAR and that its complementarity determining region, CDR-H3, penetrates into the receptor. CDR-H3 is located in a similar position to the G-protein C-terminal fragment in the active opsin structure1 and to the CDR-3 of the nanobody in the active β2 adrenergic receptor structure2 but locks the A2AAR in an inactive conformation. These results shed light on a novel strategy to modulate GPCR activity.
The GPCR structures in an inactive conformation solved recently3-12 largely advance our understanding of the molecular signalling mechanisms of the receptors. The first details of GPCR activation were provided by the structure of bovine opsin in an active conformation complexed with a G-protein C-terminal peptide (GαCT)1. Most recently, Kobilka and colleagues obtained the crystal structures of β2AR in an active state with a camelid antibody fragment (nanobody, Nb80)2 and with a heterotrimeric Gs-protein13. In these structures, the complementarity-determining region (CDR-3) of Nb80 and C-terminal α-helix of a subunit (Gαs) of Gs-protein were located in the same pocket as for GαCT in the opsin structure. They showed that Nb80 and Gs protein change the conformational equilibrium of β2AR toward the active state in a similar manner, thereby substantially increase their agonist affinities2,13.
A2AAR is responsible for regulating blood flow to the cardiac muscle and is important in the regulation of glutamate and dopamine release in the brain14. Caffeine is a well-known antagonist of this receptor. Strong epidemiological evidence indicates that coffee drinkers have a lower risk of Parkinson’s disease15. The structure of A2AAR has been reported9,16 as a complex with both an antagonist (ZM241385) and an agonist (UK-432097). These structures reveal the molecular framework of the receptor; however, in both cases the intracellular loop 3 (ICL3), critical for G-protein binding, has been replaced by T4-lysozyme (T4L).
Here, we report the crystal structure of A2AAR with complete ICL3 in complex with a mouse monoclonal-antibody Fab-fragment, Fab2838. A2AAR was expressed in Pichia pastoris and the antibody was raised to the purified receptor with antagonist (ZM241385) bound using the conventional mouse-hybridoma system combined with improved immunisation and screening methods (for details, see Methods). Fab2838, a Fab fragment generated from one (IgG2838) of the obtained antibodies completely inhibited binding of the agonist [3H]-NECA but did not affect binding of the antagonist [3H]-ZM241385 (Fig. 1a,d and Supplementary Fig. 2). The results were confirmed by competition binding assays (for details, see Supplementary Discussion and Fig. 1). These findings suggest that Fab2838 induces an inactive conformation, (i.e. to which agonist cannot bind) of the A2AAR ligand-binding pocket without blocking the ligand-binding site.
Figure 1. Effect of Fab2838 on A2AAR -ligand binding.

a, Saturation binding curves for an antagonist [3H]-ZM241385 binding to A2AAR with (open circle) or without (closed circle) Fab2838. b and c, Inhibition of [3H]-ZM241385 binding by the antagonists, theophylline (b) and SCH442416 (c) with (open circles) and without (closed circles) Fab2838. The binding of [3H]-ZM241385 in the absence of competitor was set at 100%. d, Similar to a, but for the agonist [3H]-NECA. e and f, Similar to c and d but for the agonists, adenosine (e) and NECA (f), respectively. All data are the mean ± standard estimated errors (SEM) of three independent experiments performed in duplicate.
We crystallised A2AAR with Fab2838 in the presence of ZM241385 and solved the structure at a resolution of 2.7 Å (Supplementary Table 2). Since the occupancy of ZM241385 in the structure was low for unknown reasons, we repeated the experiments and obtained a higher occupancy structure at 3.1 Å (Supplementary Table 2 and Supplementary Fig. 3 and 4). Except for the occupancy of the ligand, the two structures are almost identical (RMSD of Cα; 0.57 Å) (Supplementary Table 2). ZM241385 occupies the ligand-binding pocket on the extracellular side by making hydrophobic interactions with F1685.29 and I2747.39, and hydrogen-bonds with N2536.55 as observed in the A2AAR-T4L structure (Supplementary Fig. 4). While the overall structure of A2AAR in the A2AAR-Fab2838 complex is similar to that of the T4L construct (PDB; 3EML) (RMSD of Cα; 0.85 Å), there is a major difference around the intracellular portions of helices V and VI, which are connected by ICL3, where T4L is inserted in A2AAR-T4L (Supplementary Fig. 5). In our structure, ICL3 forms two regular helices, effectively continuations of helices V and VI respectively, connected by a short turn (Supplementary Fig. 6a).
The A2AAR–Fab2838 structure has a modified ‘ionic lock’ where E2286.30 (helix VI) and R1023.50 of the D(E)RY motif (helix III) interact via a water molecule (W1; Fig. 2c,d). In the inactive bovine rhodopsin structure, the equivalent residues form a direct salt-bridge3 (Supplementary Fig. 7). R1023.50 of A2AAR-Fab2838 forms salt-bridges/hydrogen-bonds with D1013.49, Y112 in ICL2 and T412.39 as observed in the A2AAR-T4L structure (Supplementary Fig. 5b). Because of the insertion of the water molecule, E2286.30 shifts towards the cytoplasmic space, as compared to the equivalent residue in rhodopsin (E2476.30), resulting in the formation of a salt bridge with R220 in the short helical turn of ICL3. This interaction may be important in the formation of the helical structure in ICL3. The ‘ionic lock’ has not been observed in the crystal structures of other inactive GPCRs6-11, including A2AAR-T4L, except for the D3 dopamine receptor12. This may be because the ICL3s in the other structures were modified to stabilise the protein. While this manuscript was in review, the crystal structures of thermostabilised A2AAR mutants with native ICL3 were published17,18. The antagonist-bound inactive structures have the ‘ionic lock’18. Thus, the ‘ionic lock’ of A2AAR seems to stabilise the inactive conformation of the protein, which is why the receptor has a low basal activity.
Figure 2. Structure of the A2AAR complex with an antibody Fab2838 fragment.

a, Overall structure viewed parallel to the membrane. A2AAR and the Fab light and heavy chains are shown in blue-grey, cyan, and magenta, respectively. The three disulfide bonds in the ECLs are represented by yellow sticks. The bound antagonist ZM241385 in the ligand-binding pocket is shown as a space-filling model. The complementarity-determining regions (CDRs) of Fab2838 are as follows: CDR-H1, yellow; CDR-H2, orange; CDR-H3, red; CDR-L1, green; CDR-L2, purple; CDR-L3, marine. b, Surface representation of the interface between A2AAR (top) and Fab2838 (bottom). Compared to a, A2AAR has been rotated 90° around a horizontal axis, whereas Fab2838 is shown in the same orientation. c, View of the A2AAR (green residues) and CDR-H3 (orange residues) interface. Red dotted lines indicate polar interactions. d, Schematic representation of the A2AAR and CDR-H3 interface.
Fab2838 binds on the intracellular side of the receptor (Fig. 2a). CDR-H3 of Fab2838 is unusually long and penetrates into a pocket formed by helices II, III, VI and VII (Fig. 2b). CDR-H3 interacts with the surrounding helices by forming 6 hydrogen bonds and 8 van der Waals contacts (Fig. 2c,d). The most extensive interactions are with helix II (mainly through hydrogen bonds) and helix VI (mainly through van der Waals contacts). In addition, a hydrogen-bond network including 2 water molecules is observed between CDR-H3 and helices III and VI (Fig. 2c,d). This hydrogen-bond network together with the van der Waals interactions seem to stabilise the modified ‘ionic lock’ interaction between E2286.30 (helix VI) and R1023.50 (helix III) discussed above. Other CDRs further stabilise the A2AAR–Fab2838 complex by forming 14 hydrogen bonds with helices VI and VIII and ICLs 1, 2, and 3 (Fig. 2b). The extensive interactions explain the high affinity of Fab2838 (KD = 4.4 nM) (Supplementary Fig. 8).
The Fab2838 CDR-H3 binding site in A2AAR is similar to those for Nb80 CDR-3 in β2AR2 and for GαCT in opsin1. A critical difference is that Fab2838 stabilises an inactive conformation whereas the others recognise active conformations of the receptors. These structures are compared in Figure 3. In the opsin structure, GαCT, which forms a short α-helix, fits into a large pocket formed by helices II, III, V, VI, and VII interacting with the Arg residue of the D(E)RY motif in helix III (Fig. 3, left panels). CDR-3 of Nb80 in the β2AR structure binds in a similar position to GαCT although CDR-3 forms a β-hairpin1 (Fig. 3, middle panels). Interestingly, CDR-H3 of Fab2838 also forms a β-hairpin but induces a differently shaped binding-pocket (Fig. 3c). In the β2AR structure, CDR-3 of Nb80 is positioned between helices III and VI, whereas in the A2AAR structure CDR-H3 of Fab2838 is ~ 6 Å closer to helices II and VII (Fig. 3b and Supplementary Fig. 9). This allows the close association of helices III and VI and the formation of the modified ‘ionic lock’ between R1023.50 in helix III and E2286.30 in helix VI, consequently stabilising the inactive conformation. In the β2AR-Gs protein complex structure, the C-terminal α-helix (α5) of Gαs also binds in a similar position to CDR-H313 (supplementary Fig. 10). The conformational changes of α5 together with the Gαs N-terminal region induced by the activated receptor was proposed to lead a nucleotide exchange from GDP to GTP in Gαs and to subsequent dissociation of the subunit from the receptor19. Thus, the binding pocket formed by helices II, III, VI, and VII seems to be the key site for the signal transfer between GPCR and G-protein.
Figure 3. Comparison of the structures of opsin-GαCT, β2AR-Nb80 and A2AAR-Fab2838 complexes.

Left, middle and right panels show the structures of an active form of opsin with GαCT (opsin in green and GαCT in yellow), an active form of β2AR with Nb80 CDR-3 (β2AR in brown and Nb80 CDR-3 in blue) and an inactive form of A2AAR with Fab2838 CDR-H3 (A2AAR in blue-grey and Fab2838 CDR-H3 in red). a, Views parallel to the membrane. Bound ligands are shown as stick models in β2AR and A2AAR. The residues involved in the ‘ionic lock’ formation are also shown. Nitrogen and oxygen atoms are in blue and red, respectively. b, Cytoplasmic views of the complexes. c, Surface representations of cytoplasmic surfaces of the receptors. Surfaces within 4 Å of GαCT/CDR-3/CDR-H3 are red.
A possible inactivation mechanism of A2AAR by Fab2838 is summarised as follows. Agonist binding induces large displacements of the intracellular ends of helices III, VI, and VII16,17, which are essential to form the G-protein binding-pocket13,19 (Supplementary Fig. 1). This indicates that the signal from the ligand-binding pocket is transferred through these helices and the conformations of the two pockets are strongly coupled. Our agonist and antagonist binding experiments indicate that this coupling also allows signal transfer in a reverse direction, from the G-protein binding to the ligand-binding pockets (Fig. 1). CDR-H3 of Fab2838 locks the positions of helices III, VI, and VII from the cytoplasmic side, leading to an inactive conformation of the extracellular ligand-binding pocket to which agonists cannot bind probably because of the rearrangement of the side chains at the bottom of the ligand-binding pocket including W2466.48, the toggle switch for activation (for details, see Supplementary Fig. 1 and 11). A similar conceptual model on the β2AR activation was reported by Kobilka, Sunahara, and colleagues20. In the case of β adrenergic receptors, the conformational coupling of the ligand and G-protein binding pockets seems less strict as demonstrated in the structures of β1AR-agonist complexes21 and β2AR-irreversible agonist complex22. This may be because the A2AAR and β1/β2AR agonists interact with different helices in the binding pockets (for details, see Supplementary Discussion).
Antibody fragments (and nanobodies) such as Nb80 and Fab2838 that recognise conformational epitopes of GPCRs have great potential for GPCR studies in vitro and in vivo. Although antibodies recognising the intracellular surface are not suitable for direct therapeutic use, the CDR structures should provide useful information to design peptides or small-molecule compounds against their clearly defined pockets to control the activation states of GPCRs. The antibody-fragments will be also useful tools to study ligand-binding kinetics of GPCRs because they can separate ligand-binding from equilibrium-shifts between different activation states of the receptors. Our approach based on the conventional mouse-hybridoma system allows us to raise antibodies against various receptors in 3-4 months using standard laboratory equipment.
METHODS SUMMARY
Expression and purification
A2AAR N154Q (residues 1-316) was expressed in P. pastoris as described previously23 and purified as described in Methods.
Antibody generation
MRL/lpr mice were immunised with the purified A2AAR with the antagonist ZM241385. Antibodies were raised to recognise conformational epitopes of A2AAR using the conventional mouse-hybridoma system24 combined with new screening methods as described in Methods. The Fab fragments were obtained by papain cleavage and purified by anion-exchange column chromatography.
Crystallisation, data collection, and structure determination
Purified A2AAR was mixed with the Fab fragment and the A2AAR -Fab complex was purified twice by gel filtration chromatography. Crystals were grown by vapour diffusion under the conditions described in the Methods. Diffraction data were collected from a single cryo-cooled crystal on beamline I24 at the Diamond Light Source Ltd., UK. The structures were solved by molecular replacement using the receptor from the A2AAR-T4L structure (PDB code, 3EML) and an antibody Fab-fragment structure (PDB code 1P7K) as search models. Data collection and refinement statistics are summarised in Supplementary Table 2.
Supplementary Material
Acknowledgements
This work was supported by a grant from the ERATO Human Receptor Crystallography Project from the Japan Science and Technology Agency, by the Targeted Proteins Research Program from the Ministry of Education, Culture, Sports, Science and Technology, and by Development of New Functional Antibody Technologies of the New Energy and Industrial Technology Development Organization (NEDO), Japan. It was also partly funded by the Biotechnology and Biological Sciences Research Council (BBSRC) (BB/G023425/1). The work was partly performed in the Membrane Protein Laboratory funded by the Wellcome Trust (grant 062164/Z/00/Z) at the Diamond Light Source Limited. Data were collected at Diamond Light Source ID24 with the assistance of the beam line scientists, Gwyndaf Evans, Robin Owen, Danny Axford and Jun Aishima.
APPENDIX
METHODS
Construction of A2AAR expression vectors for Pichia pastoris
The coding sequence of A2AAR from residues 1 to 316 including the N-terminal 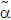 factor, FLAG-tag sequence, and C-terminal His (10)-tag was synthesised by optimisation of codon usage for P. pastoris (TAKARA bio Inc.). In the construct, N154 was also replaced by Gln to eliminate N-linked glycosylation. The DNA fragment was inserted into the multiple cloning site of the pPIC9K vector, and the linearised vector was transformed into the P. pastoris strain SMD1163 (Invitrogen) as described previously23. The transformed cells were stored as glycerol stocks at −80°C.
factor, FLAG-tag sequence, and C-terminal His (10)-tag was synthesised by optimisation of codon usage for P. pastoris (TAKARA bio Inc.). In the construct, N154 was also replaced by Gln to eliminate N-linked glycosylation. The DNA fragment was inserted into the multiple cloning site of the pPIC9K vector, and the linearised vector was transformed into the P. pastoris strain SMD1163 (Invitrogen) as described previously23. The transformed cells were stored as glycerol stocks at −80°C.
Expression and purification of A2AAR
A2AAR was expressed in P. pastoris as described previously23. Cells were suspended in buffer A (50 mM Sodium Phosphate, 100 mM NaCl, 5% glycerol, 2 mM EDTA, protease inhibitor cocktail (Roche); pH 7.4) and disrupted with glass beads (0.5 mm Biospec) by vigorous agitation at 350 rpm for 2 h at 4°C. Following removal of unbroken cells and cell debris at 10,000 × g, membranes were isolated by ultracentrifugation at 100,000 × g for 45 min. Membranes were resuspended in buffer B (20 mM Hepes, 500 mM NaCl, 30 % glycerol, EDTA-free protease inhibitor cocktail (Roche); pH 7.0) and solubilised by 1% n-dodecyl β-D-maltoside (DDM; Anatrace) containing 0.2% cholesterol hemi succinate (CHS; Sigma) in the presence of 4 mM theophylline (antagonist) for 1-2 h at 4°C. After ultracentrifugation, the supernatant was supplemented with solid imidazole to a final concentration of 40 mM and incubated overnight with a TALON immobilised metal ion affinity chromatography resin (Clonetech) at 4°C with gentle rotation (1 mL of TALON resin/150 mg of total protein). The resin was washed with buffer C (20 mM Hepes, 250 mM NaCl, 10% glycerol, protease inhibitor cocktail, 0.05% DDM, 0.01% CHS; pH 7.0) containing 20 mM imidazole, and the bound A2AAR was eluted with buffer C containing 300 mM imidazole. The purified sample was incubated overnight with ConA resin at 4°C to remove contaminating glycosylated proteins and was collected in the flow-through fraction. The final purified sample was dialysed against buffer C and concentrated to approximately 20 mg/mL by ultrafiltration (ULTRA-4 100 K, Millipore).
Construction, expression, and purification of A2AAR-T4L
A2AAR-T4L is a variant of A2AAR in which the ICL3 region is replaced with a bacteriophage T4 lysozyme (T4L): N2 to Y161 of T4L were inserted between L208 and R222 within the ICL3 region replacing residues K209 to A221. A2AAR-T4L was expressed in P. pastoris and purified as described above.
Antibody generation
All animal experiments described in this study conformed to the guidelines outlined in the Guide for the Care and Use of Laboratory Animals of Japan and were approved by the University of Tokyo Animal Care Committee (approval no. RAC07101). To raise antibodies against conformational epitopes of A2AAR, we modified existing protocols for immunisation and screening of mouse monoclonal antibodies. A detailed description of these modified protocols will be published elsewhere. Briefly, MRL/lpr mice were immunised with 0.1 mg purified A2AAR-antagonist ZM241385 complex 3 times with 2 week intervals. The immunised mice were sacrificed and single cell suspensions were prepared from the spleen. These cells were fused with NS-1 myeloma cells using polyethylene glycol (PEG) according to conventional methods24. Primary screening of hybridoma culture supernatants was performed using the liposome ELISA assay. To screen antibodies that specifically recognise native receptors, we developed a novel ELISA method using proteoliposomes. For ‘liposome-ELISA’, we used purified A2AAR reconstituted into liposomes containing biotinyl phosphatidylethanolamine (Avanti) to maintain the protein in its native conformation and effectively immobilise liposomes onto streptavidin-coated plates (Nunc). To eliminate antibodies recognising flexible loops, N (and C)-termini or unstructured regions of A2AAR, we performed ELISA using A2AAR denatured with 1% sodium dodecyl sulphate. Denatured ELISA-negative cells were collected and evaluated using a BIAcore T100 (GE Healthcare) as described below. The selected cells were isolated by limiting dilution to establish monoclonal hybridoma cell lines producing antibodies against A2AAR. For large-scale antibody production, the monoclonal hybridoma cells were inoculated into BALB/c athymic nude mice. IgG was collected from mouse ascites fluid by precipitating twice with 50% ammonium sulphate and purified using Melon Gel (Thermo) according to the manufacturer’s protocol. Fab fragments were obtained by proteolytic cleavage of IgG with papain (Worthington) and purified by anion-exchange column chromatography (DEAE 5-PW, TOSOH). The sequences of Fab fragments were determined according to the standard 5′-RACE method using total RNA isolated from hybridoma cells.
Binding assay by surface plasmon resonance
The BIAcore T100 system and reagents, including sensor chips and amine coupling kit, were obtained from GE Healthcare. Monoclonal anti-mouse Fc antibody (200 μg/mL; Millipore) was immobilised on a CM5 sensor chip using the amine coupling kit according to the manufacturer’s instructions. Antibodies in hybridoma culture supernatants (50 μL) or purified monoclonal antibodies (50 μg/mL) were tightly trapped by the Fc-antibody fixed on the sensor chip. The antibodies bound tightly so as not to be released from the surface when washing with buffer D (20 mM Hepes, 100 mM NaCl, 0.05% DDM, 0.01% CHS; pH 7.0). Purified A2AAR (or A2AAR-T4L) was passed over the surface and the specific binding was monitored for 2 min at 20°C. Subsequently, the sensor surface was washed with buffer D and the dissociation was monitored for 6 min at 20°C. Association and dissociation rate constants (kon and koff) were determined using a curve-fitting protocol as implemented in the BIAevaluation software (Version 1.1, GE Healthcare) based on the Langmuir isotherm model assuming 1:1 binding stoichiometry.
Ligand binding assays
Ligand binding assays were performed using radioligands of the antagonist [3H]-ZM241385 and the agonist [3H]-NECA (GE Healthcare). For single-point binding assays, 5 nM [3H]-ZM241385 or 5 μM [3H]-NECA was incubated in 50 μL of buffer D containing 5 nM or 50 nM purified A2AAR with or without 500 nM antibody for 1 h on ice. For saturation-binding assays, varying concentrations of [3H]-ZM241385 or [3H]-NECA were incubated in 50 μL of buffer D containing 5 nM or 50 nM purified A2AAR with or without 500 nM antibody (Fab2838) for 1 h on ice. Receptor-bound ligands were separated by gel filtration25 and radioactivity was measured using a LS6500 scintillation counter (Beckman). Data were analysed by a nonlinear-regression-fitting program using the GraphPad Prism software. Competition assays with antagonists (SCH442416, theophylline) and agonists (NECA, adenosine) were performed in the presence of 1.0 nM [3H]-ZM241385 for A2AAR or 1.5 nM for A2AAR-Fab (corresponding to the respective KDs).
Purification and crystallisation of the A2AAR-Fab complex
Purified A2AAR and the Fab fragments were mixed at 1:1.2 molar ratio and were incubated on ice for 1 h. The mixture was loaded onto a Superdex 10/300 column (GE Healthcare) equilibrated with buffer C and eluted using the same buffer. The gel filtration step was repeated twice to ensure successful crystallisation of the A2AAR-Fab complex. Fractions containing the complex were concentrated to approximately 20 mg/mL by ultrafiltration (ULTRA-4 100 K, Millipore). Initial crystals were obtained using MemGold (Molecular Dimensions Ltd.). After optimisation, well-diffracting crystals were obtained in hanging drops by vapour diffusion at 20°C with the protein solution containing 0.3-0.6% octyl-thioglucoside and the reservoir solution (1 μL) containing 30% PEG400, 0.1 M MES (pH 6.5) and 0.2 M MgCl2. Crystals appeared after 1 day and grew to maximum dimensions in 1 week before being flash-frozen and stored in liquid nitrogen.
Data collection and structure determination
Diffraction data were collected from single cryo-cooled crystals (100K) on beamline I24 at Diamond Light Source Ltd., UK using a 10-μm focused beam (wavelength 0.9795 Å) and a pilatus 6 M detector (Dectris Ltd., Switzerland). Data were processed using MOSFLM and SCALA from the CCP4 program suite26. The structure was initially solved using the 2.7 Å data. Molecular replacement was carried out with PHASER27 using the receptor from the A2AAR-T4 lysozyme fusion structure (PDB code 3EML) and an antibody fragment (PDB code 1P7K) as search models. Iterative cycles of model building and structure refinement were performed using Coot28, Refmac529 and phenix.refine in the PHENIX program package30. The final model from this refinement was used as the initial model for refinement against the 3.1 Å data. The refinement was carried out similarly to above. Model validation was performed using Procheck31 and MolProbity32. The resulting crystallographic and refinement statistics are summarised in Supplementary Table 2. Disordered region of A2AAR was predicted by the RONN program33. Figures were prepared using PyMOL (The PyMOL Molecular Graphics System, Version 1.3, Schrodinger, LLC.).
- 25.Warne T, Chirnside J, Schertler GFX. Expression and purification of truncated, non-glycosylated turkey beta-adrenergic receptors for crystallization. Biochim. Biophys. Acta. 2003;1610:133–140. doi: 10.1016/s0005-2736(02)00716-2. [DOI] [PubMed] [Google Scholar]
- 26.Collaborative Computational Project N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 27.McCoy AJ, et al. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 29.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 30.Afonine PV, Grosse-Kunstleve RW, Adams PD. A robust bulk-solvent correction and anisotropic scaling procedure. Acta Crystallogr. D. 2005;61:850–855. doi: 10.1107/S0907444905007894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laskowski RA, MacArthur MW, Thornton JM. Validation of protein models derived from experiment. Curr. Opin. Struct. Biol. 1998;8:631–639. doi: 10.1016/s0959-440x(98)80156-5. [DOI] [PubMed] [Google Scholar]
- 32.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang ZR, Thomson R, McNeil P, Esnouf RM. RONN: the bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics. 2005;21:3369–3376. doi: 10.1093/bioinformatics/bti534. [DOI] [PubMed] [Google Scholar]
Footnotes
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
References
- 1.Scheerer P, et al. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 2.Rasmussen SGF, et al. Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palczewski K, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 4.Shimamura T, et al. Crystal structure of squid rhodopsin with intracellularly extended cytoplasmic region. J. Biol. Chem. 2008;283:17753–17756. doi: 10.1074/jbc.C800040200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murakami M, Kouyama T. Crystal structure of squid rhodopsin. Nature. 2008;453:363–367. doi: 10.1038/nature06925. [DOI] [PubMed] [Google Scholar]
- 6.Warne T, et al. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rasmussen SGF, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 8.Cherezov V, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaakola V-P, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu B, et al. Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shimamura T, et al. Structure of the human histamine H1 receptor complex with doxepin. Nature. 2011;475:65–70. doi: 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chien EYT, et al. Structure of the Human Dopamine D3 Receptor in Complex with a D2/D3 Selective Antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rasmussen SGF, et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fredholm BB, Chen JF, Masino SA, Vaugeois JM. Actions of adenosine at its receptors in the CNS: insights from knockouts and drugs. Annu. Rev. Pharmacol. Toxicol. 2005;45:385–412. doi: 10.1146/annurev.pharmtox.45.120403.095731. [DOI] [PubMed] [Google Scholar]
- 15.Müller CE, Jacobson KA. Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim. Biophys. Acta. 2011;1808:1290–1308. doi: 10.1016/j.bbamem.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu F, et al. Structure of an Agonist-Bound Human A2A Adenosine Receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lebon G, et al. Agonist-bound adenosine A(2A) receptor structures reveal common features of GPCR activation. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doré AS, et al. Structure of the adenosine A(2A) receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure. 2011;19:1283–1293. doi: 10.1016/j.str.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chung KY, et al. Conformational changes in the G protein Gs induced by the β2 adrenergic receptor. Nature. 2011;477:611–615. doi: 10.1038/nature10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yao XJ, et al. The effect of ligand efficacy on the formation and stability of a GPCR-G protein complex. Proc. Natl Acad. Sci. USA. 2009;106:9501–9506. doi: 10.1073/pnas.0811437106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Warne T, et al. The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature. 2011;469:241–244. doi: 10.1038/nature09746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosenbaum DM, et al. Structure and function of an irreversible agonist-beta(2) adrenoceptor complex. Nature. 2011;469:236–240. doi: 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yurugi-Kobayashi T, et al. Comparison of functional non-glycosylated GPCRs expression in Pichia pastoris. Biochem. Biophys. Res. Commun. 2009;380:271–276. doi: 10.1016/j.bbrc.2009.01.053. [DOI] [PubMed] [Google Scholar]
- 24.Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.