

Abstract
Advanced glycation endproducts (AGEs) and a receptor for AGEs (RAGE) have been linked in the pathogenesis of diabetic nephropathy. RAGE is usually localized to podocytes and is increased in diabetes. RAGE activation increases reactive oxygen species production, which mediates hyperglycemia-induced podocyte apoptosis in early diabetic nephropathy. Here, we examined the interaction of AGE and RAGE on podocyte apoptosis. When we exposed murine cultured podocytes to bovine serum albumin (BSA) that was modified by AGEs or to carboxymethyl-lysine BSA, more apoptosis was found when compared with unmodified BSA. Similarly, more podocytes underwent detachment and apoptosis when cultured on AGE-modified collagen IV than on native collagen IV. AGEs isolated from sera of patients with chronic kidney disease also caused apoptosis of podocytes. Apoptosis was diminished by small interference RNA (siRNA) for RAGE in podocytes exposed to AGE-BSA, but not to AGE-modified collagen IV. Both AGE- and carboxymethyl-lysine modified-BSA activated p38MAP kinase and inhibition of this kinase reduced the apoptotic effect of AGE-BSA. Exposure to AGE-BSA was associated with Akt dephosphorylation and FOXO4 transcriptional activation leading to an increase in the expression of an effector protein of apoptosis, Bim. siRNA for FOXO4 abolished AGE-BSA-induced apoptosis of podocytes. Our study suggests that an AGE–RAGE interaction contributes to podocyte apoptosis by activation of the FOXO4 transcription factor.
Keywords: phosphorylation, RAGE, Akt, advanced glycation endproducts, transcription factor
Diabetic nephropathy is the major cause of chronic and end-stage renal disease in the United States.1 None of the currently available pharmacologic interventions for diabetic nephropathy can completely forestall or reverse the progression of the disease. Much is known of the progression of glomerulosclerosis and tubulointerstitial fibrosis in diabetic nephropathy after the onset of albuminuria as mediated by TGF-β, angiotensin II, and advanced glycation endproducts (AGEs).2,3 However, cellular events preceding the onset of albuminuria in diabetic nephropathy have not been well characterized.
The podocyte is a terminally differentiated cell with a limited capacity to proliferate and regenerate after injury. Any disease process that causes podocyte loss, either from detachment or apoptosis, will lead to a decrease in the density of podocytes. Reduction in podocyte density is an important determinant of progressive diabetic nephropathy4 and precedes the development of renal dysfunction and albuminuria in diabetic patients and animal models of diabetes mellitus.4-6 There is a body of emerging literature, suggesting that podocyte loss is an early finding in diabetic nephropathy.7 A recent study by Susztak et al. showed that hyperglycemia causes podocyte apoptosis through a reactive oxygen species (ROS)-mediated pathway during the early stage of diabetic nephropathy.6 Our knowledge of the extracellular signals and intracellular responses leading to podocyte apoptosis in the diabetic milieu, however, remains limited.
One of the changes that occur as a result of elevated blood glucose in diabetes mellitus is the generation and accumulation of AGEs. Both AGEs and their receptors have been shown to play a key role in the pathogenesis of diabetic nephropathy.3,8-12 The receptor for AGEs (RAGE) is a member of the immunoglobulin superfamily of cell surface molecules.13 Ligand binding to RAGE by AGEs or members of the S100/calgranulin family triggers the activation of key cell signaling pathways, such as p21ras, mitogen-activated protein kinases (MAPKs), and nuclear factor-κB (NF-κB), leading to the activation of proinflammatory responses.14-16 The expression of RAGE is localized predominately to the podocyte and further increased in diabetes.3 Wautier et al.17 showed that AGEs initiate a vicious cycle of inflammation involving the generation of ROS via the activation of NADPH oxidase with further amplification of AGE production and inflammatory reaction. This observation was corroborated by our recent study showing that AGEs enhance ROS production.18
In diabetes, the extracellular matrix of the glomerular basement is modified by AGEs. The accumulation of AGEs and subsequent expansion of the glomerular basement membrane occur before the onset of albuminuria in the early phase of diabetic nephropathy.19 Podocytes cover the glomerular basement membrane and constitute an integral component of the glomerular filtration barrier. They are exposed to both circulating and glomerular-basement-membrane-bound AGEs. In this study, we tested the hypothesis that soluble and matrix-bound AGEs contribute to podocyte apoptosis via the activation of the AGE–RAGE axis, the FOXO4 transcription factor, and ROS production.
RESULTS
AGE exposure causes podocyte apoptosis
We examined the effects of soluble AGEs (CML-bovine serum albumin (BSA) and AGE-BSA) and matrix-bound AGEs (AGE-modified collagen IV) on podocyte apoptosis. Podocytes exposed to either Carboxymethyl-lysine-BSA (CML-BSA) or AGE-BSA underwent apoptosis at significantly higher rates than BSA (Figure 1). There was a two- to threefold increase in apoptosis when podocytes were cultured on AGE-modified collagen IV as compared with native collagen IV (Figure 1c). A similar increase in apoptosis was also noted when podocytes were cultured on AGE-modified collagen I as compared with native collagen I (data not shown). Effects of soluble (CML-BSA and AGE-BSA) and matrix-bound (AGE-modified collagen IV) AGEs on apoptosis were additive (Figure 1c). The apoptotic response of podocytes to AGE-BSA exposure occurred in a dose-dependent manner (Figure 2). Significant podocyte necrosis occurred at 100 μg/ml of AGE-BSA (Figure 2).
Figure 1. Podocyte apoptosis with soluble (CML-BSA and AGE-BSA) and matrix-bound AGE (AGE-modified collagen IV) exposures.
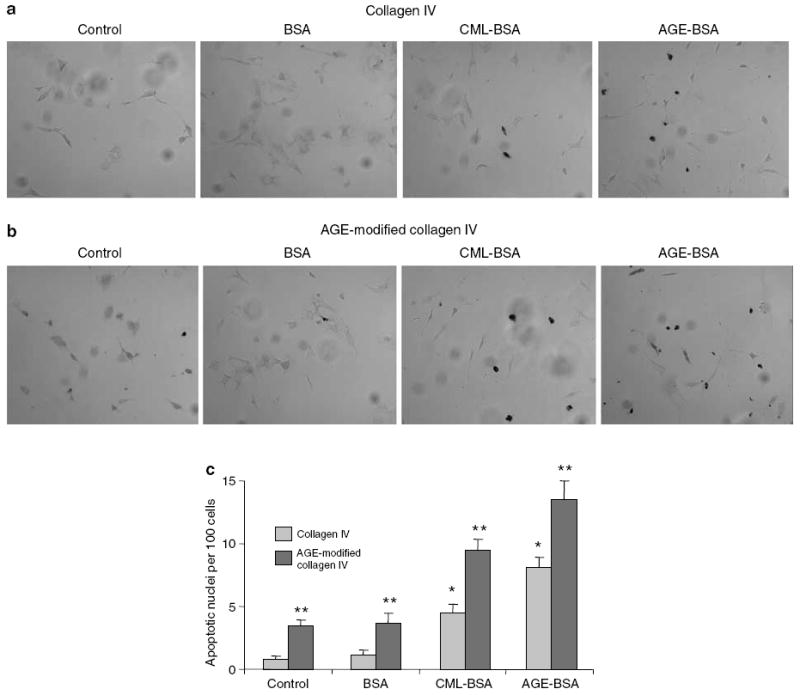
Differentiated podocytes cultured on coverslips coated with either collagen IV or AGE-modified collagen IV were exposed to 50 μg/ml of BSA, AGE-BSA, or CML-BSA for overnight. Apoptosis of podocytes was determined by TUNEL staining. (a, b) Representative photographs of TUNEL staining are presented. (c) Bar graph representing the means±s.e.m. of the number of apoptotic nuclei per 100 cells in five fields at original magnification x 400. *P<0.01 as compared with BSA exposure on collagen-IV-coated coverslips (n =4). **P<0.01 as compared with corresponding collagen-IV-coated coverslips (n =4).
Figure 2. Podocytes undergo apoptosis after AGE-BSA exposure in a dose-dependent manner.
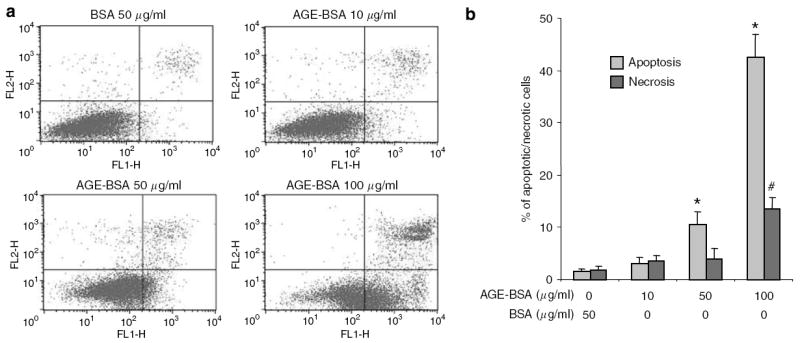
Apoptosis and necrosis were quantified by FACS after Annexin V-FITC and PI labeling. (a) Representative FACS data for podocytes treated with BSA and AGE-BSA. The abscissa and ordinate represent the fluorescence intensity of Annexin V-FITC and PI, respectively. (b) Bar graph represents the mean percentages of apoptosis and necrosis±s.e.m. (n =3). *P<0.05 versus apoptosis of BSA 50 μg/ml. #P<0.05 versus necrosis of BSA 50 μg/ml.
Effect of soluble and matrix-bound AGEs on podocyte detachment, apoptosis, and necrosis
To investigate podocyte detachment, apoptosis, and necrosis after exposure to soluble and matrix-bound AGEs, we treated podocytes cultured on plates coated with either collagen IV or AGE-modified collagen IV with AGE-BSA. The absolute number and percentage of cells in each group were determined and presented as means±s.e.m. (n = 3). Statistical analyses were performed on the percentages of cells relative to the total number of cells. Although the total cell number and total adherent cell number did not differ between all conditions, a higher proportion of podocytes cultured on AGE-modified collagen IV became detached as compared with collagen IV (Table 1). All the detached cells were non-viable by Trypan blue staining. Exposure to increasing doses of AGE-BSA did not cause a significant change in the percentages of detached cells for both collagen-IV-and AGE-modified collagen-IV-coated plates. However, there was a dose response increase in the apoptosis of adherent cells with AGE-BSA treatment.
Table 1.
Effect of soluble and matrix-bound AGEs on podocyte detachment, anoikis, and apoptosis
| BSA 50 μg/ml
|
AGE-BSA 10 μg/ml
|
AGE-BSA 50 μg/ml
|
AGE-BSA 100 μg/ml
|
|||||
|---|---|---|---|---|---|---|---|---|
| Cell number | % Of total | Cell number | % Of total | Cell number | % Of total | Cell number | % Of total | |
| Collagen IV coated | ||||||||
|
| ||||||||
| Detached | 54 900±7600 | 4.2±0.5 | 60 200±8500 | 4.370.6 | 91 800±8800 | 6.5±0.9 | 80 300±6600 | 5.7±0.7 |
| Adherent apoptotic | 41 700±3700 | 3.2±0.3 | 50 500±5200 | 3.670.7 | 114 000±4300 | 8.2±1.4*,# | 238 000±18 000 | 16.8±1.7* |
| Adherent necrotic | 47 400±2100 | 3.6±0.2 | 48 400±3400 | 3.570.5 | 57 000±6900 | 4.1±0.4 | 166 000±14 000 | 11.7±1.2*,Δ |
| Total adherent | 1 260 000±7200 | 95.8±0.3 | 1 360 000±97 000 | 95.770.6 | 1 300 000±64 000 | 93.5±0.6 | 1 350 000±71 000 | 94.3±0.5 |
| Total | 1 310 000±1500 | 1 420 000±100 000 | 1 390 000±56 000 | 1 430 000±65 000 | ||||
| AGE-modified collagen IV coated | ||||||||
| Detached | 137 000±9200 | 10.4±0.9φ | 137 000±5200 | 10.270.4φ | 157 000±6200 | 12.8±0.3φ | 147 000±6100 | 12.3±0.4φ |
| Adherent apoptotic | 65 800±2900 | 5.0±0.2φ | 86 300±6400 | 6.570.7φ | 143 000±1300 | 11.7±0.9*,#,φ | 334 000±38 000 | 27.9±2.6*,Δ,φ |
| Adherent necrotic | 45 400±5400 | 3.4±0.4 | 47 600±4400 | 3.670.4 | 66 600±7700 | 5.4±0.7 | 91 700±11 000 | 7.7±0.9* |
| Total adherent | 1 180 000±39 000 | 89.6±1.0 | 1 210 000±50 000 | 89.870.4 | 1 070 000±16 000 | 87.2±0.3 | 1 050 000±73 000 | 87.7±0.1 |
| Total | 1 320 000±31 000 | 1 350 000±53 000 | 1 220 000±21 000 | 1 200 000±79 000 | ||||
AGE, advanced glycation endproduct; BSA, bovine serum albumin;
P<0.05 versus BSA 50 μg/ml in the corresponding group.
P<0.05 versus AGE-BSA 10 μg/ml in the corresponding group.
P<0.05 versus AGE-BSA 50 μg/ml in the corresponding group.
P<0.05 versus the corresponding group with collagen IV coating.
The role of RAGE in AGE-BSA-induced podocyte apoptosis
To determine the role of RAGE in AGE-BSA-induced podocyte apoptosis, we knocked down RAGE expression by transfecting podocytes with a RAGE-specific small interference RNA (siRNA). The efficiency of transfection was demonstrated by concurrent transfection with a green fluorescent protein (GFP)-vector (Figure 3a). The siRNA was able to reduce the RAGE mRNA level and protein expression by 60–70% (Figure 3b and c). We found that podocytes transfected with the RAGE-specific siRNA had a significantly diminished apoptosis when exposed to 10 and 50 μg/ml of AGE-BSA, but only partially when exposed to 100 μg/ml (Figure 4a). RAGE-specific gene knockdown did not significantly reduce the apoptosis of podocytes cultured on AGE-modified collagen IV (Figure 4b), although a trend towards a reduction was observed. These results suggest that the AGE–RAGE interaction played a major role in the apoptosis of podocytes triggered by soluble AGE, but not by matrix-bound AGE.
Figure 3. Targeted gene knockdown of RAGE with siRNA.
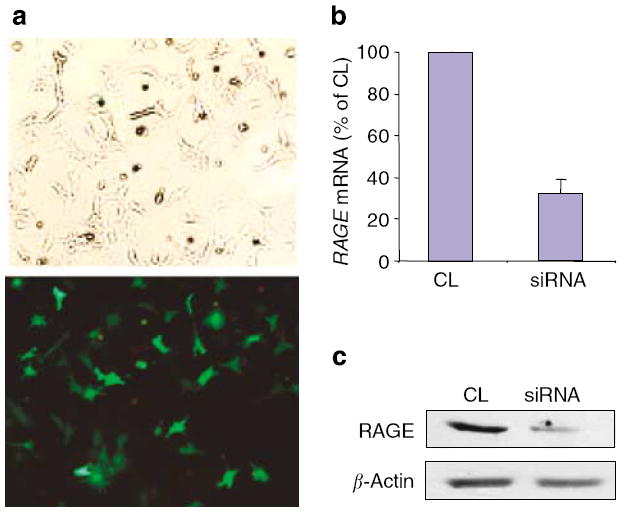
Transient transfection of podocytes with a siRNA for RAGE or the miRIDIAN microRNA Mimic negative control sequence (CL) was carried out using the Amaxa Nucleofection Device. Transfection efficiency was measured by the percentage of cells expressing GFP after transfection with pmaxGFP™. (a) Representative photographs of pmaxGFP™-transfected podocytes under light (top) and epifluorescence (bottom) microscopy. Effects of siRNA for RAGE and CL on RAGE mRNA level (b) and protein expression (c) were analyzed by real-time polymerase chain reaction and Western blotting, respectively. Representative data from three experiments are shown.
Figure 4. Knockdown of RAGE in podocytes mitigates the apoptotic effect of AGE-BSA, but not AGE-modified collagen IV.
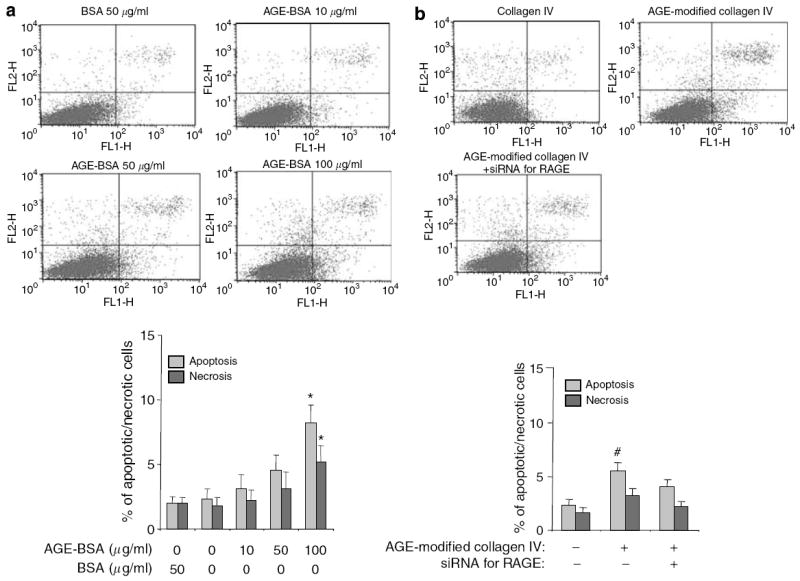
Apoptosis and necrosis were determined by FACS. Representative FACS data and bar graph summarizing the FACS results are shown. (a) Summary of FACS for apoptosis and necrosis of podocytes transfected with the siRNA for RAGE and treated with either BSA or AGE-BSA are shown. *P<0.05 versus BSA 50 μg/ml (n =3). (b) Apoptosis of podocytes transfected with the siRNA for RAGE and cultured on AGE-modified collagen IV. #P<0.05 versus the percentage of apoptosis in the other two groups (n =3).
AGEs isolated from patients with renal impairment caused podocyte apoptosis in vitro
Patients with chronic kidney disease (CKD) have higher serum levels of AGEs. To ensure that the apoptosis we observed in podocytes exposed to AGE-BSA and CML-BSA was not an artifact of the in vitro AGE preparation process, we isolated AGEs from sera of patients with CKD. The concentration of AGEs from patients with CKD was 46-fold higher than patients without CKD (55 versus 1.2 U/mg protein as measured by enzyme-linked immunosorbent assay). AGEs isolated from patients with stages 3 and 5 CKD caused a three and 4.6-fold increase in podocyte apoptosis, respectively, as compared with those from normal patients (Figure 5). We were able to abrogate the apoptotic effect of AGEs isolated from patients with CKD by pretreating the cultured podocytes with the siRNA for RAGE (Figure 5). These results suggest that the serum fraction extracted by a lysozyme (LZ) column caused the apoptosis of cultured podocytes via a pathway involving RAGE. Although we cannot exclude other co-extracted proteins contributing to the apoptosis of podocytes, it appears likely that AGE-like structures are the cause.
Figure 5. AGEs isolated from sera of CKD patients induce podocyte apoptosis.
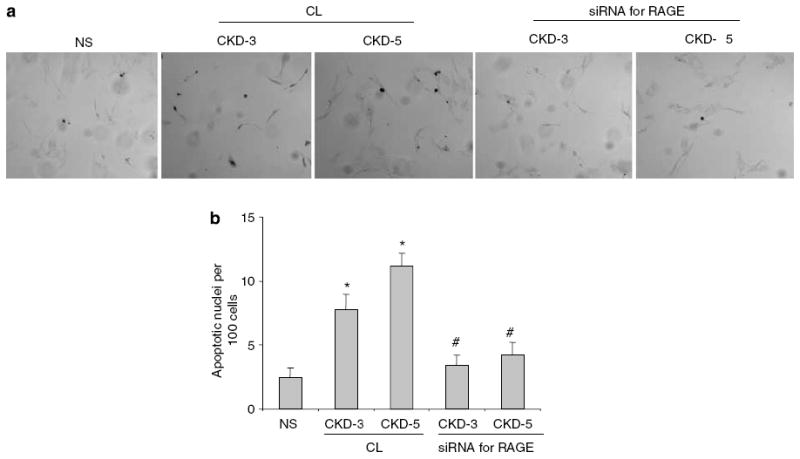
The effect of AGEs isolated from sera of patients with stages 3 and 5 CKD-3 and CKD-5 on apoptosis was compared with those of normal subjects (NS). (a) Representative fields of TUNEL at original magnification x 400. (b) Podocytes were transfected with either a negative control oligonucleotide (CL) or the siRNA for RAGE. The mean percentages of apoptotic cells after exposure to AGEs from groups CKD-3 and CKD-5 are presented in the bar graph. *P<0.01 versus NS. (n =3) #P<0.01 versus the corresponding group treated with CL (n =3).
AGE-BSA triggers ROS production leading to podocyte apoptosis
AGE–RAGE interaction leading to an increase in ROS production has been shown to cause apoptosis of fibroblasts and neuronal cells.20,21 To test whether ROS production is triggered by AGE–RAGE interaction in podocytes, we transfected podocytes with a RAGE-specific siRNA and measured the production of ROS after AGE-BSA stimulation. There was a dose-dependent increase in ROS production with AGE-BSA exposure, which was diminished in podocytes transfected with the siRNA for RAGE (Figure 6a). To verify that ROS production by AGE-BSA stimulation was responsible for podocyte apoptosis, we incubated podocytes with a ROS scavenger, N-acetyl-cysteine (NAC). Treatment with NAC reduced AGE-BSA-induced apoptosis (Figure 6b and c).
Figure 6. ROS production and apoptosis induced by AGE-BSA are diminished by a ROS scavenger, NAC.
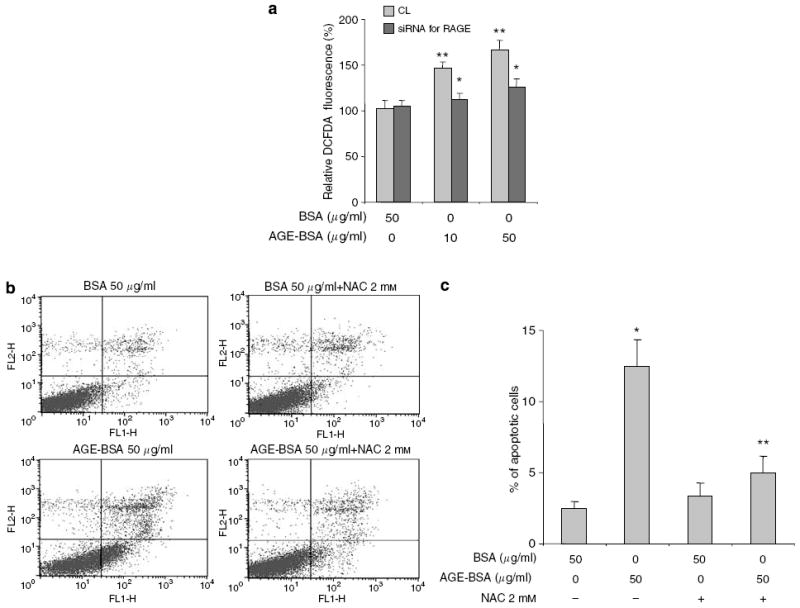
ROS production was measured by 2′,7′-dichlorofluorescein diacetate fluorescence. (a) ROS production triggered by AGE-BSA stimulation was diminished in podocytes transfected with the siRNA for RAGE. (b) Representative FACS data showing that pretreatment with NAC protected podocytes from AGE-BSA induced apoptosis. (c) FACS data from three different sets of experiments are summarized in a bar graph. *P<0.05 versus BSA alone. ** P<0.01 versus AGE-BSA alone.
Activation of proapoptotic intracellular signaling pathways by AGEs
Using Panomics TranSignal DNA/protein arrays as an approach to profile multiple transcription factors simultaneously, we identified FOXO4 as the only transcription factor from the forkhead family that was activated by AGE-BSA and CML-BSA stimulation. To determine the regulation of FOXO4 by upstream signaling pathways after AGE exposure, we performed Western blotting of proteins prepared from podocytes stimulated with BSA, CML-BSA, or AGE-BSA (Figure 7). The Akt signaling pathway is a known upstream regulator of FOXO4 transcription factors. Akt-mediated phosphorylation of FOXO4 promotes nuclear exclusion and transcriptional deactivation.22 Immunoblotting results showed that both CML-BSA and AGE-BSA diminished phosphorylation of Akt without a significant change of phosphatas and tensin homologue levels (Figure 7a). After 60 min of incubation with AGE-BSA and CML-BSA, the level of phosphorylated FOXO4 was decreased, likely due to a diminished Akt activity (Figure 7a). Dephosphorylation of FOXO4 is known to cause transcriptional activation of FOXO4, which is consistent with our observation from the Panomics TranSignal DNA/protein arrays.
Figure 7. AGE-BSA and CML-BSA activate p38MAPK and reduce Akt phosphorylation.
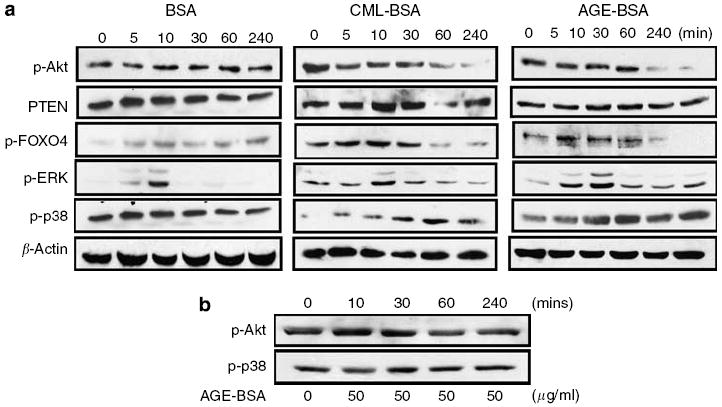
(a) A profile of signaling pathway activation in podocytes after exposure to BSA (50 μg/ml), CML-BSA (50 μg/ml), or AGE-BSA (50 μg/ml) at time 0, 5, 10, 30, 60, and 240 min. (b) Targeted-gene knockdown of RAGE prevented AGE-BSA-mediated p38MAPK phosphorylation and Akt de-phosphorylation. Representative blots of at least three independent experiments are shown.
Hyperglycemia and puromycin have been reported to cause podocyte apoptosis through activation of the p38MAP kinase (p38MAPK) pathway.6,23 We found that both AGE-BSA and CML-BSA caused a transient activation of extracellular signal-regulated kinase1/2 at 10 min and a more sustained activation of p38MAPK from 30 min to 2 h after AGE-BSA and CML-BSA stimulations (Figure 7a). c-Jun NH2-terminal kinase (JNK) phosphorylation with or without AGE-BSA and CML-BSA stimulation was too weak for detection (data not shown). Targeted gene knock down of RAGE in podocytes prevented AGE-BSA-induced Akt dephosphorylation and p38MAPK phosphorylation (Figure 7b). To confirm that p38MAPK contributes to AGE-BSA-mediated podocyte apoptosis, we inhibited p38MAPK in podocytes before AGE-BSA exposure. AGE-BSA-induced podocyte apoptosis was partially diminished by inhibition of p38MAPK with a specific inhibitor (Figure 8).
Figure 8. Partial protection from AGE-BSA-induced apoptosis by P38MAPK inhibition.
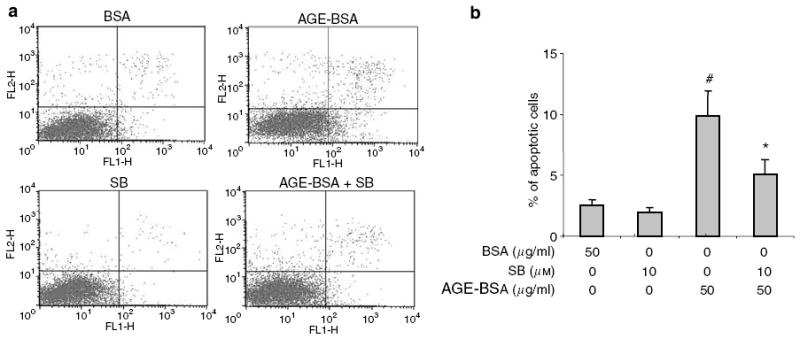
SB203580 (SB), a specific inhibitor of P38MAPK, partially reduced the apoptosis of podocytes treated with AGE-BSA. (a) Representative FACS data of apoptosis. (b) Summary of FACS data showing that #P<0.01 versus BSA 50 μg/ml and SB 10 μm + AGE-BSA 50 μg/ml (n =3). *P<0.05 versus BSA 50 μg/ml and AGE-BSA 50 μg/ml (n =3).
Role of transcription factor FOXO4 in AGE-induced podocyte apoptosis
It is known that dephosphorylation of FOXO transcription factors leads to nuclear translocation and activation of gene transcription. To further confirm our Panomics TranSignal DNA/protein array and Western blot result on FOXO4 activation by AGE-BSA, we examined the nuclear translocation of FOXO4. As demonstrated by immunofluorescence staining for FOXO4, AGE-BSA stimulated nuclear translocation of FOXO4 in podocytes (Figure 9a). We also determined that the expression of Bim, a downstream effector protein of apoptosis activated by FOXO,24 was significantly increased by AGE-BSA exposure in a dose-dependent manner (Figure 9b). To assess the role of FOXO4 in AGE-induced podocyte apoptosis, we knocked down the expression of FOXO4 by more than 75% in podocytes using a FOXO4-specific siRNA (Figure 10a and b). Inhibition of the FOXO4 expression diminished AGE-BSA- and CML-BSA-induced podocyte apoptosis (Figure 10c and d), thus implicating FOXO4 as a key regulator of podocyte apoptosis due to AGE exposure.
Figure 9. AGE-BSA stimulates nuclear translocation of FOXO4 and increases Bim expression.
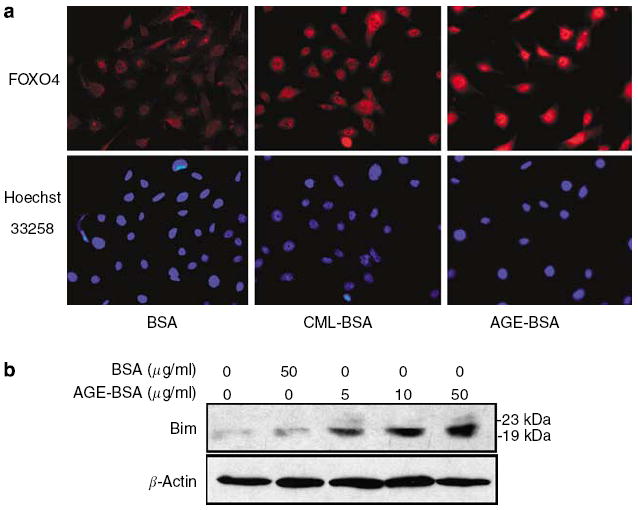
Podocytes were treated with AGE-BSA (50 μg/ml), CML-BSA (50 μg/ml), and BSA (50 μg/ml) for 2 h in RPMI medium with 1% FBS. (a) Representative photographs of immunofluorescence staining for FOXO4 and nuclear staining with Hoechst 33258 dye at original magnification x 400. (b) A representative Western blot for Bim expression after AGE-BSA or BSA incubation.
Figure 10. Suppression of FOXO4 expression abrogates AGE-BSA-induced podocyte apoptosis.
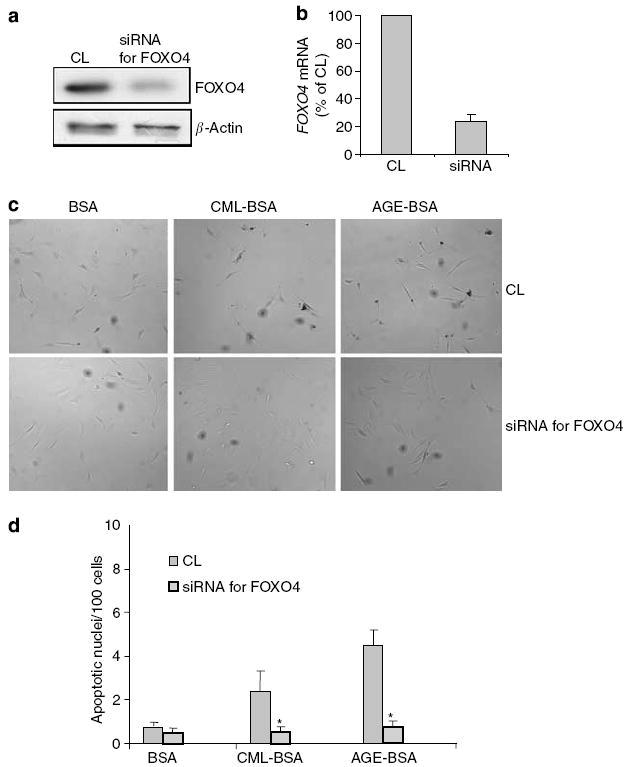
Podocytes were pretreated with either CL or the siRNA for FOXO4, and then exposed to 50 μg/ml of BSA, CML-BSA, or AGE-BSA. (a, b) Immunoblotting of cell lysates and real-time polymerase chain reaction of mRNA from podocytes transfected with siRNA for FOXO4 showed >75% knock down of expression as compared with a negative control sequence (CL). (c) Representative photographs of TUNEL staining results at original magnification x 400. (d) Bar graph summarizing the TUNEL staining results. Average rates of apoptosis per 100 nuclei±s.e.m. are presented. *P<0.01 versus the corresponding CL-treated group (n =3).
DISCUSSION
This study was designed to investigate the role of the AGE–RAGE interaction on podocyte apoptosis. We demonstrated for the first time that murine podocytes in culture undergo apoptosis when exposed to both soluble AGEs (CML-BSA and AGE-BSA) and matrix-bound AGEs (AGE-modified collagen IV). Our data suggest that AGEs play an important role in reducing the podocyte number via apoptosis, which could potentially accelerate the development and progression of diabetic nephropathy. Our findings on AGE-induced apoptosis in podocytes are consistent with previous reports showing AGE-induced apoptosis in osteo-blasts,25 fibroblasts,20 and neurons.21 Similar to our findings, increased ROS production was the mechanism that underlies the apoptosis of fibroblasts and neurons after AGE exposure.20,21
An interesting observation from this study was the additive effect of soluble AGEs (CML-BSA and AGE-BSA) and matrix-bound AGEs (AGE-modified collagen IV) on podocyte apoptosis. Methylglyoxal is an AGE with a reactive dicarbonyl moiety. Methylglyoxal modification of the argi-nine-glycine-aspartate motif of collagen IV, the major extracellular matrix protein in the glomerular basement membrane, has been shown to cause endothelial cell detachment and anoikis by interfering with binding of α1β1 integrin receptor to collagen IV.26 In our study, targeted-gene knockdown of RAGE did not significantly reduce the apoptosis of podocytes cultured on AGE-modified collagen IV. Moreover, we observed a higher percentage of detachment when podocytes were cultured on AGE-modified collagen IV versus collagen IV. These findings implied that mechanisms other than the RAGE–AGE interaction were responsible for the apoptosis of podocytes cultured on the AGE-modified substratum; and detachment/anoikis might be the mechanism of apoptosis triggered by matrix-bound AGEs. Future studies on the effect of extracellular matrix modification by AGEs on podocyte attachment and apoptosis/anoikis are warranted.
The intracellular pathways activated by oxidative stress leading to cellular responses are well known. One of these pathways involves MAP kinases (p38MAPK, extracellular signal-regulated kinase1/2, JNK). The activation of p38MAPK has been reported in AGE-induced fibroblast apoptosis,20 and now in podocyte apoptosis. Akt is a cell survival factor. We observed that Akt phosphorylation was suppressed by AGE-BSA. Akt activation by phosphorylation is an antiapoptotic signal that has been demonstrated previously in podocytes.27 Growth factors or extracellular signals lead to the activation of phosphoinositide kinase, which results in Akt phosphorylation and activation leading to cell proliferation. In podocytes, both nephrin and CD2AP are able to stimulate Akt phosphorylation,27 possibly protect podocytes from programmed cell death and promote survival. Reduction of nephrin expression has been reported in human diabetic nephropathy.28 An area of interest for future studies would be determining if the phosphoinositide kinase-Akt pathway is defective in podocytes of diabetic patients and whether overexpression of nephrin can rescue podocytes from apoptosis.
Forkhead transcription factors (FOXO1, FOXO3, and FOXO4) are a family of proteins that function as sensors of signaling pathways and modulate apoptosis, cell cycle, and metabolism through regulation of gene expression.29 Activated-Akt phosphorylates and inactivates FOXO proteins.30 In the absence of Akt inhibition, FOXO is translocated to the nucleus leading to gene activation. FOXO transcription factors activate three major groups of genes: anti-oxidant genes, cell cycle arrest genes, and apoptotic genes.22 Our data suggest that AGEs activate FOXO4 leading to apoptosis in podocytes. Similarly, Alikhani et al.20 have demonstrated that another FOXO factor, FOXO1, is involved in AGE-induced apoptosis of fibroblasts. Consistent with prior reports on the activation of effector proteins of apoptosis (Bim) by FOXO3a,24 we observed enhanced expression of Bim after AGE-BSA exposure. The specificity of FOXO transcriptional activity is influenced by Sirtuin 1 mediated acetylation and JNK-dependent phosphorylation.31,32 In this study, we did not examine the expression of Sirtuin 1 with AGE exposure. Sirtuin 1 is a potential target for pharmacologic modulation of the FOXO-mediate activation of apoptosis that deserves further investigation. Stress-activated JNK modulates FOXO activity by phosphorylation leading to the expression of apoptosis-related genes.32 In our study, we were unable to show activation of JNK by AGEs in podocytes. However, AGE activation of p38MAPK could regulate FOXO transcriptional activity through a similar pathway, which will need further studies for confirmation.
In summary, we have shown that AGEs are one of the risk factors for podocyte depletion in diabetic nephropathy. AGE-induced podocyte apoptosis is mediated via the activation of RAGE, ROS production, and FOXO4, providing a novel mechanism of podocyte injury in diabetic nephropathy. Our data suggest that FOXO transcriptional activity is an important determinant of podocyte apoptosis. The role of FOXO4 in podocytes deserves further investigation in diabetic animal models. This study provides additional evidence that the reduction of ROS production, AGE burden, and RAGE expression are important therapeutic approaches to slowing the progression of diabetic nephropathy. These findings may provide us with not only the mechanism of podocyte injury but also new targets of therapy for diabetic nephropathy.
MATERIALS AND METHODS
AGE-preparations
Endotoxin-free AGE-BSA was prepared from endotoxin-free, lyophilized BSA (fraction IV; Sigma, St Louis, MO, USA), and glucose, as described previously.18 AGE-modified collagen IV and I were prepared by incubating endotoxin-free, sterile type IV, and I collagens (BD Bioscience, Bedford, MA, USA) with glucose similar to the preparation of AGE-BSA. An endotoxin-binding affinity column (Pierce, Rockford, IL, USA) was used to remove endotoxin from AGE-BSA, AGE-collagens, and native BSA. The absence of endotoxin was confirmed by a Limulus amebocyte lysate assay (BioWhittaker, Walkerville, MD, USA). Endotoxin levels in preparations were further determined by Associates of Cape Cod (Falmouth, MA, USA) and were found to be less than 2 endotoxin units/mg of protein. CML-BSA was a gift from Dr H Vlassara’s laboratory (Mount Sinai Medical Center, New York, NY, USA).
Cell culture
Conditionally immortalized murine podocytes were obtained from Dr Peter Mundel (Mount Sinai Medical Center, New York). Podocytes were cultivated in RPMI 1640 (RPMI) medium containing 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mm l-glutamine. To permit immortalized growth, the culture medium was supplemented with 10 U/ml of recombinant mouse γ-interferon to induce the expression of T antigen and cells were cultured at 33°C (permissive conditions). To induce differentiation, cells were cultured on type I or collagen IV at 37°C without γ-interferon for at least 10 days. We confirmed the degradation of the T antigen under nonpermissive condition (37°C) by Western blot analysis. Differentiated podocytes were used in the study. For apoptosis detection by terminal deoxynucleotidyl transferase-mediated dUTP nick end-labelling (TUNEL) staining, podocytes were cultured on microscope coverslips.
Determination of podocyte apoptosis, necrosis, and detachment
For all apoptosis detection experiments, differentiated podocytes were cultured in RPMI medium supplemented with 1% FBS for overnight along with BSA, AGE-BSA, or CML-BSA at the indicated concentrations before apoptosis detection. Apoptosis was assessed by TUNEL staining according to the manufacturer’s protocol (DeadEnd Colorimetric TUNEL System, Promega, Madison, WI, USA). Apoptotic cells with nuclei staining dark brown were counted by light microscopy. The percentage of apoptotic cells was quantified by counting five different fields at 400 x magnification with two independent investigators. Apoptosis was expressed as the ratio of apoptotic cells to the total number of cells in the field of view. Fluorescence-activated cell sorting (FACS) was used for the quantification of apoptotic and necrotic podocytes after Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) labeling as described in the manufacturer’s protocol (Annexin V-FITC Apoptosis Detection Kit I, BD Bioscience, San Jose, CA, USA). All FACS analyses for apoptosis detection were performed in triplicates.
Equal number of podocytes were cultivated on 6-cm tissue culture plates coated with either collagen IV or AGE-modified collagen IV in RPMI medium with 10% FBS for 4–6 h. Cells were washed with phosphate-buffered saline (PBS), then exposed to either BSA or AGE-BSA for overnight in RPMI medium with 1% FBS. Each experimental condition was performed in triplicate. After overnight treatment, cell culture medium was collected and saved. Cells were washed once with 1.5 ml of PBS. PBS used for washing was combined with the saved culture medium. The concentration of detached cells was determined using a hemocytometer. Adherent cells remaining on the plate were recovered by incubation with 1 × trypsin/ethylenediaminetetraacetic acid solution. Each sample was counted three times. The percentages of apoptosis and necrosis in the adherent-cell fraction were determined by FACS after Annexin V-FITC and PI labeling. The number of apoptotic and necrotic cells were calculated by multiplying the percentages of apoptosis and necrosis as determined by FACS to the total number of adherent cells. The total number of cells was defined as the number of detached cells plus the number of adherent cells.
Isolation of AGE-modified serum proteins using a LZ-linked Sepharose 4B column
Sera of patients with CKD stages 3 and 5, as defined by Kidney Disease Outcomes Initiative guidelines, were obtained and pooled as part of a previous study (J Uribarri, manuscript submitted). Patients were recruited from the Mount Sinai School of Medicine’s renal clinic with the approval of the Institutional Review Board for human investigation. Pool sera from healthy volunteers were used as the control. AGEs were fractionated from serum with the use of an LZ-linked matrix column as described by Mitsuhashi et al.33 The LZ-linked column was prepared by conjugating LZ, a known AGE-binding protein, to cyanogen bromide-activated Sepharose 4B beads according to the manufacturer’s instructions. A control column was prepared by conjugating BSA to Sepharose 4B beads following the same procedure. One-milliliter samples of sera were diluted 1:5 with PBS just before loading on LZ columns (bed volume, 2 ml). After loading the diluted serum samples and collecting pass-through fractions, LZ columns were extensively washed with PBS. Then the LZ-bound fractions (0.5 ml per fraction) were eluted with 0.1 n NaOH and neutralized immediately with HCl. AGE concentration of these aliquots was determined by enzyme-linked immunosorbent assay, as described previously.34
Western blotting
Podocytes were lysed with a buffer containing 1% NP40, a protease inhibitor cocktail and tyrosine and serine-threonine phosphorylation inhibitors. After determination of protein concentration, cell lysates were subjected to 8–12% sodium dodecyl sulfate-polyacry-lamide gel electrophoresis before transfer to polyvinylidine difluoride membranes. Immunoblottings were performed using the following specific antibodies from Cell Signaling Laboratory (Beverly, MA, USA): phospho-Akt (ser308), phospho-p38, phospho-extracellular signal-regulated kinase1/2, phosphatas and tensin homologue, phospho-FOXO4 (ser193), and total FOXO4. Antibodies against β-actin, Bim, and RAGE were purchased from Sigma, Chemicon (Temecula, CA, USA), and Affinity BioReagents (Golden, CO, USA), respectively.
Inhibition of p38MAPK
For specific inhibition of p38MAPK, podocytes were pretreated with 10 μm of SB203580 (Calbiochem, San Diego, CA, USA) for 2 h in RPMI medium with 1% FBS before overnight AGE-BSA (50 μg/ml) stimulation. Experiments were carried out in triplicates.
Quantitative one step real-time polymerase chain reaction
Ribonucleic acid from cultured podocytes was extracted using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s protocol. Qiagen OneStep RT-PCR Kit (Qiagen, Hilden, Germany) was used for quantitative one-step real-time polymerase chain reaction. The final reaction volume was 20 μl containing 500 ng of total RNA, 1 × QuantiTect SYBR Green RT-PCR Master Mix, 0.2 μl of QuantiTect RT Mix, and 1.0 μm each of forward (5′-AGGAGCGTGCAGAACTGAAT-3′) and reverse (5′-TTGGCAAGGTGGGGTTATAC-3′) primers for RAGE. The primer set used for the determination of FOXO4 mRNA level was purchased from Qiagen (QuantiTect Primers). RNA was reversed transcribed and directly amplified in the same glass capillary of a LightCycler™ system (Roche Diagnostics, Indianapolis, IN, USA). Cycler condition was reverse transcription for 20 min at 50°C, and then HotStarTaq DNA polymerase activation for 15 min at 95°C. This was followed by 45 sequential cycles of denaturation (15 s of 94°C), annealing (30 s of 55°C), and extension (30 s at 72°C). Online fluorescence of SYBR Green I was monitored by the LightCycler for the determination of crossing point. We used the 2−ΔΔCp method for the analysis of relative gene expression as described previously.35
Panomics TranSignal DNA/protein arrays
TranSignal transcription factor array profiles multiple transcription factor–DNA interactions simultaneously. Array profiling was carried out according to the protocols provided by the manufacturer. Briefly, nuclear proteins were extracted from cell lysates. The quality of nuclear proteins was verified using Western blotting for a cytoplasmic protein, Raf1 (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) and a nuclear protein, cAMP-responsive element-binding protein (Cell Signaling Laboratory). Nuclear proteins were incubated with transcription-factor-specific biotinylated DNA oligonucleotides. The protein–DNA complexes were purified and separated from free probes by spin column separation. After hybridization of the purified oligonucleotides to the array, positive spots were visualized by enhanced chemiluminescence. This assay system examines over 350 transcription factors. Experiments were performed in triplicates. Podocytes were exposed to AGE-BSA (50 μg/ml), CML-BSA (50 μg/ml), or BSA (50 μg/ml) for 2 h in RPMI medium with 1% FBS.
Determination of ROS production
After treatment with BSA or AGE-BSA for overnight in RPMI medium with 1% FBS, podocytes were washed twice with warm RPMI medium. Podocytes were incubated for 45 min at 37°C with 10 μmol/l of 2′,7′-dichlorofluorescein diacetate (Invitrogen) in RMPI medium. Cells were washed twice with ice-cold PBS, scraped, and lysed with PBS containing 0.1% Triton-100, and 0.5 mm ethylenediaminetetraacetic acid. Luminescence spectrometer Perkin-Elmer LS50B (Perkin-Elmer, Wellesley, MA, USA) was used for the measurement of emitted fluorescence at 530 nm with the excitation wavelength of 485 nm.
Inhibition of ROS production with NAC
NAC (Sigma) is a free radical scavenger. Differentiated podocytes were pretreated with 2 mm of NAC for 2 h, then incubated with AGE-BSA (50 μg/ml) in 1% FBS for overnight before labeling with Annexin IV-FITC and PI.
Immunofluorescence of FOXO4 in podocytes
Podocytes cultured on coverslips were exposed to either BSA or AGE-BSA at 50 μg/ml in RPMI with 1% FBS for 2 h and then fixed in 4% paraformaldehyde at room temperature for 15 min. After permeabilization in 1% Triton, cells were incubated with anti-FOXO4 polyclonal antibody (Cell Signaling Laboratory) at room temperature for 1 h. After washing, cells were incubated with fluorescence-labeled secondary antibodies from Invitrogen (Alexa Fluor 488 anti-rabbit immunoglobulin G or Alexa Fluor 568 anti-rabbit immunoglobulin G). Hoechst 33258 dye (Invitrogen) was used for nuclear staining. After mounting, slides were examined using a fluorescent microscope.
Target gene knockdown using gene-specific siRNAs
Podocytes were transiently transfected using the Amaxa Nucleofection technology (Amaxa Biosystems, Gaithersburg, MD, USA), which utilizes electroporation for gene transfer. We tested several protocols based on the manufacturer’s instructions. After optimization, we were able to achieve 80–90% transfection efficiency based on GFP expression using Amaxa’s Nucleofection kit and Program T-20 (Figure 3a). Transfection efficiency was determined by co-transfection of podocytes with a vector (pmaxGFP™) encoding the GFP and counting the percentage of GFP-positive cells. We used a technique combining the Dharmacon On TargetPlus SMARTpool siRNAs and Amaxa RNAi Nucleofection kit to successfully introduce siRNAs into podocytes. Two million podocytes grown to 70–80% confluence and 1.5 μg of Dharmacon On Target Plus SMARTpool siRNA were used for each Nucleofection reaction. Cell survival after transfection was about 50–60%. Transfected podocytes were cultivated for at least 72 h in RPMI medium with 10% FBS before treatment with BSA, AGE-BSA, or CML-BSA in RPMI medium with 1% FBS. miRIDIAN microRNA Mimic Negative Control (CL) sequence based on C. elegans miRNA from Dharmacon Corporation (Lafayette, CO, USA) was used as negative experimental controls in mammalian cells (5′-UCACAACCUCCUAGAAAGAGUAGA-3′). The specific siRNAs targeting FOXO4 and RAGE were purchased from Dharmacon Corporation (Dharmacon On Target Plus SMARTpool siRNAs).
Statistical analysis
The data were presented as mean±s.e.m. For Western blot analyses, all experiments were repeated at least three times. Statistically significant differences between the means were determined by the unpaired t-test or the Mann–Whitney U-test where appropriate. Significance was defined as a P<0.05.
Acknowledgments
We thank Dr Helen Vlassara’s laboratory for measurement of AGEs in patients’ sera. JH is supported by a K08 award (DK-65495).
References
- 1.USRDS: the United States Renal Data System. Am J Kidney Dis. 2003;43:1–230. [PubMed] [Google Scholar]
- 2.Ziyadeh FN. Mediators of diabetic renal disease: the case for TGF-beta as the major mediator. J Am Soc Nephrol. 2004;15:S55–S57. doi: 10.1097/01.asn.0000093460.24823.5b. [DOI] [PubMed] [Google Scholar]
- 3.Wendt TM, Tanji N, Guo J, et al. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am J Pathol. 2003;162:1123–1137. doi: 10.1016/S0002-9440(10)63909-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pagtalunan ME, Miller PL, Jumping-Eagle S, et al. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest. 1997;15:342–348. doi: 10.1172/JCI119163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drummond K, Mauer M. the early natural history of nephropathy in type 1 diabetes. II. Early renal structural changes in type 1 diabetes. Diabetes. 2002;51:1580–1587. doi: 10.2337/diabetes.51.5.1580. [DOI] [PubMed] [Google Scholar]
- 6.Susztak K, Raff AC, Schiff M, et al. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55:225–233. [PubMed] [Google Scholar]
- 7.Shankland SJ. The podocyte’s response to injury: role in proteinuria and glomerulosclerosis. Kidney Int. 2006;69:2131–2147. doi: 10.1038/sj.ki.5000410. [DOI] [PubMed] [Google Scholar]
- 8.Vlassara H, Striker LJ, Teichberg S, et al. Advanced glycation end products induce glomerular sclerosis and albuminuria in normal rats. Proc Natl Acad Sci USA. 1994;91:11704–11708. doi: 10.1073/pnas.91.24.11704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng F, He C, Cai W, et al. Prevention of diabetic nephropathy in mice by a diet low in glycoxidation products. Diabetes Metab Res Rev. 2002;18:224–237. doi: 10.1002/dmrr.283. [DOI] [PubMed] [Google Scholar]
- 10.Soulis T, Cooper ME, Vranes D, et al. Effects of aminoguanidine in preventing experimental diabetic nephropathy are related to duration of treatment. Kidney Int. 1996;50:627–634. doi: 10.1038/ki.1996.358. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto Y, Kato I, Doi T, et al. Development and prevention of advanced diabetic nephropathy in RAGE-overexpressing mice. J Clin Invest. 2001;108:261–268. doi: 10.1172/JCI11771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neeper M, Schmidt AM, Brett J, et al. Cloning and expression of RAGE: a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem. 1992;267:14998–15004. [PubMed] [Google Scholar]
- 13.Schmidt AM, Vianna M, Gerlach M, et al. Isolation and characterization of binding proteins for advanced glycosylation endproducts from lung tissue which are present on the endothelial cell surface. J Biol Chem. 1992;267:14987–14997. [PubMed] [Google Scholar]
- 14.Ehlermann P, Eggers K, Bierhaus A, et al. Increased proinflammatory endothelial response to S100A8/A9 after preactivation through advanced glycation end products. Cardiovasc Diabeto. 2006;5:6. doi: 10.1186/1475-2840-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan SD, Schmidt AM, Anderson GM, et al. Enhanced cellular oxidant stress by the interaction of advanced glycation endproducts with their receptors/binding proteins. J Biol Chem. 1994;269:9889–9897. [PubMed] [Google Scholar]
- 16.Lander HL, Tauras JM, Ogiste JS, et al. Activation of the receptor for advanced glycation endproducts triggers a MAP kinase pathway regulated by oxidant stress. J Biol Chem. 1997;272:17810–17814. doi: 10.1074/jbc.272.28.17810. [DOI] [PubMed] [Google Scholar]
- 17.Wautier MP, Chappey O, Corda S, et al. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab. 2001;280:E685–E694. doi: 10.1152/ajpendo.2001.280.5.E685. [DOI] [PubMed] [Google Scholar]
- 18.Cai W, He JC, Zhu L, et al. Advanced glycation end product (AGE) receptor 1 suppresses cell oxidant stress and activation signaling via EGF receptor. Proc Natl Acad Sci USA. 2006;103:13801–13806. doi: 10.1073/pnas.0600362103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drummond KN, Kramer MS, Suissa S, et al. International Diabetic Nephropathy Study Group. Effects of duration and age at onset of type 1 diabetes on preclinical manifestations of nephropathy. Diabetes. 2003;52:1818–1824. doi: 10.2337/diabetes.52.7.1818. [DOI] [PubMed] [Google Scholar]
- 20.Alikhani M, Maclellan C, Raptis M, et al. Advanced glycation endproducts induce apoptosis in fibroblasts through activation of ROS MAP kinases and FOXO1 transcription factor. Am J Physiol Cell Physiol. 2007;292:C850–C856. doi: 10.1152/ajpcell.00356.2006. [DOI] [PubMed] [Google Scholar]
- 21.Kikuchi S, Shinpo K, Moriwaka F, et al. Neurotoxicity of methylglyoxal and 3-deoxyglucosone on cultured cortical neurons: synergism between glycation and oxidative stress, possibly involved in neurodegenerative diseases. J Neurosci Res. 1999;57:280–289. doi: 10.1002/(SICI)1097-4547(19990715)57:2<280::AID-JNR14>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 22.Accili D, Arden K. FoxOs at the crossroads of cellular metabolism, differentiation, transformation. Cell. 2004;117:421–426. doi: 10.1016/s0092-8674(04)00452-0. [DOI] [PubMed] [Google Scholar]
- 23.Koshikawa M, Mukoyama M, Mori K, et al. Role of p38 mitogen-activated protein kinase activation in podocyte injury and proteinuria in experimental nephrotic syndrome. J Am Soc Nephrol. 2005;16:2690–2701. doi: 10.1681/ASN.2004121084. [DOI] [PubMed] [Google Scholar]
- 24.Dijkers PF, Birkenkamp KU, Lam EW, et al. FKHR-L1 can act as a critical effector of cell death induced by cytokine withdrawal: protein kinase B-enhanced cell survival through maintenance of mitochondrial integrity. J Cell Biol. 2002;156:531–542. doi: 10.1083/jcb.200108084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alikhani M, Alikhani Z, Boyd C, et al. Advanced glycation end products stimulate osteoblast apoptosis via the MAP kinase and cytosolic apoptotic pathways. Bone. 2007;40:345–353. doi: 10.1016/j.bone.2006.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pedchenko VK, Chetyrkin SV, Chuang P, et al. Mechanism of perturbation of integrin-mediated cell-matrix interactions by reactive carbonyl compounds and its implication for pathogenesis of diabetic nephropathy. Diabetes. 2005;54:2952–2960. doi: 10.2337/diabetes.54.10.2952. [DOI] [PubMed] [Google Scholar]
- 27.Huber TB, Hartleben B, Kim J, et al. Nephrin and CD2AP associate with phosphoinositide 3-OH kinase and stimulate Akt-dependent signaling. Mol Cell Biol. 2003;23:4917–4928. doi: 10.1128/MCB.23.14.4917-4928.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doublier S, Salvidio G, Lupia E, et al. Nephrin expression is reduced in human diabetic nephropathy: evidence for a distinct role for glycated albumin and angiotensin II. Diabetes. 2003;52:1023–1030. doi: 10.2337/diabetes.52.4.1023. [DOI] [PubMed] [Google Scholar]
- 29.Furukawa-Hibi Y, Kobayashi Y, Chen C, et al. FOXO transcription factors in cell-cycle regulation and the response to oxidative stress. Antiox Redox Signaling. 2005;7:752–760. doi: 10.1089/ars.2005.7.752. [DOI] [PubMed] [Google Scholar]
- 30.Kops GJ, Burgering BM. Forkhead transcription factors: new insights into protein kinase B (c-akt) signaling. J Mol Med. 1999;77:656–665. doi: 10.1007/s001099900050. [DOI] [PubMed] [Google Scholar]
- 31.Brunet A, Sweeney LB, Sturgill JF, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 32.Smith WW, Norton DD, Gorospe M, et al. Phosphorylation of p66Shc and forkhead proteins mediates A-beta toxicity. J Cell Biol. 2005;169:331–339. doi: 10.1083/jcb.200410041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitsuhashi T, Li YM, Fishbane S, et al. Depletion of reactive advanced glycation endproducts from diabetic uremic sera using a lysozyme-linked matrix. J Clin Invest. 1997;100:847–854. doi: 10.1172/JCI119600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Makita Z, Vlassar A, Cerami A, et al. Immunochemical detection of advanced glycation end products in vivo. J Biol Chem. 1992;267:5133–5138. [PubMed] [Google Scholar]
- 35.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]