

Abstract
IL-6 and downstream JAK-dependent signaling pathways have critical roles in the pathophysiology of multiple myeloma. We investigated the effects of a novel small-molecule JAK inhibitor (AZD1480) on IL-6/JAK signal transduction and its biological consequences on the human myeloma-derived cell lines U266 and Kms.11. At low micromolar concentrations, AZD1480 blocks cell proliferation and induces apoptosis of myeloma cell lines. These biological responses to AZD1480 are associated with concomitant inhibition of phosphorylation of JAK2, STAT3 and MAPK signaling proteins. In addition, there is inhibition of expression of STAT3 target genes, particularly Cyclin D2. Examination of a wider variety of myeloma cells (RPMI 8226, OPM-2, NCI-H929, Kms.18, MM1.S, IM-9) as well as primary myeloma cells showed that AZD1480 has broad efficacy. By contrast, viability of normal PBMCs and CD138+ cells derived from healthy controls was not significantly inhibited. Importantly, AZD1480 induces cell death of Kms.11 cells grown in the presence of HS-5 bone marrow-derived stromal cells and inhibits tumor growth in a Kms.11 xenograft mouse model, accompanied with inhibition of phospho-FGFR3, phospho-JAK2, phospho-STAT3 and Cyclin D2 levels. In sum, AZD1480 blocks proliferation, survival, FGFR3 and JAK/STAT3 signaling in myeloma cells cultured alone or co-cultured with bone marrow stromal cells and in vivo. Thus, AZD1480 represents a potential new therapeutic agent for patients with multiple myeloma.
Keywords: myeloma, JAK2, STAT3, FGFR3
Introduction
Multiple myeloma is a malignancy of plasma cells that responds to a limited set of treatment options and is an often incurable disease with a short survival time, especially in older adults(1). During the past decade new multiple myeloma drugs have been developed and clinical trials with new therapies are ongoing(2-4). These new agents and their combinations with chemotherapies have resulted in highly effective regimens, with increased response rates in both the frontline setting for patients not eligible for high-dose therapy/stem cell transplantation and for patients whose disease has relapsed or become resistant to conventional therapy(5, 6). However, some of these new agents exhibit significant toxicity and eventually patients develop resistance to these drugs(7, 8). Therefore, there is the need to add more targeted approaches for treatment in order to improve the anti-myeloma efficacy and enhance the safety and tolerability of these regimens.
IL-6 and the downstream activation of JAK-dependent and JAK-independent signaling pathways have a critical role in the pathophysiology of multiple myeloma by acting as a potent proliferation, survival, and drug resistance determinant for myeloma cells(9, 10). Among the major signaling pathways downstream of IL-6 are the JAK/STAT3 and Ras/MAPK proteins, which are implicated in survival and proliferation of myeloma cells, respectively(11, 12). Thus, a small-molecule inhibitor of JAK and downstream signaling could provide clinical benefits in multiple myeloma. There is no JAK targeted therapy currently available for patients with multiple myeloma. Compounds including curcumin, atiprimod, the tyrosine kinase inhibitor AG490 and the pan-JAK inhibitors pyridone 6 and INCB20 lead to inhibition of IL-6-induced MM cell survival associated with inhibition of STAT3 activity(11, 13-17). However, none of these agents is currently approved for treatment of MM.
AZD1480 is a potent, ATP competitive, small-molecule inhibitor of JAK2 kinase(18), which is in early phase clinical trials for treatment of myelofibrosis. In the present study, we investigated the effect of AZD1480 on IL-6/JAK2 downstream effectors and its biological consequences on human myeloma-derived cell lines. These model cell lines (U266 and Kms.11) express constitutively-activated STAT3 and are IL-6 growth stimulated. Kms.11 cells over-express FGFR3, which is frequently translocated in MM patients. We show that AZD1480 is a potent JAK2 inhibitor that can suppress growth, survival, as well as FGFR3 and STAT3 signaling and downstream targets including Cyclin D2 in human multiple myeloma cells.
Materials and Methods
Drugs and cytokines
AZD1480 was provided by AstraZeneca (Waltham, MA). For in vitro experiments, AZD1480 was dissolved in 100% DMSO to prepare a 10 mM stock and stored at -20°C. For in vivo experiments, AZD1480 was formulated daily in purified, sterile water supplemented with 0.5% Hypromellose and 0.1% Tween 80. Doxorubicin was provided by Sigma and melphalan was obtained from a pharmacy; both drugs were dissolved in RPMI-1640 medium to prepare mM range stocks and stored at 4°C or -20°C, respectively. IL-6 (R&D Systems) was reconstituted in sterile 1× PBS containing 0.1% BSA to prepare a 10 μg/mL stock and stored at -20°C. NF449 and JAK2 inhibitor IV were purchased from Calbiochem (San Diego, CA); NF007 and FGFR inhibitor were purchased from Tocris Bioscience (Ellisville, MO).
Cell lines and cell culture conditions
Human myeloma U266, RPMI 8226, MM1.S, IM-9, NCI-H929 cell lines as well as bone marrow stromal cells were obtained from the American Type Culture Collection (Manassas, VA). The OPM-2 cells were purchased from the European Collection of Cell Culture (ECACC). The Kms.11 and Kms.18 cells were a gift from Dr. P. L. Bergsagel. Cells were maintained in RPMI-1640 medium containing 10% fetal bovine serum and 50 units/mL penicillin and streptomycin at 37°C in an atmosphere of 5% CO2 and passaged twice a week.
Isolation of CD138+ cells from primary PBMCs and BMMCs
Bone marrow (BM) aspirates were collected from 4 patients with multiple myeloma and peripheral blood (PB) samples were collected from 5 healthy donors. BM mononuclear cells (BMMCs) and PB mononuclear cells (PBMCs) were isolated with Ficoll-Hypaque sedimentation (Sigma-Aldrich, St. Louis, MO) and enriched for CD138-positive cells by immunomagnetic nanoparticles positive selection method using the EasySep kit (StemCell Technologies, Vancouver, British Columbia, Canada). The yield of MM cells was high (>95%). Viability of the MM-cell enriched fractions was 99%. All donors and patients had given informed consent for sample acquisition as a part of a protocol approved by the local Institutional Review Board.
Cocultures with BMSCs
Bone marrow stromal layers were established by plating HS.5 cells at a density of 250 000 cells/well in 12-well plates for 24 h. For experiments to calculate the percentage of tumor cell death, Kms.11 cells were first labeled with 5 μM CellTrace™ CFSE (Molecular Probes, Eugene, OR), resuspended in serum-free medium and then applied to the wells containing the HS.5 stromal cells layer at a concentration of 250 000 cells/mL. After 24 h of coculture, AZD1480 was added to the coculture media. Following incubation for 48 h with AZD1480, Kms.11 cells were separated from the stromal layer by carefully pipetting twice with ice-cold PBS. Tumor cells were stained with DAPI and analyzed using a flow cytometer with excitation and emission wavelengths appropriate for fluorescein and DAPI. The % cell death was calculated based on all the DAPI-positive cells after gating on CFSE-positive Kms.11 cells.
Analysis of primary cells by DIMSCAN
Primary cells were cultured at a density of 25 000 cells/well in 96-well plates with RPMI-1640 medium containing 10% fetal bovine serum and 50 units/mL penicillin and streptomycin, and treated with different doses of AZD1480 up to 48 h. To determine the cytotoxicity of AZD1480, cells were assessed by the fluorescence-based DIMSCAN, which uses digital imaging microscopy to quantify viable cells that selectively accumulate fluorescein diacetate.
Cell viability assays
Cells were seeded in 96-well plates at a density of 10 000 cells/well. After 24, 48 or 72 h, cell viability was determined by assaying with MTS [(3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt] assay (Promega, Madison, WI). The MTS assay was performed according to instructions from the supplier. Absorbance was measured at 490 nm with a Chameleon plate reader (Bioscan, Washington DC).
Apoptosis assay by flow cytometry
Untreated and drug-treated cells were stained with Annexin V and Propidium Iodide (PI) using Annexin V-FITC Apoptosis Detection Kit I (BD Biosciences Pharmingen, San Diego, CA). The percentage of apoptotic and nonviable cells was determined by flow cytometry. At least 50 000 cells were collected with a CyAn ADP Violet (Dako) cytometer and calculated using the Summit software (Dako). Percent apoptosis was calculated based on all the Annexin V-positive plus the Annexin V/PI-positive cells. The % loss of cell viability was calculated considering all the Annexin V-positive plus the PI-positive and the Annexin V/PI-positive cells.
Western blot analysis
Cells were washed with ice-cold PBS containing 0.1 mM sodium orthovanadate and total proteins were isolated using RIPA lysis buffer, which included protease inhibitors (leupeptin, antipain, and aprotinin), 0.5 mM PMSF and 0.2 mM sodium orthovanadate. Protein amounts were quantified using the Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA). Equal amounts of proteins were loaded onto a SDS-PAGE gel, transferred onto nitrocellulose membrane, and probed with the indicated antibody: rabbit polyclonal anti-phospho-JAK2 (Chemicon International); rabbit polyclonal anti-STAT3, anti-phospho-STAT3 (Tyr705), anti-phospho-STAT3 (Ser727), anti-p44/42 MAPK and anti-phospho-p44/42 MAPK (Cell Signaling Technology, Danvers, MA); mouse monoclonal anti-c-Myc, rabbit polyclonal anti-Mcl-1, rabbit polyclonal anti-Cyclin-D2 (Santa Cruz Biotechnology, Santa Cruz, CA); mouse monoclonal Bcl-xL antibody (Calbiochem); and mouse monoclonal β-actin antibody (Sigma-Aldrich, St Louis, MO). Membranes were then washed, reprobed with appropriate horseradish peroxidase-conjugated secondary antibodies (Amersham Biosciences, Buckinghamshire, UK), and developed with SuperSignal chemiluminescent substrate (Pierce Biotechnology, Rockford, IL).
Protein immunoprecipitation
Cell lysates (250 μg protein) were incubated with JAK1, JAK2 antibodies (Cell Signaling Technology, Danvers, MA) or FGFR3 antibody (Novus Biologicals, Littleton, CO) overnight at 4°C. To this mixture, washed protein A beads were added and incubated for 1 h at 4°C. Next, the immunoprecipitates were washed five times with the lysis buffer and proteins were eluted with the SDS sample buffer, loaded on 12% SDS-PAGE gels and analyzed by Western blotting analysis using phospho-JAK1, phospho-JAK2 antibodies (Cell Signaling Technology) or phospho-Tyr antibody (Millipore, Temecula, CA).
Kinase assay
Cell-free kinase assays were performed as previously described(19). Briefly, assays were carried-out in 50 μl of kinase buffer (60 mM HEPES-NaOH pH 7.5, 3 mM MgCl2, 3 mM MnCl2, 3 μM Na3VO4), for 30 minutes at 30°C in the presence of 2.5 μg of polyethylene glycol, 10 μM ATP, 200 ng of recombinant STAT1 (Prolias, Rockville, MD) as a substrate, 200 ng of recombinant kinase, and inhibitors added directly to the kinase reaction. Recombinant FGFR3, JAK2 and JAK3 were purchased from SignalChem (Richmond, Canada). The following antibodies were used: STAT1, P-STAT1 (Y701), JAK2, JAK3 (Cell Signaling Technology, Beverly, MA); FGFR3 (Santa Cruz Biotechnology, Santa Cruz, CA); 4G10 (Upstate Biotechnology, Lake Placid, NY).
Transfection
Transfections of Kms.11 cells were performed with the Nucleofector Kit C, program X-005 (Amaxa Biosystems, Gaithersburg, MD). siRNAs (200 nM) and plasmids (2 μg) were used in each transfection with 2 million cells. The 27mer dicer substrate STAT3 siRNA was designed using the City of Hope siRNA Site Selector/Duplex-End Energy Difference Calculator(20, 21). Cy3-labeled STAT3 siRNA was synthesized in the City of Hope DNA/RNA synthesis laboratory. The Cy3-labeled 27mer dicer substrates FGFR3 and negative control siRNA were obtained from IDT (Coralville, IA). Twenty-four hours after transfection with siRNAs, Cy3-positive cells were sorted with MoFlo MLS sorter (Beckman Coulter, Fort Collins, CO) and calculated with the software Summit version 4.3. The Cy3 label was excited by 514-nm laser and the signal was detected with a 600/30-nm bandpass filter. Constitutive activated STAT3 expression plasmid vector, pRC/CMV-STAT3c-Flag was a generous gift from J. Darnell(22). For establishing the stable STAT3c-expressing cells, plasmids pRC/CMV-vector and pRC/CMV-STAT3c-Flag were transected into Kms.11 cells. Twenty-four hours after transfection, 0.5 mg/ml G418 (Invitrogen) was added for selection. Resistant pools of cells were characterized by western blot and maintained in medium containing 0.2 mg/ml G418. Kms.11 cells were also transiently transfected with the pRK7 vectors carrying FLAG-tagged wild-type and Y373C-FGFR3 constructs, which were described previously(19). The vector containing no insert was pcDNA3.
Animal model studies
Kms.11 cells (1×107) were resuspended in RPMI medium and injected subcutaneously into the flank of four to six week old NOD/SCID IL2Rγ-null mice (Jackson Laboratories, Bar Harbor, ME). When tumors reached approximately 65 mm3, mice were divided into one control group and one treated group (10/group) which were dosed orally (p.o.) with the vehicle or AZD1480 (30 mg/kg), respectively. Mice were dosed twice a day for seven days a week. At the dose of AZD1480 indicated, no lethal toxicity or weight loss (greater than 10% body weight) was observed amongst treated animals. Tumors were measured every 3-4 days with vernier calipers, and tumor volumes were calculated by the following formula: 0.5 × (larger diameter) × (smaller diameter). All mice were maintained under specific pathogen-free conditions and were used in compliance with protocols approved by the local Institutional Animal Care and Use Committee.
Statistical analysis and software
The data shown represent mean values of at least three independent experiments and expressed as mean±SD. Statistical analysis was performed by the Student's t-test, using the statistical software GraphPad Prism 4. Statistical significance was set at a level of P<0.05. Four patients were selected based on the criteria that this is a sufficient sample size to rule out lack of drug action if there is consistent drug activity demonstrated in all samples (p<0.02). The Mann Whitney test was used to calculate the P value in Figure 1C and Figure 9. The statistical analysis in Figure 8A was performed with Two-way ANOVA. The statistical significance of the downregulation of Cyclin D2 in Figure 8B was calculated using the IDV values normalized to those of β-actin, which were determined by the densitometry software AlphaImager. The IC50 values in Table 1 and the combination index (CI) values in Table 3 were calculated by the software Compusyn; the descriptions in Table 3 are based on the ranges of CI refined from those described by Chou(23).
Figure 1. AZD1480 induces apoptosis of myeloma cells but does not affect the viability of normal peripheral blood mononuclear cells.
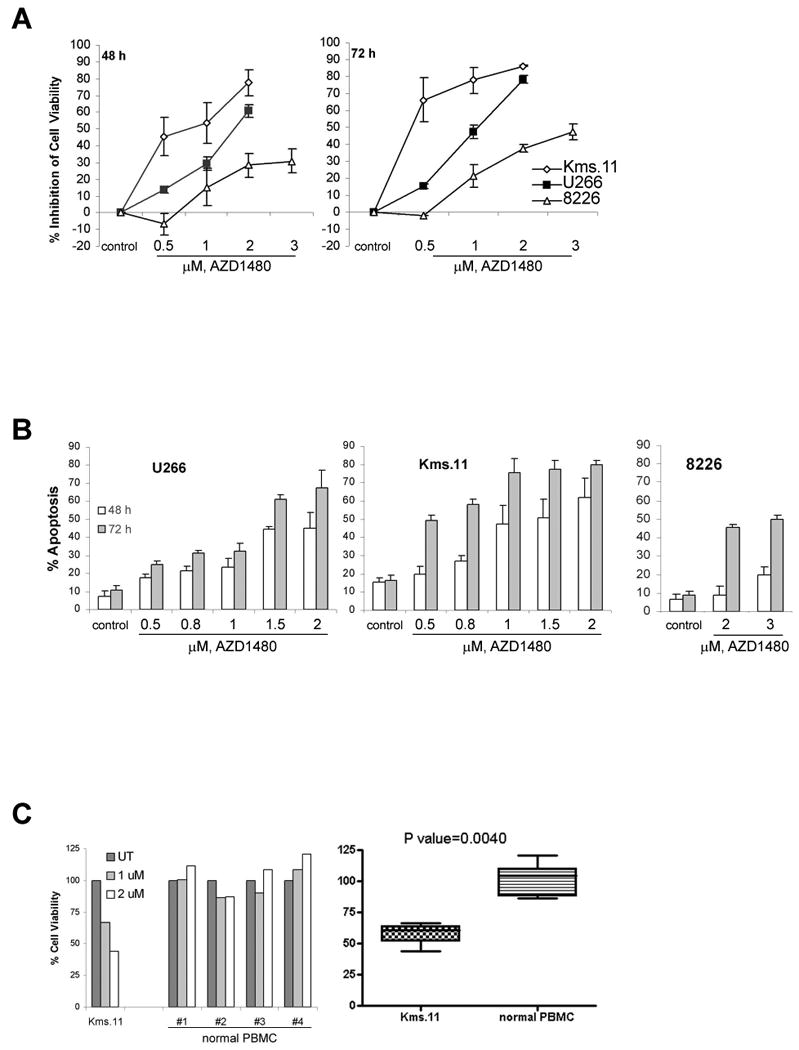
(A) Cells were treated with the indicated concentrations of AZD1480 for 48 or 72 h. Following this, the percentage of cell viability inhibition was determined by MTS assay. Values represented as graphs are the mean of 3 independent experiments with standard deviation. (B) Cells were treated with the indicated concentrations of AZD1480 for 48 or 72 h. Following this, the percentage of apoptotic cells was determined by flow cytometry using Annexin V/Propidium Iodide staining. Values represented as bar graphs are the mean of 3 independent experiments with the standard deviation. (C) Samples of primary PBMCs cells from healthy donors were treated in culture with the indicated concentrations of AZD1480 for 48 h. The percentage of viable cells was determined by DIMSCAN analysis. Values represent the percentages of cell viability normalized to that of the untreated cells and are the mean of three separate treatments.
Figure 9. AZD1480 does not affect the viability of normal CD138+ cells.
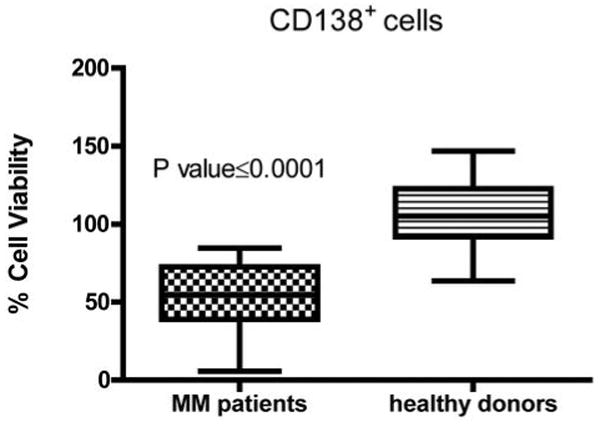
Primary CD138+ cells isolated from the peripheral blood of 5 healthy donors and from the bone marrow of 4 MM patients were treated in culture with the indicated concentrations of AZD1480 for 48 h. The percentage of viable cells was determined by DIMSCAN analysis. Values represent the percentages of cell viability normalized to that of the untreated cells. Values from two separate treatments were used to generate the graph.
Figure 8. AZD1480 reduces the tumor growth of Kms.11 xenograft in mice associated with inhibition of JAK2/STAT3 and FGFR3 signaling and Cyclin D2 in vivo.
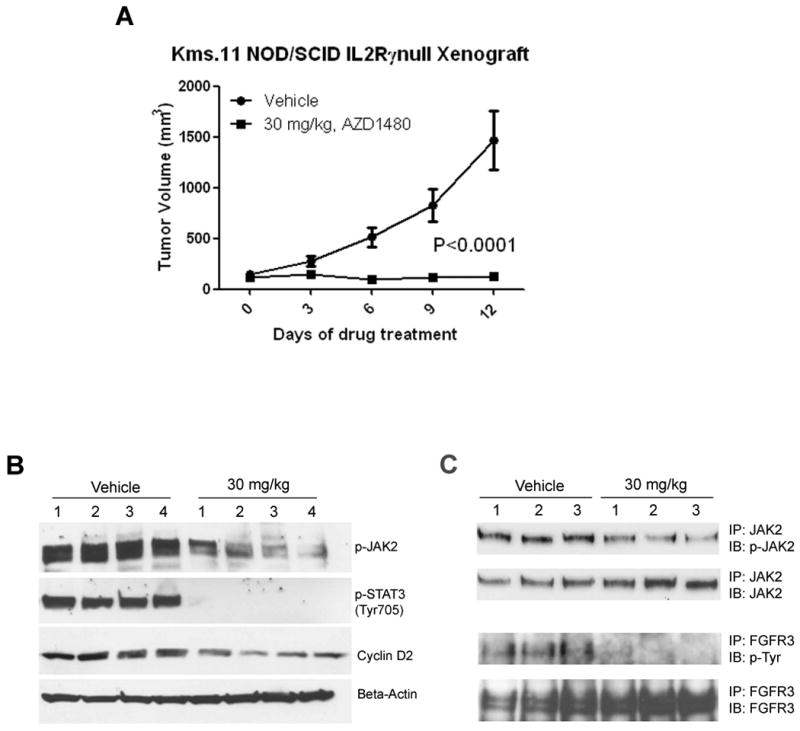
(A) Kms.11 tumor-bearing mice were treated with vehicle or AZD1480 at 30 mg/kg twice a day for two weeks. (B) A separate cohort of mice was treated once a day with vehicle or 30 mg/kg AZD1480. When the tumor size of vehicle group and drug group reached an average volume of 1467 and 162 mm3, respectively, four tumor samples from each group were harvested 2 h post dosing; the weight average was 1.52 and 0.24 g in the vehicle and drug group mice, respectively. Whole-cell lysates were prepared and subjected to Western blot analysis or immunoprecipitation followed by immunoblot (C). Equal amounts of protein were analyzed.
Table 1. The cytotoxic effect of AZD1480 on MM cell lines correlates with the status of STAT3 and FGFR3.
Cells were treated with AZD1480 for 48 h. Following this, the percentage of cell viability inhibition was determined by MTS assay and IC50 values were calculated.
| Cell line | IC50 (uM) | Constitutively active STAT3 | Translocated FGFR3 t(4;14) | Translocated FGFR3 t(4;14) with not activating mutation | Translocated FGFR3 t(4;14) with activating mutation | Reference |
|---|---|---|---|---|---|---|
| U266 | 1.59 | ✓ | (54) | |||
| Kms.11 | 0.68 | ✓ | ✓ | ✓ Y373C | (55) | |
| RPMI 8226 | 7 | (54) | ||||
| OPM-2 | 1.62 | ✓ | ✓ | ✓ K650E | (55), (56) | |
| NCI-H929 | 6.17 | (54) | ||||
| Kms.18 | 4.87 | ✓ | ✓ G384D | (55) | ||
| MM1.S | 6 | (57) | ||||
| IM-9 | 2.81 | ✓ | (58) |
Table 3. AZD1480 enhances sensitivity to melphalan and doxorubicin of myeloma cells cultured alone or in the presence of bone marrow stromal cells.
CFSE-labeled Kms.11 cells were plated on an established bone marrow stromal layer. After 24 h of coculture, cells were incubated for 48 h with AZD1480, doxorubicin, melphalan or a combination of these drugs. Cells were then separated from the stromal layer, stained with DAPI and analyzed by flow cytometry. The percentage of cell death was calculated based on all DAPI-positive cells after gating on CFSE-positive Kms.11 cells. In cocultures, incubation with 1 μM AZD1480, 5 μM melphalan or 100 nM doxorubicin alone induced 20%, 20% and 10% cell death, respectively; combination of 1 μM AZD1480 with 5 μM melphalan or 1 μM AZD1480 with 100 nM doxorubicin induced 50% and 40% cell death, respectively. Descriptions of combination effect are represented as tabular material.
| Culture condition/treatment | Combination Index | Description |
|---|---|---|
| Culture alone | ||
| 1 μM AZD1480 + 5 μM Melphalan | 0.89261 | Slight synergism |
| 2 μM AZD1480 + 5 μM Melphalan | 0.72982 | Moderate synergism |
| 1 μM AZD1480 + 0.1 μM Doxorubicin | 0.68701 | Synergism |
| 2 μM AZD1480 + 0.1 μM Doxorubicin | 0.69247 | Synergism |
| Cocultured with with stromal cells | ||
| 1 μM AZD1480 + 5 μM Melphalan | 0.78307 | Moderate synergism |
| 2 μM AZD1480 + 5 μM Melphalan | 0.94190 | Nerly additive |
| 1 μM AZD1480 + 0.1 μM Doxorubicin | 0.79610 | Moderate synergism |
| 2 μM AZD1480 + 0.1 μM Doxorubicin | 0.99562 | Nerly additive |
Results
AZD1480 inhibits proliferation and induces apoptosis of human myeloma cell lines
We investigated the effects of treatment with AZD1480 on a panel of human myeloma cell lines. We first determined the effect of AZD1480 on the proliferation of U266, Kms.11 and 8226 cells (Figure 1A). MTS assays showed that AZD1480 markedly inhibits the growth of U266, Kms.11 and 8226 cells in a time- and dose-dependent manner. The data also indicate that AZD1480 is more potent in U266 and Kms.11 cells than in 8226 cells for inhibiting proliferation. The 50% inhibitory concentration (IC50) value for inhibition of proliferation at 48 h is approximately 2 μM for U266 cells and approximately 1 μM for Kms.11 cells; in the same cell lines the IC50 at 72 h is approximately 1 μM and 0.5 μM, respectively. 8226 cells require higher concentrations of AZD1480 with an IC50 at 72 h of approximately 3 μM. The IC50 values were determined for a wider variety of myeloma cell lines. Table 1 shows that AZD1480 has broad efficacy, which correlates not only with the presence of activated STAT3 but also with the expression of FGFR3. Cells lacking both phospho-STAT3 and FGFR3 are less sensitive than cells possessing activated STAT3 or overexpressing FGFR3. The levels of FGFR3 and phospho-STAT3 detected by western blot analysis (Supplemental Figure 1) are in agreement with the references shown in Table 1.
The apoptotic effect of AZD1480 was determined in the two more sensitive cell lines, U266 and Kms.11 (Figure 1B). Exposure to AZD1480 induces apoptosis of both U266 and Kms.11 cells in a time- and dose-dependent manner. The IC50 in terms of apoptosis at 72 h is approximately 1.5 μM for both U266 and Kms.11 cells. Representative flow cytometry plots for other cell lines are shown in Supplemental Figure 2, indicating that the apoptotic effects correlate with the IC50 values. In contrast, no cytotoxic effects were observed in normal cells. Figure 1C shows that the viability of human peripheral blood mononuclear cells was not affected at concentrations in the IC50 range for inducing apoptosis of Kms.11 cells.
Because IL-6 has a prominent role in MM(9, 10) we evaluated whether AZD1480 suppresses the IL-6-induced proliferation and survival of myeloma cells that have been reported to be growth stimulated by IL-6(13, 24). The MTS assay (Figure 2A) showed that 0.5 μM AZD1480 at 48 h completely inhibits IL-6-induced cell proliferation in U266 cells and inhibits approximately 50% of IL-6-induced cell proliferation in Kms.11 cells. U266 and Kms.11 cells treated for 48 h with 2 μM AZD1480 compared with the untreated cells stimulated with IL-6 showed 70% and 50% cell proliferation inhibition, respectively. AZD1480 also inhibits the survival of both cell lines in the presence of IL-6, inducing 50% and 60% apoptosis at 2 μM at 48 h in U266 and Kms.11 cells compared with the untreated controls grown in presence of IL-6, respectively (Figure 2B). Therefore, the addition of IL-6, which induced proliferative responses in U266 and Kms.11 cells (20% and 30%, respectively), did not cause a significant shift in IC50 for U266 cells and did not protect them from AZD1480-mediated inhibition of proliferation and survival. However, Kms.11 cells are less sensitive to AZD1480 in terms of inhibition of proliferation when cultured in the presence of IL-6 but still respond to the drug with IC50 at 2 μM.
Figure 2. AZD1480 induces apoptosis of myeloma cells growth stimulated by IL-6.
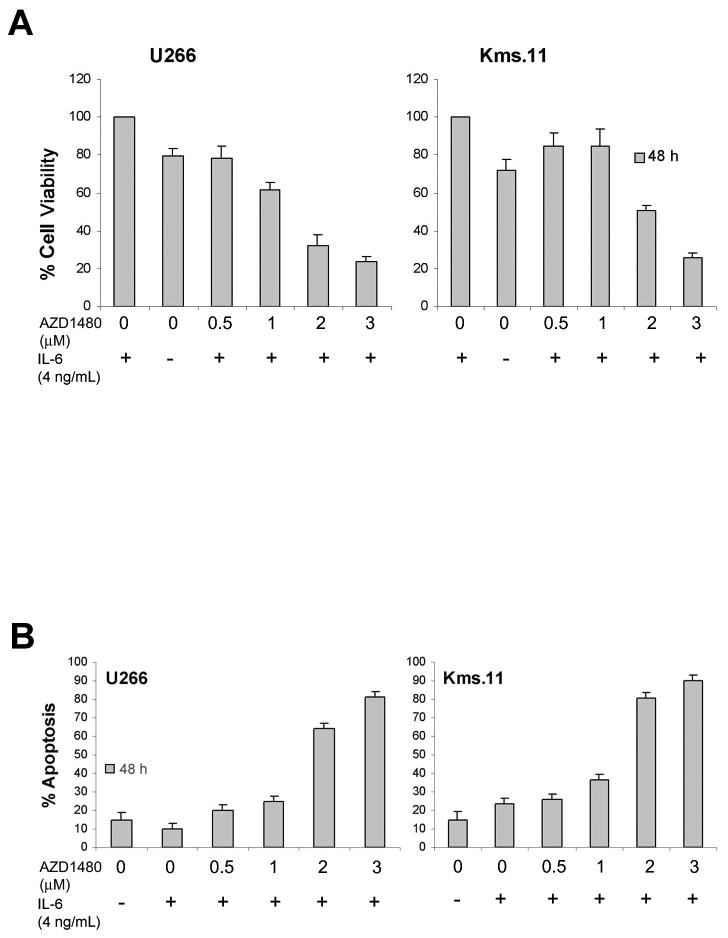
(A) Cells were grown in the presence of IL-6 for 16 h followed by treatment with the indicated concentrations of AZD1480 for 48 h. Following this, the percentage of cell viability inhibition was determined by MTS assay. Values represented as bar graphs are the mean of 3 independent experiments with the standard deviation. (B) Cells were grown in the presence of IL-6 for 16 h followed by treatment with the indicated concentrations of AZD1480 for 48 h. Following this, the percentage of apoptotic cells was determined by flow cytometry using Annexin V/Propidium Iodide staining. Values represented as bar graphs are the mean of 3 independent experiments with the standard deviation.
AZD1480 inhibits IL-6-inducible JAK2/STAT3 and MAPK signaling pathways in vitro
Because JAK family kinases are known to be activated by phosphorylation upon IL-6/gp130 engagement(25) we examined the effect of AZD1480 on IL-6-dependent JAK2 phosphorylation in myeloma cells pretreated with AZD1480 and then stimulated with IL-6. Figure 3A shows that IL-6-induced phosphorylation of JAK2 was inhibited at 0.25-0.5 μM and virtually abolished at 1 μM in both U266 and Kms.11 cells. The JAK1 phosphorylation was not inhibited at these doses.
Figure 3. AZD1480 inhibits IL-6-inducible activation of JAK2 and phosphorylation of STAT3 and MAPK in myeloma cells.
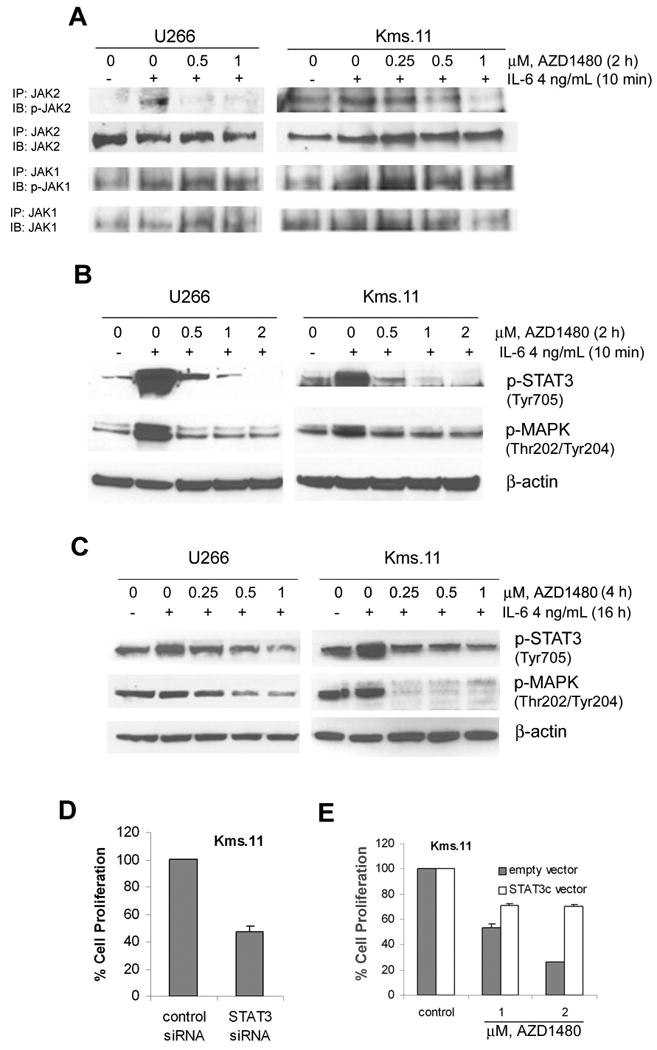
(A) Cells were incubated overnight in medium containing charcoal-stripped serum and then treated with the indicated concentrations of AZD1480 for 2 h followed by stimulation with IL-6 for 10 min. After this, immunoprecipitation with JAK1 or JAK2 antibody followed by Western blot analysis of phospho-JAK1 or phospho-JAK2 was performed on the cell lysates from U266 or Kms.11. The levels of total JAK1 or JAK2 protein served as loading controls. Results are representative of three independent experiments. (B) Cells were incubated overnight in medium containing charcoal-stripped serum and then treated with the indicated concentrations of AZD1480 for 2 h, followed by stimulation with IL-6 for 10 min. Subsequently, Western blot analysis of p-STAT3 and p-MAPK was performed on the cell lysates from U266 and Kms.11. The levels of β-actin protein served as loading controls. Results are representative of three independent experiments. (C) Cells were grown in the presence of IL-6 for 16 h followed by treatment with indicated concentrations of AZD1480 for 4 h. After this, Western blot analysis of p-STAT3 and p-MAPK was performed on the cell lysates from U266 and Kms.11. The levels of β-actin protein served as loading controls. Results are representative of three independent experiments. (D) Kms.11 cells were transfected with Cy3-labeled STAT3 siRNA or negative control siRNA and 24 h later Cy3-positive cells were sorted. The percentage of cell proliferation was determined 72 h post transfection by MTS assay. Values represented as bar graphs are the means of 3 independent experiments plus the standard deviation. (E) Cells were stable transfected with a vector expressing STAT3c or empty vector. Stable transfected cells were treated with AZD1480. After 48 h of treatment, the percentage of cell proliferation was determined by MTS assay. Both empty vector and STAT3c vector values are normalized to 100%. Values represented as bar graphs are the means of 3 independent experiments plus the standard deviation.
It is well established that IL-6 activates distinct downstream signaling pathways: JAK/STAT3 and Ras/MAPK pathways(12, 26). We therefore investigated whether the blockade of IL-6-inducible activation of JAK2 by AZD1480 would prevent the phosphorylation of STAT3 and MAPK in myeloma cell lines pretreated with AZD1480 and then stimulated with IL-6. Figure 3B shows that the phosphorylation of both STAT3 and ERK1/2 induced by IL-6 in U266 and Kms.11 cells was reduced to basal levels at 0.5-1 μM and phosphorylation of STAT3 was abolished at 2 μM. We also demonstrated that AZD1480 inhibits constitutive tyrosine phosphorylation of STAT3 but not the serine phosphorylation (data not shown).
To better mimic the in vivo condition, U266 and Kms.11 cells were cultured for 16 h in the presence of IL-6 and then treated with AZD1480. In this setting, we observed a significant decrease of the level of STAT3 and MAPK phosphorylation at 4 h (Figure 3C) and 24 h post-treatment (data not shown). We also confirmed that 8226 cells lack constitutively activated STAT3, whereas IL-6 stimulated the tyrosine phosphorylation of STAT3 and the activation of MAPK, as previously described(27). AZD1480 suppressed the IL-6-induced phosphorylation of JAK2, STAT3 and MAPK even though phospho-JAK2 levels were downregulated at higher concentrations compared to those required to downregulate phospho-JAK2 levels in U266 and Kms.11 cells (data not shown). Therefore, the inhibition of IL-6-induced growth of myeloma cells by AZD1480 correlates with induction of apoptosis and decreased JAK2, STAT3 and MAPK phosphorylation.
Our observation that the IC50 for AZD1480, in terms of inhibition of proliferation or survival, was higher than the concentration required to reduce STAT3 activation in myeloma cells may suggest that these cells may not be completely dependent on STAT3 for survival. Figure 3D demonstrates that Kms.11 cells depend on STAT3 for proliferation since downregulation of STAT3 by siRNA results in 50% inhibition of proliferation compared to cells transfected with control siRNA. Downregulation of STAT3 mRNA levels was confirmed by quantitative real-time PCR 72 hours post transfection (approximately 70% inhibition compared to control) (data not shown). To determine if STAT3 is involved in the response to AZD1480, we performed experiments in Kms.11 cells transfected with constitutive activated STAT3 (STAT3c) (Figure 3E). STAT3c rescues Kms.11 cells from AZD1480-mediated loss of viability, consistent with role of STAT3 in the biological effects mediated by AZD1480. Nevertheless, we do not exclude the possibility that other pathways may be involved in the response to AZD1480.
Because AZD1480 inhibits proliferation and induces apoptosis associated with inhibition of IL-6-inducible STAT3 phosphorylation, we investigated the effect of AZD1480 on direct or indirect STAT3 targets in MM that have key roles in cell-cycle regulation (Cyclin D2), proliferation (c-Myc) and survival (Mcl-1 and Bcl-xL)(11, 28). We first analyzed the levels of these proteins in U266 and Kms.11 cells cultured for 16 h in the presence of IL-6 and then treated with AZD1480. Figure 4A shows that IL-6 upregulates c-Myc, Cyclin D2 and Bcl-xL in U266 cells. Both c-Myc and Cyclin D2 were decreased in a dose-dependent manner by AZD1480 at 24 h, while Bcl-xL was decreased after 48 h by AZD1480 treatment. At 48 h post-treatment the inhibition of c-Myc and Cyclin-D2 was even more dramatic (Figure 4B). We did not observe IL-6-dependent upregulation of Mcl-1, which was downregulated by AZD1480. IL-6 upregulates Cyclin D2 also in Kms.11 cells and AZD1480 inhibits this upregulation in a dose-dependent manner (Figure 4A), even more so at 48 h post-treatment (Figure 4B). Cyclin D2 protein levels were decreased in U266 or Kms.11 cells treated for 24 h with AZD1480 without being precultured in the presence of IL-6 (data non shown). No change in Cyclin D2, or Mcl-1, or Bcl-xL was observed in 8226 cells prestimulated for 16 h with IL-6 and then treated with AZD1480 for 24 h (data not shown).
Figure 4. AZD1480 inhibits the IL-6-inducible upregulation of the STAT3 target genes c-Myc and Cyclin-D2.
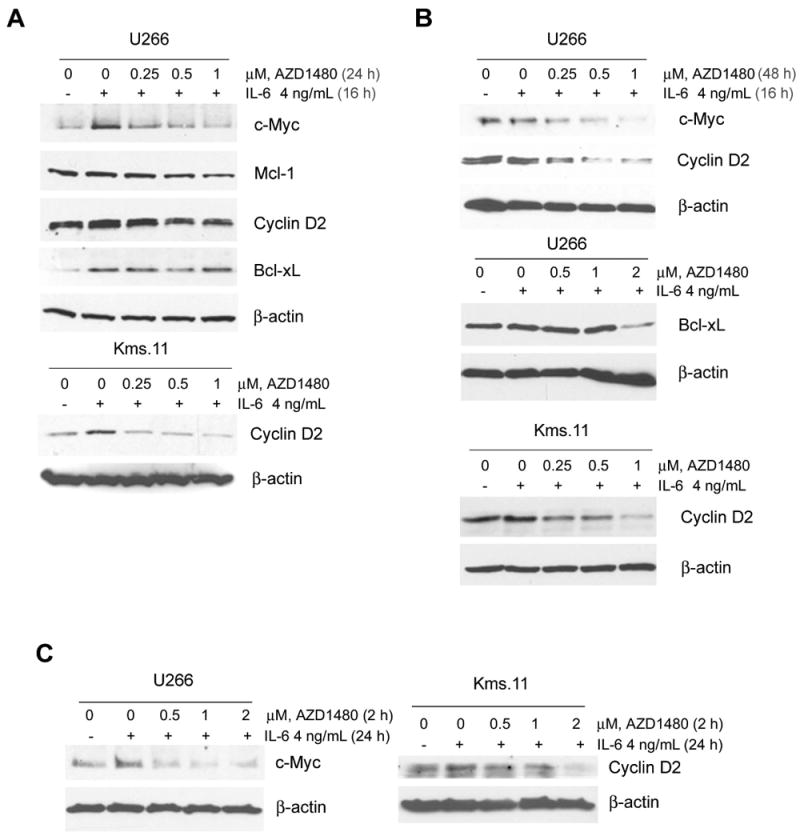
Cells were treated with the indicated concentrations of AZD1480 for 24 h (A) or 48 h (B) after a preincubation with IL-6 for 16 h. Subsequently, Western blot analysis of c-Myc, Mcl-1, Cyclin-D2 and Bcl-xL proteins was performed on the lysates from U266 and Kms.11 cells. The levels of β-actin protein served as loading controls. Results are representative of three independent experiments. (C) Cells were treated with the indicated concentrations of AZD1480 for 2 h followed by incubation with IL-6 for 24 h. Subsequently, Western blot analysis of c-Myc and Cyclin-D2 proteins was performed on the lysates from U266 and Kms.11 cells. The levels of β-actin protein served as loading controls. Results are representative of three independent experiments.
We then analyzed the protein levels of c-Myc, Mcl-1, Cyclin D2 and Bcl-xL in U266 and Kms.11 cells pretreated with AZD1480 and then stimulated with IL-6, demonstrating that AZD1480 inhibits the IL-6-dependent expression of c-Myc and Cyclin D2 in U266 and Kms.11, respectively (Figure 4C). These results suggest that AZD1480 could inhibit myeloma cell proliferation and survival through the down-regulation of c-Myc or Cyclin D2.
AZD1480 effects on myeloma cells cocultured in vitro with bone marrow stroma cells
The bone marrow stromal environment is a major factor in myeloma cell growth and resistance to chemotherapy(29, 30). We first investigated whether AZD1480 has cytotoxic effects in the human bone marrow stroma-derived HS.5 cell line. Figure 5A shows that the proliferation of HS.5 cells was not affected at concentrations in the IC50 range for Kms.11 cells. We then tested the effect of the bone marrow environment on myeloma cell response to AZD1480. Figure 5B shows that Kms.11 cells cocultured with the bone marrow stromal cells HS.5 are not resistant to AZD1480, showing the same percentage of cell death compared to the tumor cells grown alone.
Figure 5. Coculture of myeloma cells with bone marrow stroma-derived cells does not confer resistance to AZD1480.
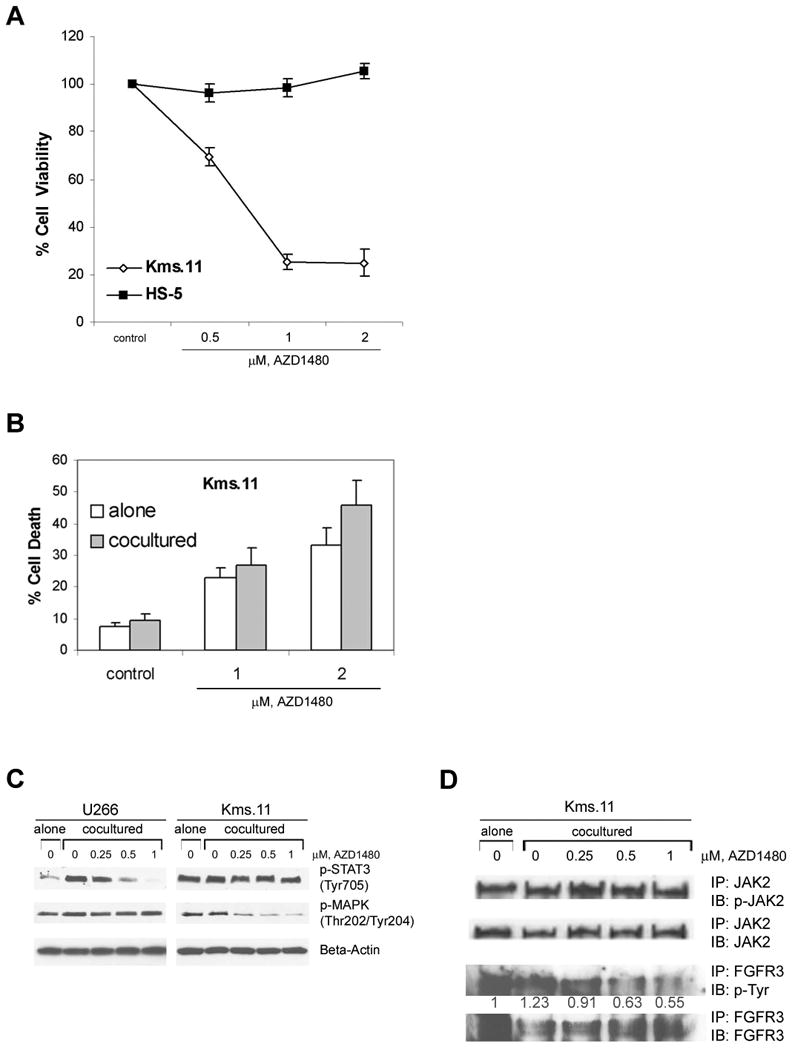
(A) HS-5 or Kms.11 cells were treated with the indicated concentrations of AZD1480 for 48 h. Following this, the percentage of cell viability inhibition was determined by MTS assay. Values represented as graphs are the mean of 3 independent experiments with the standard deviation. (B) CFSE-labeled Kms.11 cells were plated on an established bone marrow stromal layer. After 24 h of coculture, cells were treated with AZD1480 for 48 h. Subsequentially, cells were separated from the stromal layer, stained with DAPI and analyzed by flow cytometry. The percentage of cell death was calculated based on all DAPI-positive cells after gating on CFSE-positive Kms.11 cells. Values represented as bar graphs are the mean of 3 independent experiments with the standard deviation. (C) Tumor cells were kept overnight in medium containing charcoal-stripped serum and then plated on an established bone marrow stromal layer. After 48 h of coculture, cells were incubated with AZD1480 for 2 h. Tumor cells were then separated from the stromal layer and Western blot analysis of p-STAT3 and p-MAPK was performed on the cell lysates. The levels of β-actin protein served as loading controls. Results are representative of three independent experiments. (D) Tumor cells were kept overnight in medium containing charcoal-stripped serum and then plated on an established bone marrow stromal layer. After 48 h of coculture, cells were incubated with AZD1480 for 2 h. Tumor cells were then separated from the stromal layer and immunoprecipitation with JAK2 or FGFR3 antibody followed by Western blot analysis of phospho-JAK2 or phospho-Tyr was performed on the cell lysates from Kms.11 cells. The levels of total JAK2 or FGFR3 protein served as loading controls. The values shown below p-Tyr immunoblot represent the relative expression of p-FGFR3, calculated using the IDV values normalized to those of FGFR3. Results are representative of three independent experiments.
To determine if STAT3 and MAPK signaling pathways are downregulated by AZD1480 in myeloma cells cocultured with bone marrow stromal cells, we treated U266 and Kms.11 cell lines for 2 h after being cocultured for two days with HS.5 cells monolayer (Figure 5C). In this setting, we observed a significant decrease in the level of stroma-induced STAT3 phosphorylation in U266 cells, although stroma-induced phosphorylation of MAPK was minimal. In contrast, we did not observe any significant increase in stroma-induced phospho-JAK2/STAT3 levels in the Kms.11 cell line and AZD1480 had no apparent effect on JAK2/STAT3 phosphorylation in these cells; instead there was a decrease in phospho-MAPK levels, suggesting that stromal cells are modulating AZD1480 responses differently compared to cells cultured with IL-6.
AZD1480 inhibits both JAK2 and FGFR3 activity in vitro
Recurrent translocations have been identified in approximately 50% of primary MM patient samples; the most frequent of these translocations involves the receptor tyrosine kinase FGFR3(31, 32). Because Kms.11 cells possess translocated FGFR3 and AZD1480 has activity against FGFR1(18), combined with the finding that Kms.11 cocultured with bone marrow stromal cells are sensitive to AZD1480 without showing inhibition of phospho-STAT3, we hypothesized that AZD1480 may inhibit FGFR3 signaling in these settings. (Figure 5D) demonstrates that AZD1480 inhibits phosphorylation of FGFR3 in Kms.11 cells cocultured with HS.5 bone marrow stromal cells. These results suggest that inhibition of FGFR3 signaling is very likely driving sensitivity of Kms.11 cells, rather than JAK/STAT signaling, in these settings.
To confirm the finding that AZD1480 inhibits FGFR3 signaling we examined the effect of AZD1480 on b-FGF-dependent FGFR3 phosphorylation in Kms.11 cells pretreated with AZD1480 and then stimulated with b-FGF. Figure 6A shows that b-FGF-induced phosphorylation of FGFR3 was inhibited in Kms.11 cells at 0.25 μM and virtually abolished at 1 μM, which are similar doses that inhibit IL-6-induced-phosphorylation of STAT3 in U266 and Kms.11 cells. We postulated that Kms.11 cells may be sensitive to downregulation of FGFR3 by siRNA. Figure 6B demonstrates that Kms.11 cells depend on FGFR3 for proliferation since downregulation of FGFR3 by siRNA results in 40% inhibition of proliferation compared to cells transfected with control siRNA. Downregulation of FGFR3 mRNA levels was confirmed by quantitative real-time PCR 72 hours post transfection (approximately 60% inhibition compared to control) (data not shown). To demonstrate if FGFR3 is involved in the response to AZD1480, we performed experiments in Kms.11 cells transfected with WT FGFR3 or mutant activated FGFR3 (Figure 6C). Both WT and activated FGFR3 rescue Kms.11 cells from AZD1480-mediated loss of viability, indicating that inhibition of FGFR3 may contribute to AZD1480 efficacy in Kms.11 cells. In vitro kinase assays (Figure 7) confirmed that AZ1480 inhibits both JAK2 and FGFR3 activity, consistent with role of JAK2/STAT3 and FGFR3 in the biological effects mediated by AZD1480. Nevertheless, we do not exclude the possibility that other pathways may be involved in the response to AZD1480. Figure 7 shows also that AZD1480 is effective against JAK2 and FGFR3 activity at lower doses than another JAK2 or FGFR3 inhibitor. Compared with a commercially available JAK2 inhibitor, AZD1480 is even more potent in terms of cytotoxicity (Supplemetal Figure 3).
Figure 6. AZD1480 inhibits b-FGF-inducible activation of FGFR3 in Kms.11 cells.
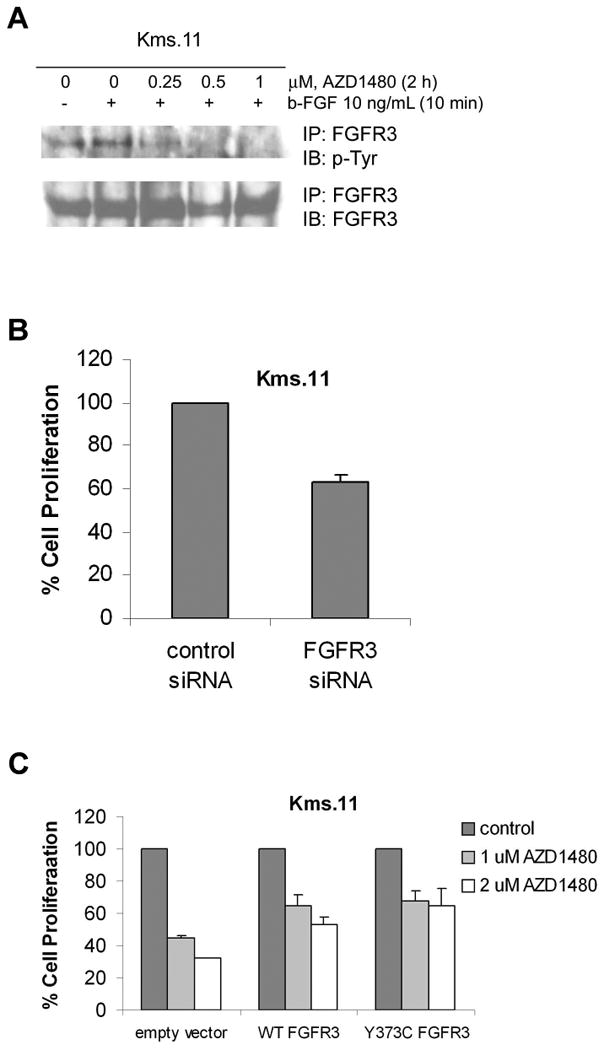
(A) Cells were incubated overnight in medium containing charcoal-stripped serum and then treated with the indicated concentrations of AZD1480 for 2 h followed by stimulation with b-FGF for 10 min. After this, immunoprecipitation with FGFR3 antibody followed by Western blot analysis of phospho-Tyr was performed on the cell lysates from Kms.11 cells. The levels of total FGFR3 protein served as loading controls. Results are representative of three independent experiments. (B) Kms.11 cells were transfected with Cy3-labeled FGFR3 siRNA or negative control siRNA and 24 h later Cy3-positive cells were sorted. The percentage of cell proliferation was determined 72 h post transfection by MTS assay. Values represented as bar graphs are the means of 3 independent experiments plus the standard deviation. (C) Cells were transiently transfected with vectors expressing wild-type (WT) or Y373C-FGFR3 constructs or empty vector. Transfected cells were treated with AZD1480. After 48 h of treatment, the percentage of cell proliferation was determined by MTS assay. Empty vector and FGFR3 vectors values are normalized to 100%. Values represented as bar graphs are the means of 3 independent experiments plus the standard deviation.
Figure 7. AZD1480 inhibits both JAK2 and FGFR3 activity in a cell-free kinase assay.
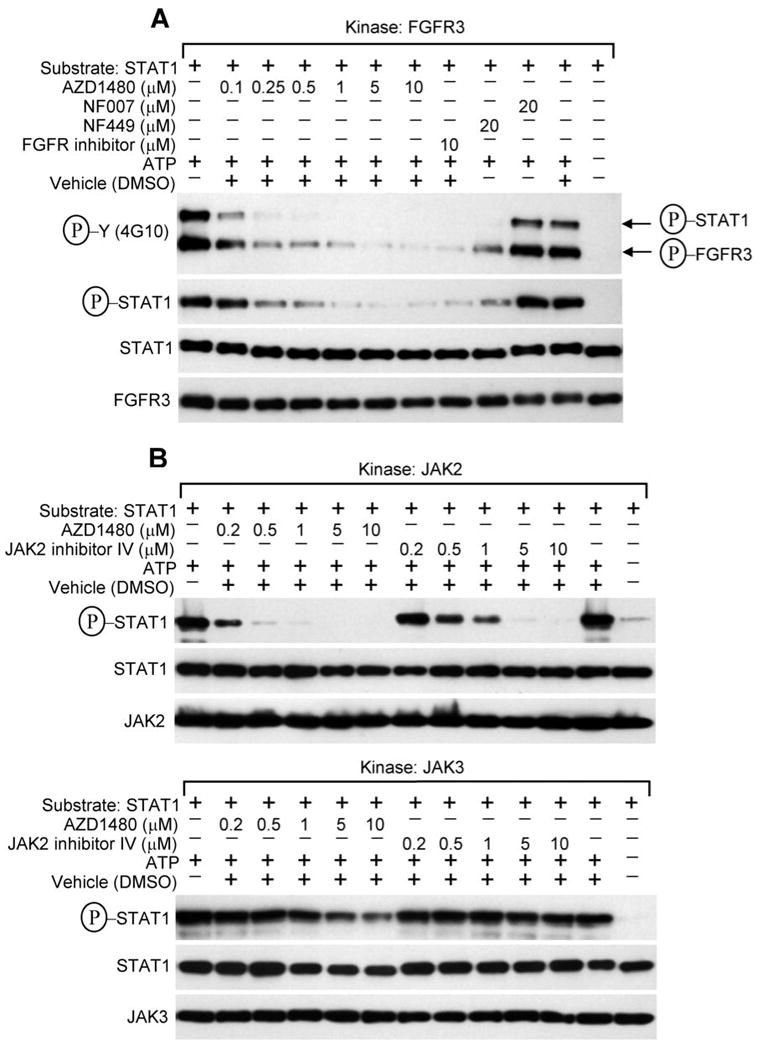
(A-B) Cell-free kinase assays were carried-out as described in the Experimental procedures, with the recombinant FGFR3, JAK2 and JAK3 as kinases, recombinant STAT1 as a substrate, and inhibitors added directly to the kinase reactions. Kinase-mediated phosphorylation of STAT1 was detected by Western blotting with P-STAT1 (Y701) antibody or 4G10 P-Y antibody. The membranes probed for FGFR3, JAK2, JAK3 and STAT1 serve as controls for kinase and substrate quantity, respectively. Sample with ATP omitted serves as a negative control for the kinase reaction. (A) Note that both FGFR3 autophosphorylation and FGFR3-mediated phosphorylation of STAT1 are inhibited by low concentrations of AZD1480, as compared to FGFR inhibitor or NF449. NF007 serves as negative control for FGFR3 inhibition (53). (B) Note the potent inhibitory activity of AZD1480 against JAK2, as compared to JAK inhibitor (upper panel). Also note the poor inhibitory activity of AZD1480 against JAK3 (lower panel).
AZD1480 blocks growth of Kms.11 xenografts associated with inhibition of JAK2/STAT3 and FGFR3
The presence of translocated FGFR3 in patients is associated with shorter duration of remission, higher relapse rate, and shorter survival(33). Thus, the Kms.11 cell line represents more aggressive disease, and for this reason we considered Kms.11 as a good model to study the effect of AZD1480 in vivo. In NOD/SCID IL2Rγ-null mice treated twice a day with 30 mg/kg AZD1480, we observed statistically significant tumor growth inhibition (Figure 7A). After only six days from the first treatment, we observed regression of the tumor in 80% of the mice for as much as 40% volume reduction compared with the initial tumor volume prior to the first treatment. The average tumor size of the treated group was approximately six and 12 times smaller than the average of tumor size of control vehicle group 6 and 12 days post treatment, respectively. Similar data were observed in a separate experiment with athymic nude mice (data not shown). Regression of the tumor was associated with complete inhibition of STAT3 and FGFR3 phosphorylation and modest inhibition of Cyclin D2 (approximately 50% reduction, P=0.0286) in tumors harvested 2 h after dosing (Figure 7B-C). However, we did not observe consistent inhibition of phospho-MAPK among tumors treated with AZD1480 (data not shown).
AZD1480 causes loss of viability of primary MM cells cultured in vitro and enhances the sensitivity to chemotherapy
We next determined whether AZD1480 treatment could also be active against primary plasma cells isolated from bone marrow samples of multiple myeloma patients. Table 2 indicates that all 4 patient samples analyzed responded to the AZD1480 treatment with an increase in the percentage of nonviable cells in a dose-dependent manner. Importantly, one of these samples was treated with AZD1480 in the presence of bone marrow stromal cells and responded even better than when it was treated in the absence of stroma. In contrast, no cytotoxic effects were observed in 5 normal CD138+ samples at the same doses (Figure 9).
Table 2. AZD1480 causes loss of viability of primary multiple myeloma cells.
Samples of primary MM or normal CD138+ cells were treated in culture with the indicated concentrations of AZD1480 for 48 h. The percentage of nonviable cells was determined by DIMSCAN. The values represent the percentages of loss of viability normalized to that of the untreated cells and are the means of two separate treatments. The percentage of loss of cell viability is indicated as 0% when the percentage of cell viability was 100% or higher than 100%. *Patient sample 2 was treated with AZD1480 in the presence of bone marrow stromal cells.
| Loss of Cell Viability, % | ||
|---|---|---|
| Patient sample | AZD1480, 1 μM | AZD1480, 2 μM |
| 1 | 15.5 | 55.7 |
|
| ||
| 2 | 16.6 | 42.9 |
|
| ||
| 3 | 31.1 | 38.5 |
|
| ||
| 4 | 60.3 | 94.3 |
|
| ||
| 2* | 47.8 | 63.3 |
|
| ||
| Healthy donor sample | ||
|
| ||
| 1 | 11.4 | 14.8 |
|
| ||
| 2 | 3.6 | 0 |
|
| ||
| 3 | 2.5 | 0 |
|
| ||
| 4 | 0 | 0 |
|
| ||
| 5 | 0 | 0 |
The bone marrow stromal environment is a major factor in myeloma cell resistance to chemotherapeutic agents such as dexamethasone(34), melphalan and mitoxantrone(30, 35). We tested the effect of the bone marrow environment on myeloma cell response to AZD1480 in combination with doxorubicin and melphalan in terms of induction of cell death (data not shown). Coculturing of Kms.11 cells with human bone marrow stromal HS-5 cells does not protect the myeloma cells to doxorubicin compared with the controls that were not cocultured. Protection from melphalan treatment was also not observed. AZD1480 potentiated the cell death induced by doxorubicin and melphalan in Kms.11 grown alone and even more if cocultured with HS.5 cells. This enhanced response with combination treatment represents an additive to synergistic response (Table 2).
Discussion
In the present study, we investigated the biologic mechanism of action of the novel small-molecule JAK kinase inhibitor AZD1480 on human myeloma cells. We found that AZD1480, at low micromolar concentrations, inhibits the viability of cell lines that express constitutively activated STAT3 (U266, IM-9), or translocated FGFR3 (Kms.18) or both (Kms.11, OPM-2). By contrast, higher concentrations of AZD1480 are required to inhibit cells (8226, NCI-H929, MM1.S) that lack both FGFR3 overexpression and activated STAT3 and are not growth stimulated by IL-6(13, 24). Moreover, the lack of inhibition of proliferation and viability of bone marrow stromal cells and PBMC derived from healthy donors indicates that the drug may not have cytotoxic effects in normal cells. Anti-tumor activity of AZD1480 was observed also in myeloma cells growth stimulated by IL-6. We found that AZD1480 abrogates IL-6-induced activation of JAK2, tyrosine phosphorylation of STAT3 and phosphorylation of MAPK. AZD1480 suppresses the proliferative response to IL-6 with concomitant decreases in the protein levels of Cyclin D2 in these cells. Cyclin D2 is known to be important in the growth of MM cells and has been shown to be regulated by STAT3 in MM(36, 37).
That IL-6 activates MAPK through a Ras-dependent cascade is well established(12, 38). Our finding that AZD1480 inhibits IL-6-induced phosphorylation of MAPK suggests that the drug may also act through inhibition of JAK1 activity, which in turn is not able to activate JAK2. This is consistent with potency of AZD1480 for JAK1 in enzymatic assays(18), and prior data indicating that JAK1 has a major role in IL-6 mediated activation of STAT3(39). Whether AZD1480 is acting mainly through JAK1 or JAK2 requires further investigation.
Translocations involving FGFR3 do not directly target a cyclin D gene, but they are associated with a high level of Cyclin D2 expression(36). Cyclin D2 leads to growth promotion and survival in MM(40). We show that AZD1480 downregulates Cyclin D2 in both U266 and Kms.11 cells cultured in the presence or absence of IL-6, suggesting that Cyclin D2 is a major downstream target of AZD1480-induced inhibition of STAT3 activity in an IL-6 dependent and independent manner. AZD1480 may downregulate Cyclin D2 by inhibiting FGFR3 and/or inhibiting STAT3 binding to the c-maf promoter(37), since Cyclin D2 is a target of c-maf(40). 8226 cells, which among the cell lines analyzed here are the least sensitive to AZD1480 in terms of viability inhibition, do not exhibit downregulation of Cyclin D2, suggesting that in those cells Cyclin D2 may be regulated by different pathways. This finding supports the conclusion that Cyclin D2 could be a major mediator in the response to AZD1480. We also confirmed that c-Myc, a common secondary translocation partner in MM(41), is upregulated upon IL-6 stimulation of MM cells(42) and we found that AZD1480 downregulates c-Myc expression in an IL-6 dependent manner.
Mcl-1 and Bcl-xL are implicated in the survival of myeloma cells(43, 44), and expression of these proteins can be selectively down-regulated by dominant/negative STAT3 or JAK2 inhibitors(11, 45). We did not observe IL-6-inducible upregulation of Mcl-1, in contrast to what has been shown in different MM cell lines(28, 46) and in CD45+ U266 cells(47), but consistent with other cells that exhibited high levels of Mcl-1 expression and were unaffected by IL-6(48, 49). Nevertheless, we observed that Mcl-1 is downregulated by AZD1480. We also observed that IL-6 induces upregulation of Bcl-xL in U266 and AZD1480 downregulates Bcl-xL at higher concentrations associated with complete inhibition of STAT3 phosphorylation.
Bone marrow stroma cells induce activation of pathways involved in proliferation, survival, migration and drug-resistance in MM cells(29, 50). Moreover, MM cells may become independent of the STAT3 pathway in the presence of BM stroma(30, 51). We found that myeloma cells are equally sensitive to AZD1480 regardless of whether they were cultured alone or in the presence of bone marrow stromal cells. However, STAT3 and MAPK pathways are differentially deregulated in the presence of stromal cells compared to what occurs in cells cultured in the presence of IL-6. In the presence of IL-6, both U266 and Kms.11 cell lines treated with AZD1480 exhibit downregulation of IL-6-induced phosphorylation of STAT3 and MAPK. When co-cultured with stromal cells, the two cell lines exhibit differential inhibition of STAT3 or MAPK activity in U266 or Kms.11, respectively, demonstrating that under these conditions combined disruption of both MAPK and STAT3 pathways is not required to induce MM cell apoptosis. Therefore, in this setting, FGFR3 signaling inhibition may be the key driver of response to AZD1480 for Kms.11 cells rather than STAT3 inhibition; these data are supported by the finding that AZD1480 inhibits b-FGF-mediated phosphorylation of FGFR3. It is possible that the release of b-FGF from bone marrow stromal cells is predominant over IL-6 release, and hence Kms.11 cells may become more dependent on b-FGF/FGFR3 signaling than IL-6/JAK2/STAT3 in the presence of stromal cells.
Importantly, we observed strong inhibition of both STAT3 and FGFR3 activity in Kms.11 bearing-mice treated with AZD1480, suggesting that the in vivo microenvironment influences signaling in the tumor in a different manner than in vitro conditions in which tumor cells are cocultured with bone marrow stromal cells. Inhibition of constitutive STAT3 activity sensitizes MM cells to apoptosis induced by conventional chemotherapy(52). Notably, our results indicate that AZD1480 sensitizes myeloma cells to doxorubicin and melphalan, regardless of whether they were cultured alone or in the presence of bone marrow stromal cells. These findings raise the possibility that combination with AZD1480 may further enhance clinical response to conventional chemotherapy in MM patients.
Our transfection experiments indicate that both STAT3 and FGFR3 constructs protect Kms.11 cells from AZD1480-induced loss of viability, suggesting that inhibition of STAT3 as well as inhibition of FGFR3 contribute to efficacy of AZD1480 in those cells. These results indicate that both STAT3 and FGFR3 may be necessary for response of Kms.11 cells to AZD1480, and the inhibition of only one of these pathways may be not sufficient for induction of viability loss in response to AZD1480 treatment. It is possible that both STAT3 and FGFR3 regulate the expression of Cyclin D2, and this may explain why Kms.11 cells are more sensitive than U266 cells in terms of apoptosis and inhibition of proliferation. Our kinase assays demonstrate that AZD1480 is active against both JAK2 and FGFR3, possibly explaining why cells that possess constitutively activated STAT3 or translocated FGFR3 are more sensitive than those that lack of both; cells expressing both activated STAT3 and translocated FGFR3 are the most sensitive. AZD1480 activity on JAK2 and FGFR3 is even greater than other JAK2 and FGFR3 inhibitors analyzed in our kinase assays. These observations do not exclude the possibility that AZD1480 inhibits other kinases, but support the potential of AZD1480 as a dual JAK/FGFR inhibitor for targeting myeloma cells.
In sum, AZD1480 suppresses the JAK2/STAT3 and FGFR3 signaling pathways, down-regulates Cyclin D2 protein expression, and inhibits growth and survival of human myeloma cells in vitro, in vivo and in coculture with bone marrow stroma. Combined with its potential to enhance sensitivity to chemotherapy, our findings provide a compelling rationale for clinical trials of AZD1480 in patients with multiple myeloma.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institute of Health (R01 CA-055652) and AstraZeneca to R.J., W. M. Keck Foundation and Tim Nesvig Lymphoma Fellowships to A.S., Ministry of Education, Youth and Sports of the Czech Republic (MSM0021622430) and Grant Agency of the Czech Republic (301/09/0587) to P.K.. We thank Drs. Dennis Huszar and Michael Zinda from AstraZeneca R&D Boston for providing AZD1480 and valuable suggestions, members of our laboratories for stimulating discussion and the Analytical Cytometry Core at City of Hope.
Support for the work: This work was supported by a grant from the National Institutes of Health (R01 CA-055652) and AstraZeneca; W. M. Keck Foundation and Tim Nesvig Lymphoma Fellowships to A.S.; Ministry of Education, Youth and Sports of the Czech Republic (MSM0021622430) and Grant Agency of the Czech Republic (301/09/0587) to P.K.
Footnotes
Supplementary Information accompanies the paper on the Leukemia website (http://www.nature.com/leu)
Conflict of interest: This work was partially supported by AstraZeneca whose product, AZD1480, was studied in the present work.
References
- 1.Kyle RA, Rajkumar SV. Multiple myeloma. Blood. 2008 Mar 15;111(6):2962–2972. doi: 10.1182/blood-2007-10-078022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fonseca R, Stewart AK. Targeted therapeutics for multiple myeloma: the arrival of a risk-stratified approach. Mol Cancer Ther. 2007 Mar;6(3):802–810. doi: 10.1158/1535-7163.MCT-06-0620. [DOI] [PubMed] [Google Scholar]
- 3.San-Miguel J, Harousseau JL, Joshua D, Anderson KC. Individualizing treatment of patients with myeloma in the era of novel agents. J Clin Oncol. 2008 Jun 1;26(16):2761–2766. doi: 10.1200/JCO.2007.15.2546. [DOI] [PubMed] [Google Scholar]
- 4.Ocio EM, Mateos MV, Maiso P, Pandiella A, San-Miguel JF. New drugs in multiple myeloma: mechanisms of action and phase I/II clinical findings. Lancet Oncol. 2008 Dec;9(12):1157–1165. doi: 10.1016/S1470-2045(08)70304-8. [DOI] [PubMed] [Google Scholar]
- 5.Vallet S, Palumbo A, Raje N, Boccadoro M, Anderson KC. Thalidomide and lenalidomide: Mechanism-based potential drug combinations. Leuk Lymphoma. 2008 Jul;49(7):1238–1245. doi: 10.1080/10428190802005191. [DOI] [PubMed] [Google Scholar]
- 6.Richardson PG, Mitsiades C, Schlossman R, Ghobrial I, Hideshima T, Munshi N, et al. Bortezomib in the front-line treatment of multiple myeloma. Expert Rev Anticancer Ther. 2008 Jul;8(7):1053–1072. doi: 10.1586/14737140.8.7.1053. [DOI] [PubMed] [Google Scholar]
- 7.Argyriou AA, Iconomou G, Kalofonos HP. Bortezomib-induced peripheral neuropathy in multiple myeloma: a comprehensive review of the literature. Blood. 2008 Sep 1;112(5):1593–1599. doi: 10.1182/blood-2008-04-149385. [DOI] [PubMed] [Google Scholar]
- 8.Palumbo A, Rajkumar SV, Dimopoulos MA, Richardson PG, San Miguel J, Barlogie B, et al. Prevention of thalidomide- and lenalidomide-associated thrombosis in myeloma. Leukemia. 2008 Feb;22(2):414–423. doi: 10.1038/sj.leu.2405062. [DOI] [PubMed] [Google Scholar]
- 9.Bommert K, Bargou RC, Stuhmer T. Signalling and survival pathways in multiple myeloma. Eur J Cancer. 2006 Jul;42(11):1574–1580. doi: 10.1016/j.ejca.2005.12.026. [DOI] [PubMed] [Google Scholar]
- 10.Kawano MM, Ishikawa H, Tsuyama N, Abroun S, Liu S, Li FJ, et al. Growth mechanism of human myeloma cells by interleukin-6. Int J Hematol. 2002 Aug;76 1:329–333. doi: 10.1007/BF03165278. [DOI] [PubMed] [Google Scholar]
- 11.Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999 Jan;10(1):105–115. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- 12.Ogata A, Chauhan D, Teoh G, Treon SP, Urashima M, Schlossman RL, et al. IL-6 triggers cell growth via the Ras-dependent mitogen-activated protein kinase cascade. J Immunol. 1997 Sep 1;159(5):2212–2221. [PubMed] [Google Scholar]
- 13.Bharti AC, Donato N, Aggarwal BB. Curcumin (diferuloylmethane) inhibits constitutive and IL-6-inducible STAT3 phosphorylation in human multiple myeloma cells. J Immunol. 2003 Oct 1;171(7):3863–3871. doi: 10.4049/jimmunol.171.7.3863. [DOI] [PubMed] [Google Scholar]
- 14.Amit-Vazina M, Shishodia S, Harris D, Van Q, Wang M, Weber D, et al. Atiprimod blocks STAT3 phosphorylation and induces apoptosis in multiple myeloma cells. Br J Cancer. 2005 Jul 11;93(1):70–80. doi: 10.1038/sj.bjc.6602637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Vos J, Jourdan M, Tarte K, Jasmin C, Klein B. JAK2 tyrosine kinase inhibitor tyrphostin AG490 downregulates the mitogen-activated protein kinase (MAPK) and signal transducer and activator of transcription (STAT) pathways and induces apoptosis in myeloma cells. Br J Haematol. 2000 Jun;109(4):823–828. doi: 10.1046/j.1365-2141.2000.02127.x. [DOI] [PubMed] [Google Scholar]
- 16.Pedranzini L, Dechow T, Berishaj M, Comenzo R, Zhou P, Azare J, et al. Pyridone 6, a pan-Janus-activated kinase inhibitor, induces growth inhibition of multiple myeloma cells. Cancer Res. 2006 Oct 1;66(19):9714–9721. doi: 10.1158/0008-5472.CAN-05-4280. [DOI] [PubMed] [Google Scholar]
- 17.Burger R, Le Gouill S, Tai YT, Shringarpure R, Tassone P, Neri P, et al. Janus kinase inhibitor INCB20 has antiproliferative and apoptotic effects on human myeloma cells in vitro and in vivo. Mol Cancer Ther. 2009 Jan;8(1):26–35. doi: 10.1158/1535-7163.MCT-08-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009 Dec 8;16(6):487–497. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krejci P, Salazar L, Kashiwada TA, Chlebova K, Salasova A, Thompson LM, et al. Analysis of STAT1 activation by six FGFR3 mutants associated with skeletal dysplasia undermines dominant role of STAT1 in FGFR3 signaling in cartilage. PloS one. 2008;3(12):e3961. doi: 10.1371/journal.pone.0003961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heale BS, Soifer HS, Bowers C, Rossi JJ. siRNA target site secondary structure predictions using local stable substructures. Nucleic acids research. 2005;33(3):e30. doi: 10.1093/nar/gni026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scherer LJ, Yildiz Y, Kim J, Cagnon L, Heale B, Rossi JJ. Rapid assessment of anti-HIV siRNA efficacy using PCR-derived Pol III shRNA cassettes. Mol Ther. 2004 Sep;10(3):597–603. doi: 10.1016/j.ymthe.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 22.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. 1999 Aug 6;98(3):295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 23.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacological reviews. 2006 Sep;58(3):621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 24.Ogata A, Chauhan D, Urashima M, Teoh G, Treon SP, Anderson KC. Blockade of mitogen-activated protein kinase cascade signaling in interleukin 6-independent multiple myeloma cells. Clin Cancer Res. 1997 Jun;3(6):1017–1022. [PubMed] [Google Scholar]
- 25.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003 Aug 15;374(Pt 1):1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998 Sep 1;334(Pt 2):297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang B, Fenton RG. Proliferation of IL-6-independent multiple myeloma does not require the activity of extracellular signal-regulated kinases (ERK1/2) J Cell Physiol. 2002 Oct;193(1):42–54. doi: 10.1002/jcp.10148. [DOI] [PubMed] [Google Scholar]
- 28.Puthier D, Bataille R, Amiot M. IL-6 up-regulates mcl-1 in human myeloma cells through JAK / STAT rather than ras / MAP kinase pathway. Eur J Immunol. 1999 Dec;29(12):3945–3950. doi: 10.1002/(SICI)1521-4141(199912)29:12<3945::AID-IMMU3945>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 29.Meads MB, Hazlehurst LA, Dalton WS. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin Cancer Res. 2008 May 1;14(9):2519–2526. doi: 10.1158/1078-0432.CCR-07-2223. [DOI] [PubMed] [Google Scholar]
- 30.Nefedova Y, Landowski TH, Dalton WS. Bone marrow stromal-derived soluble factors and direct cell contact contribute to de novo drug resistance of myeloma cells by distinct mechanisms. Leukemia. 2003 Jun;17(6):1175–1182. doi: 10.1038/sj.leu.2402924. [DOI] [PubMed] [Google Scholar]
- 31.Chesi M, Brents LA, Ely SA, Bais C, Robbiani DF, Mesri EA, et al. Activated fibroblast growth factor receptor 3 is an oncogene that contributes to tumor progression in multiple myeloma. Blood. 2001 Feb 1;97(3):729–736. doi: 10.1182/blood.v97.3.729. [DOI] [PubMed] [Google Scholar]
- 32.Chesi M, Nardini E, Brents LA, Schrock E, Ried T, Kuehl WM, et al. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat Genet. 1997 Jul;16(3):260–264. doi: 10.1038/ng0797-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fonseca R, Blood E, Rue M, Harrington D, Oken MM, Kyle RA, et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood. 2003 Jun 1;101(11):4569–4575. doi: 10.1182/blood-2002-10-3017. [DOI] [PubMed] [Google Scholar]
- 34.Grigorieva I, Thomas X, Epstein J. The bone marrow stromal environment is a major factor in myeloma cell resistance to dexamethasone. Exp Hematol. 1998 Jul;26(7):597–603. [PubMed] [Google Scholar]
- 35.Nefedova Y, Cheng P, Alsina M, Dalton WS, Gabrilovich DI. Involvement of Notch-1 signaling in bone marrow stroma-mediated de novo drug resistance of myeloma and other malignant lymphoid cell lines. Blood. 2004 May 1;103(9):3503–3510. doi: 10.1182/blood-2003-07-2340. [DOI] [PubMed] [Google Scholar]
- 36.Bergsagel PL, Kuehl WM, Zhan F, Sawyer J, Barlogie B, Shaughnessy J., Jr Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood. 2005 Jul 1;106(1):296–303. doi: 10.1182/blood-2005-01-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang Y, Ochando J, Yopp A, Bromberg JS, Ding Y. IL-6 plays a unique role in initiating c-Maf expression during early stage of CD4 T cell activation. J Immunol. 2005 Mar 1;174(5):2720–2729. doi: 10.4049/jimmunol.174.5.2720. [DOI] [PubMed] [Google Scholar]
- 38.Schaper F, Gendo C, Eck M, Schmitz J, Grimm C, Anhuf D, et al. Activation of the protein tyrosine phosphatase SHP2 via the interleukin-6 signal transducing receptor protein gp130 requires tyrosine kinase Jak1 and limits acute-phase protein expression. Biochem J. 1998 Nov 1;335(Pt 3):557–565. doi: 10.1042/bj3350557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guschin D, Rogers N, Briscoe J, Witthuhn B, Watling D, Horn F, et al. A major role for the protein tyrosine kinase JAK1 in the JAK/STAT signal transduction pathway in response to interleukin-6. Embo J. 1995 Apr 3;14(7):1421–1429. doi: 10.1002/j.1460-2075.1995.tb07128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hurt EM, Wiestner A, Rosenwald A, Shaffer AL, Campo E, Grogan T, et al. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell. 2004 Feb;5(2):191–199. doi: 10.1016/s1535-6108(04)00019-4. [DOI] [PubMed] [Google Scholar]
- 41.Shou Y, Martelli ML, Gabrea A, Qi Y, Brents LA, Roschke A, et al. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc Natl Acad Sci U S A. 2000 Jan 4;97(1):228–233. doi: 10.1073/pnas.97.1.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wen XY, Stewart AK, Sooknanan RR, Henderson G, Hawley TS, Reimold AM, et al. Identification of c-myc promoter-binding protein and X-box binding protein 1 as interleukin-6 target genes in human multiple myeloma cells. Int J Oncol. 1999 Jul;15(1):173–178. doi: 10.3892/ijo.15.1.173. [DOI] [PubMed] [Google Scholar]
- 43.Wuilleme-Toumi S, Robillard N, Gomez P, Moreau P, Le Gouill S, Avet-Loiseau H, et al. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia. 2005 Jul;19(7):1248–1252. doi: 10.1038/sj.leu.2403784. [DOI] [PubMed] [Google Scholar]
- 44.Zhang B, Gojo I, Fenton RG. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood. 2002 Mar 15;99(6):1885–1893. doi: 10.1182/blood.v99.6.1885. [DOI] [PubMed] [Google Scholar]
- 45.Oshiro MM, Landowski TH, Catlett-Falcone R, Hazlehurst LA, Huang M, Jove R, et al. Inhibition of JAK kinase activity enhances Fas-mediated apoptosis but reduces cytotoxic activity of topoisomerase II inhibitors in U266 myeloma cells. Clin Cancer Res. 2001 Dec;7(12):4262–4271. [PubMed] [Google Scholar]
- 46.Jourdan M, Veyrune JL, De Vos J, Redal N, Couderc G, Klein B. A major role for Mcl-1 antiapoptotic protein in the IL-6-induced survival of human myeloma cells. Oncogene. 2003 May 15;22(19):2950–2959. doi: 10.1038/sj.onc.1206423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou Q, Yao Y, Ericson SG. The protein tyrosine phosphatase CD45 is required for interleukin 6 signaling in U266 myeloma cells. Int J Hematol. 2004 Jan;79(1):63–73. doi: 10.1007/BF02983536. [DOI] [PubMed] [Google Scholar]
- 48.Muto A, Hori M, Sasaki Y, Saitoh A, Yasuda I, Maekawa T, et al. Emodin has a cytotoxic activity against human multiple myeloma as a Janus-activated kinase 2 inhibitor. Mol Cancer Ther. 2007 Mar;6(3):987–994. doi: 10.1158/1535-7163.MCT-06-0605. [DOI] [PubMed] [Google Scholar]
- 49.Zhang B, Potyagaylo V, Fenton RG. IL-6-independent expression of Mcl-1 in human multiple myeloma. Oncogene. 2003 Mar 27;22(12):1848–1859. doi: 10.1038/sj.onc.1206358. [DOI] [PubMed] [Google Scholar]
- 50.Podar K, Chauhan D, Anderson KC. Bone marrow microenvironment and the identification of new targets for myeloma therapy. Leukemia. 2009 Jan;23(1):10–24. doi: 10.1038/leu.2008.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chatterjee M, Honemann D, Lentzsch S, Bommert K, Sers C, Herrmann P, et al. In the presence of bone marrow stromal cells human multiple myeloma cells become independent of the IL-6/gp130/STAT3 pathway. Blood. 2002 Nov 1;100(9):3311–3318. doi: 10.1182/blood-2002-01-0102. [DOI] [PubMed] [Google Scholar]
- 52.Alas S, Bonavida B. Inhibition of constitutive STAT3 activity sensitizes resistant non-Hodgkin's lymphoma and multiple myeloma to chemotherapeutic drug-mediated apoptosis. Clin Cancer Res. 2003 Jan;9(1):316–326. [PubMed] [Google Scholar]
- 53.Krejci P, Murakami S, Prochazkova J, Trantirek L, Chlebova K, Ouyang Z, et al. NF449 is a novel inhibitor of fibroblast growth factor receptor 3 (FGFR3) signaling active in chondrocytes and multiple myeloma cells. J Biol Chem. 2010 Jul 2;285(27):20644–20653. doi: 10.1074/jbc.M109.083626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nelson EA, Walker SR, Kepich A, Gashin LB, Hideshima T, Ikeda H, et al. Nifuroxazide inhibits survival of multiple myeloma cells by directly inhibiting STAT3. Blood. 2008 Dec 15;112(13):5095–5102. doi: 10.1182/blood-2007-12-129718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ronchetti D, Greco A, Compasso S, Colombo G, Dell'Era P, Otsuki T, et al. Deregulated FGFR3 mutants in multiple myeloma cell lines with t(4;14): comparative analysis of Y373C, K650E and the novel G384D mutations. Oncogene. 2001 Jun 14;20(27):3553–3562. doi: 10.1038/sj.onc.1204465. [DOI] [PubMed] [Google Scholar]
- 56.Ramakrishnan V, Timm M, Haug JL, Kimlinger TK, Wellik LE, Witzig TE, et al. Sorafenib, a dual Raf kinase/vascular endothelial growth factor receptor inhibitor has significant anti-myeloma activity and synergizes with common anti-myeloma drugs. Oncogene. Feb 25;29(8):1190–1202. doi: 10.1038/onc.2009.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li P, Harris D, Liu Z, Liu J, Keating M, Estrov Z. Stat3 activates the receptor tyrosine kinase like orphan receptor-1 gene in chronic lymphocytic leukemia cells. PloS one. 5(7):e11859. doi: 10.1371/journal.pone.0011859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chebel A, Kagialis-Girard S, Catallo R, Chien WW, Mialou V, Domenech C, et al. Indirubin derivatives inhibit malignant lymphoid cell proliferation. Leuk Lymphoma. 2009 Dec;50(12):2049–2060. doi: 10.3109/10428190903288449. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.