

Abstract
G-protein-coupled receptors (GPCRs) represent an important group of targets for pharmaceutical therapeutics. The completion of the human genome revealed a large number of putative GPCRs. However, the identification of their natural ligands, and especially peptides, suffers from low discovery rates, thus impeding development of therapeutics based on these potential drug targets. We describe the discovery of novel GPCR ligands encrypted in the human proteome. Hundreds of potential peptide ligands were predicted by machine learning algorithms. In vitro screening of selected 33 peptides on a set of 152 GPCRs, including a group of designated orphan receptors, was conducted by intracellular calcium measurements and cAMP assays. The screening revealed eight novel peptides as potential agonists that specifically activated six different receptors in a dose-dependent manner. Most of the peptides showed distinct stimulatory patterns targeted at designated and orphan GPCRs. Further analysis demonstrated a significant in vivo effect for one of the peptides in a mouse inflammation model.
G-protein-coupled receptors (GPCRs)2 represent a large group of receptors that are directly involved in cellular signaling networks and are considered to be an important family of targets for pharmacological intervention (reviewed in Ref. 1). It is estimated that approximately one-third of therapeutic development in the pharmaceutical industry is targeted toward drug discovery in the area of GPCRs (2). Recent analysis of the human genome identified over 800 putative members of the GPCR superfamily, over half of which are thought to be involved in sensory mechanisms and are not considered to be drug targets. Among the known GPCRs, ∼300 are considered potential pharmaceutical targets (3). Out of this group around 150 GPCRs have no designated natural/endogenous ligand or activator. These receptors are termed “orphan GPCRs” and have recently become very attractive to many industrial and academic researchers as they hold great potential as targets for novel drug discovery (4).
Lately, a major effort was made by many academic institutes as well as biotech and pharmaceutical companies to identify both natural and surrogate ligands for both known and orphan GPCRs (5, 6). The most common method to screen for novel GPCR activators is through large scale screening of hundreds and thousands of compounds on many GPCRs, testing for in vitro activation of any given receptor by assessing its signaling response of an expressing cell to binding of a specific compound or tissue extract. This method, called “reverse pharmacology” (7, 8), gave rise in the past decade to over 30 new receptor/ligand pairs and many previously known or novel ligands, most of them peptides (5). However, this method is very expensive and time-consuming and might lead to many potential ligands either being missed or falsely annotated (6, 9). To increase the feasibility of this type of testing, it is necessary to pre-screen any potential group of ligands and focus on high chance, low risk candidates. This might allow more thorough screening and validation of true novel activators with better chances of becoming therapies.
Computational screening can supplement experimental efforts as a preliminary screening tool for efficient identification of natural ligands, especially peptides (10). Bioinformatics can serve as a powerful tool that provides reliable predictive measures to select for the high potential candidates and provide a spotlight pointed at potential new candidates for experimental discovery, thereby enabling higher success rates in identification of novel GPCR ligands (11).
In this study, we describe the experimental validation of computationally discovered novel GPCR-activating peptides. A subgroup of 33 potential candidate peptides was experimentally screened. Here we report the in vitro validation of eight novel GPCR peptide activators (hit rate of ∼25%) as potential candidates for related clinical utilities. One of the examined peptides was tested in vivo and was found effective in a mouse inflammation model.
EXPERIMENTAL PROCEDURES
Data Set Preparation for the Proteolytic Site Predictor—All mammalian proteins (28,780) were downloaded from the Swiss Protein Database (release 43) (12). Of these, 11,705 proteins were classified as secreted or membrane proteins according to Swiss-Prot annotation. From these 11,705 proteins, 553 experimentally validated 18-mer peptide convertase proteolytic sites were extracted based on Swiss-Prot FT annotation lines. This subset of sequences was used as a learning set for Breiman's random forest classifier algorithm (13), whereas the negative set (non proteolytic sites) consisted of all extracellular 18-mers, excluding the boundaries of pro-peptides, chains, and peptides, according to Swiss-Prot annotations.
Creation of a Predicted Human Peptidome Dataset—A predicted human secretome was generated by analyzing human proteins from the following two sources: (i) the Uniprot/Swiss Protein Database (version 47) and (ii) Refseq sequences from the NCBI nr protein data base (GenBank™ version 149). There were 29,530 proteins from NCBI nr, including proteins with various validation annotations, reviewed, predicted, and modeled. The annotation for Uniprot proteins was used to filter out proteins that do not have a signal peptide, validated or predicted. For NCBI nr proteins, the SignalP 3.0 software (14) was used to predict the existence and location of a signal peptide.
For each protein precursor sequence, the following procedure was used to create the potential peptide products of the precursor. (The full cleavage prediction concept is detailed in Kliger et al. (15).) The predicted signal peptide was removed, and potential cleavage sites were scored based on the outcome of the machine learning algorithm used for cleavage site prediction. Based on precision/recall analysis, each putative cleavage site was assigned a score value between 0 and 10, where 0 represents a known cleavage site (as annotated on Uniprot) and 10 corresponds to unlikely cleavage sites. Sites with a score of 4 or less, and sites that contained two consecutive basic amino acids (e.g. Arg or Lys), regardless of their score, were selected. Peptides whose end points were either a selected cleavage site or an end point of the precursor (signal peptide cleavage site at the N terminus or the C terminus of the precursor) were created. Peptides longer than 100 amino acids were discarded. It should be noted that different peptides from the same precursor may have an overlapping sequence, and one can be a sub-sequence of another. The peptide set was extended by sequentially removing C-terminal basic amino acids and removing an additional C-terminal glycine, leaving an amide attached to the C terminus of the peptide (this mimics a known natural peptide maturation process called “glycine-directed peptide amidation”). All the intermediate peptides resulting from this extension were considered as separate peptide products. The complete list of predicted peptides (the “peptidome”) contained hundreds of thousands of peptides, which were then analyzed as a source data by the GPCR-activating ligand predictor.
GPCR Ligand Predictor—The set of known GPCR peptide ligands was extracted from Uniprot, using the Uniprot annotation (SRS from Swiss-Prot and by relevant keywords from GPCRDB (16)) and was manually curated based on literature search. The sequence lengths and positions of the peptides were extracted from the Swiss-Prot feature table. The learning data set contained 64 precursors and 123 peptides. Of these, 87 did not overlap with another peptide of the same precursor. Several known GPCR ligands were removed from the learning set (for example, the chemokine family and the hormone proteins that are larger than 100 amino acids) giving an average peptide size of 26 amino acids. The learning process also required a negative set of peptides that are not known as GPCR peptide ligands. Two negative sets of peptides were constructed. The first set was extracted based on Uniprot using annotations to filter peptides that are not GPCR peptide ligands. The second set was a collection of 3000 randomly chosen peptides from the novel peptide data base (peptidome) described above.
Using the generated learning set, a classifier based on the “Random Forest” algorithm (13) was implemented to select peptides that are likely to be GPCR ligands. Following a thorough optimization process, the classifier was programmed to analyze the following index parameters. The length of the peptide (in amino acids). The frequency of each amino acid in the peptide relative to the peptide length (21 parameters, one for each amino acid and an extra parameter for those that are not conventional amino acids). The amino acids in proximity to the N-terminal cleavage site from both sides as follows: the first four amino acids in the N terminus of the peptide and the last four amino acids of the precursor preceding the cleavage site (eight parameters, and each can be one of 21 values, one for each amino acid and an extra value for other letters or for amino acids beyond the signal peptide cleavage site). This parameter (four amino acids) was found most effective in the optimization process. The amino acids in proximity to the C-terminal cleavage site from both sides are as follows: the last three amino acids in the C terminus of the peptide and the first three amino acids of the precursor following the cleavage site (six parameters, each can be one of 21 values, one for each amino acid and an extra value for other letters or for amino acids beyond the precursor end). This parameter (3 amino acids) was found most effective in the optimization process. The last entry is the number of coding exons in the gene coding for of the precursor.
Based on these numeric parameters the classifying program was trained to distinguish between the learning set of known ligands (true set) and the negative (false) set. This program was then used to give each predicted peptide a score signifying the likelihood of being a GPCR-activating ligand. At the end of the process, a dataset of 100 peptides with the highest classifier scores was created. These peptides underwent a manual expert analysis process and literature review by a few different biologists. The selection of GPCR-activating peptides was based on the following criteria: expression profile and tissue specificity of the precursor, with relation to the receptor; comparison of the cleavage and GPCR classifier scores of the candidate peptides and their mouse orthologs; conservation of the sequence and cleavage sites of the peptide in the precursor proteins of all orthologs; position of the peptide within the precursor with respect to known domains and features (as a negative rule); and the number of cysteine residues and disulfide bridge annotations.
The peptides were also checked for their novelty status based upon the comparison with sequences appearing in patent data bases with very strict exclusion parameters. This process resulted in a subset of 35 confirmed novel peptides most likely to be GPCR ligands.
Peptide Synthesis—For each peptide, from the subset of 35 peptides, a number of modifications were made to enhance stability and functionality. In peptides containing a single cysteine residue, the cysteine was replaced by either a serine or a valine depending on the hydrophobicity and amino acid content of the peptide. For peptides containing a glycine residue at the C terminus, the glycine was replaced by an amide. Peptides were synthesized (10 mg) by Pepscan Inc. (The Netherlands) and purified by high pressure liquid chromatography to >90% (confirmed by mass spectrometry). Apart from one peptide that failed synthesis, and one insoluble peptide, which was discarded, all remaining 33 peptides were soluble in PBS or in Lab-grade purified water.
G-protein-coupled Receptor Selection—The list of receptors was generated by ranking all human (GPCRDB (16)) receptors according to their clinical relevance. All known odorant and taste receptors were omitted. GPCRs known to be activated by non-peptide ligands and small molecules (such as dopamine, serotonin, purines, etc.) were removed from the list together with receptors for proteins (size over 50 amino acids) such as chemokines. The final list of receptors contained mainly peptide-activated GPCRs together with receptors annotated as orphan, with a predicted high probability of being activated by a peptide (based upon sequence similarity and evolutionary relation to GPCRs activated by peptides and literature searches). Each receptor was also assessed for its most potent known ligand as a positive control for the screening experiments.
GPCR Screening for Ca2+ Activation by the Predicted Peptides—The experimental screening was done by Applied Cell Sciences, Inc., on all the selected GPCRs by utilizing the promiscuous Gα16 to divert signaling to the Gq pathway, thus enabling readout of GPCR activation by testing for Ca2+ flux as described (17). Peptides were diluted in PBS containing 0.1% bovine serum albumin. All plates were stored at –80 °C until used. cDNA clones of the GPCRs were commercially obtained in one of the following expression vectors: pcDNA3.1, pCMV6, or MO2.
Transient transfections were performed using CHO-K1 cells as host cells. Cells (12 millions) were plated into T75 flasks on the day preceding transfection. Cells were transfected with a GPCR DNA and Gα16 using a lipid technique according to the manufacturer's recommendation. Cells were transfected for 5 h, then re-plated into 96-well dishes (60,000 cells per well), and grown overnight.
On the day of the experiment, cells were loaded with Fluo4-NW (Invitrogen) according to the manufacturer's recommendation. Plates were loaded into a FlexStation™ (Molecular Devices) plate reader, and fluorescence was monitored. Seventeen seconds following initiation of reading, cells were stimulated with the indicated agonist/compound at final concentration of 1 μm. Each 96-well plate contained each of the examined peptides in triplicate. We defined a hit as a peptide that elicited a clear and distinct increase in intracellular calcium that is clearly visible and statistically significant (using a t test comparing the levels of calcium before and after peptide addition with p value lower than 0.001) upon examination of the calcium trace for at least two repeats.
GPCR Screening for Cyclic AMP Accumulation with Candidate Peptides—Applied Cell Sciences transiently transfected 13 different GPCRs included within the list of screened receptors into CHO-K1 cells as described previously. Transfected cells were plated into 24 wells of a 96-well plate. Cells were pretreated with 0.5 mm isobutylmethylxanthine (stimulation buffer) for 10 min at 37 °C, then stimulated with either a positive control or a candidate peptide (for Gs functional examination), followed by stimulation or a preincubation with 10 μm forskolin (for Gi functional examination). Following a 20-min stimulation with 1 μm of either positive control or the tested peptides, either with or without forskolin, intracellular cAMP was assayed using the Hit Hunter cAMP kit (DiscoveRx Corp.), according to the manufacturer's recommended protocol. Data were converted to nanomoles of cAMP by running a standard cAMP curve.
Dose Response of GPCR Activation and Affinity (EC50)
Measurements—Cells were transfected as described above. Each
peptide was added, in triplicate, according to functional assay selected
(either Ca2+ or cAMP) in final concentrations of 1, 3, 10, 30, 100,
and 300 nm and 1 and 3 μm. All peptides were examined
and compared with similar concentrations of a selected positive control (as
described in Table 1).
EC50 best fit values (representing affinity) were calculated by
nonlinear regression of sigmoidal dose-response curves, using Prism version 4
(GraphPad Software Inc., San Diego). The formula for the sigmoidal
dose-response curves was defined as
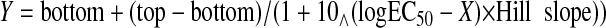 .
.
TABLE 1.
Novel peptides that were found to activate GPCRs
Description of the precursor proteins and peptides that were found to functionally activate GPCRs in the screening process (at 1 μm) and in dose-response activation assays.
| Peptide name | Protein name of precursor | Swiss-Prot ID | Sequence |
|---|---|---|---|
| P33 | HWKM1940 | Q6UWF9_HUMAN | SMCHRWSRAVLFPAAHRPa |
| P58 | B9 | Q9UPM9_HUMAN | TIPMFVPESTSKLQKFTSWFMGb |
| P58-5 | LQKFTSWFMGb | ||
| P58-4 | FTSWFMGb | ||
| P59 | Complement C1q tumor necrosis factor-related protein 8 (Precursor) | C1QT8_HUMAN | GQKGQVGPPGAACRRAYAAFSVGa,b |
| P74 | Complement C1q tumor necrosis factor-related protein 8 (Precursor) | C1QT8_HUMAN | GQKGQVGPPGAACRRAYAAFSVGRRAYAAFSVGa, b |
| P60 | Uncharacterized protein C5orf29 | CE029_HUMAN | GIGCVWHWKHRVATRFTLPRFLQc |
| P61 | TIMM9 | Q6UXN7_HUMAN | FLGYCIYLNRKRRGDPAFKRRLRDc |
| P63 | Bone morphogenetic protein 3b (Precursor) | BMP3B_HUMAN | AHAQHFHKHQLWPSPFRALKPRPGb |
| P94 | Bone morphogenetic protein 3b (Precursor) | BMP3B_HUMAN | AAQATGPLQDNELPGLDERPPRAHAQHFHKHQLWPSPFRALKPRPGb |
Underlined Cys was replaced by Val for the validation experiment.
C-terminal glycine was replaced by an amide.
Underlined Cys was replaced by Ser for the validation experiment.
In Vivo Activity of P58 and Its Derivates—Male out-bred Swiss albino mice were purchased from Harlan, UK (T.O. strain). All animals were housed for 7 days prior to experimentation to allow body weight to reach ∼25 g on the day of the experiment. Air pouches were formed by injecting 2.5 ml of sterile air subcutaneously to the back of the mice 3 and 6 days before the experiment. Before use, peptides were dissolved with pyrogen-free PBS (Invitrogen, catalog number 14190-094). Peptides or vehicle were administered intravenously at a final volume of 200 μl, at the indicated doses immediately before intra-pouch injection of 1 mg of zymosan A (Sigma). Four hours post-treatment, air pouches were washed with 2 ml of ice-cold PBS containing 3 mm EDTA and 25 units/ml of heparin and kept on ice. Polymorphonuclear neutrophil content was determined by staining an aliquot of the lavage with phycoerythrin-conjugated anti-GR-1 monoclonal antibody (eBiosciences). An irrelevant phycoerythrin-conjugated rat IgG2b antibody served as isotype control. Samples were analyzed by FACScan (BD Biosciences). For determination of total leukocytes, lavages diluted 1:10 in Turk's solution (0.01% crystal violet in 3% acetic acid) were analyzed using Neubauer hemocytometer and light microscope (Olympus B061). Statistical differences were determined by analysis of variance, plus Student Newman Keuls test. A p value <0.05 was taken as significant.
RESULTS
Generation of in Silico Predicted Human GPCR-activating Peptides—A list of the 100 highest scoring peptides as potential GPCR ligands were manually examined and ranked according to additional peptide characteristics (see “Experimental Procedures”). Finally, 33 peptides were synthesized and screened against 152 GPCRs.
Screening Results, Ca2+ Flux Assay—Screening was carried out on all types of GPCRs by utilizing the promiscuous Gα16 to divert signaling to the Gq pathway, thus enabling readouts of Gs,Gi, and Gq for the GPCR activation by testing for Ca2+ flux (17). The efficiency of this method in diverting either cAMP signaling into Ca2+ flux was estimated at ∼70% for Gi/Gs-activating receptors.
Of the 152 receptors screened 54 were orphan receptors (according to the literature), and 98 GPCRs had known activating ligands (non-orphan). Of the 98 non-orphan receptors that were screened, 68 (∼70%) showed a response to a known agonist stimulation.
Out of the 152 receptors screened by the Ca2+ flux activation assay, four receptors (MRGX1, MRGX2, MAS1, and FPRL1) showed distinct activation with at least one of the novel peptides added at a concentration of 1 μm. Six peptides (peptides P33, P58, P60, P61, P63, and P94; see Table 1) that showed either a moderate or a strong hit for either receptor were further examined by dose response on the specific GPCRs they activated. Curve analysis and affinity (EC50) calculation of the dose-response experiments were compared with the most potent activator for the receptor as described in the literature, and the results are illustrated in Table 2 and Fig. 1. In all cases, we were able to demonstrate a dose-dependent activation of Ca2+ flux with good correlation to the positive control used (Table 2).
TABLE 2.
GPCRs activation and specificity by screened peptides
Positive hits and calculated EC50 values for Compugen's peptides and receptor-specific positive controls. Each hit is indicated by a “+” mark as well as a calculated EC50 value (μm). The observed affinity (EC50) value is also indicated for the positive control peptides (with the exception of Ang 1–7, where no activation of the MAS1 receptor was observed, NA). ND means no data.
| Receptor | Positive controls (EC50 μm) | P60 | P61 | P94 | P63 | P58 | P33 | P59 | P74 | Other GPCRsaactivated by same positive control |
|---|---|---|---|---|---|---|---|---|---|---|
| MRGX1 | BAM22 (0.08) | + (0.3) | OPRM, OPRK, OPRL, and OPRD | |||||||
| MRGX2 | Cort14 (0.61) | + (1.0) | + (0.9) | + (0.6) | + (0.5) | SSTR (1-5) | ||||
| MAS1 | (Ang1-7) (NA) | + (0.57) | + (1.0) | AGTR1, AGTR 2 | ||||||
| FPRL1 | fMLP (0.85) | + (1.3) | + (0.55) | + (2.0) | FPR1 | |||||
| LGR7 (RXFP1)b | Relaxin (ND) | + (ND) | + (ND) | GPR135 (RXFP3) | ||||||
| LGR8 (RXFP2)b | Relaxin (ND) | + (ND) | + (ND) | GPR135 (RXFP3) |
Receptors were included in the screening. No activation by Compugen peptides was observed. Activation by positive control is indicated from the literature.
Data were examined by cAMP inhibition assay. No dose response was performed (ND).
FIGURE 1.

Dose-response activation measured for Ca2+ flux-activating peptides. Peptides that showed significant results in the screening assay were tested for dose-dependent activation on the activated receptors (MRGX1, MRGX2, FPRL1, and MAS1, counterclockwise). Dose-response activation for positive control peptides are indicated with a thick line and solid circles. Dose-response curves for the tested peptides are indicated by thin lines and open squares. The best fit curves and EC50 were calculated as described under “Experimental Procedures” and are presented in Table 1. For the MAS1 receptor, no activation was observed by the suggested positive control (Ang 1–7).
We observed activation of MRGX1 with our peptide P60 (Tables 1 and 2). According to the dose-response experiments we conducted (Fig. 1), P60 activates MRGX1 in a dose-dependent manner, with affinity estimated within the nanomolar range, slightly lower than that of the positive control used (BAM22) (18). In addition, further experimental results indicate that P60 and BAM22 differ in their activation of opioid-related receptors (19). Although BAM22 activates the latter, P60 is specific to MRGXs (activating MRGX1 and weakly activating MRGX2), and it did not activate any of the known opioid receptors (D1, M1, L1, and K1) that were included in the screen.
For MRGX2, we identified several potential peptides (Table 2). According to dose-response experiments, the affinity of two of the novel peptides, P94 and P63 (Table 1), was similar to that of the positive control, cortistatin-14 (18) (Fig. 1). However, unlike cortistatin-14, none of the peptides had any effect upon the receptors of the somatostatin family (SSTR 1–5) that are known to be activated by cortistatin-14.
The FPRL1 (ALX) receptor is involved in regulation of inflammation by controlling leukocytes activities such as chemotaxis and phagocytosis (20, 21). We identified several peptides that activated this receptor, P58, P94, and P33 (Tables 1 and 2). The leading hit, peptide P58, exhibited in dose-response experiments higher potency and higher affinity than formyl-methionyl-leucyl-phenylalanine, which was used as a positive control, although it is not the most potent agonist for this receptor (22) (Fig. 1). Importantly, P58, as well as none of the other peptides examined, did not activate another family member, FPR1. However, a recent experiment3 indicates a dose-dependent activation of FPRL2, a closer family member (data not shown).
We also identified two peptides, P61 and P33 (Table 1), that dose-dependently activated the MAS1 receptor (Table 2 and Fig. 1). In agreement with previous publications, we were unable to demonstrate calcium flux induction by the positive control (Ang 1–7), a known ligand for MAS1 (23). However, our peptides did activate the receptor at concentrations similar to that found for dynorphin A (an established surrogate, lower affinity activator of MAS1). Our peptides were specific to MAS1 and did not activate AT1 or AT2, angiotensin II receptors which are known to be weakly activated by Ang 1–7 (Table 2).
Cyclic AMP Screening Assay Results—Because the promiscuous Gα16 does not always divert a cAMP-related signal to Ca2+ accumulation, a subset of 13 GPCRs with very low activation by either the positive controls or any of the novel examined peptides was selected to be tested for Gi/Gs activation by 2 of the 33 examined peptides. These receptors were screened for cAMP accumulation (Gs) or inhibition (Gi) as described below.
Two of the peptides (P59 and P74, see Table 1) showed distinct Gi-dependent activation of two related GPCRs (LGR7 (RXFP1) and LGR8 (RXFP2), see Table 2 and Fig. 2). Both of these receptors were recently identified as being activated by Relaxin and INSL3, respectively (24). A much weaker hit on LGR4, an orphan family member (data not shown), was also observed. Specificity was examined by GPR135 (RXFP3), a receptor for Relaxin 3, which was not activated by peptide P59 (Table 2).
FIGURE 2.

Inhibition of cAMP in forskolin-stimulated cells by activation of LGR7 and LGR8. cAMP was stimulated by incubation for 10 min in the presence of 10 μm forskolin, as described above. The peptide was added at a final concentration of 1μm, and the sample was incubated for 20 min following the readout of the end point cAMP concentration (measured in arbitrary fluorescence units (AFU)). NT represents addition of the buffer only as a negative control. Forskolin with no added peptide serves as a positive control.
In Vivo Activation of the FPRL1 Receptor by P58—FPRL1 activation promotes resolution of inflammation, an active and tightly synchronized process, involving counter-regulation of leukocytes, which leads to a prominent anti-inflammatory effect (21). To study the in vivo activity of our novel FPRL1 peptide agonist, P58, and two of its derivatives (P58-4 and P58-5), we used a model of acute inflammation, involving zymosan-induced leukocyte recruitment into the murine dorsal air pouch (see “Experimental Procedures”). The novel peptide P58 as well as P58-4 and P58-5 were tested at doses of 20 and 80 nmol per mouse and were given intravenously at the time of zymosan A injection (time 0). These doses correspond to 50 and 200 μg of P58 per mouse, respectively; and equivalent doses were used for the shorter peptides. Cell recruitment into the air pouches was determined at the 4-h time point, using both light microscopy (for total leukocyte count) and fluorescence-activated cell sorter analysis for Gr1-stained cells (for assessment of PMNs). A significant inhibition of about 50% was observed in the infiltration of PMNs into the air pouch with P58 at 20 nmol/mouse (Fig. 3). Out of the two derivatives tested, P58-4 displayed a 47% inhibition of Gr1+ cell migration into air pouches at the highest dose tested (80 nmol/mouse). P58-5 produced a discrete effect at the highest dose that was not statistically significant. Similar effects were observed on total leukocyte counts (data not shown).
FIGURE 3.

Effects of P58 and its derivatives on zymosan-induced PMN infiltration into murine air pouch. Mice were treated intravenously with the peptides (20 or 80 nmol per mouse) or vehicle, immediately followed by an intra-pouch challenge with zymosan A at time 0. Four hours after the zymosan A injection, pouch cavities were washed, and the leukocytes recovered in the lavage fluids were stained with an anti-GR-1 antibody and analyzed by fluorescence-activated cell sorter. Data are shown as GR-1+ cells (×105) per mouse (mean ± S.E.). *, p < 0.05 versus vehicle group.
DISCUSSION
In this work we describe the results of a screen for novel peptide GPCR agonists. A subset of the predicted candidate peptides was screened on a large group of GPCRs, and eight peptide agonists were identified for six different GPCRs. The peptides activity was further validated experimentally using established biochemical assays such as dose-dependent induction of calcium flux and by assessing the efficacy in vivo in one case.
By utilizing bioinformatic capabilities and machine learning, tools both related to identifying all potential secreted proteins (the secretome), and predicting all potential Arg/Lys cleavage sites, we were able to create a comprehensive peptidome that includes hundreds of thousands of potential peptides, ranked and prioritized by computational scores. Sequences of the predicted peptidome were further analyzed for features and assigned a probability score of being GPCR ligands by a machine learning algorithm. This collection contained hundreds of candidate peptides that were scored and ranked by distinct parameters and expert examination. The highest scoring candidates were checked for novelty and synthesized for further screening for activation on a group of target GPCRs. The screening included 33 peptides that were screened for activation of 152 GPCRs selected according to relevance and availability.
Out of the 33 peptides screened, 8 (∼25%) showed a distinct activation of at least one receptor. The ratio of successful candidates is remarkable when compared with the average ratio of novel GPCR peptide ligand discovery rate, which is on average around two to three new candidates per year in the past decade (25). All of these peptides are novel, and most of them are derived from hypothetical protein precursors. Most of the peptides that showed initial hits also displayed dose-response activation. Furthermore, one of the peptides, designated peptide P58 (now named CGEN855A), which showed a dose-dependent activation of the FPRL1 receptor, was further examined for in vivo activation of the FPRL1 receptor using a murine model of acute inflammation. The activation of FPRL1, as determined by inhibition of PMNs infiltration, was found to be significant. Moreover, out of two subsequent peptides derived from P58, P58-4 (now CGEN-855B) and P58-5 (now CGEN-855C), at least one (P58-4) was found to invoke an in vivo response mediated by the FPRL1 receptor, similar to the original P58 peptide. An examination of the activation of the receptors by the novel peptides shows cross-hits of the same peptides on several different GPCRs. This does not seem surprising because some of the receptors, such as the two relaxin-related GPCRs, namely RXFP1 (LGR7) and RXFP2 (LGR8), belong to the same family, whereas the others (MAS1, MRGX1, MRGX2, and FPRL1) belong to the same evolutionary branch (according to Ref. 26).
Even though no sequence homology was found between the newly discovered peptides and the known GPCR ligands, we predict that other structural properties (such as secondary structure) might show a mechanistic resemblance between the novel and known peptides. A preliminary computational structural prediction of the eight novel peptides by using the PSIPRED (27) secondary structure predictor revealed that all of them contain at least one helical stretch, a feature common in many known peptide GPCR ligands (data not shown).
We conclude that we were able to show efficacy and a proof of concept for our discovery platform of GPCR peptide agonists. We believe that we will be able to broaden our analysis and discover additional novel peptide GPCR ligands by this method. We further predict that more of the newly discovered and validated activators found for these GPCRs will be further investigated for their potential as therapeutic compounds targeted at receptors involved in distinct diseases and conditions.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: GPCR, G-protein-coupled receptor; PBS, phosphate-buffered saline; PMN, polymorphonuclear leukocyte; Ang, angiotensin.
I. Hecht, J. Rong, A. L. F. Samapio, C. Hermesh, C. Rutledge, R. Shemesh, A. Toporik, M. Beiman, L. Dassa, H. Niv, G. Cojocaru, A. Zuberman, G. Rotman, M. Perretti, J. Vinten-Johansen, and Y. Cohen, manuscript in preparation.
References
- 1.Imming, P., Sinning, C., and Meyer, A. (2006) Nat. Rev. Drug Discov. 5 821–834 [DOI] [PubMed] [Google Scholar]
- 2.Hill, S. J. (2006) Br. J. Pharmacol. 147 Suppl. 1, 27–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin, S. H., and Civelli, O. (2004) Ann. Med. 36 204–214 [DOI] [PubMed] [Google Scholar]
- 4.Leifert, W. R., Aloia, A. L., Bucco, O., Glatz, R. V., and McMurchie, E. J. (2005) J. Biomol. Screen. 10 765–779 [DOI] [PubMed] [Google Scholar]
- 5.Kutzleb, C., Busmann, A., Wendland, M., and Maronde, E. (2005) Curr. Protein Pept. Sci. 6 265–278 [DOI] [PubMed] [Google Scholar]
- 6.Jiang, Z., and Zhou, Y. (2006) Curr. Protein Pept. Sci. 7 459–464 [DOI] [PubMed] [Google Scholar]
- 7.Howard, A. D., McAllister, G., Feighner, S. D., Liu, Q., Nargund, R. P., Van der Ploeg, L. H., and Patchett, A. A. (2001) Trends Pharmacol. Sci. 22 132–140 [DOI] [PubMed] [Google Scholar]
- 8.Civelli, O. (2005) Trends Pharmacol. Sci. 26 15–19 [DOI] [PubMed] [Google Scholar]
- 9.Eglen, R. M. (2005) Comb. Chem. High Throughput Screen. 8 311–318 [DOI] [PubMed] [Google Scholar]
- 10.Bock, J. R., and Gough, D. A. (2005) J. Chem. Inf. Model. 45 1402–1414 [DOI] [PubMed] [Google Scholar]
- 11.Huang, E. S. (2005) Drug Discov. Today 10 69–73 [DOI] [PubMed] [Google Scholar]
- 12.Bairoch, A., Apweiler, R., Wu, C. H., Barker, W. C., Boeckmann, B., Ferro, S., Gasteiger, E., Huang, H., Lopez, R., Magrane, M., Martin, M. J., Natale, D. A., O'Donovan, C., Redaschi, N., and Yeh, L. S. (2005) Nucleic Acids Res. 33 D154–D159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Breiman, L. (2001) Machine Learning 45 5–32 [Google Scholar]
- 14.Bendtsen, J. D., Nielsen, H., von Heijne, G., and Brunk, S. (2004) J. Mol. Biol. 340 783–795 [DOI] [PubMed] [Google Scholar]
- 15.Kliger, Y., Gofer, E., Wool, A., Toporik, A., Apatoff, A., and Olshansky, M. (2008) Bioinformatics (Oxf.) 24 1049–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horn, F., Bettler, E., Oliveira, L., Campagne, F., Cohn, F. E., and Vriend, G. (2003) Nucleic Acids Res. 31 294–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu, A. M., Ho, M. K., Wong, C. S., Chan, J. H., Pau, A. H., and Wong, Y. H. (2003) J. Biomol. Screen. 8 39–49 [DOI] [PubMed] [Google Scholar]
- 18.Burstein, E. S., Ott, T. R., Feddock, M., Ma, J. N., Fuhs, S., Wong, S., Schiffer, H. H., Brann, M. R., and Nash, N. R. (2006) Br. J. Pharmacol. 147 73–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wollemann, M., and Benyhe, S. (2004) Life Sci. 75 257–270 [DOI] [PubMed] [Google Scholar]
- 20.Iribarren, P., Zhou, Y., Hu, J., Le, and Wang, J. M. (2005) Immunol. Res. 31 165–176 [DOI] [PubMed] [Google Scholar]
- 21.Perretti, M., and D'Acquisto, F. (2006) Inflamm. Allergy Drug Targets 5 107–114 [DOI] [PubMed] [Google Scholar]
- 22.VanCompernolle, S. E., Claek, K. L., Rummel, K. A., and Todd, S. C. (2003) J. Immunol. 171 2050–2056 [DOI] [PubMed] [Google Scholar]
- 23.Santos, R. A., Simoes e Silva, A. C., Maric, C., Silva, D. M., Machado, R. P., de Buhr, I., Heringer-Walther, S., Pinheiro, S. V., Lopes, M. T., Bader, M., Mendes, E. P., Lemos, V. S., Campagnole-Santos, M. J., Schultheiss, H.-P., Speth, R., and Walther, T. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 8258–8263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Halls, M. L., Bathgate, R. A., and Summers, R. J. (2006) Mol. Pharmacol. 70 214–226 [DOI] [PubMed] [Google Scholar]
- 25.Katugampola, S., and Davenport, A. (2003) Trends Pharmacol. Sci. 24 30–35 [DOI] [PubMed] [Google Scholar]
- 26.Joost, P., and Methner, A. (2002) Genome Biol. 3 RESEARCH0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McGuffin, L. J., Bryson, K., and Jones, D. T. (2000) Bioinformatics (Oxf.) 16 404–405 [DOI] [PubMed] [Google Scholar]