

SUMMARY
Increasing antiviral drug resistance is a major concern for treating influenza, especially in a pandemic setting when the availability of a protective vaccine is uncertain. Resistance is often an issue with drugs directed at viral proteins and for small RNA viruses; there are also a limited number of viral proteins that are amenable to inhibition by a small molecule. A new approach that is gaining support is that cellular proteins, which facilitate virus replication, may be used as alternative targets. Whereas drugs directed at viral proteins tend to be virus‐specific, drugs directed at host targets have the potential to have broad‐spectrum antiviral activity as many viruses may share a dependency on that host function. For influenza virus, we have very limited knowledge of which cellular factors are involved in virus replication, let alone which of these have suitable properties to serve as drug targets. Through the use of high‐throughput RNA interference screens, several studies have addressed this gap in our knowledge. The resulting datasets provide new insight into host pathways that are involved in the influenza virus replication cycle and identify specific host factors in these pathways that may serve as potential targets for future antiviral drug development. Copyright © 2011 John Wiley & Sons, Ltd.
Abbreviations used
- M1
matrix protein
- NEP
nuclear export protein
- RNAi
RNA interference
- siRNA
small inhibitory RNA
- vRNPs
viral ribonucleoprotein complexes
INTRODUCTION
Influenza viruses belong to the Orthomyxoviridae family, whose members are defined by a segmented, single‐stranded, negative‐sense RNA genome (which is replicated in the nucleus), and an envelope that is derived from the host cell 1. Influenza A viruses have eight genome segments that encode for 10 or 11 viral proteins, depending on the strain. Nine of these proteins are found in the virion. These include the following: the HA, NA, and M2 proteins that are all inserted into the lipid envelope; the matrix (M1) protein that lies beneath the membrane; the NP that coats the viral genome; the polymerase complex (PB1, PB1, and PA) that is associated with the encapsidated genome; and the nuclear export protein (NEP). The remaining viral proteins, NS1 and PB1‐F2, are expressed in infected cells but are not packaged into the virus particle.
Influenza virus initiates infection via attachment of HA to sialic acid‐containing proteins on the host cell membrane (Figure 1). The virus particle then enters the cell by pH‐dependent endocytosis, although there appears to be flexibility in the pathway that is used, with an estimated two thirds using a clathrin‐dependent pathway and the remaining third entering via an undefined pathway that is independent of both clathrin and caveolin 2. Once in the acid environment of the late endosome, the HA undergoes a conformational change and drives fusion of the viral envelope with that of the endosome 3. In addition, the M2 protein, which has ion channel activity 4, pumps H+ ions into the interior of the virion and this dissociates M1 from the viral ribonucleoprotein complexes (vRNPs). The released vRNPs enter the cytoplasm through the fusion pore and are transported into the nucleus via interaction of NP with karyopherin alpha proteins 5, which are part of the nuclear import machinery. Once in the nucleus, the incoming viral polymerase complex initiates genome transcription. In a process known as “cap‐snatching”, the PB2 protein binds to the 5′ methyl cap of cellular pre‐mRNAs, and the PA protein cleaves the pre‐mRNA to produce a capped primer that is used to start transcription 6, 7, 8. The PB1 protein contains the RNA‐dependent RNA polymerase activity, and it is also responsible for the addition of a poly(A) tail via a stuttering mechanism 9. PB1 also catalyzes genome replication, which occurs via a positive sense cRNA intermediate that is an exact copy of the vRNA. In the case of replication, it is still unclear whether initiation is primer‐dependent,—independent or a combination of both depending on anti‐genome or genome synthesis 10, 11. The newly‐synthesized vRNA, NP, and polymerase proteins are complexed together with M1 and NEP in the nucleus and via an interaction with Crm1, NEP exports the new vRNPs back out of the nucleus 12, 13. The assembly pathway for influenza virus is one of the least well‐understood stages of the viral life cycle. We know that the viral glycoproteins, HA and NA, traffic through the endoplasmic reticulum and accumulate at the plasma membrane in specific regions termed lipid raft domains 14. These serve as a virus assembly platform, and the other components of the virion are recruited via mechanisms that have yet to be fully determined. Evidence strongly suggests that HA initiates bud formation, and it has recently been shown that the M2 protein provides membrane scission activity and thereby completes bud formation 15. Finally, the neuraminidase activity of the NA protein is required to release virus from the cell by cleaving sialic acid attachments from HA and other surface glycoproteins 16.
Figure 1.
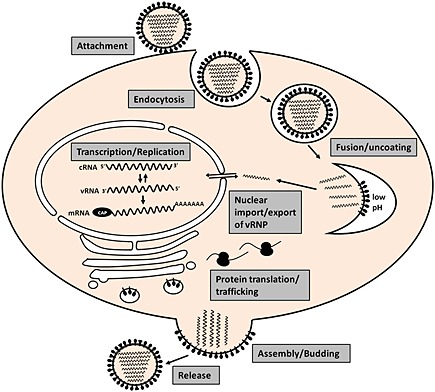
Illustration of the influenza A virus replication cycle indicating all major stages from virus attachment until release of newly formed virus particles
NS1 and PB1‐F2 are accessory proteins that promote virus replication indirectly by either subverting or promoting cellular signaling pathways. NS1 is a multifunctional protein, but its major function is to inhibit activation of the cellular innate immune response 17. In the absence of NS1 expression, influenza virus is rendered non‐pathogenic in an immune‐competent host, and thus it is classified as a viral pathogenicity factor 18. PB1‐F2 is expressed from an alternative ORF in the PB1 gene. This ORF is absent in some viruses 19, which obviously indicates that it is nonessential. However, its presence in pandemic strains or avian viruses correlates with increased lung inflammation and pathology, which contributes to increased virulence and therefore PB1‐F2 constitutes the second pathogenicity factor encoded by influenza virus 20.
In addition to the viral proteins, there are numerous cellular proteins involved at each stage of the influenza virus life cycle. A few of these are mentioned above and have been identified and characterized through detailed studies of individual viral proteins, most often interaction studies. However, these likely only represent a small fraction of required host factors and efforts to broaden our knowledge in this area will provide a much more sophisticated picture of the influenza virus replication cycle. The advent of RNA interference (RNAi) technology now allows one to query the participation of each encoded host protein in a particular function such as virus replication. A number of studies 21, 22, 23, 24, 25 have recently used this approach to unveil the human proteins that are essential for efficient influenza virus replication, and in doing so, they have considerably expanded our view of how influenza virus interacts with its host cell. This review will discuss these findings and their implication for development of new options for influenza therapy.
CURRENT OPTIONS FOR INFLUENZA THERAPY
There are four drugs currently approved for the treatment or prevention of influenza and all of these act on viral proteins (Table 1). Amantadine and rimantadine both target the M2 protein and are specific for influenza A virus. By inhibiting M2 ion channel activity, they block acidification of the incoming virus particle and therefore prevent the release of the viral genome 26. However, because of widespread resistance to these drugs in current circulating viruses, the Centers for Disease Control (CDC) has recommended that they not be used during the 2010/2011 influenza season in the USA. The drugs that are recommended for clinical use are oseltamivir and zanamivir, which target the neuraminidase activity of the NA protein and are active against both influenza A and B viruses 27. NA inhibition prevents release of the newly‐formed virus particle at the last step of the replication cycle. Although oseltamivir is an oral drug, zanamivir is administered via inhalation. Another NA inhibitor, peramivir, obtained approval from the FDA for emergency use during the 2009 pandemic, but this approval has since expired 28. Peramivir can be administered intravenously and is therefore of particular use for critically ill patients.
Table 1.
Approved antiviral drugs for the treatment of influenza
| Antiviral drug | Active against | Viral target | Route | Recommended use in 2010/2011 |
|---|---|---|---|---|
| Amantadine | Influenza A virus | M2 | Oral | Not recommended because of widespread resistance |
| Rimantadine | Influenza A virus | M2 | Oral | |
| Oseltamivir | Influenza A and B virus | NA | Oral | Treatment/prophylaxis |
| Zanamivir | Influenza A and B virus | NA | Inhalation | Treatment/prophylaxis |
| Peramivir* | Influenza A and B virus | NA | Intravenous | Awaiting permanent approval |
Approved for emergency use during the 2009 pandemic only.
One of the major issues with drugs that target viral proteins is that resistance is very likely to develop. This is particularly true of RNA viruses that have a more error‐prone polymerase and for influenza virus, the segmented nature of the genome allows for mutations to be transferred to new virus strains during reassortment. Mutations in the M2 protein that confer resistance to the adamantanes are found in nearly 100% of H3N2 influenza A viruses, as well as the pandemic H1N1 influenza A viruses that are currently circulating (CDC surveillance). In contrast, the seasonal H1N1 viruses that were circulating prior to the H1N1 pandemic remain sensitive to the adamantanes. The pattern is reversed when looking at resistance to oseltamivir, where the H3N2 viruses and pandemic H1N1 viruses are sensitive whereas the seasonal H1N1 viruses are 100% resistant. Oseltamivir resistance arose over a relatively short timescale (three seasons), which was unexpected as it was believed that oseltamivir resistant viruses carried a fitness deficit 29. However, evidence suggests that compensatory mutations, which arose before the drug resistance mutation, rescued the fitness and therefore allowed for worldwide spread of these viruses 30. Note that so far mutations that confer oseltamivir resistance confer little or no resistance to zanamivir. The co‐circulation of viruses that are resistant to either of the two classes of antiviral drugs obviously brings with it the concern that a multi‐drug resistant virus may emerge. We may have been granted a reprieve from this situation in that the prevalence of seasonal H1N1 viruses has dropped significantly since the appearance of the pandemic H1N1 virus, and therefore the oseltamivir‐resistance mutation is no longer in circulation. Nevertheless, if H1N1 viruses have a propensity for developing oseltamivir resistance and this is acquired by the pandemic H1N1 virus in addition to its existing adamantane resistance, we would have few therapeutic options at our disposal. Some examples of multi‐drug resistant viruses have been isolated from patients undergoing therapy, but so far, they have not been seen in general circulation 31, 32. Examination of these resistant H1N1 viruses in animal models for influenza virus has shown that they do not appear to be attenuated either in terms of pathogenicity or transmission 33, 34. This current status fuels the argument that new options for influenza antiviral therapy are sorely needed, and moreover, that we must start exploring different targets, including non‐viral ones.
HOST FACTORS AS POTENTIAL ANTIVIRAL DRUG TARGETS
Almost all approved antiviral drugs on the market are highly specific for a particular virus or family of viruses by virtue of the fact that they target viral proteins. Understandably, this strategy is used because of the need to establish selectivity and to not cause undue harm to the host. The downside is that resistance is far more likely to develop if the target is virus‐encoded and also that there are a limited number of viral proteins that possess properties amenable to developing pharmaceutically acceptable inhibitors (“druggable”). As described above, all approved influenza antiviral drugs are directed at either the M2 or NA proteins. For small RNA viruses in particular, the number of host functions they rely on is likely to far outnumber the viral functions, so by identifying these required host factors, we will immediately increase the number of potential drug targets.
DAS181 or Fludase is a developmental therapeutic candidate for influenza and is unique in that it targets a cellular component. DAS181 is a recombinant protein with sialidase activity and acts by removing sialic acid from proteins in the airway and thus prevents influenza virus attachment 35. Maraviroc, which was approved in 2007 as an antiretroviral drug, is another example of a host‐directed antiviral. Maraviroc targets one of the HIV‐1 co‐receptors, C‐C chemokine receptor type 5 (CCR5) 36, and thereby prevents virus entry. Support for the value of pursuing CCR5 as a drug target came from individuals who have a deletion in their CCR5 gene leading to loss of function and who are more resistant to HIV‐1 infection 37. Admittedly, maraviroc has also taught us that antiviral drugs directed at cellular targets are not immune to resistance, as maraviroc‐resistance has been observed both in the laboratory and in patients. Interestingly, resistance seen in vitro results from the virus adapting to use the drug‐bound form of CCR5, while in patients, the drug selects for the growth of viruses that use the alternative receptor, C‐X‐C chemokine receptor type 4, which are therefore insensitive to a CCR5‐inhibitor 38. This only emphasizes the close interplay between virus and host cell and how the role of any required cellular protein will have to be carefully dissected in order to validate it as a drug target. One should also distinguish between chronic and acute infections, as the length of drug treatment for acute infections such as influenza will usually be limited to about a week. Therefore, inhibition of a cellular function will be temporary, and perhaps, there is greater opportunity to explore the potential for host‐directed antiviral therapies in this setting, rather than with a chronic infection.
It is also possible that a particular host function is required for the replication of multiple viruses, which opens the door to the development of an antiviral drug with broad‐spectrum activity. This would be particularly useful for the treatment of diseases caused by emerging or neglected viruses for which there is little incentive for specific drug development by the pharmaceutical industry.
GENOME‐WIDE RNAI SCREENS UNVEIL THE HOST FACTORS REQUIRED BY INFLUENZA VIRUS
Genome‐wide RNAi screens are proving to be a vital tool in our efforts to gather more information on host‐pathogen interactions because they provide a global view of all host factors that are required by a particular pathogen. For influenza virus, five studies have used this approach to define the cellular network that is critical for efficient replication of the virus. In addition to learning how these host factors facilitate virus replication, it is hoped that we can use this information to pursue new targets for antiviral therapy.
Description of the RNAi screens for influenza virus
The first RNAi screen for influenza virus was performed by Hao et al. 22. This was done in Drosophila cells, as at that time the RNAi systems and tools were more advanced in this species. Drosophila cells are not permissive for influenza virus infection, so the authors generated a modified influenza virus. To enable the virus to enter, they replaced the HA gene with that of the glycoprotein from vesicular stomatitis virus. In the absence of HA, NA is not essential, so they also replaced the NA with a gene encoding Renilla luciferase and this served as a convenient marker for virus gene expression. The Drosophila cells were shown to support entry and gene expression of this recombinant influenza A/WSN/33‐based virus, however, virus assembly was defective due to lack of expression of certain influenza virus proteins. Thus, this system could be used for detecting the stages of the influenza virus life cycle that cover post‐entry events up to and including gene expression. A library of dsRNA (Ambion) targeting 13 071 Drosophila genes was screened for those that would reduce luciferase expression in cells infected with the recombinant influenza virus. The authors describe 176 primary hits, of which 110 could be confirmed with alternate dsRNAs. The human homologues for three selected genes (ATP6V0D1, COX6A1, and NXF1) were confirmed to be required for influenza infection of human cells, indicating that data from Drosophila screens can be translated to the human system.
The second, third, and fourth screens are probably the most similar in that each is a genome‐wide siRNA screen performed in human cells. Yet, all used distinct approaches. Brass et al. 21 infected osteosarcoma (U2OS) cells with influenza A/PR/8/34 virus for 12 h and monitored infection by staining for cell surface expression of HA. This captures all stages of the life cycle up to and including HA trafficking to the cell surface. The authors targeted 17 877 genes using the Dharmacon siRNA library that comes in pools of four siRNAs per gene. They identified 312 pools that reduced HA surface expression, and of these, 129 genes were confirmed with at least two individuals siRNAs per gene. These represent the genes required by the virus. Another four genes (IFITM3, PUSL1, TPST1, and WDR33) were identified as restriction factors because infection was enhanced in their absence. The first of these has been characterized in more detail along with other IFITM family members and shown to block entry of not only influenza virus but also West Nile virus, dengue virus, HIV‐1, filoviruses, and SARS coronavirus 21, 39, 40. König et al. 24 used lung epithelial carcinoma (A549) cells for their genome‐wide RNAi screen. Using a similar strategy to that of Hao et al. 22, they also engineered influenza A/WSN/33 virus so that it expressed Renilla luciferase in place of HA. This recombinant virus was only capable of multi‐cycle growth in HA‐complementing cells and when used to infect A549, as done in the screen, it established a single round of infection. Thus, their screen captured everything from attachment up until viral gene expression, which was monitored via luciferase activity. They targeted 19 628 genes with an siRNA library from Qiagen and reported 295 primary hits for which at least two siRNAs per gene reduced luciferase expression. Of these, 219 genes were confirmed to be required in the context of wild‐type influenza A/WSN/33 virus infection. Karlas et al. 23 also used A549 cells and the Qiagen siRNA library (22,843 genes targeted) but they used a two‐step infection process that allowed them to capture the entire influenza virus life cycle. The siRNA‐transfected A549 cells were infected with influenza A/WSN/33 virus and stained for NP expression at 24 h post‐infection, which captured the early to mid stages of the life‐cycle. The supernatants from this infection were then transferred onto 293T cells, which contained an influenza‐specific luciferase reporter that is activated upon influenza virus infection. siRNAs that resulted in reduced luciferase expression here but did not affect the NP levels in the first round would indicate that the targeted gene was required for the assembly or release of virus particles, which was a unique aspect of this study. The authors report 287 primary hits from this screening approach, with 168 of these confirmed to reduce replication of either influenza A/WSN/33 or A/Hamburg/04/2009 (H1N1) viruses with at least two siRNAs per gene. Of note, 72 of these genes were commonly required by both influenza virus strains, and König et al. also showed that 12 out of 12 selected hits were also required by influenza A/Netherlands/602/2009 (H1N1) virus, whereas only two of the 12 were required by vesicular stomatitis virus. This suggests that these RNAi screens are able to identify host factors that are specifically required by influenza viruses, as well as those that may be required for efficient replication of multiple viruses. The latter would of course be potential targets for antiviral agents with broad‐spectrum activity.
The fifth screen by Shapira et al. 25 used a very different approach from the other four. They did not perform a genome‐wide RNAi screen, but instead selected genes to target based on protein interaction and transcriptional response datasets. From yeast‐2‐hybrid screens performed with 10 influenza A/PR/8/34 virus proteins (all except PB1‐F2), they selected 259 genes that encode either direct interacting proteins or their first neighbors in the interaction network. A further 1056 genes were selected because they were shown to be transcriptionally regulated in response to influenza virus infection. Finally, 504 genes that encode proteins involved in several pathways found to be over‐represented in the datasets were selected for inclusion. In total, 1745 genes were targeted with siRNA pools from Dharmacon. The siRNAs were transfected into primary human bronchial epithelial cells, and growth of influenza A/PR/8/34 virus in these cells was monitored by transferring the supernatants onto 293T cells that contained an influenza polymerase‐driven reporter, similar to that used by Karlas et al. 23. This captures the entire life cycle from attachment until virus budding and release. In addition, these authors monitored the involvement of the selected host factors in the ability of the cells to induce interferon‐β in response to (i) infection with PR8 virus lacking NS1 expression and (ii) transfection with viral RNA. A total of 616 genes scored as hits in one of these assays with 220 of these being involved in influenza virus replication.
Comparison of the results of all influenza RNAi screens
Although each of these studies analyzed their own datasets in detail and provided certain levels of validation for their hits, this review will focus more on comparing the results and discussing the power and limitations of the RNAi approach. From the descriptions above, it should already be clear that each study employed distinct assays that include differences in cell types, virus strain, siRNA library, and assay readout. Despite this, it is still surprising that not one hit was common to all five screens, although a similar low level of overlap was observed between three HIV‐1 RNAi screens 41. To look at this in more detail, a list of hits was generated composed of the primary hits from all five screens. For the Drosophila screen, 130 human homologues could be identified from the list of 176 Drosophila hits. From Shapira et al., the list of 220 hits required for virus replication was taken into consideration and for Brass et al., 249 of the initial 312 hits were used as these confirmed with at least one siRNA from the pool. The hits considered for the König and Karlas screens were those validated with two or more siRNAs. Analysis of this combined list of 1077 reveals 992 genes that are unique to one of the screens, whereas 85 genes are found in two or more of the screens (Figure 2). More specifically, 72 genes are common to two of the five screens, eight are common to three screens and five are common to four of the screens. Within this small group of 13 genes found in three or more screens, those encoding subunits of the Vacuolar‐type H+‐ATPase (vATPase) complex and the members of the coatomer or COPI complex clearly stand out, leaving no doubt that these cellular complexes are required during the influenza virus life‐cycle. vATPase activity is known to be required for acidification of the endosome and has previously been implicated in influenza virus entry 42, 43, so this finding was not unexpected. The involvement of the COPI complex, however, was a new finding, and we await a thorough analysis of its role in influenza virus replication. Initial characterization by two of the groups suggests potential roles in both virus entry 24 and HA trafficking 21.
Figure 2.
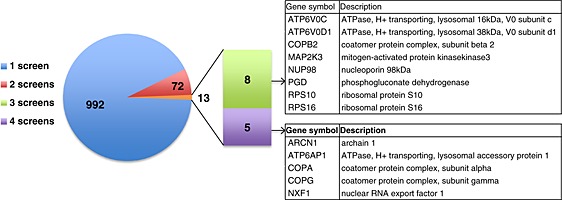
Representation of the number of common hits among five influenza virus RNAi screens 21, 22, 23, 24, 25. 992 genes were unique to one of the screens, 72 were identified in two screens, 8 were identified in three screens and 5 were identified in four screens. The genes in the latter two categories are shown in the tables on the right
To delve further into the other host functions that are most strongly supported by these multiple RNAi screens, all genes that were identified in two or more screens were considered further. When this list of 85 genes is analyzed in terms of function and gene classification, there are six categories that are enriched and represented by at least five genes (Table 2). In addition to the aforementioned COPI and vATPase complexes, which are ranked second and third, the ribosome (ranked first), spliceosome, nuclear pore/envelope, and kinase signaling categories are also enriched. Dependency on ribosomes is not unexpected for a virus that replicates fast and must express its proteins rapidly. Neither is the involvement of the splicing machinery or elements that control nuclear trafficking, as both are known to be fundamental to influenza virus replication. However, the identification of specific factors involved in these processes in the infected cell, may point towards interfaces with the virus, for example, interactions with viral proteins. As an example, NXF1 is described to be required for the efficient export of specific influenza virus mRNAs 44 and to interact with the NS1 protein 45. Kinase signaling is obviously a broader category and is likely to be involved at multiple stages of the virus life‐cycle, however, the kinases shown in Table 2 that were identified by at least two studies, represent top candidates for further study.
Table 2.
Over‐represented functional categories of 85 genes found in two or more screens
| Rank# | Gene classification cluster | Enrichment score* | Genes |
|---|---|---|---|
| 1 | Ribosome | 6.12 | RPS4X, RPS5, RPS10, RPS14, RPS16, RPS20, RPL13A, FAU |
| 2 | COPI vesicle | 3.72 | ARCN1, COPA, COPB1, COPB2, COPG |
| 3 | Proton‐transporting V‐type ATPase complex | 2.87 | ATP6V1A, ATP6V1B2, ATP6V0C, ATP6V0B, ATP6AP1, ATP6V0D1 |
| 4 | Spliceosome | 2.66 | NHP2L1, PRPF8, SF3B1, SF3A1, SNRP70 |
| 5 | Nuclear pore/envelope | 2.41 | KPNB1, NUP98, NUP153, NXF1, NUP205 |
| 6 | Kinase/signaling | 1.16 | CLK1, PLK3, FGFR2, MAP2K3, TNK2, DCLK2 |
Ranked according to enrichment score.
Enrichment score indicates the degree to which genes associated with a particular function/pathway are over‐represented in this list relative to their representation in the entire genome.
One of the possible reasons that there is not more overlap in the hits identified by the different screens is that each RNAi library contains siRNAs with varying efficacies for particular targets. (It should be noted that due to the use of multiple siRNAs per target, off‐target effects are not considered to be a major contributing factor). Those that made it to the hit‐list in each screen, will be those for which efficient knockdown was achieved in order for the biological effect on influenza virus replication to be observed. Yet, cellular proteins rarely act alone and if the virus is dependent on particular pathways or complexes, it is likely that depletion of any vital factor in that pathway will affect the virus. Therefore, despite the lower confidence level, useful information can still be obtained by analyzing the hits that were only identified in one screen. When combined with the list of 85 higher confidence hits, and analyzed for over‐represented functions or classifications, these unique hits change the ranking of enriched categories (Table 3). Factors involved in kinase signaling are now the most highly enriched, indicating that the addition of the unique hits has extended the support for this category. There are also some new categories that score highly, such as mitosis/cell cycle, translation initiation, phosphoinositide metabolism, and several others. This stresses the point that comparison of these screens at the pathway level rather than at the gene level will reveal more functional overlap and may also provide new information that would not be observed if studying the results of each screen in isolation.
Table 3.
Over‐represented functional categories of 1077 genes found in any of the five screens
| Rank# | Gene classification cluster | Enrichment score* | No of genes |
|---|---|---|---|
| 1 | Kinase/signaling | 29.52 | 134 |
| 2 | Mitosis/cell cycle | 16.24 | 9 |
| 3 | Translation initiation | 12.69 | 12 |
| 4 | Ribosome | 11.54 | 29 |
| 5 | Phosphoinositide metabolic process | 10.34 | 7 |
| 6 | mRNA processing | 9.66 | 44 |
| 7 | ABC transporter | 9.26 | 5 |
| 8 | Protein transport | 8.6 | 4 |
| 9 | Nucleus | 8.04 | 7 |
| 10 | Regulation of ubiquitin‐protein ligase activity | 8.01 | 23 |
| 11 | PI3 kinase | 5.21 | 4 |
| 12 | Ras small GTPase, Rab type | 5.03 | 10 |
| 13 | Nuclear pore/envelope | 4.59 | 15 |
| 14 | ATP biosynthetic process | 4.49 | 16 |
| 15 | Protein‐tyrosine phosphatase, dual specificity | 4.31 | 5 |
| 16 | COPI vesicle | 3.87 | 14 |
| 17 | Mitochondrial membrane | 3.61 | 10 |
| 18 | Ubl conjugation pathway | 3.35 | 16 |
| 19 | Transcription regulation | 2.91 | 46 |
| 20 | Actin‐binding | 2.81 | 6 |
| 21 | Transcription factor activity | 2.68 | 21 |
| 22 | Intracellular protein transport | 2.51 | 5 |
| 23 | Nucleosome | 2.38 | 4 |
| 24 | Tetratricopeptide repeat | 2.2 | 4 |
| 25 | Regulation of Ras GTPase activity | 1.7 | 6 |
| 26 | Golgi apparatus | 1.66 | 10 |
| 27 | Leucine‐rich repeat | 1.66 | 4 |
| 28 | WD40 repeat | 1.35 | 17 |
Ranked according to enrichment score.
Enrichment score indicates the degree to which genes associated with a particular function/pathway are over‐represented in this list relative to their representation in the entire genome.
SELECTION OF NEW TARGETS FOR THERAPEUTIC INTERVENTION
Faced with this vast amount of new data, how does one progress to the identification of host factors that would make suitable targets for anti‐influenza virus drugs? Beyond anything else, it is important to gather more supporting information that the host factor plays a critical role in influenza virus replication. The precise mechanism of action will have to be elucidated, and it must be determined whether the required function is something that is amenable to inhibition by a small molecule. In some cases, small molecule inhibitors of the host factors already exist and can be used as validation for the requirement of that cellular function. For example, König et al. 24 identified the calcium/calmodulin‐dependent kinase, CAMK2B, in their screen, and they showed that a specific inhibitor of this kinase, KN93, could inhibit influenza virus replication in a dose‐dependent manner. Similarly, Karlas et al. 23 showed that TG003, an inhibitor of CDC‐like kinase 1 (CLK1), can reduce influenza virus growth. When interpreting these results, one has to ensure that the inhibitor is specific for the intended target, as it is often the case that small molecules can inhibit multiple related proteins to varying extents. Apart from chemical inhibitors, it is also possible to test whether enzymatic activity is required by expressing dominant negative forms of the enzyme and determining whether this reduces influenza virus replication. Knock‐out mice, when available, are particularly beneficial in terms of examining whether the loss of the cellular factor has an impact on influenza virus replication in vivo and if this translates into reduced pathogenicity. This was demonstrated for one of the hits identified in the screens, where p27‐deficient mice were shown to have lower viral titers in their lungs compared with wild‐type mice 23. In addition, knock‐out mice provide evidence that the cellular factor is not required for viability. Although this is a comforting finding, it is unclear whether it is entirely necessary in order to support a case for a host factor as a drug target. With drug‐mediated inhibition, especially if treating an acute disease such as influenza, the loss of cellular function is temporary and it may be quite possible for the host to withstand this without deleterious consequences.
Of the 85 cellular factors that were identified in two or more of the influenza virus screens, 50 are considered to have druggable properties (Table 4), according to the Integrated Druggable Genome Database available from Sophic (http://www.sophicalliance.com/). Of these, 21 have been confirmed to be required for replication of wild‐type influenza virus (underlined in Table 4), so could serve as top candidates for further exploration.
Table 4.
Fifty Druggable genes found in two or more influenza virus RNAi screens
| Gene Symbol | Gene ID | Name |
|---|---|---|
| ATP6AP1 | 537 | ATPase, H+ transporting, lysosomal accessory protein 1 |
| ATP6AP2 | 10159 | ATPase, H+ transporting, lysosomal accessory protein 2 |
| ATP6V1A | 523 | ATPase, H+ transporting, lysosomal 70 kDa, V1 subunit A |
| ATP6V1B2 | 526 | ATPase, H+ transporting, lysosomal 56/58 kDa, V1 subunit B2 |
| BUB3 | 9184 | Budding uninhibited by benzimidazoles 3 homolog (yeast) |
| BZRAP1 | 9256 | Benzodiazapine receptor (peripheral) associated protein 1 |
| CD81 | 975 | CD81 molecule |
| CLIC4 | 25932 | Chloride intracellular channel 4 |
| CLK1 | 1195 | CDC‐like kinase 1 |
| COPB2 | 9276 | Coatomer protein complex, subunit beta 2 (beta prime) |
| COPG | 22820 | Coatomer protein complex, subunit gamma |
| DCLK2 | 166614 | Doublecortin‐like kinase 2 |
| EIF4A2 | 1974 | Eukaryotic translation initiation factor 4A, isoform 2 |
| FAU | 2197 | Finkel–Biskis–Reilly murine sarcoma virus (FBR‐MuSV) ubiquitously expressed |
| FBXW2 | 26190 | F‐box and WD repeat domain containing 2 |
| FGFR2 | 2263 | Fibroblast growth factor receptor 2 |
| FKBP8 | 23770 | FK506 binding protein 8, 38 kDa |
| HAND2 | 9464 | Heart and neural crest derivatives expressed 2 |
| IFNGR2 | 3460 | Interferon gamma receptor 2 (interferon gamma transducer 1) |
| IL17RA | 23765 | Interleukin 17 receptor A |
| IRF2 | 3660 | Interferon regulatory factor 2 |
| JUN | 3725 | Jun oncogene |
| KPNB1 | 3837 | Karyopherin (importin) beta 1 |
| KRAS | 3845 | v‐Ki‐ras2 Kirsten rat sarcoma viral oncogene homolog |
| LRP1B | 53353 | Low density lipoprotein‐related protein 1B (deleted in tumors) |
| LY6G6C | 80740 | Lymphocyte antigen 6 complex, locus G6C |
| MAP2K3 | 5606 | Mitogen‐activated protein kinase kinase 3 |
| MDM2 | 4193 | Mdm2 p53 binding protein homolog (mouse) |
| MYC | 4609 | v‐myc myelocytomatosis viral oncogene homolog (avian) |
| NTSR1 | 4923 | Neurotensin receptor 1 (high affinity) |
| NUP153 | 9972 | Nucleoporin 153 kDa |
| NUP98 | 4928 | Nucleoporin 98 kDa |
| OSMR | 9180 | Oncostatin M receptor |
| PEPD | 5184 | Peptidase D |
| PGD | 5226 | Phosphogluconate dehydrogenase |
| PLK3 | 1263 | Polo‐like kinase 3 (Drosophila) |
| PPP1R14D | 54866 | Protein phosphatase 1, regulatory (inhibitor) subunit 14D |
| PRPF8 | 10594 | PRP8 pre‐mRNA processing factor 8 homolog (S. cerevisiae) |
| PSENEN | 55851 | Presenilin enhancer 2 homolog (C. elegans) |
| PTPRN | 5798 | Protein tyrosine phosphatase, receptor type, N |
| PTS | 5805 | 6‐pyruvoyltetrahydropterin synthase |
| RAB10 | 10890 | RAB10, member RAS oncogene family |
| RAB5A | 5868 | RAB5A, member RAS oncogene family |
| RACGAP1 | 29127 | Rac GTPase activating protein 1 pseudogene; Rac GTPase activating protein 1 |
| RBCK1 | 10616 | RanBP‐type and C3HC4‐type zinc finger containing 1 |
| RPS14 | 6208 | Ribosomal protein S14 |
| RUNX1 | 861 | Runt‐related transcription factor 1 |
| SF3A1 | 10291 | Splicing factor 3a, subunit 1, 120 kDa |
| SLC1A3 | 6507 | Solute carrier family 1 (glial high affinity glutamate transporter), member 3 |
| TNK2 | 10188 | Tyrosine kinase, non‐receptor, 2 |
The underlined factors are those for which it has been shown that knockdown reduces replication of wild‐type influenza virus.
Further insight into the role of the cellular protein in the influenza virus life cycle is crucial in order to comprehend the consequences of manipulating its function. For this, we will rely on integrating the data from the RNAi screens with those studies that use alternative approaches to address virus‐host interactions. For example, protein interaction studies such as the yeast‐two‐hybrid assay employed by Shapira et al. 25 and gene expression data that may indicate whether these pathways are regulated during influenza virus infection. It is expected that those host factors for which we obtain multiple lines of evidence for an involvement in the influenza virus life cycle will naturally attract more research interest, and that in turn will foster their development as potential drug targets.
CONCLUSION
In summary, the use of RNAi screens has expanded our knowledge of the number of cellular proteins potentially involved in the replication of influenza virus by several fold. The fact that multiple such studies were published over a short time span has provided us the opportunity to assess the usefulness of the approach and more importantly to learn how one should interpret such data. Clearly, if we restrict ourselves to analyzing the overlapping hits at the level of gene name, we will limit the pool of required host factors to a few key members. However, if a cross‐comparison is performed at the level of gene function, the multiple studies provide support for the participation of distinct cellular pathways or protein complexes, some of which are not seen if analyzing the studies on an individual basis. Overall, these studies provide strong evidence that the vATPase and COPI complexes, the ribosomal, mRNA splicing and nuclear trafficking machinery, and kinase‐regulated signaling are all required for efficient replication of influenza A virus. Of these, the COPI complex is the best example of a new cellular function that was uncovered by the use of RNAi screening and this is sure to spur further research to understand how it facilitates influenza virus replication. The screens also present new avenues to explore in terms of potential targets for antiviral drugs. Some of these cellular factors may be specifically required by influenza virus, whereas others may also play a role in other virus infections, which presents the opportunity for development of a host‐directed drug with broad‐spectrum activity.
CONFLICT OF INTEREST
The authors have declared that there is no conflict of interest.
ACKNOWLEDGEMENTS
The author thanks Peter Palese for critically reading the manuscript and acknowledges funding from the National Institutes of Health (HHSN272200900032C, R21 AI083673, and U01 AI1074539).
REFERENCES
- 1. Palese P, Shaw ML. Orthomyxoviridae: the viruses and their replication In Fields Virology, Knipe DM, Howley PM. (eds). Lippincott Williams & Wilkins: Philadelphia, 2007; 1647–1689. [Google Scholar]
- 2. Rust MJ, Lakadamyali M, Zhang F et al Assembly of endocytic machinery around individual influenza viruses during viral entry. Nature Structural and Molecular Biology 2004; 11: 567–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annual Review of Biochemistry 2000; 69: 531–569. [DOI] [PubMed] [Google Scholar]
- 4. Pinto LH, Holsinger LJ, Lamb RA. Influenza virus M2 protein has ion channel activity. Cell 1992; 69: 517–528. [DOI] [PubMed] [Google Scholar]
- 5. Cros JF, Garcia‐Sastre A, Palese P. An unconventional NLS is critical for the nuclear import of the influenza A virus nucleoprotein and ribonucleoprotein. Traffic 2005; 6: 205–213. [DOI] [PubMed] [Google Scholar]
- 6. Li ML, Ramirez BC, Krug RM. RNA‐dependent activation of primer RNA production by influenza virus polymerase: different regions of the same protein subunit constitute the two required RNA‐binding sites. EMBO Journal 1998; 17: 5844–5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fechter P, Brownlee GG. Recognition of mRNA cap structures by viral and cellular proteins. Journal of General Virology 2005; 86: 1239–1249. [DOI] [PubMed] [Google Scholar]
- 8. Dias A, Bouvier D, Crepin T et al The cap‐snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 2009; 458: 914–918. [DOI] [PubMed] [Google Scholar]
- 9. Poon LL, Pritlove DC, Fodor E et al Direct evidence that the poly(A) tail of influenza A virus mRNA is synthesized by reiterative copying of a U track in the virion RNA template. Journal of Virology 1999; 73: 3473–3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Deng T, Vreede FT, Brownlee GG. Different de novo initiation strategies are used by influenza virus RNA polymerase on its cRNA and viral RNA promoters during viral RNA replication. Journal of Virology 2006; 80: 2337–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Perez JT, Varble A, Sachidanandam R et al Influenza A virus‐generated small RNAs regulate the switch from transcription to replication. Proceedings of the National Academy of Sciences of the United States of America 2010; 107: 11525–11530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. O'Neill RE, Talon J, Palese P. The influenza virus NEP (NS2 protein) mediates the nuclear export of viral ribonucleoproteins. EMBO Journal 1998; 17: 288–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Neumann G, Hughes MT, Kawaoka Y. Influenza A virus NS2 protein mediates vRNP nuclear export through NES‐independent interaction with hCRM1. EMBO Journal 2000; 19: 6751–6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Leser GP, Lamb RA. Influenza virus assembly and budding in raft‐derived microdomains: a quantitative analysis of the surface distribution of HA, NA and M2 proteins. Virology 2005; 342: 215–227. [DOI] [PubMed] [Google Scholar]
- 15. Rossman JS, Lamb RA. Influenza virus assembly and budding. Virology 2011; 411: 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Palese P, Tobita K, Ueda M et al Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology 1974; 61: 397–410. [DOI] [PubMed] [Google Scholar]
- 17. Hale BG, Randall RE, Ortin J et al The multifunctional NS1 protein of influenza A viruses. Journal of General Virology 2008; 89: 2359–2376. [DOI] [PubMed] [Google Scholar]
- 18. Garcia‐Sastre A, Egorov A, Matassov D et al Influenza A virus lacking the NS1 gene replicates in interferon‐deficient systems. Virology 1998; 252: 324–330. [DOI] [PubMed] [Google Scholar]
- 19. Chen W, Calvo PA, Malide D et al A novel influenza A virus mitochondrial protein that induces cell death. Nature Medicine 2001; 7: 1306–1312. [DOI] [PubMed] [Google Scholar]
- 20. Conenello GM, Zamarin D, Perrone LA et al A single mutation in the PB1‐F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathogens 2007; 3: 1414–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brass AL, Huang IC, Benita Y et al The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 2009; 139: 1243–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hao L, Sakurai A, Watanabe T et al Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature 2008; 454: 890–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Karlas A, Machuy N, Shin Y et al Genome‐wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 2010; 463: 818–822. [DOI] [PubMed] [Google Scholar]
- 24. Konig R, Stertz S, Zhou Y et al Human host factors required for influenza virus replication. Nature 2010; 463: 813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shapira SD, Gat‐Viks I, Shum BO et al A physical and regulatory map of host‐influenza interactions reveals pathways in H1N1 infection. Cell 2009; 139: 1255–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pinto LH, Lamb RA. Understanding the mechanism of action of the anti‐influenza virus drug amantadine. Trends in Microbiology 1995; 3: 271. [DOI] [PubMed] [Google Scholar]
- 27. Gubareva LV, Kaiser L, Hayden FG. Influenza virus neuraminidase inhibitors. Lancet 2000; 355: 827–835. [DOI] [PubMed] [Google Scholar]
- 28. Hernandez JE, Adiga R, Armstrong R et al Clinical experience in adults and children treated with intravenous peramivir for 2009 influenza a (H1N1) under an emergency IND program in the United States. Clinical Infectious Diseases 2011; 52: 695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ives JA, Carr JA, Mendel DB et al The H274Y mutation in the influenza A/H1N1 neuraminidase active site following oseltamivir phosphate treatment leave virus severely compromised both in vitro and in vivo. Antiviral Research 2002; 55: 307–317. [DOI] [PubMed] [Google Scholar]
- 30. Bloom JD, Gong LI, Baltimore D. Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. Science 2010; 328: 1272–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Memoli MJ, Hrabal RJ, Hassantoufighi A et al Rapid selection of oseltamivir‐ and peramivir‐resistant pandemic H1N1 virus during therapy in 2 immunocompromised hosts. Clinical Infectious Diseases 2010; 50: 1252–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Graitcer SB, Gubareva L, Kamimoto L et al Characteristics of patients with oseltamivir‐resistant pandemic (H1N1) 2009, United States. Emerging Infectious Diseases 2011; 17: 255–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Seibert CW, Kaminski M, Philipp J et al Oseltamivir‐resistant variants of the 2009 pandemic H1N1 influenza A virus are not attenuated in the guinea pig and ferret transmission models. Journal of Virology 2010; 84: 11219–11226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Memoli MJ, Davis AS, Proudfoot K et al Multidrug‐resistant 2009 pandemic influenza A(H1N1) viruses maintain fitness and transmissibility in ferrets. Journal of Infectious Diseases 2011; 203: 348–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Malakhov MP, Aschenbrenner LM, Smee DF et al Sialidase fusion protein as a novel broad‐spectrum inhibitor of influenza virus infection. Antimicrobial Agents and Chemotherapy 2006; 50: 1470–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. MacArthur RD, Novak RM. Reviews of anti‐infective agents: maraviroc: the first of a new class of antiretroviral agents. Clinical Infectious Diseases 2008; 47: 236–241. [DOI] [PubMed] [Google Scholar]
- 37. Dean M, Carrington M, Winkler C et al Genetic restriction of HIV‐1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia growth and development study, multicenter AIDS cohort study, multicenter hemophilia cohort study, San Francisco City cohort, ALIVE study. Science 1996; 273: 1856–1862. [DOI] [PubMed] [Google Scholar]
- 38. Moore JP, Kuritzkes DR. A piece de resistance: how HIV‐1 escapes small molecule CCR5 inhibitors. Current Opinion in HIV and AIDS 2009; 4: 118–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huang IC, Bailey CC, Weyer JL et al Distinct patterns of IFITM‐mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathogens 2011; 7: e1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lu J, Pan Q, Rong L et al The IFITM proteins inhibit HIV‐1 infection. Journal of Virology 2011; 85: 2126–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bushman FD, Malani N, Fernandes J et al Host cell factors in HIV replication: meta‐analysis of genome‐wide studies. PLoS Pathogens 2009; 5: e1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Perez L, Carrasco L. Involvement of the vacuolar H(+)‐ATPase in animal virus entry. Journal of General Virology 1994; 75(Pt 10): 2595–2606. [DOI] [PubMed] [Google Scholar]
- 43. Guinea R, Carrasco L. Requirement for vacuolar proton‐ATPase activity during entry of influenza virus into cells. Journal of Virology 1995; 69: 2306–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Read EK, Digard P. Individual influenza A virus mRNAs show differential dependence on cellular NXF1/TAP for their nuclear export. Journal of General Virology 2010; 91: 1290–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Satterly N, Tsai PL, van Deursen J et al Influenza virus targets the mRNA export machinery and the nuclear pore complex. Proceedings of the National Academy of Sciences of the United States of America 2007; 104: 1853–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]