

Abstract
The MYST protein lysine acetyltransferases are evolutionarily conserved throughout eukaryotes and acetylate proteins to regulate diverse biological processes including gene regulation, DNA repair, cell-cycle regulation, stem cell homeostasis and development. Here, we demonstrate that MYST protein acetyltransferase activity requires active site lysine autoacetylation. The X-ray crystal structures of yeast Esa1 (yEsa1/KAT5) bound to a bisubstrate H4K16CoA inhibitor and human MOF (hMOF/KAT8/MYST1) reveal that they are autoacetylated at a strictly conserved lysine residue in MYST proteins (yEsa1-K262 and hMOF-K274) in the enzyme active site. The structure of hMOF also shows partial occupancy of K274 in the unacetylated form, revealing that the side chain reorients to a position that engages the catalytic glutamate residue and would block cognate protein substrate binding. Consistent with the structural findings, we present mass spectrometry data and biochemical experiments to demonstrate that this lysine autoacetylation on yEsa1, hMOF and its yeast orthologue, ySas2 (KAT8) occurs in solution and is required for acetylation and protein substrate binding in vitro. We also show that this autoacetylation occurs in vivo and is required for the cellular functions of these MYST proteins. These findings provide an avenue for the autoposttranslational regulation of MYST proteins that is distinct from other acetyltransferases but draws similarities to the phosphoregulation of protein kinases.
Keywords: acetylation, HATs, MYST proteins
Introduction
Histone acetyltransferase (HAT) enzymes play important roles in the regulation of chromatin assembly, RNA transcription, DNA repair and other DNA-templated reactions through the lysine side-chain acetylation of histones and other transcription factors (Marmorstein, 2001; Wang et al, 2008). HATs fall into at least four different families based on sequence conservation within the HAT domain (Marmorstein and Trievel, 2009). This includes Gcn5/PCAF, p300/CBP, Rtt109 and MYST (named for the founding members MOZ, Ybf2/ Sas3, Sas2 and Tip60) families. The different HAT families contain a structurally conserved central region associated with acetyl-Coenzyme A (Ac-CoA) cofactor binding but distinct catalytic mechanisms and structurally divergent flanking regions that mediate different chromatin regulatory functions (Marmorstein and Trievel, 2009). Several recent proteomics studies also reveal that protein acetylation extends beyond histones to other nuclear proteins and even cytoplasmic proteins to regulate diverse biological processes including the regulation of the cell cycle, vesicular trafficking, cytoskeleton reorganization and metabolism (Choudhary et al, 2009; Smith and Workman, 2009; Spange et al, 2009; Wang et al, 2010; Zhao et al, 2010). In this way, protein acetylation may rival protein phosphorylation to mediate signal transduction pathways that control key cellular activities.
The MYST proteins represent the largest family of HATs. They are conserved from yeast to man and mediate diverse biological functions including gene regulation, DNA repair, cell-cycle regulation, stem cell homeostasis and development (Sapountzi and Cote, 2010). MYST proteins have also been shown to acetylate several non-histone substrates as highlighted by a recent protein acetylation microarray screen, revealing that the yeast Esa1 (yEsa1) containing NuA4 complex acetylates several cytoplasmic non-chromatin substrates known to be involved in the regulation of metabolism, the cell cycle and response to stress (Lin et al, 2009). MYST proteins have also been implicated in several forms of cancer and are particularly associated with chromosomal translocations in leukaemias (Avvakumov and Cote, 2007), also making them attractive drug targets.
As with protein kinase enzymes (Morgan, 1995; Nolen et al, 2004), the activities and substrate selectivities of several MYST acetyltransferases are modulated by the binding of regulatory protein subunits. For example, yeast Sas2 requires binding to Sas4 and Sas5 for catalytic activity (Sutton et al, 2003), yEsa1 is found in the NuA4 complex and in a smaller Piccolo NuA4 complex to acetylate specific or global genomic locations, respectively (Allard et al, 1999; Boudreault et al, 2003), and human MOF (hMOF) is found in at least two distinct complexes, MSL and MOF-MSL1v1, that have indistinguishable activity on histone H4 K16, but differ dramatically in acetylating the non-histone substrate p53 (Li et al, 2009). Given the large number and diversity of MYST protein substrates and their involvement in various cellular processes, coupled with their modulation by other protein cofactors, we investigated the possibility that, also like protein kinases, the MYST proteins might be autoregulated by posttranslational modification.
Results
Structure of yEsa1 bound to a bisubstrate inhibitor reveals acetylation of a conserved lysine in the active site
We previously reported on X-ray crystal structures of the HAT domain of the yEsa1 MYST protein bound to acetyl-CoA and CoA (Yan et al, 2000, 2002). In order to obtain insights into protein substrate binding by MYST proteins, we now report on the X-ray crystal structure of yEsa1 in complex with an H4K16CoA bisubstrate inhibitor (Wu et al, 2009) to 2.1 Å resolution (Figure 1A; Table I). The yEsa1 and CoA portion of the complex superimpose well with the previously reported yEsa1/CoA complex, although additional continuous electron density was observed emanating from the tip of CoA, which was used to model the linkage and H4K16 lysine side chain. Continuous electron density corresponding to backbone and side chain residues flanking H4K16 was not observed and we presume that these regions of the bisubstrate inhibitor are disordered in our structure. The H4K16 lysine side chain sits in a fairly open, largely hydrophobic, groove between the α2–β7 and β10–α4 loops flanking the Esa1 active site (Figure 1A).
Figure 1.

Structure of MYST protein active sites. (A) Superimposed structures of yEsa1 (pink) and hMOF (hMOF, green). The active sites are highlighted showing catalytic residues (glutamate and cysteine), the autoacetylated lysine and the H4K16CoA inhibitor bound to yEsa1. Numbering of residues corresponds to the yEsa1 sequence. (B) Structure of the yEsa1 active site. The simulated annealing omit map for K262Ac and the H4K16CoA inhibitor (1.5 sigma) is shown. Side chains that play catalytic roles and that make interactions with K262Ac are included as stick figures. The omit maps were generated by omitting a 10-Å radius around the selected atoms. (C) Sequence alignment of MYST protein active site residues and residues proximal to the autoacetylated lysine residues of yEsa1 and hMOF. Numbering above the sequences corresponds to the yEsa1 sequence. The conserved autoacetylated lysine residue is indicated with an inversed triangle and residues that interact with this lysine are highlighted with a star above the sequence. Only a selected set of MYST proteins are shown. (D) Structure of the hMOF active site. This figure is analogous to (B) except that corresponding hMOF residues are highlighted and the two alternate conformations of the α2–β7 loop harbouring the acetylated (K274Ac) and unacetylated (K274) residues, respectively, are highlighted with their corresponding simulated annealing omit maps (1.0 sigma). (E) Superposition of the yEsa1 and hMOF active sites. Numbering of residues corresponds to the hMOF sequence. (F) LC–MS/MS analysis of recombinant yEsa1 (upper panel) and hMOF (lower panel) tryptic peptides containing K262Ac and K274Ac, respectively. The extracted ion chromatogram (XIC) of the indicated peptide is shown on the left. A corresponding MS/MS spectrum of the 2+ charge state precursor is shown on the right with major b- and y-ions labelled.
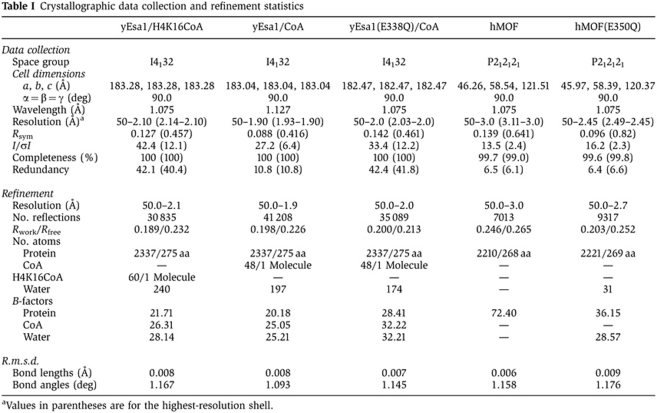
Table 1. Crystallographic data collection and refinement statistics.
| yEsa1/H4K16CoA | yEsa1/CoA | yEsa1(E338Q)/CoA | hMOF | hMOF(E350Q) | |
|---|---|---|---|---|---|
| Data collection | |||||
| Space group | I4132 | I4132 | I4132 | P212121 | P212121 |
| Cell dimensions | |||||
| a, b, c (Å) | 183.28, 183.28, 183.28 | 183.04, 183.04, 183.04 | 182.47, 182.47, 182.47 | 46.26, 58.54, 121.51 | 45.97, 58.39, 120.37 |
| α=β=γ (deg) | 90.0 | 90.0 | 90.0 | 90.0 | 90.0 |
| Wavelength (Å) | 1.075 | 1.127 | 1.075 | 1.075 | 1.075 |
| Resolution (Å)a | 50–2.10 (2.14–2.10) | 50–1.90 (1.93–1.90) | 50–2.0 (2.03–2.0) | 50–3.0 (3.11–3.0) | 50–2.45 (2.49–2.45) |
| Rsym | 0.127 (0.457) | 0.088 (0.416) | 0.142 (0.461) | 0.139 (0.641) | 0.096 (0.82) |
| I/σI | 42.4 (12.1) | 27.2 (6.4) | 33.4 (12.2) | 13.5 (2.4) | 16.2 (2.3) |
| Completeness (%) | 100 (100) | 100 (100) | 100 (100) | 99.7 (99.0) | 99.6 (99.8) |
| Redundancy | 42.1 (40.4) | 10.8 (10.8) | 42.4 (41.8) | 6.5 (6.1) | 6.4 (6.6) |
| Refinement | |||||
| Resolution (Å) | 50.0–2.1 | 50.0–1.9 | 50.0–2.0 | 50.0–3.0 | 50.0–2.7 |
| No. reflections | 30 835 | 41 208 | 35 089 | 7013 | 9317 |
| Rwork/Rfree | 0.189/0.232 | 0.198/0.226 | 0.200/0.213 | 0.246/0.265 | 0.203/0.252 |
| No. atoms | |||||
| Protein | 2337/275 aa | 2337/275 aa | 2337/275 aa | 2210/268 aa | 2221/269 aa |
| CoA | — | 48/1 Molecule | 48/1 Molecule | — | — |
| H4K16CoA | 60/1 Molecule | — | — | — | — |
| Water | 240 | 197 | 174 | — | 31 |
| B-factors | |||||
| Protein | 21.71 | 20.18 | 28.41 | 72.40 | 36.15 |
| CoA | 26.31 | 25.05 | 32.22 | — | — |
| Water | 28.14 | 25.21 | 32.21 | — | 28.57 |
| R.m.s.d. | |||||
| Bond lengths (Å) | 0.008 | 0.008 | 0.007 | 0.006 | 0.009 |
| Bond angles (deg) | 1.167 | 1.093 | 1.145 | 1.158 | 1.176 |
| aValues in parentheses are for the highest-resolution shell. | |||||
A more detailed analysis of the electron density map proximal to the bisubstrate inhibitor in the active site revealed unmodelled density at the tip of the K262 side chain in the active site, located in the 14-residue α2–β7 loop. We modelled an acetyl group in this position and subsequent crystallographic refinement and visualization of a simulated annealing omit map reinforced the presence of acetylated K262 (K262Ac) (Figure 1B). To confirm that the observed acetylation of K262 was not an artefact of cocrystallizing Esa1 bound to the H4K16Ac bisubstrate inhibitor, we determined the crystal structure of Esa1 bound to CoA to 1.9 Å resolution (Table I; Supplementary Figure S1A) and again observed electron density consistent with acetylated K262 (Supplementary Figure S1A). In addition, we also observed well-formed electron density in place of the lysine portion of the bisubstrate inhibitor that we have modelled and refined as a cacodylate buffer molecule containing disordered oxygen ligands as observed in other structures (Raman et al, 2001) (Supplementary Figure S1A).
Inspection of the environment proximal to K262Ac reveals that the acetyl group is buried in the active site of the enzyme, with the methyl group making van der Waals interactions with phenylalanine residues 271 and 273 of β7 and the carbonyl moiety hydrogen bonding to Y289 and S291 of β8 of the structurally conserved HAT core region (Figure 1B). Correlating with the significance of these interactions and its generality to other MYST proteins, K262, F271, F273, Y289 and S291 are highly conserved throughout the MYST family of proteins (Figure 1C). The acetyl group of K262Ac is also ∼7 Å from the sulphur atom of the bisubstrate inhibitor and ∼5 Å and 8 Å from the C304 and E338 catalytic residues, respectively (Figure 1B).
Structure of hMOF reveals a conformational change upon active site acetylation
To establish if analogous lysine acetylation might extend to MYST proteins in other species, we determined the crystal structure of hMOF (Figure 1A; Table I). Inspection of the electron density map proximal to K274 in hMOF, the analogous lysine to yEsa1 K262, revealed that, the α2–β7 loop bearing K274 adopts two conformations. In one of these conformations, K274 is acetylated and is oriented towards the active site of the enzyme, interacting with residues that are conserved and making analogous interactions with yEsa1 (F283, F285, Y301 and S303 in hMOF) (Figure 1D). In the second α2–β7 loop conformation, K274 is not acetylated and flipped out of the active site by ∼90° and pointing across the cognate protein lysine binding site (Figure 1D) to make a long H-bond (3.4 Å) with the E350 catalytic base residue located on the α3–α4 loop that flanks the opposite side of the cognate protein lysine binding site. A superposition of the hMOF and yEsa1/H4K16CoA structures (Figure 1E) also reveals that the α2–β7 loop has a more varied conformation than other protein elements surrounding the cognate protein lysine binding site and that the position of the unacetylated K274 of hMOF overlaps with the backbone of the lysine portion of the H4K16CoA bisubstrate inhibitor of yEsa1. These structural observations suggest that the unacetylated lysine adopts a conformation that inhibits cognate protein lysine acetylation, possibly by directly blocking substrate binding.
To further confirm the presence of K262Ac and K274Ac in yEsa1 and hMOF, respectively, we subjected both recombinant proteins to LC–MS/MS and unambiguously identified the presence of peptides consistent with acetylation at these positions (Figure 1F; Table II). A weak MS signal for the unacetylated LFLDHK peptide was detected, indicating a low level of unacetylated K274 in wild-type (WT) hMOF (Table II). These results, together with the conservation of this lysine throughout the MYST family, suggest that analogous lysine acetylation occurs throughout this family of HAT proteins.
Table 2. Mass spectrometry analysis of recombinant yEsa1 and hMOF proteinsa.
Active site acetylation by MYST proteins occurs by autoacetylation
To demonstrate that the acetylation of recombinant yEsa1 and hMOF proteins was mediated by autoacetylation, we prepared the recombinant yEsa1 and hMOF HAT domains in bacteria and then treated them with the relatively promiscuous yeast Hst2 deacetylase plus NAD+ cofactor to deacetylate them. We then added nicotinamide to inhibit Hst2 activity and Ac-CoA to provide a cofactor for any potential autoacetylation activity. We subjected both the untreated and Ac-CoA treated HAT domains to LC–MS/MS to survey for acetylated and unacetylated peptides containing K262 and K274 of yEsa1 and hMOF, respectively. As shown in Table II, these studies revealed that Hst2-mediated deacetylation of the recombinant yEsa1 and hMOF samples reduced the acetylation levels of K262 and K274 to <1% and 10%, respectively, based upon normalized integrated areas of the acetylated tryptic peptides. Concurrently, a strong signal for the unacetylated smaller tryptic peptide was observed in these samples. Reacetylation upon Ac-CoA addition increased the extent of acetylation as indicated by the marked reduction of the unacetylated peptide for both proteins. These data are consistent with the conclusion that active site acetylation of yEsa1, hMOF, and likely other MYST proteins occurs by autoacetylation.
To further characterize whether autoacetylation occurs in cis (intramolecularly) or trans (intermolecularly), we subjected the Hst2-treated yEsa1 to reacetylation with saturating concentrations of Ac-CoA and increasing concentrations of yEsa1 and measured the amount of autoacetylated yEsa1 using filter binding and scintillation counting (Yan et al, 2002). As can be seen in Figure 2A, these studies reveal that the rate of yEsa1 autoacetylation is largely first order with respect to protein concentration consistent with autoacetylation in cis. Autoacetylation in cis is also consistent with the structural observation that the autoacetylated lysine is located within the enzyme active site and about 5 Å away from the catalytic cysteine residue (C304 in yEsa1 and C316 in hMOF) that would transfer the acetyl group via a ping-pong catalytic mechanism (Yan et al, 2002) and about 7 Å from the CoA sulphur atom that might transfer the acetyl group via a ternary complex mechanism (Berndsen et al, 2007). Therefore, a relatively minor movement of the acetylated lysine side chain (e.g. a change of rotamer) and/or acetyl donors could easily accommodate autoacetylation in cis.
Figure 2.

Activity of MYST proteins harbouring mutation of the conserved acetyllysine residue. (A) Autoacetylation of the yEsa1 HAT domain as a function of protein concentration. Each data point was obtained in triplicate with standard deviations indicated with error bars. (B) Catalytic activities of the yEsa1, hMOF HAT domains and the ySas2/4/5 complex and of their corresponding KAc-R mutants. Activity is indicated along the ordinate as the radioactivity (in CPM) incorporated into a 21-residue N-terminal histone H4 peptide from [14C] Ac-CoA as a function of protein (along the abscissa). (C) Histone peptide quenching of tryptophan fluorescence of hMOF-wt and the hMOF-K274R and hMOF-K274A mutants. The fluorescence emission spectra with 0 or 7 μM of H4p21 peptide are shown. The insert shows the quenching percentage as a function of H4p21 peptide concentration. F is the fluorescence intensity of hMOF with H4p21 peptide and F0 is the fluorescence intensity of hMOF in the absence of peptide.
Previous studies on MYST proteins revealed that a conserved glutamate residue (E338 in yEsa1 and E350 in hMOF) functions as a general base for cognate protein lysine substrate acetylation and mutation of this residue to glutamine results in near background levels of cognate protein acetyltransferase activity (Yan et al, 2000). In order to test if autoacetylation also requires this conserved glutamate residue, we prepared the HAT domains of yEsa1 and hMOF harbouring glutamate-to-glutamine mutations in these residues for crystal structure determination. The structure of yEsa1-E338Q revealed that K262 was still fully acetylated (Table I; Supplementary Figure S1B), and the structure of hMOF-E350Q revealed that the α2–β7 loop still adopted the same two conformations containing the acetylated and unacetylated K274 forms of the WT enzyme (Table I; Supplementary Figure S1C). This was also confirmed by LC–MS/MS mass spectrometry (Supplementary Figure S1D; Table II). These structural findings suggest that autoacetylation of the MYST proteins do not require, or are less dependent on, the conserved glutamate general base residue that is required for cognate protein lysine substrate acetylation.
To more directly establish the dependence of MYST autoacetylation on the glutamate general base residue, we determined rate curves as a function of Ac-CoA concentration for the deacetylated yEsa1-wt and yEsa1-E338Q proteins (Supplementary Figure S1E). As can be seen from these rate curves, relative to yEsa1-wt, the yEsa1-E338Q mutant still showed appreciable activity with a reduction in Vmax of less than two-fold and an elevation in Km of less than five-fold for autoacetylation. This is in contrast to the effect of the E338Q mutation on cognate protein lysine acetylation, which shows near background levels of acetylation (Yan et al, 2000). These data demonstrate that MYST autoacetylation is significantly less dependent on the conserved glutamate general base residue than MYST cognate protein lysine acetylation.
Autoacetylation is required for HAT activity and histone substrate binding in vitro
To directly test the hypothesis that acetylation of K262 and K274 in yEsa1 and hMOF, respectively, contributes to cognate protein lysine acetylation, we prepared recombinant HAT domains containing the corresponding arginine mutations (yEsa1-K262R and hMOF-K274R) and measured their catalytic activities towards a histone H4 tail peptide substrate. These mutant proteins eluted from gel filtration at the same position as the WT protein (Supplementary Figure S2A and B), suggesting that they were properly folded. Interestingly, enzymatic analysis of these mutant proteins showed that they were catalytically inactive (Figure 2B). We also prepared the WT and corresponding K-to-R mutation of the autoacetylation site of yeast Sas2(K168R) and assembled these proteins into the catalytic Sas2/Sas4/Sas5 ternary complex by bacterial coexpression and purification (Sutton et al, 2003). While the ternary complex containing the Sas2(K168R) mutant elutes from gel filtration at the same position as the WT complex (Supplementary Figure S2C), the mutant complex also displayed background levels of HAT activity (Figure 2B).
To probe the consequence of other mutations of yEsa1-K262, hMOF-K274 and Sas2-K168, we also mutated these residues to alanine and glutamine (a possible acetyllysine mimic). Like the corresponding lysine-to-arginine mutants, no activity could be detected for either of the alanine mutants or the hMOF and Sas2 glutamine mutants, although the yEsa1 glutamine mutant retained about 25% of WT activity (Supplementary Figure S3). Taken together, these mutational data suggest that active site lysine autoacetylation is important for MYST cognate protein substrate acetylation.
To test the hypothesis that the conserved acetyllysine residue in the MYST proteins directly disrupts cognate protein lysine binding, we exploited the presence of an endogenous tryptophan residue (W192) near the cognate protein binding site of hMOF to monitor the binding of an H4K16 peptide as a function of WT lysine or mutant arginine or alanine at position 274. As shown in Figure 2C, titration of the H4K16 peptide substrate into the WT enzyme results in a dramatic quenching of the fluorescence while a similar titration into the hMOF mutant proteins results in a significantly reduced fluorescence quenching (Figure 2C). These studies are consistent with a defect in histone binding of the hMOF-K274R and hMOF-K274A mutants and the conclusion that autoacetylation of hMOF K274, and likely other MYST proteins, is important for cognate protein lysine binding.
Autoacetylation of lysine 262 on Esa1 occurs in vivo and is essential for cell viability
To determine if lysine 262 autoacetylation of Esa1 is relevant to its in vivo function, we first isolated the native 13-subunit NuA4 complex from yeast cells using a TAP-tagged Epl1 subunit and subjected this complex to LC–MS/MS to survey for acetylated or unacetylated peptides containing K262. As shown in Figure 3A, this resulted in the unambiguous identification of a doubly charged semi-tryptic peptide (257-LFLDHKacTLY-265) containing Esa1-K262Ac. Peptides containing unacetylated K262 were not detected. This result demonstrates that lysine 262 autoacetylation of Esa1 occurs in vivo.
Figure 3.

Characterization of yEsa1 autoacetylation in vivo. (A) LC–MS/MS analysis of Esa1 prepared from yeast cells. The native NuA4 complex was purified through the TAP-tagged Epl1 subunit and the Esa1 subunit analysed by LC–MS/MS. (Right panel) Extracted ion chromatogram (XIC) of a doubly charged Esa1 semi-tryptic peptide (257-LFLDHKTLY-265) containing Esa1-K262Ac. (Left panel) ESI-MS/MS spectrum of the doubly charged precursor peptide. The m/z difference between the b5 and b6 fragments is consistent with K262 acetylation. (B) Growth of yeast cells as a function of yEsa1 K262 acetylation. (Left) Serial 10-fold dilution of Δesa1 cells expressing WT ESA1 from an ARS/CEN/URA3 plasmid, and transformed with pSAPE (ARS/CEN LEU2) plasmid expressing HA-tagged versions of WT ESA1, esa1K262R, esa1K262Q or no ORF (–) were spotted on SC plates in the absence or presence of 5′-FOA. Cells were grown at 30 °C for 2–3 days. (C) Anti-HA western blot to verify HA-Esa1 expression in the cells used for the spot assay in (B).
To further assess the biological importance of this autoacetylation activity, we transformed a Δesa1 deletion strain of yeast with N-terminal haemagglutinin (HA) epitope-tagged ESA1, HA alone (ORF−) or ESA1 mutants containing K262R or K262Q a possible acetyllysine mimic (Megee et al, 1990). The essential function of Esa1 was supplied by a URA3 vector expressing WT ESA1. As shown in Figure 3B, while the expression of HA-ESA1 and HA-esa1K262Q supports growth, expression of HA-esa1K262R does not. We also used western blotting on whole-cell extracts to demonstrate that each of the proteins were expressed at comparable levels (Figure 3C). These results demonstrate that K262 on Esa1 is essential for viability in yeast and is consistent with the importance of K262 acetylation for Esa1 function in vivo.
Autoacetylation of lysine 274 on hMOF occurs in vivo and is essential for p53 and histone H4K16 substrate acetylation
hMOF acetylates several substrates in vivo (Sapountzi and Cote, 2010) including K16 on histone H4 for transcription and DNA damage repair regulation (Smith et al, 2005; Taipale et al, 2005; Sharma et al, 2010) and K120 of the p53 tumour suppressor protein to mediate p53-dependent transcription of pro-apoptotic genes (Sykes et al, 2006; Tang et al, 2006). To determine if the in vivo activities of hMOF are dependent on K274 acetylation, we first assessed whether K274 of hMOF is acetylated in human cells. To do this, an epitope-tagged allele of hMOF was ectopically expressed in the human lung cancer cell line H1299 and hMOF was isolated from these cells via affinity chromatography, resolved on SDS–PAGE and subjected to LC–MS/MS analysis (Taplin Facility-Harvard University). This analysis resulted in the identification of six acetylated lysines (113, 116, 174, 221, 274, 351) including K274, which was identified as a strong acetylation site (Figure 4A). Together, these data demonstrate that K274 of hMOF is acetylated in human cells.
Figure 4.

Characterization of hMOF and ySas2 autoacetylation and function in cells. (A) LC–MS/MS analysis of ectopically expressed, FLAG-tagged hMOF in H1299 cells. FLAG-hMOF from nuclear extracts was purified by FLAG affinity and resolved on SDS–PAGE. The hMOF doublet was excised and subjected to LC–MS/MS analysis (Taplin Facility-Harvard University). (B) Analysis of p53-K120 acetylation as a function of hMOF-K274 acetylation. The p53 null human lung cancer line H1299 was transfected with p53 and V5-hMOF expression plasmids. Cells were harvested 24 h posttransfection, lysed and immunoprecipitated with Ac-K120 antibody and then subjected to immunoblot with antibody that recognizes total p53. A blot of lysates was also probed with antibody recognizing total p53, hMOF (V5) and actin. (C) H4K16 acetylation as a function of hMOF-K274 acetylation. HeLa cells were transfected with the indicated V5-hMOF expression plasmids, γ-irradiated (10 Gy) 24 h later, allowed to recover for 2 h and then stained with DAPI, for V5 and for acetylated H4K16 (see Supplementary Figure S3) and quantitated for H416 in the nucleus. Cells were also prepared as non-irradiated controls. The fractions of cells expressing the V5-hMOF forms that also stain for H4K16Ac, and the number of cells counted (n), are shown, and P-values were calculated using Fisher's exact test. (D) (Above) Sas2-dependent telomere silencing assay. WT and sas2Δ strains carrying a URA3 reporter inserted at the XR element of telomere TEL11L were plated in 10-fold serial delution and spotted on plates containing synthetic complete media with or without 5-FOA. (Below) Sas2-dependent telomere silencing assay employing Sas2 mutants. sas2Δ strains transformed with either a vector or various SAS2 constructs were serially diluted and spotted on selective media with or without 5-FOA. (E) Western blotting analysis of Sas2 mutants. Whole-cell extracts for strains from (D) were analysed by western blotting using an anti-Myc antibody.
To determine whether hMOF-K274 acetylation has an impact on the ability of hMOF to acetylate substrates in vivo, first the WT and K274R mutant version of hMOF were expressed via plasmid transfection in H1299 cells, along with ectopic p53. As seen in Figure 4B, WT hMOF and hMOF-K274R were expressed at comparable levels in H1299 cells. However, while WT hMOF induced robust p53-K120 acetylation, the K274R mutant showed only background levels of p53-K120 acetylation. Second, WT and K274R hMOF were expressed in the same fashion in HeLa cells, and H4K16Ac levels were assessed by immunofluorescence microscopy. H4K16Ac levels varied among cells, consistent with previous studies (Vaquero et al, 2006), but the fraction of cells with high H4K16Ac levels was much greater among cells expressing plasmid-based hMOF than among cells expressing K274R hMOF (Figure 4C; Supplementary Figure S4). The difference held under basal conditions and following γ irradiation, which induces hMOF-dependent H4K16 acetylation (Li et al, 2010). Collectively, these studies support the model that hMOF-K274 acetylation is required for cognate substrate acetylation in vivo.
Autoacetylation of lysine 168 on yeast Sas2 is required for silencing in vivo
Yeast Sas2 is a member of the MYST HAT family, is involved in transcriptional silencing at all silent loci in Saccharomyces cerevisiae and is thought to be the functional orthologue of hMOF (Lafon et al, 2007). To test whether autoacetylation of yeast Sas2 is required for in vivo function, we tested if mutation of K168 in Sas2 can rescue the silencing defect caused by deletion of SAS2 in a strain bearing an insertion of a URA3 reporter gene at the XR element near TEL11L where silencing of URA3 confers a resistance phenotype to 5-fluoroorotic acid (5-FOA) (Pryde and Louis, 1999). As expected, we found that a plasmid carrying a WT copy of SAS2 can restore the resistance to 5-FOA in sas2Δ cells (Figure 4D). In addition, a control K189R mutation that does not affect the catalytic activity of Sas2 also showed largely restored resistance to 5-FOA (Figure 4D). In contrast, substituting arginine (R), glutamine (Q) or methionine (M) for K168 showed 5-FOA sensitivity similar to that of sas2Δ (Figure 4E), indicating that mutations to the K168 autoacetylation site on Sas2 abolished its activity and physiological function in vivo. Importantly, the Sas2 mutants did not show significant reductions in protein abundance under the experimental conditions (Figure 4E). These results show that lysine 168 plays an important role in Sas2 function in cells and together with the biochemical data on Sas2 and the structural and biochemical data on yEsa1 and hMOF described above, strongly suggests that lysine 168 autoacetylation is required for Sas2 function in vivo.
Discussion
Here, we present structural, biochemical and cellular data demonstrating that the MYST proteins Esa1 and Sas2 from yeast and MOF from human are each acetylated on a conserved active site lysine residue that is required for catalytic activity in vitro and protein function in vivo. A comparison and contrast of the structures of hMOF in both the K274 unacetylated and acetylated form and Esa1 bound to an H4K16CoA bisubstrate inhibitor in the K262 acetylated form further suggests the molecular basis for how active site lysine acetylation promotes cognate protein lysine acetylation. Specifically, active site lysine autoacetylation appears to bury the acetyl group in a complementary pocket in the enzyme active site and to configure the active site into a relatively open conformation that is compatible for cognate protein lysine binding and acetylation (Figure 1A and B). In contrast, the unacetylated form of the hMOF enzyme reveals that K274 flips ∼90° out of the active site to a position that would block cognate protein substrate binding and engage and shield the catalytic glutamate residue (Figure 1D). We also show here that mutation of hMOF-K274, yEsa1-K262 and ySas2-K168 to either arginine or alanine is also defective in catalysis (Figure 2B; Supplementary Figure S3) and we show that hMOF-K274 mutation to arginine or alanine are defective in histone substrate binding (Figure 2C). We hypothesize that these mutants cannot be accommodated like acetyllysine in the enzyme active site and so adopt the flipped out conformation of the unacetylated lysine. Modelling of either of these amino-acid substitutions in place of K274 in the unacetylated form would form a steric clash with the cognate lysine substrate, as observed in the yEsa1 H4K16CoA bisubstrate complex (Figure 1E), consistent with the inability of these mutations to support cognate protein lysine acetylation. Taken together, it appears that active site lysine autoacetylation of hMOF, yEsa1 and ySas2 is important for proper cognate protein lysine binding, although it is possible that it might have other roles as well.
Interestingly, mutation of the active site lysine in these MYST proteins to glutamine (a possible acetyllysine mimic) also does not support activity of hMOF and ySas2, but does support about 25% of WT activity for yEsa1 in vitro (Supplementary Figure S3) and yEsa1 activity in vivo (Figure 3B). These results suggest that there may be specific differences in the mechanism of how active site lysine acetylation promotes enzyme activity within these proteins. The strict conservation of this lysine in other MYST proteins suggests that active site lysine autoacetylation is conserved among this family of enzymes (Figure 1C). This conclusion is also consistent with two recent studies. In one study, K274 acetylation was observed on recombinant drosophila MOF and mutation of this lysine to alanine was shown to abrogate catalytic activity (Kadlec et al, 2011). In another study, K274 acetylation was observed on hMOF and mutation of this lysine to arginine or glutamine was shown to decrease catalytic activity to 0.1 and 2.9% of WT catalytic activity, respectively (Sun et al, 2011). Together, these findings provide novel insight into the regulation of MYST proteins by autoposttranslational modification.
Another interesting outcome of this study is that we found that yEsa1 autoacetylation (and probably autoacetylation of other MYST proteins) does not appear to be as dependent as cognate protein lysine acetylation on the catalytic Glu338 general base residue (Supplementary Figure S1E). We propose that this is because the intramolecular autoacetylation reaction has a templating advantage over intermolecular cognate protein lysine acetylation since for autoacetylation the effective lysine substrate concentration is higher and the activation energy for appropriate lysine substrate orientation is likely lower. This is consistent with the about 15-fold higher apparent Km for cognate protein substrate acetylation of 154 μM (Yan et al, 2002) over autoacetylation (9.4 μM; Supplementary Figure S1E). The importance of substrate templating for catalysis by HAT proteins is supported by the observation that while the catalytic mechanism of the four HAT families, Gcn5/PCAF, MYST, p300 and Rtt109 differ, they contain a structurally conserved core region that plays a conserved templating function (Hodawadekar and Marmorstein, 2007; Marmorstein and Trievel, 2009). As a consequence of this, we propose that autoacetylation is less dependent on the contribution of the yEsa1-E338 general base than cognate protein lysine acetylation, although the possibility that an alternative pathway or mechanism is at play for autoacetylation cannot be ruled out.
It is possible that MYST protein autoacetylation might have additional consequences on protein activity. Consistent with this possibility, a recent report revealed that K274 acetylation of hMOF is correlated with the increased recruitment of hMOF to chromatin (Lu et al, 2011). This same study also showed that hMOF interacts with and is deacetylated by SIRT1, further suggesting that, at least in some cases, MYST protein autoacetylation might be negatively regulated by protein deacetylases. Interestingly, we have been unable to demonstrate hMOF deacetylation in the laboratory using recombinant SIRT1 (data not shown). This could imply that other cofactors are required for SIRT1-mediated hMOF deacetylation in cells. To our knowledge, there is no evidence for orthologous Sir2-mediated deacetylation of yEsa1 in cells, although as demonstrated in this study, we are able to deacetylate yEsa1 with the yHst2 deacetylase in vitro, although not to completion (Figure 2A). Clearly, the potential regulatory role of MYST protein acetylation/deacetylation requires further study.
As in the MYST proteins, lysine autoacetylation also occurs within the HAT domain of the p300/CBP (Thompson et al, 2004) and Rtt109 (Lin and Yuan, 2008; Stavropoulos et al, 2008; Tang et al, 2008) family of HATs, and in each case this autoacetylation is required to stimulate HAT activity. However, the mode of autoacetylation in the three HAT families show significant differences. While a single lysine residue within Rtt109 (K290 in yeast Rtt109) is acetylated in a buried site, the lysine residue is located in a helical segment and is about 12 Å from the sulphur atom of Ac-CoA (Lin and Yuan, 2008; Stavropoulos et al, 2008; Tang et al, 2008), nearly twice the distance as the autoacetylated lysine in the MYST proteins. In stark contrast, p300/CBP contains an ∼60 residue highly basic and proteolytically sensitive loop that harbours multiple lysine acetylation sites (Thompson et al, 2004). Taken together, the p300/CBP, Rtt109 and MYST family of HATs appear to be regulated by autoacetylation through distinct mechanisms.
The activities of protein kinases are modulated by multiple mechanisms, although nearly all share regulation by a 20–30 residue polypeptide segment located just outside of the active site cleft called the activation loop (McInnes and Fischer, 2005). This segment typically contains one or more phosphorylation sites (either serine, threonine or tyrosine residues) that are modified by either the same kinase or a different kinase. In the unphosphorylated form, the activation loop takes on an autoinhibitory conformation; while in the phosphorylated form, the activation loop adopts a conformation that facilitates either substrate binding, catalysis or both. Like the kinases, it appears that at least three families of HAT enzymes have elevated catalytic activities when they are posttranslationally modified at one or more residues. Indeed, stimulation of kinase activity and substrate binding by activation loop phosphorylation in kinases appears analogous to stimulation of acetylation activity and substrate binding by α2–β7 loop acetylation in MYST protein acetyltransferases. However, unlike the kinases, other HAT families, like p300/CBP and Rtt109 appear to be regulated by posttranslational modification through more diverse mechanisms, as described above. Taken together, the studies reported here point to an additional layer of acetyltransferase regulation by posttranslational modifications that might place them on the same playing field as protein kinases as key regulatory nodes in various signal transduction pathways.
Materials and methods
Protein preparations
The recombinant yEsa1 HAT domain (residues 160–435) was overexpressed and purified as previously described (Yan et al, 2002). The DNA encoding the HAT domain of hMOF (residues 174–458) was cloned into the pRSFDuet1 vector (Novagen) to encode an N-terminal 6xHis-tagged fusion protein with an intervening TEV protease recognition site. The overexpression plasmid was transformed into Escherichia coli BL21(DE3) codon plus RIL (Stratagene) cells and the protein was overexpressed in LB media by induction with 0.5 M IPTG and growth at 16 °C overnight. The cells were harvested and lysed by sonication in lysis buffer containing 50 mM HEPES (pH 7.5), 0.5 M NaCl, 5 mM β-mercaptoethanol and 0.1 mM phenylmethylsulfonyl fluoride. The lysate was cleared by spinning at 28 000 g for 30 min at 4 °C. The supernatant was loaded onto Nickel charged NTA resin (Qiagen) equilibrated with lysis buffer. The resin was then washed with 10 column volumes of 50 mM imidazole in lysis buffer and then eluted with 250 mM imidazole buffer. The eluted hMOF was subjected to TEV protease cleavage overnight at 4 °C and then dialysed into size exclusion buffer (20 mM HEPES, pH 7.5, 0.5 M NaCl). The 6xHis-tagged TEV protease and residual uncut hMOF was removed by incubating with Nickel charged NTA resin. The hMOF was then further purified using a HiLoad Superdex 75 16/60 gel filtration column.
To prepare the Sas2/Sas4/Sas5 complex, DNA encoding Sas2 was cloned into the pETDuet1 overexpression vector, and DNA encoding Sas4 and Sas5 were cloned into the pCDFDuet1 (Novagen) overexpression vector. These vectors were transformed into E. coli BL21(DE3) codon plus RIL cells and the proteins coexpressed in LB media by induction with 0.5 M IPTG and growth at 18 °C overnight. The intact complex was purified to homogeneity using a combination of Nickel charged NTA resin, SP sepharose cation exchange and a Superdex 200 gel filtration chromatography using the buffers described above. All mutations were introduced using the QuikChange Site-Directed Mutagenesis Kit (Stratagene).
Crystal structure determination of MYST HAT domains
All crystals were obtained by the hanging drop method at protein concentrations between 3 and 5 mg/ml. Both yEsa1(160–435)-wt and the E338Q mutant were incubated with CoA (3:1 molar ratio of CoA:protein) or yEsa1(160–435) was incubated with H4K16CoA (3:1 molar ratio of H4K16CoA:protein) before mixing 1:1 with reservoir buffer containing 0.1 M sodium cacodylate (pH 6.5) and 1.6 M ammonium sulphate (Yan et al, 2002) and incubating over reservoir buffer. H4K16CoA was prepared as described previously and contains residues 11–22 of H4) (Wu et al, 2009). Crystals of hMOF(174–458)-wt and the E350Q mutant were prepared analogously but against reservoir solutions containing 0.2 M calcium chloride and 22% PEG3350; and 0.2 M magnesium chloride, 0.1 M Bis-Tris (pH 5.5) and 25% PEG3350, respectively. Despite the presence of CoA or Ac-CoA in the hMOF cocrystallizations, they were not observed in the final electron density maps.
All native data sets were collected at the National Synchrotron Light Source beamlines X6A or X29. Diffraction data were processed using HKL2000 (Otwinowski and Minor, 1997) and the structures were determined using molecular replacement with Phaser (McCoy et al, 2007). Cycles of manual model building and refinement were carried out with Coot (Emsley and Cowtan, 2004) and Phenix (Adams et al, 2010), respectively.
Enzymatic analysis and histone-binding studies
A radioactive-based enzymatic assay was carried out for WT and mutant forms of recombinant yEsa1, hMOF and ySas2/4/5 complex as previously described (Yan et al, 2002). Briefly, 14C-labelled Ac-CoA (300 μM) was incubated with 2 mM of residues 1–21 of a H4 peptide (H4p21) and different concentrations of enzyme (shown to be in the linear range for catalysis) in reaction buffer (20 mM Tris–HCl, pH 7.6, 50 mM sodium chloride and 10 μg BSA) and incubated at 30 °C for 20 min. The reaction was then spotted onto P81 phosphocellulose filter paper (Whatman) and washed three times with 10 mM HEPES (pH 7.5) buffer for scintillation counting.
For the yEsa1 autoacetylation assay, yEsa1 was treated with Hst2 the same way as for LC–MS/MS autoacetylation analysis. Different concentrations of Hst2-treated yEsa1 was incubated with 3H-labelled Ac-CoA (300 μM) in the same reaction buffer at 30 °C for 15, 30 and 45 min. The reaction was stopped by addition of a 30-fold molar excess of CoA. To facilitate the binding of yEsa1 (pI=8.7) to P81 paper, the pH was lowered to 5.5 before spotting and the paper was washed three times with 20 mM NaCitrate (pH 5.5) before scintillation counting. Autoacetylation rates were obtained by a linear fit of radioactivity to the time.
For histone-binding studies with recombinant hMOF, fluorescence spectroscopic studies were employed that took advantage of a tryptophan residue (W192) proximal to the histone substrate binding site. Briefly, 50 μM H4p21 peptide was titrated 2 μl at a time into 500 μl of 2.5 μM hMOF-wt or the hMOF-K274R or hMOF-K274A mutants. The resulting tryptophan fluorescence emission spectra were recorded using a Varian Cary Eclipse Fluorescence Spectrophotometer at 20 °C, with excitation wavelength at 295 nm.
Mass spectrometry analysis of recombinant proteins
Proteins to be analysed were resolved on SDS–PAGE and excised from the gel and subsequently destained using 200 μl of 200 mM ammonium bicarbonate and 50% acetonitrile for 30 min with shaking at 37 °C and then dried in a Speedvac. This was followed by reduction/alkylation of the protein band by adding 100 μl 20 mM TCEP in 25 mM ammonium bicarbonate at pH 8.0, incubated for 15 min at 37 °C with shaking. The supernatant was discarded and 100 μl of 40 mM iodoacetamide in 25 mM ammonium bicarbonate at pH 8.0 was added and incubated for 30 min at 37 °C with shaking. The supernatant was again discarded and two subsequent washes of 200 μl 25 mM ammonium bicarbonate were done for 15 min each with shaking. A final wash was done under the same conditions except that 50% acetonitrile, 25 mM ammonium bicarbonate was used for the wash. The bands were dried in the speed vac and were then rehydrated with 20 μl of 0.02 μg/μl modified Trypsin (Promega) in 40 mM ammonium bicarbonate overnight with shaking at 37 °C. The next morning, the supernatant was removed to a clean tube on ice and 20 μl of 40 mM ammonium bicarbonate was added to the gel piece for 30 min w/shaking at 37 °C. The supernatants were combined and 4 μl of neat acetic acid was added to stop the digest.
For peptide identification, 2–4 μl of each tryptic digest was injected onto a micro-precolumn (C18, 5 μm, 100 Å resin, LCPackings Acclaim PepMap100) in line with a nanocapillary reverse-phase C18 150 × 0.75 mm2 column (New Objective 75 μm column terminating in a nanospray 15 μm tip, packed with Microm Magic C18, 5 μm 200 Å resin) directly coupled to a Thermo Fisher Orbitrap XL mass spectrometer. The data were acquired using a data-dependent method, where MS/MS spectra for the six most intense ions were acquired in the ion trap while a high-resolution MS scan was performed in the orbitrap. A customized database was created, which contained the specific WT and mutant sequences as well as the Uniref human database with yeast and common contaminants (tagged). The database was indexed for partial tryptic searching and the resulting masses and MS/MS spectra from the Orbitrap XL were searched against the custom database using the SEQUEST search engine (Yates et al, 1995; Yates, 1998) through the BioWorks software version 3.3.1 SP1. The search results were filtered using 5 p.p.m., ΔCn of 0.07, and in some experiments, the data were further filtered to retain only full tryptic peptides. Extracted ion chromatograms (XIC) were obtained using Xcalibur version 2.0 SR2 software for peptides containing either yESA1 K262 or hMOF K274. The areas of three unrelated (non-acetylated) tryptic peptides from each sample were selected as internal controls to correct for sample processing variations. Sample specific response factors for each control peptide were calculated relative to the WT sample, averaged, and used to normalize the non-acetylated and acetylated peptides of interest.
For LC–MS/MS analysis of HAT proteins that were deacetylated and reacetylated, we first treated the recombinant yEsa1 and hMOF HAT domains (20 μM) with 2 μM of yeast Hst2 and 200 μM of NAD+ at room temperature for 16 h. One half of the sample was frozen at −20 °C as a ‘deacetylated’ sample for mass spectrometry. The remaining half of the sample was treated with 200 mM of nicotinamide to inhibit Hst2 activity and 200 μM of Ac-CoA was also added to facilitate autoacetylation. The treated solution was incubated at room temperature for 24 h before freezing at −20 °C as an ‘acetylated’ sample for mass spectrometry. The deacetylated and acetylated samples were resolved on SDS–PAGE and the corresponding bands were extracted for LC–MS/MS analysis as described above.
Analysis of native yEsa1
esa1 K262R and K262Q mutants were created by PCR-mediated mutagenesis on the pSAPE1 plasmid containing N-terminal HA epitope-tagged ESA1 open reading frame, under the control of its own promoter, on a ARS/CEN LEU2 vector (Selleck et al, 2005). Plasmids carrying the mutated forms of ESA1 were verified by sequencing and transformed into QY118 MATahis3Δ1 leu2Δ0 ura3Δ0 esa1Δ::KanMX covered by plasmid pLP795 (ESA1 ARS/CEN URA3; Clarke et al, 1999). Equivalent protein expression of each mutant was verified compared with WT by western blot on whole-cell extracts prepared in RIPA buffer (50 mM HEPES (pH 7.9), 2 mM EDTA, 0.25 M NaCl, 0.1% SDS, 0.1% DOC, 1% Triton X-100), separated on 10% acrylamide gels, transferred on nitrocellulose membranes and blotted with anti-HA HRP antibody (Roche; used at a dilution of 1/1000).
Viability tests were performed following a standard plasmid shuffling protocol. Exponentially growing yeasts containing the two plasmids carrying WT ESA1 (URA3) and the WT or mutant HA-tagged versions (LEU2) in medium lacking leucine and uracil were used in serial 10-fold dilution and spotted on synthetic complete media in the presence or the absence of 0.1% of 5′-FOA. Cells were grown at 30 °C for 2–4 days. The same results were obtained in two different genetic backgrounds.
Purified native NuA4 complex was isolated using a TAP-tagged Epl1 subunit as described before (Boudreault et al, 2003). The purified fraction was loaded on SDS–PAGE and the band corresponding to Esa1 was excised and analysed by LC–MS/MS. The gel bands were destained, reduced, modified with iodoacetamide and digested with trypsin using a method similar to that described above.
Peptide mixtures were analysed on an LTQ-Orbitrap XL hybrid mass spectrometer (Thermo Fisher, San Jose, CA, USA) coupled to an Eksigent nano-LC system (Eksigent, Dublin, OH, USA). Peptides were separated on a 150-μm × 10 cm analytical column packed in-house with a C18 resin (3 μm Jupiter particles; Supelco). Solvent A was 0.2% formic acid in milli-Q water and solvent B was 0.2% formic acid in 100% acetonitrile. Peptides were eluted from the column using a gradient from 5 to 80% acetonitrile over 70 min. The mass spectrometer was operated in a data-dependent mode with full scan (m/z 300–2000) MS spectra acquired at 60 000 resolution in the Orbitrap. The three most abundant precursor ions were selected for fragmentation in the LTQ using collision-induced dissociation at a CE setting of 35. Fragment ions were analysed in the ion trap. The raw centroid data were searched against a custom database with Mascot search engine v2.10 (Matrix Science). All spectra containing Esa1 lysine 262 were manually confirmed from the raw data.
Analysis of hMOF
For analysis of the acetylation state of hMOF in human cells, H1299 cells were transfected with pcDNA3.1-FLAG-hMOF, generated by PCR amplification of the hMOF cDNA and subsequently ligated into the pcDNA3.1-TOPO-TA cloning vector (Sykes et al, 2006). FLAG-epitope-tagged hMOF was affinity purified from nuclear extracts and eluted with FLAG-peptide. Eluted hMOF protein was separated by SDS–PAGE and excised for mass spectrometry analysis (Taplin Facility-Harvard University).
For the analysis of hMOF and the hMOF-K274R mutant in vivo, pcDNA4 V5/His-hMOF plasmid was obtained as a gift from Dr Kaoru Tominaga and pRC-p53 was a gift from Dr Maureen Murphy. Quick change site-directed mutagenesis (Stratagene) was used to create the K274R single amino-acid substation in hMOF. H1299 cells were maintained in DMEM (Mediatech) with 10% FBS (Foundation) and cultures were kept at 37 °C under 5% CO2. HeLa cells were maintained in DMEM (Invitrogen) with 15% FBS (Thermo Scientific) and grown at 37 °C, under 6% CO2 and 3% O2. Transfections of V5/His-hMOF plasmids were performed using Lipofectamine 2000 (Invitrogen), according to the manufacturer's guidelines. Twenty-four hours posttransfection, cells were washed with cold PBS and lysated in 10 pellet volumes of buffer containing 20 mM NaH2PO4, 150 mM NaCl, 0.5% IGEPAL (NP-40), 30 mM sodium pyrophosphate, 10% glycerol supplemented with 30 mM sodium butyrate and protease inhibitor cocktail. To detect acetylated p53, 500 μg of lysate were immunoprecipitated with a modification-specific antibody directed against acetyl-K120 on p53. Total p53, hMOF and actin levels were quantitated by western blotting of lysates. The antibodies used were as follows: p53 DO1 (sc-126), p53 FL393G (sc-6243) and actin C-2 (sc-8432) from Santa Cruz and V5 (46-0720) from Invitrogen. For immunofluorescence microscopy-based detection of H4K16Ac and V5-hMOF in HeLa cells, 24 h posttransfection, cells were not treated or were subjected to 10 Gy γ irradiation in a Cs-137 irradiator. The cells were then incubated for an additional 2 h at 37 °C, then fixed in 3.7% formaldehyde, permeabilized in 0.1% Triton X-100 and blocked in 1% BSA. Costaining of V5-hMOF variants and acetylated H4K16, respectively, was performed using the following primary antibodies: anti-V5 (mouse, Invitrogen, R960-25, 1:500) and anti-H4K16Ac (rabbit, Active Motif, 39167, 1:400), and DNA was stained with DAPI. Cells were imaged using a Zeiss Axioplan 2 widefield fluorescence microscope and the accompanying AxioVision (Rel. 4.5) software. Statistical analyses of the resulting data were performed using a two-tailed Fisher's exact test.
Analysis of native yeast Sas2
Plasmid pAE778 carrying N-terminally Myc-tagged WT SAS2 clone was obtained from Ann Ehrenhofer-Murray. Substitution mutations were generated on pAE778 by QuikChange (Stratagene). Strain FEP210b containing a URA3 insertion in the TEL11L-XR element was obtained from Ed Louis. YWD565 bearing sas2Δ::kanMX4 was generated from FEP210b by the standard protocol (Longtine et al, 1998). Telomere silencing assays were performed as described (Sanders et al, 2009). For western blotting, yeast cells from 25 ml log-phase cultures were lysed by beadbeating with silica beads (BioSpec) and sonication (Diagenode). Whole-cell extracts were analysed immediately on a 4–12% Bis-Tris SDS gel (Invitrogen) followed by western blotting as described (Dang et al, 2009). Antibodies specific to c-myc (Sigma), β-actin (Abcam) and histone H3 (Abcam) were used in western blot detection.
Accession codes
The atomic coordinates and structure factors for yEsa1/CoA (3T07), yEsa1(E338Q)/CoA (3T09), yEsa1/H4K16CoA (3T06), hMOF (3TOA) and hMOF (E350Q) (3TOB) have been deposited in the Protein Data Bank.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH to RM (GM060293), RM, SLB and FBJ (AG031862), RS (GM55641 and GM88297), SBM (CA090465 and CA098172), and YGZ (GM086717); and a grant from the Canadian Institutes of Health Research to JC (MOP-14308), PT and AV This work was also supported by the Commonwealth Universal Research Enhancement Program, Pennsylvania Department of Health. We thank the Wistar Proteomics core facility funded by NIH grant CA 010815 for their help with the mass-spectrometry work presented in this study.
Author contributions: HY carried out all the crystallographic experiments with supervision from RM; HY, SH and ECD carried out all of the in vitro biochemical experiments with supervision from RM; DR carried out all of the cellular and molecular biology studies on yEsa1 under the supervision of JC; HM and JJ carried out the cellular and molecular biology studies on hMOF under the supervision of SBM and FBJ, respectively; WD, RP and MS carried out all of the cellular and molecular biology studies on ySas2 under the supervision of SLB (WD, RP) and RS (MS), respectively; KS carried out the mass spectrometry analysis of recombinant proteins under the supervision of DWS; NA carried out the mass spectrometry analysis of intact yEsa1 from the NuA4 complex under the supervision of PT and AV; JW and CY prepared the H4K16CoA inhibitor under the supervision of YGZ; RM wrote the initial draft of the manuscript and HY assembled the figures; HY prepared Figures 1A–E and 2 and Supplementary Figures S1A, B, C and E and S3 and Table I under the supervision of RM; KS prepared Figure 1F and 4A and Supplementary Figure S1D and Table II under the supervision of DWS; NA prepared Figure 3A under the supervision of PT and AV; DR prepared Figure 3B and C under the supervision of JC; HM prepared Figure 4B under the supervision of SBM; JJ prepared Figure 4C and Supplementary Figure S4 under the supervision of FBJ; WD and RP prepared Figure 4D and E under the supervision of SLB; all authors reviewed and edited the manuscript.
Footnotes
The authors declare that they have no conflict of interest.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allard S, Utley RT, Savard J, Clarke A, Grant P, Brandl CJ, Pillus L, Workman JL, Cote J (1999) NuA4, an essential transcription adaptor/histone H4 acetyltransferase complex containing Esa1p and the ATM-related cofactor Tra1p. EMBO J 18: 5108–5119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avvakumov N, Cote J (2007) Functions of myst family histone acetyltransferases and their link to disease. Subcell Biochem 41: 295–317 [PubMed] [Google Scholar]
- Berndsen CE, Albaugh BN, Tan S, Denu JM (2007) Catalytic mechanism of a MYST family histone acetyltransferase. Biochemistry 46: 623–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreault AA, Cronier D, Selleck W, Lacoste N, Utley RT, Allard S, Savard J, Lane WS, Tan S, Cote J (2003) Yeast enhancer of polycomb defines global Esa1-dependent acetylation of chromatin. Genes Develop 17: 1415–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325: 834–840 [DOI] [PubMed] [Google Scholar]
- Clarke AS, Lowell JE, Jacobson SJ, Pillus L (1999) Esa1p is an essential histone acetyltransferase required for cell cycle progression. Mol Cell Biol 19: 2515–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, Shilatifard A, Kaeberlein M, Kennedy BK, Berger SL (2009) Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 459: 802–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Hodawadekar SC, Marmorstein R (2007) Chemistry of acetyl transfer by histone modifying enzymes: structure, mechanism and implications for effector design. Oncogene 26: 5528–5540 [DOI] [PubMed] [Google Scholar]
- Kadlec J, Hallacli E, Lipp M, Holz H, Sanchez-Weatherby J, Cusack S, Akhtar A (2011) Structural basis for MOF and MSL3 recruitment into the dosage compensation complex by MSL1. Nat Struct Mol Biol 18: 142–149 [DOI] [PubMed] [Google Scholar]
- Lafon A, Chang CS, Scott EM, Jacobson SJ, Pillus L (2007) MYST opportunities for growth control: yeast genes illuminate human cancer gene functions. Oncogene 26: 5373–5384 [DOI] [PubMed] [Google Scholar]
- Li X, Corsa CA, Pan PW, Wu L, Ferguson D, Yu X, Min J, Dou Y (2010) MOF and H4 K16 acetylation play important roles in DNA damage repair by modulating recruitment of DNA damage repair protein Mdc1. Mol Cell Biol 30: 5335–5347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wu L, Corsa CA, Kunkel S, Dou Y (2009) Two mammalian MOF complexes regulate transcription activation by distinct mechanisms. Mol Cell 36: 290–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Yuan YA (2008) Structural insights into histone H3 lysine 56 acetylation by Rtt109. Structure 16: 1503–1510 [DOI] [PubMed] [Google Scholar]
- Lin YY, Lu JY, Zhang J, Walter W, Dang W, Wan J, Tao SC, Qian J, Zhao Y, Boeke JD, Berger SL, Zhu H (2009) Protein acetylation microarray reveals that NuA4 controls key metabolic target regulating gluconeogenesis. Cell 136: 1073–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14: 953–961 [DOI] [PubMed] [Google Scholar]
- Lu L, Li L, Lv X, Wu XS, Liu DP, Liang CC (2011) Modulations of hMOF autoacetylation by SIRT1 regulate hMOF recruitment and activities on the chromatin. Cell Res 21: 1182–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein R (2001) Structure and function of histone acetyltransferases. Cell Mol Life Sci 58: 693–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein R, Trievel RC (2009) Histone modifying enzymes: structures, mechanisms, and specificities. Biochim Biophys Acta 1789: 58–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40: 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes C, Fischer PM (2005) Strategies for the design of potent and selective kinase inhibitors. Curr Pharm Des 11: 1845–1863 [DOI] [PubMed] [Google Scholar]
- Megee PC, Morgan BA, Mittman BA, Smith MM (1990) Genetic analysis of histone H4: essential role of lysines subject to reversible acetylation. Science 247: 841–845 [DOI] [PubMed] [Google Scholar]
- Morgan DO (1995) Principles of CDK regulation. Nature 374: 131–134 [DOI] [PubMed] [Google Scholar]
- Nolen B, Taylor S, Ghosh G (2004) Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell 15: 661–675 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. In Methods Enzymol, Vol. 276, part A edn, pp 307–326. New York: Academic Press [DOI] [PubMed] [Google Scholar]
- Pryde FE, Louis EJ (1999) Limitations of silencing at native yeast telomeres. EMBO J 18: 2538–2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman CS, Li H, Martasek P, Southan G, Masters BS, Poulos TL (2001) Crystal structure of nitric oxide synthase bound to nitro indazole reveals a novel inactivation mechanism. Biochemistry 40: 13448–13455 [DOI] [PubMed] [Google Scholar]
- Sanders BD, Jackson B, Brent M, Taylor AM, Dang W, Berger SL, Schreiber SL, Howitz K, Marmorstein R (2009) Identification and characterization of novel sirtuin inhibitor scaffolds. Bioorg Med Chem 17: 7031–7041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapountzi V, Cote J (2010) MYST-family histone acetyltransferases: beyond chromatin. Cell Mol Life Sci 68: 1147–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selleck W, Fortin I, Sermwittayawong D, Cote J, Tan S (2005) The Saccharomyces cerevisiae Piccolo NuA4 histone acetyltransferase complex requires the Enhancer of Polycomb A domain and chromodomain to acetylate nucleosomes. Mol Cell Biol 25: 5535–5542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma GG, So S, Gupta A, Kumar R, Cayrou C, Avvakumov N, Bhadra U, Pandita RK, Porteus MH, Chen DJ, Cote J, Pandita TK (2010) MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double-strand break repair. Mol Cell Biol 30: 3582–3595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ER, Cayrou C, Huang R, Lane WS, Cote J, Lucchesi JC (2005) A human protein complex homologous to the Drosophila MSL complex is responsible for the majority of histone H4 acetylation at lysine 16. Mol Cell Biol 25: 9175–9188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KT, Workman JL (2009) Introducing the acetylome. Nat Biotechnol 27: 917–919 [DOI] [PubMed] [Google Scholar]
- Spange S, Wagner T, Heinzel T, Kramer OH (2009) Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol 41: 185–198 [DOI] [PubMed] [Google Scholar]
- Stavropoulos P, Nagy V, Blobel G, Hoelz A (2008) Molecular basis for the autoregulation of the protein acetyl transferase Rtt109. Proc Natl Acad Sci USA 105: 12236–12241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Guo S, Tang Q, Li C, Zeng R, Xiong Z, Zhong C, Ding J (2011) Regulation of the histone acetyltransferase activity of hMOF via autoacetylation of Lys274. Cell Res 21: 1262–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton A, Shia WJ, Band D, Kaufman PD, Osada S, Workman JL, Sternglanz R (2003) Sas4 and Sas5 are required for the histone acetyltransferase activity of Sas2 in the SAS complex. J Biol Chem 278: 16887–16892 [DOI] [PubMed] [Google Scholar]
- Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB (2006) Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell 24: 841–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale M, Rea S, Richter K, Vilar A, Lichter P, Imhof A, Akhtar A (2005) hMOF histone acetyltransferase is required for histone H4 lysine 16 acetylation in mammalian cells. Mol Cell Biol 25: 6798–6810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Holbert MA, Wurtele H, Meeth K, Rocha W, Gharib M, Jiang E, Thibault P, Verreault A, Cole PA, Marmorstein R (2008) Fungal Rtt109 histone acetyltransferase is an unexpected structural homolog of metazoan p300/CBP. Nat Struct Mol Biol 15: 738–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Luo J, Zhang W, Gu W (2006) Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell 24: 827–839 [DOI] [PubMed] [Google Scholar]
- Thompson PR, Wang D, Wang L, Fulco M, Pediconi N, Zhang D, An W, Ge Q, Roeder RG, Wong J, Levrero M, Sartorelli V, Cotter RJ, Cole PA (2004) Regulation of the p300 HAT domain via a novel activation loop. Nat Struct Mol Biol 11: 308–315 [DOI] [PubMed] [Google Scholar]
- Vaquero A, Scher MB, Lee DH, Sutton A, Cheng HL, Alt FW, Serrano L, Sternglanz R, Reinberg D (2006) SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Develop 20: 1256–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Tang Y, Cole PA, Marmorstein R (2008) Structure and chemistry of the p300/CBP and Rtt109 histone acetyltransferases: implications for histone acetyltransferase evolution and function. Curr Opin Struct Biol 18: 741–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, Ning ZB, Zeng R, Xiong Y, Guan KL, Zhao S, Zhao GP (2010) Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327: 1004–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Xie N, Wu Z, Zhang Y, Zheng YG (2009) Bisubstrate inhibitors of the MYST HATs Esa1 and Tip60. Bioorg Med Chem 17: 1381–1386 [DOI] [PubMed] [Google Scholar]
- Yan Y, Barlev NA, Haley RH, Berger SL, Marmorstein R (2000) Crystal structure of yeast Esa1 suggests a unified mechanism of catalysis and substrate binding by histone acetyltransferases. Mol Cell 6: 1195–1205 [DOI] [PubMed] [Google Scholar]
- Yan Y, Harper S, Speicher DW, Marmorstein R (2002) The catalytic mechanism of the ESA1 histone acetyltransferase involves a self-acetylated intermediate. Nature Struct Biol 9: 862–869 [DOI] [PubMed] [Google Scholar]
- Yates JR III (1998) Database searching using mass spectrometry data. Electrophoresis 19: 893–900 [DOI] [PubMed] [Google Scholar]
- Yates JR III, Eng JK, McCormack AL, Schieltz D (1995) Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal Chem 67: 1426–1436 [DOI] [PubMed] [Google Scholar]
- Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q et al. (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327: 1000–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.