

Abstract
Helicases must unwind DNA at the right place and time to maintain genomic integrity or gene expression. Biologically critical XPB and XPD helicases are key members of the human TFIIH complex; they anchor CAK kinase (cyclinH, MAT1, CDK7) to TFIIH and open DNA for transcription and for repair of duplex distorting damage by nucleotide excision repair (NER). NER is initiated by arrested RNA polymerase or damage recognition by XPC-RAD23B with or without DDB1/DDB2. XP helicases, named for their role in the extreme sun-mediated skin cancer predisposition xeroderma pigmentosum (XP), are then recruited to asymmetrically unwind dsDNA flanking the damage. XPB and XPD genetic defects can also cause premature aging with profound neurological defects without increased cancers: Cockayne syndrome (CS) and trichothiodystrophy (TTD). XP helicase patient phenotypes cannot be predicted from the mutation position along the linear gene sequence and adjacent mutations can cause different diseases. Here we consider the structural biology of DNA damage recognition by XPC-RAD23B, DDB1/DDB2, RNAPII, and ATL, and of helix unwinding by the XPB and XPD helicases plus the bacterial repair helicases UvrB and UvrD in complex with DNA. We then propose unified models for TFIIH assembly and roles in NER. Collective crystal structures with NMR and electron microscopy results reveal functional motifs, domains, and architectural elements that contribute to biological activities: damaged DNA binding, translocation, unwinding, and ATP driven changes plus TFIIH assembly and signaling. Coupled with mapping of patient mutations, these combined structural analyses provide a framework for integrating and unifying the rich biochemical and cellular information that has accumulated over forty years of study. This integration resolves puzzles regarding XP helicase functions and suggests that XP helicase positions and activities within TFIIH detect and verify damage, select the damaged strand for incision, and coordinate repair with transcription and cell cycle through CAK signaling.
INTRODUCTION
DNA-based information, which forms the foundation of all cell biology, depends critically upon the ability of cells to constantly and accurately repair their DNA [1-4]. Nucleotide excision repair (NER) is one of the classical general damage repair pathways. NER detects helix distorting lesions regardless of their nature, opens the DNA flanking the damage, removes a patch containing the damage, and replaces the sequence by a find, open, verify, cut, and patch mechanism [5, 6]. The inherited defect xeroderma pigmentosum (XP) in NER was first described by Hebra & Kaposi in 1874, and the roles of both inheritance and sunlight were clearly recognized [7] with intensive research beginning in the early 1970’s[8, 9]. Over forty years of research on NER has produced rich biochemical and cellular information, and more recently, structural biology has provided key insights for not only atomic mechanisms but for disease phenotypes.
Herein we focus on the structural biochemistry of initial NER damage recognition and helix opening. This focus centers on the XP helicases in the light of human disease-causing mutations, and on the results from powerful prokaryotic systems that provide comparative understanding of conserved and variable features. Putting together structures for a DNA repair pathway, as was first done for the base excision repair (BER) pathway [10] helps to understand the mechanistic foundation for pathway steps and coordination. Here we use this approach for XPB and XPD to identify conserved themes of DNA damage detection, verification, and removal. We also examine motifs, domains, and conformational controls that are required for functional interactions within the DNA repair and transcription factor, TFIIH, that are essential for efficient recruitment to sites of damage and CAK-mediated signaling events that trigger transcription, cell cycle checkpoints, and DNA repair [11]. Our analyses suggest that recruitment of bacterial and eukaryotic NER helicases to DNA lesions occurs by common mechanisms through evolutionarily related activities. We furthermore suggest general principles for damage recognition and repair pathway coordination. Collective data supports the concept that DNA linked XPB-XPD-CAK interactions form a keystone complex that orchestrates DNA damage recognition and verification by TFIIH to coordinate repair with transcription and cell cycle.
In general, NER processes are exciting exemplary systems not only for characterizing the interplay of repair with transcription but also for revealing how such knowledge can provide a deepened understanding and even prediction of biological outcome [12-14]. Furthermore, we now understand that combined structural, biochemical, and genetic methods can provide both detailed structural information and useful insights into functionally important dynamics that impact outcomes [15]. This level of mechanistic understanding has real biological relevance and provides the foundation to develop approaches to medical interventions that promise to be robust and practical.
Excision repair pathways in eukaryotes and bacteria are valuable comparative systems
A powerful understanding of eukaryotic NER came from the breakthrough ability to reconstitute the required proteins in vitro, so the key players of NER are known [5, 16, 17]. Furthermore, a key concept for understanding NER is the importance of sequential assembly rather than having all components present as a preassembled repairosome [18, 19]. NER that occurs globally in the genome (GG-NER) or during transcription (transcription coupled or TC-NER) differ only in their initial recognition of helix-distorting DNA damage by XPC either with or without DDB1/DDB2 for GC-NER and by RNA polymerase for TC-NER [20, 21] (Fig. 1 and considered in more detail below). Following initial damage recognition, XPA, RPA, XPG, and the TFIIH complex including the XPB and XPD helicases are recruited. TFIIH is a ten subunit complex (containing XPB, p62, p52, p44, p34, TTDA (p8), and XPD) plus the Cdk-activating-kinase (CAK) (cdk7, cyclinH and MAT1) [22]. XPB, with 3’-5’ polarity, and XPD, with 5’-3’ polarity, open the DNA helix to form a 27-nucleotide bubble asymmetrically flanking the damage (22 nts in 5’ and 5 nts in 3’) [23], verify the damage [24, 25], and interact with the CAK kinase to signal transcription and repair status [26, 27]. Following helix opening, XPF in complex with ERCC1 makes an incision 5’ to the damage, and the structure-specific endonuclease XPG makes an incision 3’ to the damage [28]. This dual incision by XP nucleases leads to the removal of ~30 nucleotides (nts) resulting in a single strand gap [29]. This gap, which is the same size for GG-NER and TC-NER, is filled by DNA polymerase δ and κ, or ε [30], which use the undamaged strand as a template, and rejoined by ligase. Although the patch is relatively short compared to DNA replication, proliferating cell nuclear antigen (PCNA) coordinates DNA synthesis and ligation and replication protein A (RPA) protects the ssDNA strand from degradation. This mechanism of eukaryotic NER roughly resembles excision repair in prokaryotes—removal of diverse bulky and helix-distorting lesions from DNA by the coordinated actions of damage detection, helix opening, and gap excision followed by repair synthesis (Fig. 1). In fact, functional similarities between NER in bacteria and eukaryotes prompt us to herein compare the structural biochemistry for the key helicases in bacteria and humans.
Figure 1. Structural basis for early NER steps in humans (right) and bacteria (left).
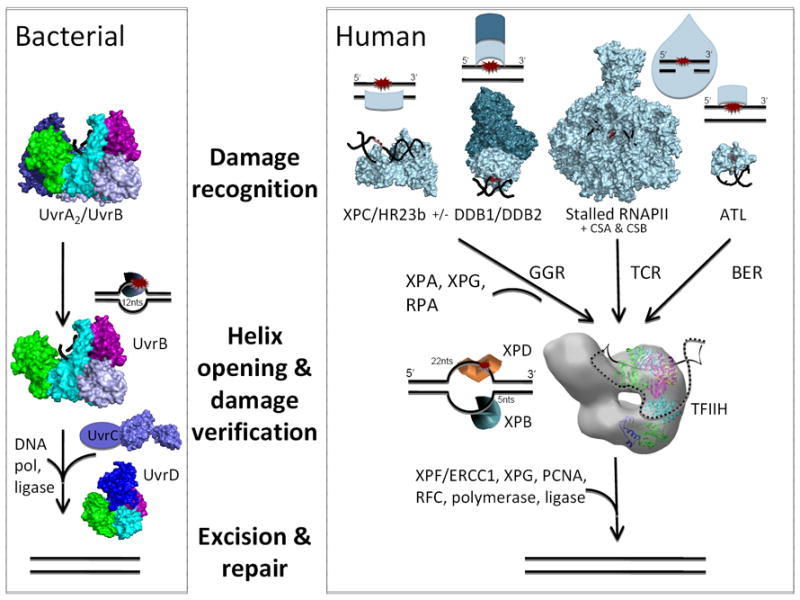
During human global genome repair (GGR), DNA damage is recognized by the XPC/RAD23B complex (Rad4/Rad23 pdb 2QSG). CPD (dotted line) was not visible in the crystal [60]. For some damage, DDB1/DDB2 (pdb 3EI1 bound to 6-4PP [63]) complex aids recognition. During transcription, the RNA polymerase (RNAPII pdb 2JA7 with CPD [68]) stalls at a lesion, recruiting CSB and CSA. Lesions that cannot be repaired by base excision repair (BER) can be shuttled to NER by the ATL protein (pdb 3GYH shown bound to cigarette smoke derived lesion O(6)-4-(3-pyridyl)-4-oxobutylguanine [47]). For clarity, cartoons next to the crystal structure molecular surfaces show which strand is bound. Regardless of recognition method, XPA, XPG, RPA, and TFIIH bind such that the XPD (pdb 3CRV [79]) and XPB (pdb 2FWR [78]) helicases can open dsDNA into a 27-nt bubble suitable for excision by XPF/ERCC1 and XPG nucleases. The gap is then filled by DNA polymerase delta and kappa, or epsilon bound to PCNA, which is loaded onto DNA by RFC and ligated. In bacteria, the UvrB helicase (pdb 2FDC with DNA [86]) together with a dimer of UvrA (pdb 2R6F [177]) recognize the lesion. UvrB opens the helix and the recruited nuclease UvrC (pdb 2NRT C-terminal domain [178]) cuts on both sides of the lesion. The UvrD (pdb 2IS6 [84]) helicase removes the excised product and the gap is filled by DNA pol I.
In prokaryotes, UvrA, UvrB and UvrC orchestrate the NER damage recognition and incision steps (Fig. 1) [31, 32]. UvrA, as of an A2B or A2B2 complex, scans DNA to recognize defects, allowing UvrB to find and verify the damage site. ATP hydrolysis is required for UvrA disassociation and UvrB becomes stably bound to the lesion promoting local destabilization of the dsDNA. In this complex, UvrB C-terminal domain is exposed to form a UvrBC-complex that makes dual incisions first on the 3’-side and then on the 5’-side of the lesion. UvrB evidently employs its ATPase activity to place the DNA in a strained conformation suitable to be recognized and incised by UvrC. The UvrD helicase unwinds the 12-nucleotide tract containing the lesion with 3’-5’ polarity after the incisions have occurred. Pol I then fills the gap and removes UvrB from the DNA and ligase seals the gap. The structures and biochemistry of these prokaryotic systems provide valuable comparative results for a mechanistic foundation critical for understanding and interventions.
NER diseases, cancer, and aging
Tragic failures in NER are associated with extreme sun sensitivity, cancer risks, neurodegeneration, and premature aging; however, these defects also provide deep insights into cell biology and hope for future knowledge-based interventions [13, 33-35]. XP helicase mutations associated with disease evidently result in deficiencies of the TFIIH complex, and disease phenotypes are complicated by TFIIH’s dual role in both NER and transcription [36, 37]. Emerging roles for XPD that are independent of TFIIH further complicate but enlighten our understanding of human disease [38]. However, considerable insight into human NER processes has come from examination of multiple rare, autosomal disorders that result from defects in NER recognition, duplex opening and excision processes: xeroderma pigmentosum (XP), Cockayne Syndrome (CS), trichothiodystrophy (TTD), and cerebro-oculo-facio-skeletal syndrome (COFS) [34, 39-42] (see also articles by Diderich et al. and Gregg et al. in this issue of DNA Repair). XP is characterized by solar hypersensitivity, extreme predisposition for cancers on areas exposed to sunlight, and neurological abnormalities in some patients. XP can be caused by mutations in one of eight genes (XPA-G and XPV). Most CS patients have defects in the CSA or CSB genes, but in rare cases, some XP-B, XP-D, and XP-G patients have XP combined with CS. CS patients also have sun sensitivity, but without increased skin cancer risk. Also in contrast to XP, CS patients have reduced stature, mental retardation, and segmental aging features such as cachexia (loss of subcutaneous fat tissue), sensorineural deafness, retinal degeneration, and calcification of the central nervous system [39]. COFS shares clinical similarities with CS but is of prenatal onset [41] and is caused by defects in XPG, XPD, or CSB. Defects in XPB, XPD, and TTDA (p8) cause TTD, which has the hallmark characteristic of sulfur-deficient brittle hair that shows a tiger tail pattern under polarized light. TTD patients may have sun sensitivity but do not have increased cancer risk and have short stature, progressive cognitive impairment, and abnormal face shape [39]. Like CS, TTD is considered a segmental premature aging syndrome. Although seen in some patients, premature aging features such as reduced lifespan, cachexia, osteoporosis, kyphosis (hunchback posture), and early graying are most clearly seen in TTD mouse models [43]. In extremely rare cases, mutations in XPD can cause XP combined with TTD [44].
Eukaryotic NER presents puzzles for structural biology
GG-NER is most efficient at targeting bulky lesions that disrupt or distort the DNA double helix, such as pyrimidine dimers (cyclobutane pyrimidine dimers and 6-4 photoproducts) caused by the UV component of sunlight [45]. Yet, NER can also be initiated during transcription by stalled RNAPII [46] or even by other proteins such as ATL that recognizes and stays bound to alkylation damage [47], thus expanding the spectrum of lesions that NER can act upon. In fact, TC-NER can repair abasic (AP) sites generated by glycosylases [48] as the RNA polymerase is arrested by AP sites on the transcribed strand [49]. Other NER substrates include bulky chemical adducts, oxidative damage [50, 51], and DNA intra-strand cross-links [52, 53]. Nevertheless the hallmark of lesions preferentially recognized by NER is that they cause both a modification of DNA chemistry and a distortion of DNA double helical structure, which places special requirements on the structural biochemistry of NER damage recognition and opening for excision. These initial NER steps present six key puzzles for the structural biology that we consider throughout this review: 1) how damage is recognized, 2) why the need for two SF2 helicases (XPB and XPD) to open a bubble around the damage, 3) why the repair bubble is 27 nt and asymmetric to the lesion 4) how damage is verified, 5) how the presence of this damage is signaled to downstream processes, and 6) how repair enzyme products are handed off to avoid release of toxic and mutagenic intermediates. We address these puzzles by integrating structural, cellular, and biochemical results into a unified bind-pry-unwind model for the initial steps of NER.
Damage recognition – insights from XPC, DDB, RNAPII, and ATL
Damage recognition for NER involves proteins that bind to either the damaged site or to the strand opposite the damage (Fig.1). Different proteins act in the recognition of the DNA damage in GG-NER and TC-NER. In GG-NER, the XPC-RAD23B complex acts in distortion recognition, and DDB1 and DDB2 (XPE) can also recognize lesions caused by UV light [54]. In TC-NER, RNA polymerase II acts in initial damage recognition instead of XPC-RAD23B. If RNA polymerase II encounters damage and is arrested, CSB recognizes the stalled polymerase and recruits CSA, chromatin remodelers, and NER proteins including XPG and TFIIH for efficient repair [20]. In vitro evidence suggests that recognition of the stalled polymerase is coordinated by both CSB and XPG [55]. Recent evidence suggests that NER factors may be recruited to certain kinds of weakly distorting lesions such as O6-alkylG by the presence of bound proteins such as ATL [47, 56].
The identification of XPC as one of the earliest acting GG-NER factors gives insights into the order of NER component actions [18]. XPC likely acts as the initial sensor for unpaired bases for the global NER pathway. XPC forms a heterotrimeric complex with RAD23B and centrin 2 that binds NER-specific lesions, bubbles, and loop structures. DNA is continually scanned for lesions that cause helix distortions by XPC-RAD23B [57] and by the UV-DNA-damage binding (UV-DDB or DDB1-DDB2) complex [58, 59]. The crystal structure of the yeast XPC orthologue Rad4 with yeast Rad23 bound to DNA containing a single CPD lesion surprisingly revealed that Rad4/Rad23 do not bind the damaged strand, but recognize local destabilization of base pairing and inserts a hairpin motif into the DNA helix [60] (Fig. 1). This insertion causes the two damaged nucleotides to flip out of the double helix in a conformation that places the CPD lesion too far from Rad4 to make any direct contacts and is disordered in the crystal structure [60]. This is distinct from base flipping in BER by DNA glycosylases that flips a single base into the enzyme active site [61, 62]. Rather, Rad4 binds the DNA through three beta hairpin domains (BHD1-3) and a transglutaminase-homology domain (TGD). BHD1 and TGD bind an 11-bp region of fully paired, undamaged double-stranded DNA. Several nucleotides away, BHD3 inserts a β-hairpin feature through the Watson-Crick helix to flip the damaged nucleotides out, while a groove in BHD2 and BHD3 holds the corresponding undamaged nucleotides like the palm of a hand [60]. By binding the strand opposite the lesion, Rad4 (XPC) can accommodate the wide range of bulky lesions that are targets of NER (for a review focusing on the damage recognition in NER see review by Naegeli and Sugasawa in this issue of DNA Repair).
In contrast to Rad4/Rad23, structures of DDB1–DDB2 bound to DNA containing a 6-4 pyrimidine-pyrimidone photodimer (6-4PP) lesion or an abasic site mimic show that DDB2 mainly binds the damaged strand (Fig. 1) and flips the damaged bases into a shallow binding pocket [63]. DDB2 (XPE) is a WD40 repeat β propeller protein and the full diameter of the β propeller is used to bind DNA [63]. The larger DDB1 protein does not participate in DNA binding and binds the DDB2 β propeller on the side opposite from the DNA. DDB2 inserts a hairpin into the minor groove, extrudes the photodimer into a binding pocket, and kinks the duplex. DDB2 also stabilizes the nucleotides opposite the damage resulting in a slight unwinding of the DNA around the damage [63]. The localized probing of photolesions combined with the proofreading by the photodimer pocket, allows DDB2 to detect CPD lesions that make smaller perturbations in DNA than most bulky lesions and explains the more limited range of lesions that DDB2 binds versus XPC [63]. In BER, structures of the Endonuclease V–DNA complex reveal a wedge of four residues (PYIP) that inserts into the minor groove of the helix. DDB2 [64] flips the photodimer somewhere between the 90° rotation of the base lesion in EndoV and the precision extrusion of a single nucleotide of almost 180° by DNA glycosylases [65].
In some bacteria and eukaryotes alkyltransferase-like (ATL) proteins provide a non-classical means to initiate NER. ATL protects DNA from alkylation damage, and shares functional motifs with chemotherapy target O6-alkylguanine DNA-alkyltransferase [47, 66, 67]. Yet, ATLs lack alkyltransferase activity but employ non-enzymatic nucleotide flipping plus DNA distortion to convert an NER invisible lesion into a visible one by forming a stable, distorted ATL-DNA base lesion complex suitable for functional interaction with NER proteins. DNA-protein contacts in ATL1-DNA complex structures are with the damaged strand (Fig. 1), analogously to DDB2, but would allow simultaneous binding by XPC-RAD23B which binds the opposite undamaged DNA strand [60]. These results suggest that the functional connection of base damage to NER requires only two features: 1) stable specific binding to base damage, and 2) the ability to interact functionally with NER excision partners, such as ERCC1-XPF and XPG. In fact, the ATL C-terminal loop resembles the loop that recruits the nuclease heterodimer XPF-ERCC1 in XPA, the classical NER damage binding protein [47].
During transcription, damage on the transcribed strand stalls the RNA polymerase and initiates TC-NER that preferentially repairs the transcribed strand of active genes over bulk DNA [46]. Crystal structures of yeast RNA pol II with four different CPD-containing DNA and RNA scaffolds reveal that the polymerase stalls after nucleotide incorporation opposite both CPD thymines such that the damage is found deep within the protein [68] (Fig. 1). Surprisingly, the conformation of the polymerase is the same in all of the CPD-containing structures solved, arguing that the signal for TC-NER is not an altered conformation of the polymerase itself. However, the downstream DNA was found in different positions in two of the RNAPII-CPD structures. Since XPB binds the downstream DNA during transcription initiation in both human and yeast systems [69, 70], XPB may recognize this altered downstream DNA conformation during repair and position TFIIH [68]. Furthermore, Kim et al. [69] proposed that XPB promotes DNA melting and promoter escape by rotating DNA downstream of the transcription bubble relative to the upstream DNA that is rotationally fixed by transcription factors and RNAPII. We propose to extend this model to DNA repair by suggesting that the stalled RNAPII in TC-NER and XPC-RAD23B (aided by DDB1-DDB2 or ATL when needed) in GG-NER serve to rotationally fix the DNA so that XPB can wedge open the DNA and allow NER pathway progression. Therefore, DNA damage recognition proteins not only recruit TFIIH through protein-protein interactions [71, 72], but they also provide DNA binding activity that promotes DNA opening by XPB, ensuring that DNA opening is properly linked to damage recognition.
Rad51/RecA domains provide a unifying structure for NER helicases
DNA helicases are macromolecular motors that catalyze the separation of the DNA duplex by moving along DNA powered by ATP binding and hydrolysis. Structurally, NER helicases are united by their conserved Rad51/RecA-like ATPase domains [73-75], which contain the ATPase and helicase motifs that are characteristic of superfamily 2 helicases. Helicases have seven conserved “helicase motifs” (Walker motif I, Ia, II, III, IV, V and VI) [76] and are grouped into three superfamilies (SF), SF1, SF2 and SF3, based on similarity and organization of these conserved motifs [77]. That XPB and XPD are both SF2 family helicases creates puzzles regarding why two SF2 helicases are required in TFIIH (For a review on functional aspects of TFIIH, see review by Egly and Coin in this issue of DNA Repair).
Crystal structures of Archaeoglobus fulgidus XPB (AfXPB) and archaeal XPDs reveal that in XPB and XPD, these Rad51/RecA domains, connected by a flexible hinge, share an α-β fold and are called helicase domains 1 and 2 (HD1 and HD2) [78-81](Fig. 2). HD1 and HD2 pack against each other to form an interface cleft that brings together motifs I, II, V, and VI to form a composite ATP-binding site. This architecture is consistent with an inchworm model where cycles of ATP binding, hydrolysis, and release are coupled to opening and closing of HD1 and HD2, which drives the enzyme along DNA [82]. The structural relationships of such motifs involved in converting ATP binding to conformation change are critical; for example, the structural placement of the signature motif for the ABC ATPases provided key understanding of their structure-function relationships [83]. Crystal structures of Bacillus caldotenax UvrB and Escherichia coli UvrD reveal that UvrB and UvrD also have Rad51/RecA domains with α-β folds [84-86] (Fig. 2). In all of the NER helicases, the helicase domains that drive helix unwinding are not sufficient for their biological roles in opening DNA bubbles for excision. Rather they require the presence of accessory domains that play roles in duplex splitting or damage recognition (Fig. 2).
Figure 2. NER helicase crystal structures and schematic sequence regions.
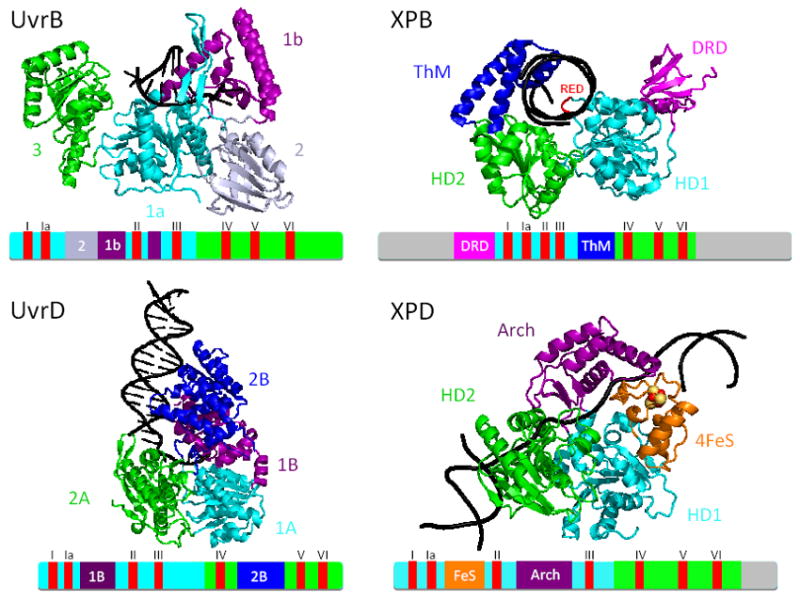
AfXPB (pdb 2FWR [78]), SaXPD (pdb 3CRV [79]), BcUvrB (pdb 2FDC [86]), and EcUvrD (pdb 2R6F [84]) folds shown as ribbons with either DNA models (cartoons for AfXPB and SaXPD) or DNA co-structures (ribbons with bases for BcUvrB and EcUvrD). AfXPB DNA was docked manually using DNA from Hel308 (pdb 2P6R [110]) and SaXPD DNA was computationally docked as described [79]. Linear schematics are shown below each structure. Rad51/RecA domains are colored in cyan (helicase domain 1) or green (helicase domain 2). Accessory domains that are insertions in HD1 are colored in purple or light purple except for XPD FeS domain which is orange and insertions in HD2 are colored blue. The AfXPB DRD domain N-terminal to HD1 is colored magenta. Grey extensions at the N- and C-termini of the XPB schematic or the C-terminus of the XPD schematic represent extensions present in the human proteins. The seven helicase motifs (I, Ia, II-VI) shared among these helicases are shown in red.
Accessory domains have diverse structures to enhance specific functions
All of the NER helicases have two functional parts: two helicase domains that use ATP binding and hydrolysis to drive conformational change and two accessory domains that help transmit helicase domain changes to the DNA and either interrupt the linear sequence of the helicase domains or are found at the N- or C-termini (Fig. 2). Their positions and structures are united only by their diversity.
In XPD, the most striking feature that emerges from first examining the new structures is that HD1 is interrupted by two additional domains, a domain containing a [4Fe-4S] cluster and an Arch-shaped domain [79] (Fig. 2). The 4FeS domains in DNA repair proteins were originally characterized in BER enzymes endonuclease III and MutY [87, 88] and identified as a DNA binding FRCL motif [89]. Despite the recognition that many proteins contain metal ions for functions [90], the discovery of the 4FeS domains in XPD, the Zn hook in Rad50, and Ni in superoxide dismutase (SOD) highlight the fact that metal sites not only play critical roles in most cellular pathways but are still often poorly characterized or even unknown [15, 91-93]. Metal ions can play direct roles in DNA binding, DNA conformational change, as well as catalysis, as seen for FEN1-DNA complexes [94]. Furthermore, loss of metal ions can promote degenerative diseases as implicated for human SOD [95]. Moreover, growing evidence supports the role of 4FeS clusters in DNA damage detection and in possible electron transfer along DNA [96, 97]. Given the emerging role of metals in diverse cellular processes, it will be important to more fully understand metals in NER.
In NER, the 4FeS domain is distinct to XPD and XPD-like helicases including FancJ and is structurally defined by the presence of a [4Fe-4S] cluster [79]. Sulfolobus acidocaldarius (SaXPD) and Thermoplasma acidophilum (TaXPD) 4FeS domains share the same overall fold with slight differences in the number of helices versus loops. In SaXPD, the 4FeS domain contains four helices connected by loops containing four Cys residues that stabilize the 4 Fe ions of the cluster [79]. The TaXPD FeS domain has six helices and three of four Fe-coordinating Cys residues are located in loops with the fourth Cys located in the middle of a helix [81]. Although the structure of Sulfolobus tokodaii (StoXPD) could be solved without an ordered 4FeS domain [80], the [4Fe-4S] cluster of SaXPD provides structural stability for the 4FeS, Arch, and HD1 domains, as chemical oxidation of the cluster in the crystal caused not only the 4FeS domain to become disordered but also part of the Arch domain and residues at the N-terminus [79]. The Arch domain forms a small interface with the 4FeS domain to form a small tunnel of between 8 and 13 Å that is large enough for ssDNA to pass [79, 81, 98] (Fig.2). The Arch represents a novel fold that consists of either a three- or four-stranded β sheet with either four or five helices, depending on the organism [79-81]. The 4FeS and Arch domains are both closely linked to HD1 by β sheet linkages. Due to the interwoven nature of the main chain hydrogen-bonding pattern, β linkages provide interstrand stability not possible with α helices. The 4FeS domain is inserted into adjacent β strands of the central HD1 β sheet while the β sheet of the Arch domain bridges between HD1 and Arch domain α helices. By physically interrupting the linear sequence of HD1, these accessory domains are closely tied to the enzyme’s ATP state, providing a mechanism linking accessory domain function to ATP binding and hydrolysis.
UvrD has an insertion into each of its helicase domains (Fig.2). Domain 1B inserts into helicase domain 1A and domain 2B inserts into helicase domain 2A [84]. Like XPD, both domains 1B and 2B are linked to the helicase domains by β strand linkages, providing a strong structural link between ATP state and the accessory domains. Both domains are inserted into the central β sheets of their respective helicase domain.
By linear sequence, the bacterial UvrB protein seems to share a similar overall architecture with XPD--two helicase domains (1a and 3) and two accessory domains (1b, 2) that are insertions in domain 1a [85] (Fig. 2). Although the two helicase domains both have α/β folds and pack up against one another with the ATP binding site shared between them, the accessory domains are topographically different from XPD. Domains 2 and 1b insert differently into domain 1a. The polypeptide chain leaves domain 1a, forms domain 2, then part of domain 1b before diving back into 1a, and then finishing domain 1b. Neither domain 2 nor 1b contact domain 3 and are on the opposite side of the protein (Fig. 2). These differences in accessory domain connections suggest that there are different ways to connect the ATP-driven actions of the helicase domains in opening DNA duplex to recognizing damage and holding open a DNA bubble (for a discussion of how differences in UvrB and XPB/XPD may affect the substrate specificity in NER, see article by Liu et al in this issue of DNA Repair). These differences and similarities imply a degree of convergent evolution for the bacterial helicases versus XPB and XPD, where domain differences may reflect alternative means to achieve similar functions and differences in their protein partners.
In XPB, the helical ThM domain is an insertion in HD2 while the DRD domain is N-terminal to HD1 (Fig. 2). The XPB ThM domain structurally resembles thumb domains of both T7 and Taq DNA polymerases that grip the minor groove of dsDNA, suggesting a DNA binding role for this domain in XPB [78]. The XPB DRD domain contains five β sheets connected by a single α helix and has structural similarity to the mismatch recognition domain of MutS, suggesting a damage recognition role for XPB [78]. Furthermore, AfXPB unwinds blunt-ended DNA containing a single UV lesion, suggesting that AfXPB can functionally interact with damaged DNA and therefore may play a role in damage verification during NER [78].
DNA binding and conformational change
Surprisingly, the NER helicases are also divergent in the structural elements used to bind DNA and whether or not DNA binding induces large structural changes. Structures of UvrB have been solved both with and without DNA, revealing that the DNA is threaded around a critical β-hairpin motif while the conformation of the rest of the protein is unchanged [86]. In contrast, UvrD undergoes a substantial domain swiveling upon DNA binding and uses a diverse set of motifs to make contacts with the DNA [84, 99]. The crystal structure of UvrB was solved with single-stranded DNA that fortuitously formed a hairpin with 3-base pair (bp) of dsDNA and a 3-bp 3’ overhang. The dsDNA is bound by domains 1a and 1b while the 3’ overhang threads behind the β-hairpin to also make contacts with domains 1a and 1b. UvrB bound to DNA has the same overall conformation as unbound UvrB with the exception of the base of the β-hairpin. The base of the β-hairpin consists of four Tyr residues (92, 93, 95, and 96 in BcUvrB) that make contact with the DNA. Tyrosine hydrogen bonding and aromatic ring structures can provide critical interaction specificity, as seen for SOD [100, 101]. In BcUvrB, Tyr96 makes π stack interactions with the first base of the 3’ overhang at the ss-ds junction [86]. Tyr96Ala mutants cannot form a stable pre-incision complex on cholesterol-containing DNA suggesting that this absolutely conserved residue stabilizes UvrB on DNA [102]. DNA filter binding assays reveal that a double Y92A/Y93A mutant does not discriminate between undamaged and damaged DNA like the wild-type protein [103]. These results imply a stacking role for Tyr in damage recognition that remains to be mechanistically defined.
Crystal structures of UvrD with DNA show that UvrD adopts a closed conformation as seen with PcrA-DNA complexes [104]. This conformation is called closed based on the two different conformations, open and closed, observed in crystal structures of the Rep helicase with DNA [99]. An open to closed conformational change is a common feature of damaged DNA binding, as first seen in UDG during BER [105, 106] and can direct macromolecular pathways and outcomes as shown for example by RFC-PCNA interactions [107]. However the open to closed conformations for NER can be much larger. The difference between the open and closed conformations in UvrD is a 130° swivel of the 2B domain to close it against the 1B domain. Although the open conformation was not observed in the crystal structures of UvrD with DNA, biochemical data supports the idea that both conformations are active for DNA unwinding, albeit by different mechanisms [84]. The closed conformation is proposed to unwind DNA by a “wrench-and-inchworm” mechanism where first the GIG motif of domain 2B binds dsDNA and motif III of domain 1A anchors the ssDNA allowing 1 bp to unwind. ATP hydrolysis and release then drives ssDNA translocation to unwind another bp. The open conformation may unwind DNA by a “strand displacement” mechanism that only requires ssDNA translocation [84].
A computational model of XPB bound to DNA similarly proposes a large conformational rotation of 170° but this change is entirely different from the UvrD swivel. In UvrD, the domain swivel brings two accessory domains together to change the mechanism of DNA unwinding. In XPB, the rotation of the hinge is necessary to bring the two helicase domains together to form the composite ATP binding site [78] (Fig. 3). An initial DNA binding event by HD1 and the DRD domain likely induces this rotation, while ATP hydrolysis facilitates the positioning of the RED motif and ThM domain to stabilize XPB on DNA [78]. This hypothesis was tested in vivo using both host-cell reactivation and UV-survival assays in CHO cells [108]. Mutations in either the ThM domain, RED motif, or the Walker A ATPase motif were defective in these assays, establishing a critical role for these domains and motifs in DNA repair [108]. Furthermore, GFP tagged mutants were defective in recruitment to sites of UV damage in CHO cells, suggesting that recruitment of XPB to damage is an active process requiring ATP hydrolysis coupled with structural stabilization of XPB on DNA by the ThM domain and RED motif [108]. In vitro experiments using mutant Sulfolobus solfataricus XPB (SsoXPB) proteins, found that the RED motif is involved but not essential for SsoXPB function while both the ThM and DRD domains were essential for XPB function [109], confirming the importance of these accessory domains and motifs for XPB function.
Figure 3. XP helicase domains and functional conformational flexibility.
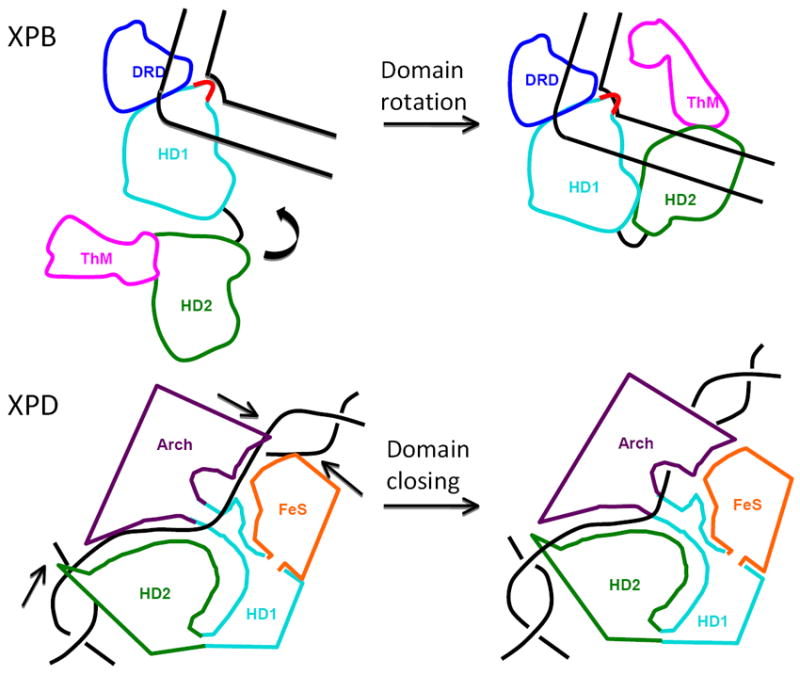
Top, proposed 170° domain rotation of XPB upon DNA binding and ATP hydrolysis [78]. This domain rotation is key to bring HD1 and HD2 together in an active conformation. Bottom, proposed XPD conformational flexibility. Loading of XPD onto DNA would require an opening of the non-covalent interactions between the Arch and FeS domain, while flexibility between HD1 and HD2 is necessary for translocation along DNA during unwinding.
Although a crystal structure of XPD with DNA has not been published, three independent computational models of XPD from three different organisms agree on the path of ssDNA binding (Fig. 2). ssDNA passes through a hole formed by the Arch, FeS, and HD1 domains, travels through a basic channel across the top of HD2, and exits near a helical wedge in HD2 [79-81]. These models require conformational flexibility at the hinge between HD1 and HD2 and between the Arch and 4FeS domains to allow for DNA loading (Fig. 3). Conformational flexibility would also explain the different hole sizes observed in the SaXPD (8 Å) and TaXPD (13 Å) structures [98]. All of these models were based on the structure of Hel308 with DNA, which is another SF2 helicase [110]. In two cases, the model was supported by crystallographic evidence of electron density in the modeled ssDNA binding channel. In the SaXPD structure, bound glycerol, isopropanol, and citrate ions from the crystallization buffer appear to mimic the DNA backbone phosphate and sugar moieties [79], while residual bound DNA seems to explain extra electron density peaks in the TaXPD structure [81]. Patches of positively charged residues on both the flat back side and on HD2 suggest the location of dsDNA interactions on either side of the molecule. In SaXPD, the most energetically favorable docking sites for dsDNA were at the HD2-Arch domain junction where a helix-loop-helix on HD2 could act as a wedge to hold open dsDNA [79]. This wedge was also observed in the TaXPD structure [81]. Although the position is different, both TaXPD and SaXPD have basic patches on the flat back suggesting a second site for dsDNA. Such positively charged patches are hallmarks for DNA binding sites in DNA repair enzymes as seen in base-excision repair, mismatch repair and double-strand break repair enzyme-DNA complexes [62, 111-115]. Such electrostatic complementarity can increase the interaction kinetics and orient macromolecules for efficient interactions [116, 117]. In XPD, the positive patches suggest a configuration consistent with XPD interacting with both sides of a DNA bubble during NER and a means for XPD to efficiently interact with DNA within the TFIIH assembly.
Active site ATP binding and hydrolysis
ATP binding and hydrolysis is critical for the function of the NER helicases. For XPB, ATPase activity seems to be more critical for its functions in transcription and NER than helicase activity [108, 118, 119]. ATP is bound between the two Rad51/RecA helicase domains through several conserved motifs. Structures of both UvrB and UvrD have been solved with Mg2+ and ATP or ATP analogs, providing a detailed picture of ATP binding and hydrolysis.
UvrB and UvrD bind both the adenine base and the phosphate moieties. The Walker A motif (motif I) coordinates the phosphates in both proteins (Fig. 4). In BcUvrB, the phosphates are bound by hydrogen bonds from backbone nitrogens of residues T41, G42, T43, and the side-chain of K45 [85]. The analogous motif I Lys in EcUvrD is K35 and it also coordinates the phosphate moieties. In addition, residues R73, R284, and R605 also bind the triphosphate [84]. In both of these structures, the adenine is sandwiched between two residues, one of which is a tyrosine (Fig. 4). Y11 and P414 in BcUvrB position the N6 and N7 of adenine for hydrogen bonding while Y283 and R37 position the adenine in EcUvrD. The Walker B (motif II) Asp and Glu residues (D338 and E339 in BcUvrB, D220 and E221 in EcUvrD) as well as the ATP β- and γ-phosphates serve to coordinate the single Mg2+ ion necessary for catalysis (Fig. 4).
Figure 4. Molecular surface pockets and important residues in ATP binding, hydrolysis, and human disease.
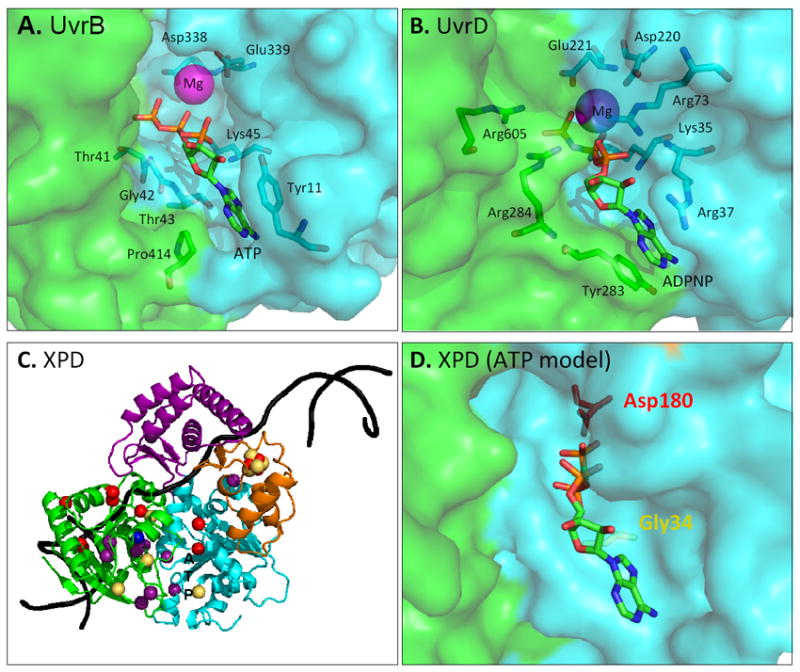
A-B. UvrB and UvrD crystal structures showing the binding pocket (transparent surface) and important coordinating residues (sticks) of Mg2+ and either ATP or ADPNP. C. Crystal structure (ribbons) of SaXPD (pdb 3CRV [79]) showing location of XP (red spheres), XP/CS (gold spheres), and TTD (purple spheres) human patient mutations. The location of an S. cerevisiae mutant that converts UV damage into replication-dependent DSBs is shown [167] (blue sphere T507). Computational DNA model (black ribbon) and position of ATP binding “ATP” are shown. D. Model of SaXPD ATP binding pocket with ATP analog, AMP-PCP or adenosine-5’;-[beta, gamma-methylene]triphosphate, from the Hjm helicase (pdb 2ZJA [179]) showing position of Gly34 Walker A residue mutated in XP/CS patients (G47R) and Asp180 Walker B residue mutated in XP patients (D234N).
In XPB and XPD, Walker A (motif I) mutants disrupt cellular functions, sometimes leading to disease. K45 of BcUvrB and K35 of EcUvrD coordinate the ATP phosphate moieties. When the analogous K346 residue is mutated in XPB, this mutant cannot support transcription or DNA repair in vitro, nor can it properly localize to DNA damage in the cell [108]. A similar mutation in XPD, K48R, supports transcription in vitro and localization in vivo, but it does not support DNA repair in vitro [108]. A mutation in the adjacent residue, G47R, leads to XP/CS in humans, supporting the importance of this motif in DNA repair in humans [120]. The Walker B (motif II) Asp residue that is important in coordinating the Mg2+ ion in UvrB and UvrD is also critical in human XPD since patients with a D234N substitution have XP. Conserved mutants in SaXPD, G34R (G47R) or D180N (D234N) (Fig. 4), are defective in ATPase and helicase activity, but can still bind DNA strongly [79].
ATP-induced conformational changes and DNA opening
ATP-binding energy is stored as conformational changes in the helicase motifs and domains described above. More specifically, the result of γ-phosphate binding and hydrolysis drives piston-like shifts in helicase motifs and rotations in domains that push the DNA strand. ATP is stabilized by specific interactions within its binding site to achieve efficient hydrolysis. An excellent model for understanding this process comes from the structure of the Hel308 bound to DNA compared to other SF2 helicases [110]. During processive translocation in the ATP-free state, phosphates of the ssDNA backbone are bound at motif Ia (position +4) on HD1 and motif IV on HD2 (position +1) and are separated by 2 intermediary bases. However, in three independent structures of SF2 DEAD-box enzymes in complex with ATP analogs and single-stranded nucleic acids, phosphates bound at motifs Ia and IV are separated by only 1 intermediary base [121-123]. Thus the implication is that ATP binding promotes a conformational change that pushes a single nucleotide across HD1. In Hel308, this ATP-driven closure of HD1 and HD2 may be triggered by binding of the conserved motif VI arginine (R369 in Hel308; R514 in SaXPD; XP/CS mutant R666W in XPD) to the ATP γ-phosphate. Upon ATP binding, XPD HD1 motif 1a may push the DNA toward HD2 motif VI. HD1 movement also necessarily pushes on the inserted 4FeS and Arch domains via its covalent connections with these domains. These ATP-driven domain movements likely result in the different hole sizes observed in the SaXPD and TaXPD structures and suggests that the hole may act as a “gripper” during DNA translocation [98]. Thus, ATP binding and hydrolysis by HD1 and the HD1-HD2 interface cause ratchet-like conformational changes that drive the directional movement of the helicase on DNA.
ATP binding and conformational change involve conserved motifs and analogous HD1-HD2 opening and closing. Yet, ATP-driven conformational changes have different impacts in the different helicases, consistent with their distinct accessory domains, divergent structural elements used to bind DNA, and distinct roles. For example, the mechanism of strand separation differs among helicases and ranges from destabilizing dsDNA by mainly binding and limiting strand separation activity (UvrB, XPB), strand displacement by translocating along ssDNA (UvrD, XPD), and unwinding by making contacts with the DNA duplex (UvrD, XPD) [78, 84, 86, 124]. Structural comparisons among helicases including NS3 and Hel308 suggest the β-hairpin is the strand separation element for these helicases [110]. In the UvrB complex with DNA, the beta hairpin is implicated in both damage recognition and strand separation. In XPB, the RED motif is proposed to introduce a wedge in the double stranded DNA that is then gripped by the ThM domain [23, 78]. UvrD has been proposed to unwind DNA by both a “wrench-and-inchworm” mechanism that involves a separation pin motif and a strand displacement mechanism that only requires translocation along ssDNA with DNA being split around a domain rather than a pin [84]. XPD may unwind DNA by such a strand displacement mechanism with the 4FeS domain playing a key role in duplex splitting [79, 81, 125]. Mutations in XPD’s FeS domain, either at sulfur-coordinating cysteines or at an arginine residue mutated in TTD patients, abolish helicase activity, establishing a role for this domain in DNA unwinding [79, 126].
The differences in the proposed DNA unwinding mechanisms and properties of XPB and XPD provide clues as to the need for two helicases in TFIIH. XPB can open bubbles from dsDNA at sites with less stable DNA duplex [78]. This may aid XPD loading by localizing TFIIH at the damaged site. The DNA binding channel of XPD extends ~10nts whereas the binding channel of XPB is shorter and ~5 nts (Fig. 2). The arch and 4FeS domains regulate DNA access to the motor, so these domains limit ssDNA movement with the probability that 4FeS sensing of damage may play a role in arresting helicase movement [127]. Assuming XPB and XPD bind to different strands, the two helicases together give TFIIH a strong hold on the bubble for repair. This model predicts that XPD is positioned to interact at the end where XPG would cut and XPB is positioned to act at the end where XPF would cut. XPD product release therefore controls repair synthesis, as it has the larger DNA binding site. This suggests a strategy for intervention by blocking XPD release, which is discussed below.
CAK structural biology and XP helicase connections
CAK is both a pivotal activator of cyclin dependent kinases (CDKs) and a component of TFIIH [128, 129]. CAK is also required for the transactivation of transcription factors and the regulation of RNA Polymerase II. CAK is itself a complex consisting of Cdk7 (Cyclin dependent kinase 7), CycH and Mat1 [130]. When CAK is bound to TFIIH, it phosphorylates the C-terminal domain of RNAPII plus various transcription factors [131]. When CAK is dissociated from TFIIH, it phosphorylates CDK1, CDK2, CDK4, CDK6, and other targets [131, 132]. Indeed, the association and dissociation of CAK from TFIIH may be an important regulator of DNA repair. Chromatin immunoprecipitation experiments have recently shown that CAK is released from core TFIIH during NER and this release is catalyzed by XPA [27]. CAK release stimulates incision and repair of the damaged DNA, while CAK reassociation with TFIIH correlates with resumption of transcription [27]. Furthermore, the CAK kinase inhibitor H-8 improved repair efficiency, indicating that CAK can negatively regulate NER by phosphorylation [16]. Both XPG and XPD are important for the association of CAK with core TFIIH as mutations from XP-G/CS patients that prevent the XPG-TFIIH association cause both CAK and XPD to dissociate from TFIIH [133]. However, in XP-G and XP-G/CS cells, XPD recruitment to damage was normal while CAK was not [134], suggesting that XPG is not required for XPD-TFIIH association. XPD may play a role in coordinating CAK activity as XPD overexpression in Drosophila negatively regulates the cell cycle function of Cdk7 and downregulation of XPD results in increased CAK activity and cell proliferation [135, 136]. During transcription, the XPB helicase is required to open the promoter around the start site; in contrast, XPD helicase activity is dispensable but XPD stimulates transcription and anchors the CAK complex to TFIIH [137], supporting the importance of connections between XPB, XPD, and CAK. MAT1 activates Cdk1 kinase activity and both XPB and XPD interact directly with the central coiled-coiled domain of the Mat1 subunit of CAK [26]. We therefore propose that MAT1 may monitor the conformational state of both helicases in TFIIH.
Structural information is available for at least part of all of the CAK subunits. Human CDK7 exhibits a typical kinase fold resembling the inactive conformation of CDK2 with two lobes—an N-terminal lobe comprised of β sheets and a C-terminal lobe of mostly α helices—with ATP bound between the two lobes [138] (Fig. 5). Human cyclin H is a regulatory subunit of CDK7 and has two α helical domains, similar to cyclin A of the CDK2 complex [139, 140]. The N-terminal RING finger domain of human MAT1 was solved by NMR and exhibits a typical RING finger βαββ topology with two zinc-binding sites [141]. This domain is important for transcriptional activation and RNAPII CTD phosphorylation, while the central coiled-coiled domain interacts with XPB and XPD, and the hydrophobic C-terminal domain stimulates CDK7 kinase activity [26]. These structures, along with the structures of other TFIIH components, have allowed us to develop a model for how the ten members of TFIIH come together and consider what insights emerge from such a composite structural model.
Figure 5. TFIIH architecture with subunit structures, proposed assembly, and functional implications.
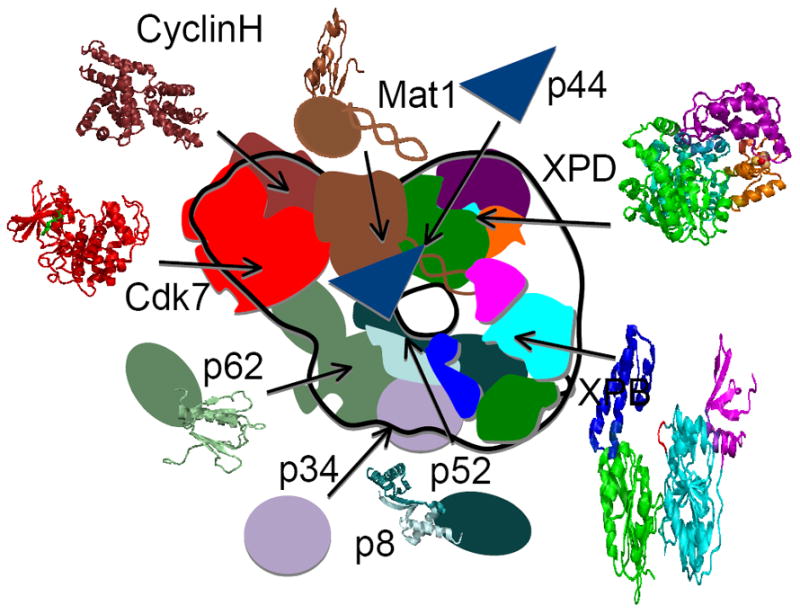
Eight of ten TFIIH subunits with structural information are shown (ribbon folds and transparent surfaces) along proteins and domains without known structures (shapes). The positions of SaXPD (pdb 3CRV [79]) and AfXPB (pdb 2FWR and 2FZL [78]) in the TFIIH EM envelope [145] were determined computationally [79]. Immunolabeling experiments positioned Cdk7 (pdb 1UA2 [138]) at the TFIIH ring protrusion [145]. Cyclin H (pdb 1KXU [140]) was positioned based on other Cdk/cyclin structures. The Mat1 N-terminal domain (pdb 1G25 [141]) is connected to the C-terminal domain (brown oval) that stimulates Cdk7 by a central coiled-coiled domain (brown coils). The Mat1 coiled-coiled domain interacts with both XPD and XPB [26], so is positioned to monitor the conformation of both helicases. P44 (blue triangle) interacts with the C-terminal extension of XPD (not shown) and stimulates its activity [180]. A crystal structure of the yeast homolog of p8 (Tfb5) was solved with the C-terminal domain of yeast p52 (Tfb2) (pdb 3DGP) [150]. P52 stimulates XPB ATPase activity and contacts XPB as P52 N- and C-terminal domains interact with XPB [118]. Although not required for TFIIH assembly, the N-terminal domain of p62 (pdb 1PFJ [154]) interacts with the NER nuclease, XPG [154]. P34 (purple circle) is a member of core TFIIH [181]. Open area (white space) within TFIIH ring between XPB and XPD is predicted to contain the human protein domain extensions absent in the archaeal enzymes.
The Tao of TFIIH structure: resolving puzzles for XP helicases and CAK functions
Lao Tzu’s Tao Te Ching (4th Century B.C.) suggests deep insights come from considering paradoxes [142]. For TFIIH the need for two SF2 helicases, the conflicting evidence for their roles in transcription and damage verification, the mechanism for generating the asymmetric bubble flanking the DNA lesion, the mysterious connections between XPD and both incision and CAK signaling, and the opposing phenotypes of excess cell death and excess cancer from adjacent XPD mutations are among the paradoxes to consider. As key insights often emerge from combining electron microscopy (EM) results with those from high-resolution X-ray and NMR structures and detailed biochemical analysis [74, 143, 144], we use this approach here to address TFIIH paradoxes.
The breakthrough EM structure of human TFIIH showed that the TFIIH core components (XPB, p62, p52, p44, p34, TTDA, and XPD) fold into a ring structure with a bulge formed by the bound CAK [145]. Since high-resolution structural information is available for eight of the ten subunits, we now have a valuable opportunity to combine EM with atomic resolution results to propose a nearly complete TFIIH assembly (Fig.5). Computational docking of our archaeal XPB and XPD structures [78, 79] found the best fit for XPB to be integrated into the ring opposite the CAK bulge while XPD fitted best into the ring between XPB and CAK [79] (Fig. 5). As XPD helps bridge the CAK complex to core TFIIH, XPD is positioned to allosterically communicate TFIIH state to the CAK kinase, which can in turn signal to partners for transcription, repair, and cell cycle regulation. Both XPB and XPD interact with the coiled-coiled domain of MAT1 [26], so we have placed the coiled-coil to contact both XPD and XPB (Fig. 5). We have positioned the hydrophobic MAT1 C-terminal domain, for which there is no structural information, towards Cdk7/Cyclin H as yeast four-hybrid experiments mapped the interaction domain there [146]. Since CAK can negatively regulate XPD unwinding activity in vitro and addition of p44 can overcome this inhibition [146], we located p44 between XPD and CAK. Thus, we propose that the coiled-coil protein MAT1, which supports CAK interaction with XPD, may be analogous to those enforcing interactions for the NHEJ pathway [147] and in the WRN helicase-nuclease and its interactions with the DNA-PK kinase, where the coiled-coil and kinase interactions stimulate processivity [148]. Such coiled-coil interactions may provide robust allosteric connections from ATPases to nucleases and kinases in many repair complexes.
Moving around the TFIIH ring (Fig. 5), p52 is known to interact with XPB and stimulate its ATPase activity [118]. Both the N- and C-terminal domains of p52 interact with XPB, suggesting a large interaction region between these two proteins [118]. The tiny TTDA (p8) protein is also known to interact with p52 and participates in p52-XPB ATPase stimulation without directly interacting with XPB [149]. The crystal structure of yeast TTDA (Tfb5) was solved with the C-terminal domain of yeast p52 (Tfb2) revealing a pseudosymmetric heterodimer that protects a hydrophobic surface of p52, suggesting a possible explanation for why TTDA patient mutations destabilize TFIIH [150]. The p62 subunit interacts with many transcription factors and tumor suppressors including TFIIE, Rb, and p53 [151, 152] as well as NER factors XPC-RAD23B and XPG [71, 153]. The interaction of XPG and p62 is mediated by the N-terminal domain of p62 that is not required for TFIIH assembly [154]. The NMR structure of the p62 N-terminal domain revealed a β-sandwich fold with seven antiparallel β strands capped by a single α helix that has structural similarity to pleckstrin homology (PH) domain found in many cytoplasmic signaling proteins [154]. Structural information is not available for p34 nor for the XPB and XPD domains that are unique to humans [78, 79].
It is important to consider that TFIIH is not static, but rather a dynamic complex where individual components shuttle in and out of the complex or undergo large conformational changes. As discussed above, the disassociation and association of the CAK complex is an important regulator of DNA repair—CAK disassociation promotes NER and downstream signaling while reassociation of CAK correlates with the resumption of transcription [27]. XPD and TTDA may also shuttle in and out of TFIIH since two different populations of XPD and TTDA have been found in cells using GFP-tagging and fluorescence recovery of photo-bleaching (FRAP) methods: a slow moving population with dynamics similar to XPB that is associated with TFIIH and another faster moving population that is thought to represent an unbound pool of these proteins [155, 156]. After UV-irradiation, both XPD and TTDA have similar dynamics to XPB, suggesting that they become more stably integrated into TFIIH during DNA repair [155]. TFIIH must also accommodate conformational changes during transcription and repair. XPB DNA binding and ATP hydrolysis promote a large conformational change that is necessary to stabilize XPB, and therefore TFIIH, on DNA [78, 108]. Furthermore, both XPB and XPD are proposed to change conformation during ATP hydrolysis and DNA translocation (Fig. 3). Therefore, accessory factors such as p52, p44, or MAT1 may act as scaffolds to maintain TFIIH architecture while catalytic domains or subunits swing or shuttle in and out of TFIIH. Flexible domains found in human but not archaeal XPB and XPD [78, 79] may act as flexible tethers that allow for catalytic movements while maintaining TFIIH connections. Such flexible tethers are seen in other DNA repair assemblies such as the Mre11-Rad50-Nbs1 complex [157, 158] or Rad51 filament interactions [74] and can pull to effect multi-protein and multi-domain allosteric signaling in response to changes such as kinase phosphorylation [159, 160]. We expect that advances in technologies such as small angle X-ray scattering (SAXS) that allow for analyses of dynamic structures in solution including flexible domains and tethers [161-165] will provide dynamic models of TFIIH that further our understanding of TFIIH function.
Consideration of overall TFIIH architecture combined with known high-resolution structures allowed us to generate a composite model that provides many insights for a unified framework to resolve TFIIH puzzles and paradoxes. We suggest that the TFIIH core can usefully be considered primarily in terms of the XP helicases and the CAK kinase enzyme conformations and activities. Other components can be viewed as scaffolding and connectors; these act analogously to the helicase accessory domains, which provide scaffolding and mechanical coupling of ATP-states to protein and DNA interactions. In these terms, allosteric changes of the enzyme components are transferred throughout TFIIH via connecting domains such that pathway coordination and biological outcomes reflect the multi-protein allostery of TFIIH, which is driven by the enzymes and their interactions with substrates and products. This more detailed view of TFIIH as a dynamic, multi-protein, allosteric machine connecting helicases and signaling kinase with XPG and XPF partner nucleases provides a foundation for a unified model for TFIIH mechanisms and functions in NER. Testable hypotheses for the integrated functions of dynamic multi-protein systems, such as TFIIH, are often best and most efficiently defined in terms of such models.
A unified model for NER
The three enzyme components of TFIIH are XPB, XPD, and CAK: these proteins are essential for the coordinated actions of damage recognition, verification, repair and signaling. Based upon the combined structural biochemistry and genetics for these enzymes, we propose here a unified, testable model of their functions in the context of TFIIH (Fig. 6). Furthermore by analogy with the MRN complex in double-strand break repair [166], these three TFIIH enzymes can be considered a keystone complex connecting damage detection, signaling, and repair. We call this a bind-pry-unwind model because it integrates DNA damage recognition (bind) with DNA opening (pry and unwind by XPB and XPD) and solves the puzzles that NER presents for structural biology we outlined at the beginning of this review.
Figure 6. Unified testable model for XPB and XPD functions within TFIIH.
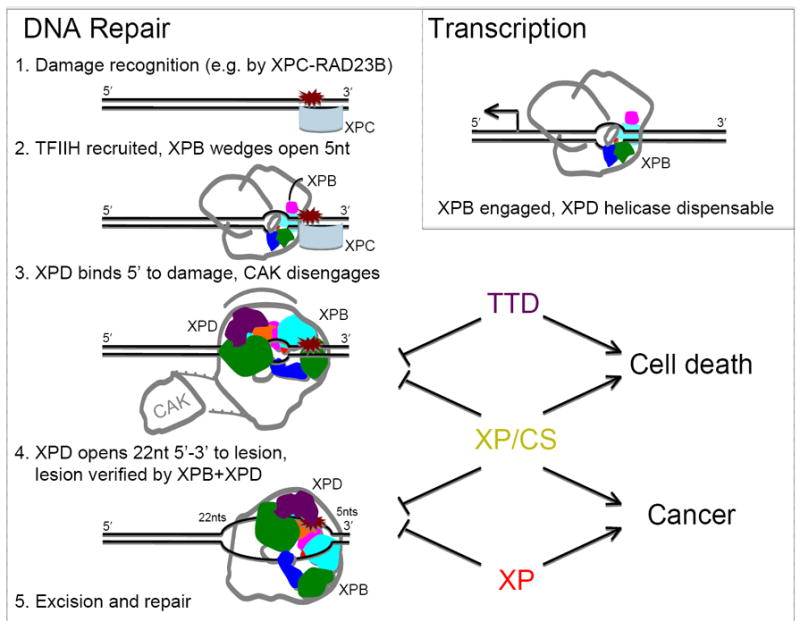
After initial damage recognition and DNA binding by XPC-RAD23B (or other proteins not shown), TFIIH is recruited (with XPA, XPG, and RPA) and XPB binds opposite the damage. DNA binding induces a conformational change in XPB. ATP hydrolysis stabilizes this interaction and may aid nuclease recruitment. Working against the grip of XPC-RAD23B binding, XPB pries open ~ 5nts. XPD binds 5’ to the lesion so the distance from XPB to XPD in the TFIIH ring defines the 27nt size of the excision bubble. Stimulated by XPA, CAK disengages from TFIIH, which then stimulates XPD to unwind in a 5’ to 3’ direction to the lesion. XPD binding anchors TFIIH at the damaged site, recruits XPG, and marks the damaged strand for incision. During transcription (Insert, top left), only XPB helicase is engaged allowing promoter opening and CAK phosphorylation of the RNAPII C-terminal domain to occur. XP/CS mutations that lock conformation and/or affect signaling within TFIIH not only disrupt repair but block downstream events leading to cell death and tissue degeneration. TTD mutations destabilize TFIIH, affecting all TFIIH activities thus leading to cell death (center, right). XP mutations in XPD and XPB that cause defects in helicase or ATPase activities disrupt repair and lead to cancer (bottom, right).
XPB engagement at promoters with its 3’ > 5’ helicase activity helps to initiate transcription (Fig. 6). XPB helicase activity (without XPD helicase activity per se but stimulated by XPD) is a critical signal for transcription: CAK remains bound to TFIIH and phosphorylates the RNAPII C-terminal domain. For DNA repair, the initial recognition of damage is by XPC-RAD23B (with or without DDB1-DDB2 or ATL) for GG-NER or RNAPII for TC-NER (Fig. 6). The damage recognition proteins not only recruit TFIIH and other downstream factors, but they also serve to rotationally fix the DNA, which we predict facilitates DNA opening by XPB analogous to the role of XPB in promoter melting and escape in transcription [69]. In this model, XPB acts as a molecular pry bar that works against the grip provided by the damage recognition proteins, thus linking damage recognition to DNA opening. This is consistent with the limited unwinding activity of XPB and its large conformational change upon DNA binding and ATP hydrolysis. Furthermore, it allows TFIIH loading directly adjacent to either XPC-RAD23B or RNAPII (during transcription initiation or stalled at damage), thus providing a shared XPB mechanism for TC-, GG-NER, and transcription. Like UvrB, XPB has a short DNA binding channel (Fig. 2) that when bound would position TFIIH perpendicular to the DNA (Fig. 6), either 5’ to XPC-RAD23B during GG-NER or downstream of RNAPII during TC-NER or transcription.
After TFIIH loading, XPB would then pry open the DNA, allowing TFIIH to tilt against the DNA and positioning XPD to bind the damaged strand 5’ to the lesion, approximately 22 nts from the lesion as defined by TFIIH architecture (Fig. 6). This binding event is monitored by MAT1 of CAK, which signals to CAK to release from TFIIH, allowing repair to proceed. XPD helicase unwinds the dsDNA until it is stopped by chemical damage reaching the 4FeS domain gateway [24, 25] (Fig. 2). We therefore propose that the span of the TFIIH ring from XPB to XPD sets the size and asymmetry of the NER bubble, as it creates the correct spacing to create 27nt bubbles (22 nts 5’ and 5 nts 3’ to the damage). After its arrest, XPD, together with XPB, anchor TFIIH and the associated NER-damage-response machinery to the repair site, providing an unambiguous platform for nuclease binding and damaged strand excision. XPD bound to the damaged DNA interacts with XPG and promotes DNA incision by XPG and XPF. Finally, removal of TFIIH is necessary for re-synthesis of the incised damaged stand and reassociation of CAK with TFIIH is necessary for resumption of cellular processes such as transcription.
After XPB binding, XPD is the anchor point for TFIIH on the bubble, so controlling XPD conformation and activity controls pathway outcome for NER. Combined structural and biochemical analyses provide a molecular framework that begins to explain how different XPD mutations lead to three different diseases, XP, XP/CS or TTD. As XP mutations (Fig. 4C red spheres) cluster along the DNA- and ATP-binding channels, they decrease helicase activity. Besides losing helicase activity as expected for XP mutations, XP/CS mutations (Fig. 4C gold spheres) also replace small flexible amino acids with larger ones and are predicted to reduce conformational flexibility. These mutations are largely found at the HD1-HD2 interface and therefore affect the conformational crosstalk between these domains to impact DNA-binding and helicase activities. TTD mutations (Fig. 4C purple spheres) are found throughout the protein and disrupt stabilizing interactions between residue side-chains and/or protein main-chains. These changes disrupt the XPD framework to reduce its structural integrity, which can affect XPD activities and/or partner interactions.
Experiments in S. cerevisiae XPD are also yielding insights into XPD disease. Surprisingly, the rad3-102 mutant (Fig. 4C blue sphere) exhibits only moderate sensitivity to UV damage, but rather causes replication-dependent double strand breaks (DSBs) that require the Mre11-Rad50-Xrs2 system for repair [167]. Evidently NER incision occurs in these cells, but postincision events are blocked due to retention of TFIIH at the site of damage, leaving a ssDNA gap. Arrival of the replication fork at the ssDNA gap causes replication fork breakage, which leads to the formation of one-ended DSBs [168]. Such one-ended breaks cannot be processed for NHEJ by the DNA-PK pathway [169] but must be processed by Mre11 complex-dependent recombination [114]. This yeast mutant evokes comparison to an XP-CS mutant (G675R) that causes aberrant DNA breaks upon UV irradiation in human cells [170]. In SaXPD, this mutant (C523R) has increased ssDNA-binding affinity [79], suggesting that increased XPD binding to DNA may be playing a role in the phenotype of these patients.
These integrated structural and biochemical insights therefore provide a framework to begin to predict why differences in single amino acids in one protein result in large phenotypic changes for an entire organism. Mutations that affect activity without affecting conformation or signaling lead to XP and cancer (Fig. 6). XP-D patients have reduced DNA repair capacity and thus cannot repair all of the damage that occurs on sunlight-exposed areas of the skin, leading to gene mutations and cancer. XP/CS patients have the same cancer susceptibility as XP patients with the added complication of having a mutation that reduces the functionally critical flexibility of XPD. This conformational restriction is predicted to alter XPD functions, partnerships, and downstream events, such a signaling to CAK (Fig. 6). This change thus compromises the cell’s ability to survive, leading to cell death premature aging [171]. TTD mutations also lead to cell death and premature aging since TTD mutations increase protein flexibility, which should both disrupt XPD interactions with other proteins and decrease stability of the TFIIH complex [172]. These changes also block downstream events (Fig. 6) despite sometimes retaining XPD activities [79].
Although specific aspects will likely need modification, this structure-based model has seven attributes we hope will prove valuable: 1) it integrates the structural biochemistry and genetics for TFIIH and its components into a single unified model, 2) it helps researchers worldwide design experiments to test separate enzymatic functions of TFIIH, 3) it provides a mechanism for creating the asymmetric 27nt bubble for repair, 4) it suggests the molecular basis for aging and cancer-causing TFIIH defects, 6) it identifies specific means and targets for therapeutic interventions, and 7) it suggests how the NER-damage response is orchestrated by the three TFIIH enzymes to connect detection of double-helix defects and chemical modifications by the XP helicases to CAK-mediated signaling.
When viewed in terms of individual TFIIH interactions, a bewildering matrix of combinations confronts the researcher. In general, the unified model proposed here (Fig. 6) implies TFIIH ring activities are controlled by allosteric conformations acting in kaleidoscopic effects that create and break specific assemblies and contacts depending upon TFIIH enzyme conformations. For example, the unequal opening flanking the DNA damage has been a mechanistic puzzle. Our TFIIH NER model suggests a simple mechanism for generating the asymmetric bubble that is entirely based upon the TFIIH architecture and the differential XPB and XPD helicase structural biochemistry. We trust the proposed unified model is sufficiently specific to allow testing of its various aspects by researchers who can bring to bear their expertise, systems, and technologies to test its merits.
Implications -- strategies for interventions
Inhibiting repair can cause genome instability [173]; yet, if done strategically then selectively inhibiting repair may allow improved interventions [174]. Indeed creating or blocking a specific protein interaction can control repair progression as shown for Rad51-BRCA2 interactions [74]. For new approaches in cancer interventions, the well-regulated NER pathway may provide a powerful strategy to target the Achilles’ heel of many cancers--defects in DNA repair and cell cycle regulation. Small molecule inhibitors that block XPD release, analogous to the yeast rad3-102 mutant that blocks NER and causes DSBs [167, 168], could potentially work for interventions in some cancers because to process such breaks requires the double-strand break machinery mutated in these cancers, such as the BRCA1 and BRCA2 breast cancers [175, 176]. In contrast to an XPD-deletion, which cannot remove the lesion and thus activates a checkpoint response, disrupting NER by blocked XPD release exemplifies a knowledge-based strategy to fool cancer cells for interventions. For example, the yeast rad3-102 mutant progresses faster through S into M-phase than normal cells and requires replication to convert NER damage to DSBs [167], causing cell death by mitotic catastrophe. This dependence on replication brings specificity for cancer cells with repair and cell cycle checkpoint defects. So small molecule ligands that bind and inactivate XPD without disrupting its ability to bind DNA and partner proteins may provide a successful cancer intervention strategy in combination with chemotherapies generating bulk lesions. This proposal suggests XPD binding is a potential target for cancer therapeutics because keeping XPD bound will block repair progression, but release CAK to allow cell cycle progression and generation of a dsDNA break at the next round of DNA replication.
TFIIH architecture enforces the spatial localization XPD, XPB and CAK such that XPD conformations can influence the binding and activities of these other enzymes. XPD conformations driven by ATP and DNA damage interactions can stabilize or destablize complexes. Such conformational changes are of great interest for controlling biological outcomes because they can also make interfaces accessible or impossible. In principle, the most extreme specificity can be achieved by sterically blocking a site so that subsequent interactions are impossible. The ability to make some interfaces impossible offers extreme specificity and a novel approach to drug design. By developing ligands that bind to and lock specific conformations for TFIIH enzymes, we expect researchers may be able to develop the ability to control interactions specifically and therefore control whether repair, signaling, or apoptosis will take place, thus limiting toxicity due to the essential role of TFIIH in transcription. As NER is more specific and less redundant than many repair responses, targeting this pathway promises to provide unique advantages for at least some cancers.
Conclusions
Collective structural, biochemical, and genetic results from bacteria to humans on NER helicases and their protein partners provide keys to understanding and ultimately controlling biological outcomes. The NER functional steps of find, open, verify, excise, signal are conserved from bacteria to humans. The non-enzyme, scaffolding components of TFIIH create a multi-protein allosteric machine where conformational changes in the enzymes are linked to partners distant from their active sites. Here we combine ideas from structure and cell biology to suggest that TFIIH enzyme structures are master keys to biological outcomes. XPB, XPD, and CAK critically connect DNA damage recognition and verification to repair processing and to signaling for coordination of transcription, repair, and cell cycle regulation.
In the broadest sense, combined studies of DNA repair mechanisms with human genetics disorders, as considered here, lay the foundation to uncover the biological basis for oncogenesis and age-related diseases. The complex molecular and cellular activities of XP helicases and their protein partners combined with the daunting complexity of disorders associated with their mutations have provided numerous puzzles plus almost overwhelming amounts of data. Yet, biology needs a unifying understanding in the face of overwhelming data. Here we show how structures are also helping to provide such a unified understanding. Based upon the structural biochemistry and genetics, we furthermore propose a simple unified model for XP helicase functions including CAK interactions and signaling. We suggest this model may help advance understanding by providing specific ideas regarding XPB and XPD structure-function relationships to be tested by researchers everywhere. We hope that structural biology will increasingly meet the challenges of large dynamic complexes and contribute to a foundation for developing new paradigms for disease prevention, susceptibility, diagnosis and interventions.
Acknowledgments
This work on cancer relevant XP helicases was supported in part by the National Institutes of Health (NIH) grant CA112093 (JAT, JOF). We thank Sophia Hartung, Justin Ishida, Susan Tsutakawa, Gareth Williams, Yuan He, Leon Mullenders, Katharina Schlacher, Walter Chazin, Priscilla Cooper, Cynthia McMurray, Orlando Schärer and members of the Structural Biology of DNA Repair Machines program for fruitful discussions. Work in the authors’ laboratory on microbial complexes is supported in part by the MAGIC component of the ENIGMA program supported by the Office of Science, Office of Biological and Environmental Research, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Friedberg EC. DNA damage and repair. Nature. 2003;421:436–440. doi: 10.1038/nature01408. [DOI] [PubMed] [Google Scholar]
- 2.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 4.Lindahl T, Wood RD. Quality control by DNA repair. Science. 1999;286:1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 5.Aboussekhra A, Biggerstaff M, Shivji MK, Vilpo JA, Moncollin V, Podust VN, Protic M, Hubscher U, Egly JM, Wood RD. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell. 1995;80:859–868. doi: 10.1016/0092-8674(95)90289-9. [DOI] [PubMed] [Google Scholar]
- 6.Sancar A. DNA excision repair. Annu Rev Biochem. 1996;65:43–81. doi: 10.1146/annurev.bi.65.070196.000355. [DOI] [PubMed] [Google Scholar]
- 7.Hebra F, Kaposi M. On Diseases of the Skin, Including Exanthemata. New Sydenham Society. 1874;3:252–258. [Google Scholar]
- 8.Cleaver JE. Defective repair replication of DNA in xeroderma pigmentosum. Nature. 1968;218:652–656. doi: 10.1038/218652a0. [DOI] [PubMed] [Google Scholar]
- 9.De Weerd-Kastelein EA, Keijzer W, Bootsma D. Genetic heterogeneity of xeroderma pigmentosum demonstrated by somatic cell hybridization. Nat New Biol. 1972;238:80–83. doi: 10.1038/newbio238080a0. [DOI] [PubMed] [Google Scholar]
- 10.Parikh SS, Mol CD, Tainer JA. Base excision repair enzyme family portrait: integrating the structure and chemistry of an entire DNA repair pathway. Structure. 1997;5:1543–1550. doi: 10.1016/s0969-2126(97)00303-1. [DOI] [PubMed] [Google Scholar]
- 11.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 12.de Boer J, Hoeijmakers JH. Nucleotide excision repair and human syndromes. Carcinogenesis. 2000;21:453–460. doi: 10.1093/carcin/21.3.453. [DOI] [PubMed] [Google Scholar]
- 13.Taylor EM, Broughton BC, Botta E, Stefanini M, Sarasin A, Jaspers NG, Fawcett H, Harcourt SA, Arlett CF, Lehmann AR. Xeroderma pigmentosum and trichothiodystrophy are associated with different mutations in the XPD (ERCC2) repair/transcription gene. Proc Natl Acad Sci U S A. 1997;94:8658–8663. doi: 10.1073/pnas.94.16.8658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 15.Perry JJ, Cotner-Gohara E, Ellenberger T, Tainer JA. Structural dynamics in DNA damage signaling and repair. Curr Opin Struct Biol. 2010;20:283–294. doi: 10.1016/j.sbi.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Araujo SJ, Tirode F, Coin F, Pospiech H, Syvaoja JE, Stucki M, Hubscher U, Egly JM, Wood RD. Nucleotide excision repair of DNA with recombinant human proteins: definition of the minimal set of factors, active forms of TFIIH, and modulation by CAK. Genes Dev. 2000;14:349–359. [PMC free article] [PubMed] [Google Scholar]
- 17.Mu D, Park CH, Matsunaga T, Hsu DS, Reardon JT, Sancar A. Reconstitution of human DNA repair excision nuclease in a highly defined system. J Biol Chem. 1995;270:2415–2418. doi: 10.1074/jbc.270.6.2415. [DOI] [PubMed] [Google Scholar]
- 18.Volker M, Mone MJ, Karmakar P, van Hoffen A, Schul W, Vermeulen W, Hoeijmakers JH, van Driel R, van Zeeland AA, Mullenders LH. Sequential assembly of the nucleotide excision repair factors in vivo. Mol Cell. 2001;8:213–224. doi: 10.1016/s1097-2765(01)00281-7. [DOI] [PubMed] [Google Scholar]
- 19.Houtsmuller AB, Rademakers S, Nigg AL, Hoogstraten D, Hoeijmakers JH, Vermeulen W. Action of DNA repair endonuclease ERCC1/XPF in living cells. Science. 1999;284:958–961. doi: 10.1126/science.284.5416.958. [DOI] [PubMed] [Google Scholar]
- 20.Fousteri M, Mullenders LH. Transcription-coupled nucleotide excision repair in mammalian cells: molecular mechanisms and biological effects. Cell Res. 2008;18:73–84. doi: 10.1038/cr.2008.6. [DOI] [PubMed] [Google Scholar]
- 21.Gillet LC, Scharer OD. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem Rev. 2006;106:253–276. doi: 10.1021/cr040483f. [DOI] [PubMed] [Google Scholar]
- 22.Tassan JP, Schultz SJ, Bartek J, Nigg EA. Cell cycle analysis of the activity, subcellular localization, and subunit composition of human CAK (CDK-activating kinase) J Cell Biol. 1994;127:467–478. doi: 10.1083/jcb.127.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oksenych V, Coin F. The long unwinding road: XPB and XPD helicases in damaged DNA opening. Cell Cycle. 2010;9:90–96. doi: 10.4161/cc.9.1.10267. [DOI] [PubMed] [Google Scholar]
- 24.Mathieu N, Kaczmarek N, Naegeli H. Strand- and site-specific DNA lesion demarcation by the xeroderma pigmentosum group D helicase. Proc Natl Acad Sci U S A. 2010;107:17545–17550. doi: 10.1073/pnas.1004339107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Naegeli H, Modrich P, Friedberg EC. The DNA helicase activities of Rad3 protein of Saccharomyces cerevisiae and helicase II of Escherichia coli are differentially inhibited by covalent and noncovalent DNA modifications. J Biol Chem. 1993;268:10386–10392. [PubMed] [Google Scholar]
- 26.Busso D, Keriel A, Sandrock B, Poterszman A, Gileadi O, Egly JM. Distinct regions of MAT1 regulate cdk7 kinase and TFIIH transcription activities. J Biol Chem. 2000;275:22815–22823. doi: 10.1074/jbc.M002578200. [DOI] [PubMed] [Google Scholar]
- 27.Coin F, Oksenych V, Mocquet V, Groh S, Blattner C, Egly JM. Nucleotide excision repair driven by the dissociation of CAK from TFIIH. Mol Cell. 2008;31:9–20. doi: 10.1016/j.molcel.2008.04.024. [DOI] [PubMed] [Google Scholar]
- 28.Evans E, Moggs JG, Hwang JR, Egly JM, Wood RD. Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J. 1997;16:6559–6573. doi: 10.1093/emboj/16.21.6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fuss JO, Cooper PK. DNA repair: dynamic defenders against cancer and aging. PLoS Biol. 2006;4:e203. doi: 10.1371/journal.pbio.0040203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ogi T, Limsirichaikul S, Overmeer RM, Volker M, Takenaka K, Cloney R, Nakazawa Y, Niimi A, Miki Y, Jaspers NG, Mullenders LH, Yamashita S, Fousteri MI, Lehmann AR. Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol Cell. 2010;37:714–727. doi: 10.1016/j.molcel.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 31.Suhasini AN, Brosh RM., Jr Mechanistic and biological aspects of helicase action on damaged DNA. Cell Cycle. 2010;9 doi: 10.4161/cc.9.12.11902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Truglio JJ, Croteau DL, Van Houten B, Kisker C. Prokaryotic nucleotide excision repair: the UvrABC system. Chem Rev. 2006;106:233–252. doi: 10.1021/cr040471u. [DOI] [PubMed] [Google Scholar]
- 33.Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol. 1987;123:241–250. doi: 10.1001/archderm.123.2.241. [DOI] [PubMed] [Google Scholar]
- 34.Lehmann AR. The xeroderma pigmentosum group D (XPD) gene: one gene, two functions, three diseases. Genes Dev. 2001;15:15–23. doi: 10.1101/gad.859501. [DOI] [PubMed] [Google Scholar]
- 35.Lehmann AR. DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie. 2003;85:1101–1111. doi: 10.1016/j.biochi.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 36.Wang Z, Svejstrup JQ, Feaver WJ, Wu X, Kornberg RD, Friedberg EC. Transcription factor b (TFIIH) is required during nucleotide-excision repair in yeast. Nature. 1994;368:74–76. doi: 10.1038/368074a0. [DOI] [PubMed] [Google Scholar]
- 37.Drapkin R, Reardon JT, Ansari A, Huang JC, Zawel L, Ahn K, Sancar A, Reinberg D. Dual role of TFIIH in DNA excision repair and in transcription by RNA polymerase II. Nature. 1994;368:769–772. doi: 10.1038/368769a0. [DOI] [PubMed] [Google Scholar]
- 38.Ito S, Tan LJ, Andoh D, Narita T, Seki M, Hirano Y, Narita K, Kuraoka I, Hiraoka Y, Tanaka K. MMXD, a TFIIH-independent XPD-MMS19 protein complex involved in chromosome segregation. Mol Cell. 2010;39:632–640. doi: 10.1016/j.molcel.2010.07.029. [DOI] [PubMed] [Google Scholar]
- 39.Cameroni E, Stettler K, Suter B. On the traces of XPD: cell cycle matters - untangling the genotype-phenotype relationship of XPD mutations. Cell Div. 2010;5:24. doi: 10.1186/1747-1028-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Emmert S, Ueda T, Zumsteg U, Weber P, Khan SG, Oh KS, Boyle J, Laspe P, Zachmann K, Boeckmann L, Kuschal C, Bircher A, Kraemer KH. Strict sun protection results in minimal skin changes in a patient with xeroderma pigmentosum and a novel c.2009delG mutation in XPD (ERCC2) Exp Dermatol. 2009;18:64–68. doi: 10.1111/j.1600-0625.2008.00763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Graham JM, Jr, Anyane-Yeboa K, Raams A, Appeldoorn E, Kleijer WJ, Garritsen VH, Busch D, Edersheim TG, Jaspers NG. Cerebro-oculo-facio-skeletal syndrome with a nucleotide excision-repair defect and a mutated XPD gene, with prenatal diagnosis in a triplet pregnancy. Am J Hum Genet. 2001;69:291–300. doi: 10.1086/321295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vermeulen W, Rademakers S, Jaspers NG, Appeldoorn E, Raams A, Klein B, Kleijer WJ, Hansen LK, Hoeijmakers JH. A temperature-sensitive disorder in basal transcription and DNA repair in humans. Nat Genet. 2001;27:299–303. doi: 10.1038/85864. [DOI] [PubMed] [Google Scholar]
- 43.Stefanini M, Botta E, Lanzafame M, Orioli D. Trichothiodystrophy: from basic mechanisms to clinical implications. DNA Repair (Amst) 2010;9:2–10. doi: 10.1016/j.dnarep.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 44.Broughton BC, Berneburg M, Fawcett H, Taylor EM, Arlett CF, Nardo T, Stefanini M, Menefee E, Price VH, Queille S, Sarasin A, Bohnert E, Krutmann J, Davidson R, Kraemer KH, Lehmann AR. Two individuals with features of both xeroderma pigmentosum and trichothiodystrophy highlight the complexity of the clinical outcomes of mutations in the XPD gene. Hum Mol Genet. 2001;10:2539–2547. doi: 10.1093/hmg/10.22.2539. [DOI] [PubMed] [Google Scholar]
- 45.Wood RD. DNA repair in eukaryotes. Annu Rev Biochem. 1996;65:135–167. doi: 10.1146/annurev.bi.65.070196.001031. [DOI] [PubMed] [Google Scholar]
- 46.Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 47.Tubbs JL, Latypov V, Kanugula S, Butt A, Melikishvili M, Kraehenbuehl R, Fleck O, Marriott A, Watson AJ, Verbeek B, McGown G, Thorncroft M, Santibanez-Koref MF, Millington C, Arvai AS, Kroeger MD, Peterson LA, Williams DM, Fried MG, Margison GP, Pegg AE, Tainer JA. Flipping of alkylated DNA damage bridges base and nucleotide excision repair. Nature. 2009;459:808–813. doi: 10.1038/nature08076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hitomi K, Iwai S, Tainer JA. The intricate structural chemistry of base excision repair machinery: implications for DNA damage recognition, removal, and repair. DNA Repair (Amst) 2007;6:410–428. doi: 10.1016/j.dnarep.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 49.Kim N, Jinks-Robertson S. Abasic sites in the transcribed strand of yeast DNA are removed by transcription-coupled nucleotide excision repair. Mol Cell Biol. 2010;30:3206–3215. doi: 10.1128/MCB.00308-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brooks PJ, Wise DS, Berry DA, Kosmoski JV, Smerdon MJ, Somers RL, Mackie H, Spoonde AY, Ackerman EJ, Coleman K, Tarone RE, Robbins JH. The oxidative DNA lesion 8,5’-(S)-cyclo-2’-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. J Biol Chem. 2000;275:22355–22362. doi: 10.1074/jbc.M002259200. [DOI] [PubMed] [Google Scholar]
- 51.Demple B, Harrison L. Repair of oxidative damage to DNA: enzymology and biology. Annu Rev Biochem. 1994;63:915–948. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- 52.De Silva IU, McHugh PJ, Clingen PH, Hartley JA. Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol Cell Biol. 2000;20:7980–7990. doi: 10.1128/mcb.20.21.7980-7990.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zamble DB, Mu D, Reardon JT, Sancar A, Lippard SJ. Repair of cisplatin--DNA adducts by the mammalian excision nuclease. Biochemistry. 1996;35:10004–10013. doi: 10.1021/bi960453+. [DOI] [PubMed] [Google Scholar]
- 54.Reardon JT, Nichols AF, Keeney S, Smith CA, Taylor JS, Linn S, Sancar A. Comparative analysis of binding of human damaged DNA-binding protein (XPE) and Escherichia coli damage recognition protein (UvrA) to the major ultraviolet photoproducts: T[c,s]T, T[t,s]T, T[6-4]T, and T[Dewar]T. J Biol Chem. 1993;268:21301–21308. [PubMed] [Google Scholar]
- 55.Sarker AH, Tsutakawa SE, Kostek S, Ng C, Shin DS, Peris M, Campeau E, Tainer JA, Nogales E, Cooper PK. Recognition of RNA polymerase II and transcription bubbles by XPG, CSB, and TFIIH: insights for transcription-coupled repair and Cockayne Syndrome. Mol Cell. 2005;20:187–198. doi: 10.1016/j.molcel.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 56.Tubbs JL, Tainer JA. Alkyltransferase-like proteins: molecular switches between DNA repair pathways. Cell Mol Life Sci. 2010;67:3749–3762. doi: 10.1007/s00018-010-0405-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sugasawa K, Ng JM, Masutani C, Iwai S, van der Spek PJ, Eker AP, Hanaoka F, Bootsma D, Hoeijmakers JH. Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol Cell. 1998;2:223–232. doi: 10.1016/s1097-2765(00)80132-x. [DOI] [PubMed] [Google Scholar]
- 58.Moser J, Volker M, Kool H, Alekseev S, Vrieling H, Yasui A, van Zeeland AA, Mullenders LH. The UV-damaged DNA binding protein mediates efficient targeting of the nucleotide excision repair complex to UV-induced photo lesions. DNA Repair (Amst) 2005;4:571–582. doi: 10.1016/j.dnarep.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 59.Sugasawa K, Okuda Y, Saijo M, Nishi R, Matsuda N, Chu G, Mori T, Iwai S, Tanaka K, Hanaoka F. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell. 2005;121:387–400. doi: 10.1016/j.cell.2005.02.035. [DOI] [PubMed] [Google Scholar]
- 60.Min JH, Pavletich NP. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature. 2007;449:570–575. doi: 10.1038/nature06155. [DOI] [PubMed] [Google Scholar]
- 61.Huffman JL, Sundheim O, Tainer JA. DNA base damage recognition and removal: new twists and grooves. Mutat Res. 2005;577:55–76. doi: 10.1016/j.mrfmmm.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 62.Slupphaug G, Mol CD, Kavli B, Arvai AS, Krokan HE, Tainer JA. A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature. 1996;384:87–92. doi: 10.1038/384087a0. [DOI] [PubMed] [Google Scholar]
- 63.Scrima A, Konickova R, Czyzewski BK, Kawasaki Y, Jeffrey PD, Groisman R, Nakatani Y, Iwai S, Pavletich NP, Thoma NH. Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell. 2008;135:1213–1223. doi: 10.1016/j.cell.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dalhus B, Arvai AS, Rosnes I, Olsen OE, Backe PH, Alseth I, Gao H, Cao W, Tainer JA, Bjoras M. Structures of endonuclease V with DNA reveal initiation of deaminated adenine repair. Nat Struct Mol Biol. 2009;16:138–143. doi: 10.1038/nsmb.1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hitomi K, DiTacchio L, Arvai AS, Yamamoto J, Kim ST, Todo T, Tainer JA, Iwai S, Panda S, Getzoff ED. Functional motifs in the (6-4) photolyase crystal structure make a comparative framework for DNA repair photolyases and clock cryptochromes. Proc Natl Acad Sci U S A. 2009;106:6962–6967. doi: 10.1073/pnas.0809180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Daniels DS, Woo TT, Luu KX, Noll DM, Clarke ND, Pegg AE, Tainer JA. DNA binding and nucleotide flipping by the human DNA repair protein AGT. Nat Struct Mol Biol. 2004;11:714–720. doi: 10.1038/nsmb791. [DOI] [PubMed] [Google Scholar]
- 67.Tubbs JL, Pegg AE, Tainer JA. DNA binding, nucleotide flipping, and the helix-turn-helix motif in base repair by O6-alkylguanine-DNA alkyltransferase and its implications for cancer chemotherapy. DNA Repair (Amst) 2007;6:1100–1115. doi: 10.1016/j.dnarep.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brueckner F, Hennecke U, Carell T, Cramer P. CPD damage recognition by transcribing RNA polymerase II. Science. 2007;315:859–862. doi: 10.1126/science.1135400. [DOI] [PubMed] [Google Scholar]
- 69.Kim TK, Ebright RH, Reinberg D. Mechanism of ATP-dependent promoter melting by transcription factor IIH. Science. 2000;288:1418–1422. doi: 10.1126/science.288.5470.1418. [DOI] [PubMed] [Google Scholar]
- 70.Miller G, Hahn S. A DNA-tethered cleavage probe reveals the path for promoter DNA in the yeast preinitiation complex. Nat Struct Mol Biol. 2006;13:603–610. doi: 10.1038/nsmb1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yokoi M, Masutani C, Maekawa T, Sugasawa K, Ohkuma Y, Hanaoka F. The xeroderma pigmentosum group C protein complex XPC-HR23B plays an important role in the recruitment of transcription factor IIH to damaged DNA. J Biol Chem. 2000;275:9870–9875. doi: 10.1074/jbc.275.13.9870. [DOI] [PubMed] [Google Scholar]
- 72.Fousteri M, Vermeulen W, van Zeeland AA, Mullenders LH. Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol Cell. 2006;23:471–482. doi: 10.1016/j.molcel.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 73.Bird LE, Subramanya HS, Wigley DB. Helicases: a unifying structural theme? Curr Opin Struct Biol. 1998;8:14–18. doi: 10.1016/s0959-440x(98)80004-3. [DOI] [PubMed] [Google Scholar]
- 74.Shin DS, Pellegrini L, Daniels DS, Yelent B, Craig L, Bates D, Yu DS, Shivji MK, Hitomi C, Arvai AS, Volkmann N, Tsuruta H, Blundell TL, Venkitaraman AR, Tainer JA. Full-length archaeal Rad51 structure and mutants: mechanisms for RAD51 assembly and control by BRCA2. EMBO J. 2003;22:4566–4576. doi: 10.1093/emboj/cdg429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Story RM, Steitz TA. Structure of the recA protein-ADP complex. Nature. 1992;355:374–376. doi: 10.1038/355374a0. [DOI] [PubMed] [Google Scholar]
- 76.Tuteja N, Tuteja R. DNA helicases: the long unwinding road. Nat Genet. 1996;13:11–12. doi: 10.1038/ng0596-11. [DOI] [PubMed] [Google Scholar]
- 77.Tanner NK. The newly identified Q motif of DEAD box helicases is involved in adenine recognition. Cell Cycle. 2003;2:18–19. doi: 10.4161/cc.2.1.296. [DOI] [PubMed] [Google Scholar]
- 78.Fan L, Arvai AS, Cooper PK, Iwai S, Hanaoka F, Tainer JA. Conserved XPB core structure and motifs for DNA unwinding: implications for pathway selection of transcription or excision repair. Mol Cell. 2006;22:27–37. doi: 10.1016/j.molcel.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 79.Fan L, Fuss JO, Cheng QJ, Arvai AS, Hammel M, Roberts VA, Cooper PK, Tainer JA. XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations. Cell. 2008;133:789–800. doi: 10.1016/j.cell.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu H, Rudolf J, Johnson KA, McMahon SA, Oke M, Carter L, McRobbie AM, Brown SE, Naismith JH, White MF. Structure of the DNA repair helicase XPD. Cell. 2008;133:801–812. doi: 10.1016/j.cell.2008.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wolski SC, Kuper J, Hanzelmann P, Truglio JJ, Croteau DL, Van Houten B, Kisker C. Crystal structure of the FeS cluster-containing nucleotide excision repair helicase XPD. PLoS Biol. 2008;6:e149. doi: 10.1371/journal.pbio.0060149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Singleton MR, Dillingham MS, Wigley DB. Structure and mechanism of helicases and nucleic acid translocases. Annu Rev Biochem. 2007;76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 83.Hopfner KP, Karcher A, Shin DS, Craig L, Arthur LM, Carney JP, Tainer JA. Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell. 2000;101:789–800. doi: 10.1016/s0092-8674(00)80890-9. [DOI] [PubMed] [Google Scholar]
- 84.Lee JY, Yang W. UvrD helicase unwinds DNA one base pair at a time by a two-part power stroke. Cell. 2006;127:1349–1360. doi: 10.1016/j.cell.2006.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Theis K, Chen PJ, Skorvaga M, Van Houten B, Kisker C. Crystal structure of UvrB, a DNA helicase adapted for nucleotide excision repair. EMBO J. 1999;18:6899–6907. doi: 10.1093/emboj/18.24.6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Truglio JJ, Karakas E, Rhau B, Wang H, DellaVecchia MJ, Van Houten B, Kisker C. Structural basis for DNA recognition and processing by UvrB. Nat Struct Mol Biol. 2006;13:360–364. doi: 10.1038/nsmb1072. [DOI] [PubMed] [Google Scholar]
- 87.Guan Y, Manuel RC, Arvai AS, Parikh SS, Mol CD, Miller JH, Lloyd S, Tainer JA. MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nat Struct Biol. 1998;5:1058–1064. doi: 10.1038/4168. [DOI] [PubMed] [Google Scholar]
- 88.Kuo CF, McRee DE, Fisher CL, O’Handley SF, Cunningham RP, Tainer JA. Atomic structure of the DNA repair [4Fe-4S] enzyme endonuclease III. Science. 1992;258:434–440. doi: 10.1126/science.1411536. [DOI] [PubMed] [Google Scholar]
- 89.Thayer MM, Ahern H, Xing D, Cunningham RP, Tainer JA. Novel DNA binding motifs in the DNA repair enzyme endonuclease III crystal structure. EMBO J. 1995;14:4108–4120. doi: 10.1002/j.1460-2075.1995.tb00083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Castagnetto JM, Hennessy SW, Roberts VA, Getzoff ED, Tainer JA, Pique ME. MDB: the Metalloprotein Database and Browser at The Scripps Research Institute. Nucleic Acids Res. 2002;30:379–382. doi: 10.1093/nar/30.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barondeau DP, Kassmann CJ, Tainer JA, Getzoff ED. Structural chemistry of a green fluorescent protein Zn biosensor. J Am Chem Soc. 2002;124:3522–3524. doi: 10.1021/ja0176954. [DOI] [PubMed] [Google Scholar]
- 92.Cvetkovic A, Menon AL, Thorgersen MP, Scott JW, Poole FL, 2nd, Jenney FE, Jr, Lancaster WA, Praissman JL, Shanmukh S, Vaccaro BJ, Trauger SA, Kalisiak E, Apon JV, Siuzdak G, Yannone SM, Tainer JA, Adams MW. Microbial metalloproteomes are largely uncharacterized. Nature. 2010;466:779–782. doi: 10.1038/nature09265. [DOI] [PubMed] [Google Scholar]
- 93.Hopfner KP, Craig L, Moncalian G, Zinkel RA, Usui T, Owen BA, Karcher A, Henderson B, Bodmer JL, McMurray CT, Carney JP, Petrini JH, Tainer JA. The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature. 2002;418:562–566. doi: 10.1038/nature00922. [DOI] [PubMed] [Google Scholar]
- 94.Tsutakawa S, Classen S, Chapados BR, Arvai AS, Finger LD, Guenther G, Tomlinson CG, Thompson P, Sarker AH, Shen B, Cooper PK, Grasby JA, Tainer JA. Human Flap Endonuclease Structures, DNA Double Base Flipping and a Unified Understanding of the FEN1 Superfamily. Cell, accepted. 2011 doi: 10.1016/j.cell.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Roberts BR, Tainer JA, Getzoff ED, Malencik DA, Anderson SR, Bomben VC, Meyers KR, Karplus PA, Beckman JS. Structural characterization of zinc-deficient human superoxide dismutase and implications for ALS. J Mol Biol. 2007;373:877–890. doi: 10.1016/j.jmb.2007.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Boon EM, Livingston AL, Chmiel NH, David SS, Barton JK. DNA-mediated charge transport for DNA repair. Proc Natl Acad Sci U S A. 2003;100:12543–12547. doi: 10.1073/pnas.2035257100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hall DB, Holmlin RE, Barton JK. Oxidative DNA damage through long-range electron transfer. Nature. 1996;382:731–735. doi: 10.1038/382731a0. [DOI] [PubMed] [Google Scholar]
- 98.Wolski SC, Kuper J, Kisker C. The XPD helicase: XPanDing archaeal XPD structures to get a grip on human DNA repair. Biol Chem. 2010;391:761–765. doi: 10.1515/BC.2010.076. [DOI] [PubMed] [Google Scholar]
- 99.Korolev S, Hsieh J, Gauss GH, Lohman TM, Waksman G. Major domain swiveling revealed by the crystal structures of complexes of E. coli Rep helicase bound to single-stranded DNA and ADP. Cell. 1997;90:635–647. doi: 10.1016/s0092-8674(00)80525-5. [DOI] [PubMed] [Google Scholar]
- 100.Guan Y, Hickey MJ, Borgstahl GE, Hallewell RA, Lepock JR, O’Connor D, Hsieh Y, Nick HS, Silverman DN, Tainer JA. Crystal structure of Y34F mutant human mitochondrial manganese superoxide dismutase and the functional role of tyrosine 34. Biochemistry. 1998;37:4722–4730. doi: 10.1021/bi972394l. [DOI] [PubMed] [Google Scholar]
- 101.Perry JJ, Shin DS, Getzoff ED, Tainer JA. The structural biochemistry of the superoxide dismutases. Biochim Biophys Acta. 2010;1804:245–262. doi: 10.1016/j.bbapap.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Skorvaga M, DellaVecchia MJ, Croteau DL, Theis K, Truglio JJ, Mandavilli BS, Kisker C, Van Houten B. Identification of residues within UvrB that are important for efficient DNA binding and damage processing. J Biol Chem. 2004;279:51574–51580. doi: 10.1074/jbc.M409266200. [DOI] [PubMed] [Google Scholar]
- 103.Moolenaar GF, Hoglund L, Goosen N. Clue to damage recognition by UvrB: residues in the beta-hairpin structure prevent binding to non-damaged DNA. EMBO J. 2001;20:6140–6149. doi: 10.1093/emboj/20.21.6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Velankar SS, Soultanas P, Dillingham MS, Subramanya HS, Wigley DB. Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell. 1999;97:75–84. doi: 10.1016/s0092-8674(00)80716-3. [DOI] [PubMed] [Google Scholar]
- 105.Parikh SS, Mol CD, Slupphaug G, Bharati S, Krokan HE, Tainer JA. Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J. 1998;17:5214–5226. doi: 10.1093/emboj/17.17.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Parikh SS, Walcher G, Jones GD, Slupphaug G, Krokan HE, Blackburn GM, Tainer JA. Uracil-DNA glycosylase-DNA substrate and product structures: conformational strain promotes catalytic efficiency by coupled stereoelectronic effects. Proc Natl Acad Sci U S A. 2000;97:5083–5088. doi: 10.1073/pnas.97.10.5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tainer JA, McCammon JA, Ivanov I. Recognition of the ring-opened state of proliferating cell nuclear antigen by replication factor C promotes eukaryotic clamp-loading. J Am Chem Soc. 2010;132:7372–7378. doi: 10.1021/ja100365x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Oksenych V, de Jesus BB, Zhovmer A, Egly JM, Coin F. Molecular insights into the recruitment of TFIIH to sites of DNA damage. EMBO J. 2009;28:2971–2980. doi: 10.1038/emboj.2009.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rouillon C, White MF. The XBP-Bax1 helicase-nuclease complex unwinds and cleaves DNA: implications for eukaryal and archaeal nucleotide excision repair. J Biol Chem. 2010;285:11013–11022. doi: 10.1074/jbc.M109.094763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Buttner K, Nehring S, Hopfner KP. Structural basis for DNA duplex separation by a superfamily-2 helicase. Nat Struct Mol Biol. 2007;14:647–652. doi: 10.1038/nsmb1246. [DOI] [PubMed] [Google Scholar]
- 111.Chapados BR, Hosfield DJ, Han S, Qiu J, Yelent B, Shen B, Tainer JA. Structural basis for FEN-1 substrate specificity and PCNA-mediated activation in DNA replication and repair. Cell. 2004;116:39–50. doi: 10.1016/s0092-8674(03)01036-5. [DOI] [PubMed] [Google Scholar]
- 112.Mol CD, Izumi T, Mitra S, Tainer JA. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination [corrected] Nature. 2000;403:451–456. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- 113.Warren JJ, Pohlhaus TJ, Changela A, Iyer RR, Modrich PL, Beese LS. Structure of the human MutSalpha DNA lesion recognition complex. Mol Cell. 2007;26:579–592. doi: 10.1016/j.molcel.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 114.Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, Groocock LM, Cahill D, Hitomi C, Guenther G, Moiani D, Carney JP, Russell P, Tainer JA. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell. 2008;135:97–109. doi: 10.1016/j.cell.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Garcin ED, Hosfield DJ, Desai SA, Haas BJ, Bjoras M, Cunningham RP, Tainer JA. DNA apurinic-apyrimidinic site binding and excision by endonuclease IV. Nat Struct Mol Biol. 2008;15:515–522. doi: 10.1038/nsmb.1414. [DOI] [PubMed] [Google Scholar]
- 116.Getzoff ED, Tainer JA, Weiner PK, Kollman PA, Richardson JS, Richardson DC. Electrostatic recognition between superoxide and copper, zinc superoxide dismutase. Nature. 1983;306:287–290. doi: 10.1038/306287a0. [DOI] [PubMed] [Google Scholar]
- 117.Roberts VA, Freeman HC, Olson AJ, Tainer JA, Getzoff ED. Electrostatic orientation of the electron-transfer complex between plastocyanin and cytochrome c. J Biol Chem. 1991;266:13431–13441. [PubMed] [Google Scholar]
- 118.Coin F, Oksenych V, Egly JM. Distinct roles for the XPB/p52 and XPD/p44 subcomplexes of TFIIH in damaged DNA opening during nucleotide excision repair. Mol Cell. 2007;26:245–256. doi: 10.1016/j.molcel.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 119.Lin YC, Choi WS, Gralla JD. TFIIH XPB mutants suggest a unified bacterial-like mechanism for promoter opening but not escape. Nat Struct Mol Biol. 2005;12:603–607. doi: 10.1038/nsmb949. [DOI] [PubMed] [Google Scholar]
- 120.Fujimoto M, Leech SN, Theron T, Mori M, Fawcett H, Botta E, Nozaki Y, Yamagata T, Moriwaki S, Stefanini M, Momoi MY, Nakagawa H, Shuster S, Moss C, Lehmann AR. Two new XPD patients compound heterozygous for the same mutation demonstrate diverse clinical features. J Invest Dermatol. 2005;125:86–92. doi: 10.1111/j.0022-202X.2005.23745.x. [DOI] [PubMed] [Google Scholar]
- 121.Andersen CB, Ballut L, Johansen JS, Chamieh H, Nielsen KH, Oliveira CL, Pedersen JS, Seraphin B, Le Hir H, Andersen GR. Structure of the exon junction core complex with a trapped DEAD-box ATPase bound to RNA. Science. 2006;313:1968–1972. doi: 10.1126/science.1131981. [DOI] [PubMed] [Google Scholar]
- 122.Bono F, Ebert J, Lorentzen E, Conti E. The crystal structure of the exon junction complex reveals how it maintains a stable grip on mRNA. Cell. 2006;126:713–725. doi: 10.1016/j.cell.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 123.Sengoku T, Nureki O, Nakamura A, Kobayashi S, Yokoyama S. Structural basis for RNA unwinding by the DEAD-box protein Drosophila Vasa. Cell. 2006;125:287–300. doi: 10.1016/j.cell.2006.01.054. [DOI] [PubMed] [Google Scholar]
- 124.Ma X, Hong Y, Han W, Sheng D, Ni J, Hou G, Shen Y. Single-stranded DNA binding activity of XPBI, but not XPBII, from Sulfolobus tokodaii causes double-stranded DNA melting. Extremophiles. 2011;15:67–76. doi: 10.1007/s00792-010-0338-z. [DOI] [PubMed] [Google Scholar]
- 125.Pugh RA, Honda M, Leesley H, Thomas A, Lin Y, Nilges MJ, Cann IK, Spies M. The iron-containing domain is essential in Rad3 helicases for coupling of ATP hydrolysis to DNA translocation and for targeting the helicase to the single-stranded DNA-double-stranded DNA junction. J Biol Chem. 2008;283:1732–1743. doi: 10.1074/jbc.M707064200. [DOI] [PubMed] [Google Scholar]
- 126.Rudolf J, Makrantoni V, Ingledew WJ, Stark MJ, White MF. The DNA repair helicases XPD and FancJ have essential iron-sulfur domains. Mol Cell. 2006;23:801–808. doi: 10.1016/j.molcel.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 127.Boal AK, Genereux JC, Sontz PA, Gralnick JA, Newman DK, Barton JK. Redox signaling between DNA repair proteins for efficient lesion detection. Proc Natl Acad Sci U S A. 2009;106:15237–15242. doi: 10.1073/pnas.0908059106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shiekhattar R, Mermelstein F, Fisher RP, Drapkin R, Dynlacht B, Wessling HC, Morgan DO, Reinberg D. Cdk-activating kinase complex is a component of human transcription factor TFIIH. Nature. 1995;374:283–287. doi: 10.1038/374283a0. [DOI] [PubMed] [Google Scholar]
- 129.Fisher RP, Morgan DO. A novel cyclin associates with MO15/CDK7 to form the CDK-activating kinase. Cell. 1994;78:713–724. doi: 10.1016/0092-8674(94)90535-5. [DOI] [PubMed] [Google Scholar]
- 130.Adamczewski JP, Rossignol M, Tassan JP, Nigg EA, Moncollin V, Egly JM. MAT1, cdk7 and cyclin H form a kinase complex which is UV light-sensitive upon association with TFIIH. EMBO J. 1996;15:1877–1884. [PMC free article] [PubMed] [Google Scholar]
- 131.Rossignol M, Kolb-Cheynel I, Egly JM. Substrate specificity of the cdk-activating kinase (CAK) is altered upon association with TFIIH. EMBO J. 1997;16:1628–1637. doi: 10.1093/emboj/16.7.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Larochelle S, Batliner J, Gamble MJ, Barboza NM, Kraybill BC, Blethrow JD, Shokat KM, Fisher RP. Dichotomous but stringent substrate selection by the dual-function Cdk7 complex revealed by chemical genetics. Nat Struct Mol Biol. 2006;13:55–62. doi: 10.1038/nsmb1028. [DOI] [PubMed] [Google Scholar]
- 133.Ito S, Kuraoka I, Chymkowitch P, Compe E, Takedachi A, Ishigami C, Coin F, Egly JM, Tanaka K. XPG stabilizes TFIIH, allowing transactivation of nuclear receptors: implications for Cockayne syndrome in XP-G/CS patients. Mol Cell. 2007;26:231–243. doi: 10.1016/j.molcel.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 134.Arab HH, Wani G, Ray A, Shah ZI, Zhu Q, Wani AA. Dissociation of CAK from core TFIIH reveals a functional link between XP-G/CS and the TFIIH disassembly state. PLoS One. 2010;5:e11007. doi: 10.1371/journal.pone.0011007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chen J, Larochelle S, Li X, Suter B. Xpd/Ercc2 regulates CAK activity and mitotic progression. Nature. 2003;424:228–232. doi: 10.1038/nature01746. [DOI] [PubMed] [Google Scholar]
- 136.Chen J, Suter B. Xpd, a structural bridge and a functional link. Cell Cycle. 2003;2:503–506. doi: 10.4161/cc.2.6.558. [DOI] [PubMed] [Google Scholar]
- 137.Tirode F, Busso D, Coin F, Egly JM. Reconstitution of the transcription factor TFIIH: assignment of functions for the three enzymatic subunits, XPB, XPD, and cdk7. Mol Cell. 1999;3:87–95. doi: 10.1016/s1097-2765(00)80177-x. [DOI] [PubMed] [Google Scholar]
- 138.Lolli G, Lowe ED, Brown NR, Johnson LN. The crystal structure of human CDK7 and its protein recognition properties. Structure. 2004;12:2067–2079. doi: 10.1016/j.str.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 139.Andersen G, Busso D, Poterszman A, Hwang JR, Wurtz JM, Ripp R, Thierry JC, Egly JM, Moras D. The structure of cyclin H: common mode of kinase activation and specific features. EMBO J. 1997;16:958–967. doi: 10.1093/emboj/16.5.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kim KK, Chamberlin HM, Morgan DO, Kim SH. Three-dimensional structure of human cyclin H, a positive regulator of the CDK-activating kinase. Nat Struct Biol. 1996;3:849–855. doi: 10.1038/nsb1096-849. [DOI] [PubMed] [Google Scholar]
- 141.Gervais V, Busso D, Wasielewski E, Poterszman A, Egly JM, Thierry JC, Kieffer B. Solution structure of the N-terminal domain of the human TFIIH MAT1 subunit: new insights into the RING finger family. J Biol Chem. 2001;276:7457–7464. doi: 10.1074/jbc.M007963200. [DOI] [PubMed] [Google Scholar]
- 142.Waley A. The Way and Its Power: Lao Tzu’s Tao Te Ching and Its Place in Chinese Thought. Grove Press; 1994. [Google Scholar]
- 143.Craig L, Taylor RK, Pique ME, Adair BD, Arvai AS, Singh M, Lloyd SJ, Shin DS, Getzoff ED, Yeager M, Forest KT, Tainer JA. Type IV pilin structure and assembly: X-ray and EM analyses of Vibrio cholerae toxin-coregulated pilus and Pseudomonas aeruginosa PAK pilin. Mol Cell. 2003;11:1139–1150. doi: 10.1016/s1097-2765(03)00170-9. [DOI] [PubMed] [Google Scholar]
- 144.Craig L, Volkmann N, Arvai AS, Pique ME, Yeager M, Egelman EH, Tainer JA. Type IV pilus structure by cryo-electron microscopy and crystallography: implications for pilus assembly and functions. Mol Cell. 2006;23:651–662. doi: 10.1016/j.molcel.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 145.Schultz P, Fribourg S, Poterszman A, Mallouh V, Moras D, Egly JM. Molecular structure of human TFIIH. Cell. 2000;102:599–607. doi: 10.1016/s0092-8674(00)00082-9. [DOI] [PubMed] [Google Scholar]
- 146.Sandrock B, Egly JM. A yeast four-hybrid system identifies Cdk-activating kinase as a regulator of the XPD helicase, a subunit of transcription factor IIH. J Biol Chem. 2001;276:35328–35333. doi: 10.1074/jbc.M105570200. [DOI] [PubMed] [Google Scholar]
- 147.Hammel M, Yu Y, Mahaney BL, Cai B, Ye R, Phipps BM, Rambo RP, Hura GL, Pelikan M, So S, Abolfath RM, Chen DJ, Lees-Miller SP, Tainer JA. Ku and DNA-dependent protein kinase dynamic conformations and assembly regulate DNA binding and the initial non-homologous end joining complex. J Biol Chem. 2010;285:1414–1423. doi: 10.1074/jbc.M109.065615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Perry JJ, Asaithamby A, Barnebey A, Kiamanesch F, Chen DJ, Han S, Tainer JA, Yannone SM. Identification of a coiled coil in werner syndrome protein that facilitates multimerization and promotes exonuclease processivity. J Biol Chem. 2010;285:25699–25707. doi: 10.1074/jbc.M110.124941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Coin F, Proietti De Santis L, Nardo T, Zlobinskaya O, Stefanini M, Egly JM. p8/TTD-A as a repair-specific TFIIH subunit. Mol Cell. 2006;21:215–226. doi: 10.1016/j.molcel.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 150.Kainov DE, Vitorino M, Cavarelli J, Poterszman A, Egly JM. Structural basis for group A trichothiodystrophy. Nat Struct Mol Biol. 2008;15:980–984. doi: 10.1038/nsmb.1478. [DOI] [PubMed] [Google Scholar]
- 151.Zurita M, Merino C. The transcriptional complexity of the TFIIH complex. Trends Genet. 2003;19:578–584. doi: 10.1016/j.tig.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 152.Di Lello P, Miller Jenkins LM, Mas C, Langlois C, Malitskaya E, Fradet-Turcotte A, Archambault J, Legault P, Omichinski JG. p53 and TFIIEalpha share a common binding site on the Tfb1/p62 subunit of TFIIH. Proc Natl Acad Sci U S A. 2008;105:106–111. doi: 10.1073/pnas.0707892105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Iyer N, Reagan MS, Wu KJ, Canagarajah B, Friedberg EC. Interactions involving the human RNA polymerase II transcription/nucleotide excision repair complex TFIIH, the nucleotide excision repair protein XPG, and Cockayne syndrome group B (CSB) protein. Biochemistry. 1996;35:2157–2167. doi: 10.1021/bi9524124. [DOI] [PubMed] [Google Scholar]
- 154.Gervais V, Lamour V, Jawhari A, Frindel F, Wasielewski E, Dubaele S, Egly JM, Thierry JC, Kieffer B, Poterszman A. TFIIH contains a PH domain involved in DNA nucleotide excision repair. Nat Struct Mol Biol. 2004;11:616–622. doi: 10.1038/nsmb782. [DOI] [PubMed] [Google Scholar]
- 155.Giglia-Mari G, Miquel C, Theil AF, Mari PO, Hoogstraten D, Ng JM, Dinant C, Hoeijmakers JH, Vermeulen W. Dynamic interaction of TTDA with TFIIH is stabilized by nucleotide excision repair in living cells. PLoS Biol. 2006;4:e156. doi: 10.1371/journal.pbio.0040156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Santagati F, Botta E, Stefanini M, Pedrini AM. Different dynamics in nuclear entry of subunits of the repair/transcription factor TFIIH. Nucleic Acids Res. 2001;29:1574–1581. doi: 10.1093/nar/29.7.1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Williams RS, Dodson GE, Limbo O, Yamada Y, Williams JS, Guenther G, Classen S, Glover JN, Iwasaki H, Russell P, Tainer JA. Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell. 2009;139:87–99. doi: 10.1016/j.cell.2009.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Williams GJ, Williams RS, Williams JS, Moncalian G, Arvai AS, Limbo O, Guenther G, Sildas S, Hammel M, Russell P, Tainer JA. ABC ATPase signature helices in Rad50 link nucleotide state to Mre11 interface for DNA repair. Nat Struct Mol Biol. 2011 doi: 10.1038/nsmb.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Engelward BP. The flap about ATM & MRE11. Cell Cycle. 2010;9:3148–3149. doi: 10.4161/cc.9.16.12815. [DOI] [PubMed] [Google Scholar]
- 160.Rahal EA, Henricksen LA, Li Y, Williams RS, Tainer JA, Dixon K. ATM regulates Mre11-dependent DNA end-degradation and microhomology-mediated end joining. Cell Cycle. 2010;9:2866–2877. doi: 10.4161/cc.9.14.12363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Classen S, Rodic I, Holton J, Hura GL, Hammel M, Tainer JA. Software for the high-throughput collection of SAXS data using an enhanced Blu-Ice/DCS control system. J Synchrotron Radiat. 2010;17:774–781. doi: 10.1107/S0909049510028566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, 2nd, Tsutakawa SE, Jenney FE, Jr, Classen S, Frankel KA, Hopkins RC, Yang SJ, Scott JW, Dillard BD, Adams MW, Tainer JA. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Methods. 2009;6:606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- 164.Rambo RP, Tainer JA. Bridging the solution divide: comprehensive structural analyses of dynamic RNA, DNA, and protein assemblies by small-angle X-ray scattering. Curr Opin Struct Biol. 2010;20:128–137. doi: 10.1016/j.sbi.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hammel M, Yu Y, Fang S, Lees-Miller SP, Tainer JA. XLF regulates filament architecture of the XRCC4.ligase IV complex. Structure. 2010;18:1431–1442. doi: 10.1016/j.str.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Williams RS, Williams JS, Tainer JA. Mre11-Rad 50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem Cell Biol. 2007;85:509–520. doi: 10.1139/O07-069. [DOI] [PubMed] [Google Scholar]
- 167.Moriel-Carretero M, Aguilera A. A postincision-deficient TFIIH causes replication fork breakage and uncovers alternative Rad51- or Pol32-mediated restart mechanisms. Mol Cell. 2010;37:690–701. doi: 10.1016/j.molcel.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 168.Moriel-Carretero M, Aguilera A. Replication fork breakage and re-start: New insights into Rad3/XPD-associated deficiencies. Cell Cycle. 2010;9:2958–2962. [PubMed] [Google Scholar]
- 169.Williams GJ, Lees-Miller SP, Tainer JA. Mre11-Rad50-Nbs1 conformations and the control of sensing, signaling, and effector responses at DNA double-strand breaks. DNA Repair (Amst) 2010;9:1299–1306. doi: 10.1016/j.dnarep.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Berneburg M, Lowe JE, Nardo T, Araujo S, Fousteri MI, Green MH, Krutmann J, Wood RD, Stefanini M, Lehmann AR. UV damage causes uncontrolled DNA breakage in cells from patients with combined features of XP-D and Cockayne syndrome. EMBO J. 2000;19:1157–1166. doi: 10.1093/emboj/19.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Niedernhofer LJ. Tissue-specific accelerated aging in nucleotide excision repair deficiency. Mech Ageing Dev. 2008;129:408–415. doi: 10.1016/j.mad.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Boyle J, Ueda T, Oh KS, Imoto K, Tamura D, Jagdeo J, Khan SG, Nadem C, Digiovanna JJ, Kraemer KH. Persistence of repair proteins at unrepaired DNA damage distinguishes diseases with ERCC2 (XPD) mutations: cancer-prone xeroderma pigmentosum vs non-cancer-prone trichothiodystrophy. Hum Mutat. 2008;29:1194–1208. doi: 10.1002/humu.20768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.McMurray CT, Tainer JA. Cancer, cadmium and genome integrity. Nat Genet. 2003;34:239–241. doi: 10.1038/ng0703-239. [DOI] [PubMed] [Google Scholar]
- 174.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 175.Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, Bishop DT, Weber B, Lenoir G, Chang-Claude J, Sobol H, Teare MD, Struewing J, Arason A, Scherneck S, Peto J, Rebbeck TR, Tonin P, Neuhausen S, Barkardottir R, Eyfjord J, Lynch H, Ponder BA, Gayther SA, Zelada-Hedman M, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium, Am J Hum Genet. 1998;62:676–689. doi: 10.1086/301749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108:171–182. doi: 10.1016/s0092-8674(02)00615-3. [DOI] [PubMed] [Google Scholar]
- 177.Pakotiprapha D, Inuzuka Y, Bowman BR, Moolenaar GF, Goosen N, Jeruzalmi D, Verdine GL. Crystal structure of Bacillus stearothermophilus UvrA provides insight into ATP-modulated dimerization, UvrB interaction, and DNA binding. Mol Cell. 2008;29:122–133. doi: 10.1016/j.molcel.2007.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Karakas E, Truglio JJ, Croteau D, Rhau B, Wang L, Van Houten B, Kisker C. Structure of the C-terminal half of UvrC reveals an RNase H endonuclease domain with an Argonaute-like catalytic triad. EMBO J. 2007;26:613–622. doi: 10.1038/sj.emboj.7601497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Oyama T, Oka H, Mayanagi K, Shirai T, Matoba K, Fujikane R, Ishino Y, Morikawa K. Atomic structures and functional implications of the archaeal RecQ-like helicase Hjm. BMC Struct Biol. 2009;9:2. doi: 10.1186/1472-6807-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Dubaele S, Proietti De Santis L, Bienstock RJ, Keriel A, Stefanini M, VanHouten B, Egly JM. Basal transcription defect discriminates between xeroderma pigmentosum and trichothiodystrophy in XPD patients. Mol Cell. 2003;11:1635–1646. doi: 10.1016/s1097-2765(03)00182-5. [DOI] [PubMed] [Google Scholar]
- 181.Egly JM. The 14th Datta Lecture. TFIIH: from transcription to clinic. FEBS Lett. 2001;498:124–128. doi: 10.1016/s0014-5793(01)02458-9. [DOI] [PubMed] [Google Scholar]