

Abstract
Raver1 is a multifunctional protein that modulates both alternative splicing and focal adhesion assembly by binding to the nucleoplasmic splicing repressor polypyrimidine tract protein (PTB) or to the cytoskeletal proteins vinculin and α-actinin. The amino-terminal region of raver1 has three RNA recognition motif (RRM1, RRM2, and RRM3) domains, and RRM1 interacts with the vinculin tail (Vt) domain and vinculin mRNA. We previously determined the crystal structure of the raver1 RRM1–3 domains in complex with Vt at 2.75 Å resolution. Here, we report crystal structure of the unbound raver1 RRM1–3 domains at 2 Å resolution. The apo structure reveals that a bound sulfate ion disrupts an electrostatic interaction between the RRM1 and RRM2 domains, triggering a large relative domain movement of over 30°. Superposition with other RNA-bound RRM structures places the sulfate ion near the superposed RNA phosphate group suggesting that this is the raver1 RNA binding site. While several single and some tandem RRM domain structures have been described, to the best of our knowledge, this is the second report of a three-tandem RRM domain structure.
Keywords: focal adhesion, actin cytoskeleton, crystallography, RNP motif, RNA binding, vinculin
Introduction
Raver1 is a ubiquitously expressed 80 kDa (748 residues) heterogeneous nuclear ribonucleoprotein (RNP) that serves as a cofactor for polypyrimidine tract-binding protein (PTB), a repressor of alternative splicing.1–3 Raver1 harbors a nuclear localization signal, a nuclear export sequence (NES), a central leucine-rich region, and three amino-terminal RNA recognition motifs (RRMs).1 The ∼90-residue RRM domains are one of the most abundant protein domains in eukaryotes, and these motifs are necessary and sufficient to bind a wide range of RNA molecules with distinct specificities and affinities.4 The RNA binding affinity of RRM domains is increased to a sub-nanomolar range by combining tandem arrays of RRM domains5 where up to six copies of RRM domains on a single polypeptide chain have been reported.5 Further, while one RRM domain binds up to six nucleotides, back-to-back RRM domains can recognize a longer continuous nucleotide sequence. RRM domains have a topology of βαββαβ that comprises two conserved six (in RNP2) to eight (in RNP1) residue-long RNP motifs that reside in the center of the RRM four-stranded β-sheet.6
Raver1 shuttles between the cytoplasm and the nucleus and can accumulate in the perinucleolar compartment, a dynamic nuclear substructure that harbors PTB.7 However, in addition to modulating PTB-mediated RNA processing, raver1 also binds to the cytoskeletal proteins α-actinin, vinculin, and metavinculin (an alternatively spliced isoform of vinculin) at adhesion complexes, particularly in differentiated muscle tissue.1 Vinculin is a large cytoskeletal protein (117 kDa)8 that functions as a critical regulator of both adherens junctions and focal adhesions,9 where it provides essential links to the F-actin cytoskeleton10 and to a large number of other adhesion proteins.11–13 Vinculin harbors five-helix bundle domains.14,15 The amino-terminal seven-helix Vh1, Vh2, and Vh3 domains are similar to the α-catenin dimerization domain,16 where two four-helix bundle subdomains share one central long α-helix. Together with the four-helix Vt2 bundle domain they form the globular vinculin head (VH) domain, that connects to the five-helix bundle vinculin tail (Vt) domain via a ∼40-residue proline-rich linker region. In its inactive, closed-clamp conformation, the binding sites of vinculin for its partners are masked by an intramolecular hydrophobic interactions of Vh1 and Vt.17 Activation of vinculin requires severing of this interaction and this can be accomplished by the binding of amphipathic α-helix vinculin binding sites (VBSs) to the vinculin Vh1 domain, which severs the head–tail interface by helix bundle conversion.14,18–22
We recently solved the crystal structures of the Vt domain in complex with the RRM1–3 domains or the RRM1 domain of raver123,24 and showed that the raver1:Vt complex can bind to vinculin mRNA (nucleotides 3089–3100). These findings suggested a feed-forward mechanism for mRNA localization during focal adhesion assembly where activated vinculin at adhesion sites binds to raver1 and its mRNA cargo for localized translation of adhesion components. Here, we report the crystal structure of the unbound raver1 RRM1–3 domains at 2 Å resolution. Comparison of the vinculin-bound and apo RRM1–3 structures reveals a large movement of the RRM1 domain that involves Arg59 of RRM1 and Glu159 of RRM2. Further, two sulfate ions are bound to the apo RRM1 structure in a locale that superimpose with the RNA binding and VBSs. Finally, the constellation of the RRM domains in raver1 is distinct from that seen in other tandem RRM structures.
Results
The crystal structure of unbound raver1 RRM1–3 domains
To further define the function of raver1, we solved the crystal structure of apo raver1 RRM1–3 domains at 2 Å resolution [Fig. 1(A)]. As seen in our Vt-bound complex structure,23 the three RRM domains exhibit the canonical RRM fold, with a four-stranded β-sheet and two α-helices. An additional β-hairpin is seen between α-helix α2 and β-strand β4 in the RRM1 and RRM2 domains. Further, α-helices that are amino-terminal to the RRM1 domain (residues 39–58) and those that are carboxy-terminal to the RRM3 domain (residues 304–321) were essential to obtain crystals (J.H.L., personal communication), suggesting roles in restraining interdomain movements.
Figure 1.

Crystal structures of the apo raver1 RRM1–3 domains versus Vt:RRM1–3. (A) Cartoon drawing of the unbound raver1 RRM1–3 structure. The amino-terminal α-helix (residues 38–58) is shown in grey, the RRM1 domain (residues 59–130) in cyan, RRM2 (residues 131–208) in yellow, RRM3 (residues 222–297) in orange, and the carboxy-terminal α-helix (residues 298–316) in green. The three RRM domains, the C-terminus, and RRM1 β-strands β1–β4 and the β-hairpin are labeled 1–4 and “β”, respectively. Sulfate ions are shown in as a ball-and-stick representation. (B) Close-up view of the sulfate ion near the RRM1–RRM2 interface, which engages in extensive interactions with the RRM1 domain of raver1 (shown in cyan). RRM2 is shown in yellow. Raver1 residues binding to the sulfate ion and the ligand are shown as ball-and-stick representation and are labeled; interactions are indicated by yellow dashes; water molecules bound to the sulfate ions are shown as red spheres. Superposition of raver1 in its Vt-bound state is shown in grey and the RRM1 residue Arg59 and RRM2 residue Glu159 engaged in electrostatic interactions (without dashes for clarity) in the Vt-bound structure only are shown in a ball-and-stick representation. (C) The sulfate ion appears to mimic the RNA binding site. Superposition of the second RRM domain of the sxl-lethal protein (shown in grey) in complex with RNA (PDB entry 1b7f) onto the raver1 RRM1 domain (shown in cyan) places the sulfate ion (indicated by the arrow) in the raver1 structure close to the RNA phosphate group in the sxl-lethal protein:RNA complex. (D) Cα-trace superposition of the two RRM2–RRM3 domains of the two Vt:RRM1–3 complex heterodimers in the asymmetric unit (chain B of PDB entry 3h2u is shown in blue and chain D in magenta) onto our unbound raver1 structure (green). The double arrowhead pointing at each Glu120 position in the respective structures indicates the large RRM1 domain movement. Glu120, Arg121, and Tyr92 that are involved in binding to Vt are shown as a ball-and-stick representation and are labeled. Tyr92 adopts a different conformation (single arrows pointing at their respective hydroxyl groups) in the apo structure (labeled in red in the unbound structure); the C-terminus is also labeled. In the unbound RRM1–3 structure, these residues are close to a symmetry-related molecule and Glu120 is in electrostatic interactions with a Lys190 of a symmetry-related molecule (not shown). As is evident, the RRM2–RRM3 domains superimpose well, with root-mean-square deviation (r.m.s.d.) of 0.6 Å–0.7 Å for residues 129–316 for 1223 or 1242 atoms for chains D or B of the Vt:RRM1–3 structure, respectively. However, superposition of all three RRM domains results in an r.m.s.d. of 2.2 Å–2.4 Å. (E) Cα-trace superposition of the RRM1 domains (chain B of PDB entry 3h2u is shown in blue; chain D in magenta; our unbound RRM1-3 domains in green). Arrows point to residues 57 and 128 near the hinge region. (F) Superposition of the RRM2 domains of Prp24 (blue) and raver1 (yellow) shows the distinct orientations of the respective RRM1 (Prp24, magenta; raver1, cyan) and RRM3 (PrP24, grey; raver1, orange) domains. For clarity, the raver1 N- and C-terminal α-helices are not shown.
We identified two electron-dense features within the RRM1 domain of the unbound RRM1–3 domains structure that were modeled as sulfate ions, given the high concentration of lithium sulfate in our crystallization conditions. Among the two sulfate ions, the binding interaction for one was near the α1–β2 loop [Fig. 1(A)] and involves contributions from the side-chain amino groups of Arg121 located on β-strand β4 and the main-chain nitrogen of Gly68 located on the α1–β2 loop. Notably, Arg121 contributes to electrostatic interactions with vinculin residues Glu932 and Asp953 in the vinculin-bound state.23 Thus, this sulfate-binding site coincides with the vinculin binding site. Notably, a sulfate ion was also found in the RRM domain of human nuclear cyclophilin 33 (PDB entry 3lpy) at an equivalent position.
A second sulfate anion located near the RRM1–RRM2 interface is directly coordinated by RRM1 residues Arg59 and Lys60 located on the linker region that connects the N-terminal α-helix and β-strand β1 and by Lys85 on β-strand β2, by water-mediated interactions, via RRM1 residues Tyr86 on β-strand β2 and Thr99 on β-strand β3, and by RRM2 residue Glu159 on β-strand β2 [Fig. 1(B)]. By contrast, raver1 Arg59 in the Vt:RRM1–3 structure engages in electrostatic interactions with Glu159, which are disrupted by the sulfate ion.
Superposition of the raver1 RRM1 domain onto the second RRM domain of the sxl-lethal protein:RNA complex (PDB entry 1b7f) places our second sulfate ion within ∼5 Å, of the nearest phosphate of the RNA that is bound to sxl-lethal [Fig. 1(C)]. This suggests that this sulfate ion occupies a position corresponding to the RNA binding site of RRM1. Indeed, inspection of the electrostatic surface potential shows a positive surface area surrounding the sulfate ion (Supporting Information Fig. S1). Moreover, in addition to the linker region (residues 208–219) that connects the RRM2 and RRM3 domains, the RRM2 and RRM3 β-sheet surfaces usually involved in RNA binding are occupied by interactions with the N- and C-terminal α-helices23,24 and, accordingly, sulfate ions are not bound to the RRM2 or RRM3 domains [Fig. 1(A)].
A C-terminal hydrophobic α-helix interacts with the RRM2 and RRM3 domains
The individual RRM domains of the Vt-bound and apo raver1 structures are very similar, with r.m.s.d. of 0.67 Å (for 497 RRM1 atoms), about 0.5 Å (for 540 RRM2 atoms), or 0.6 Å (for ∼500 RRM3 atoms), respectively. However, when superimposing tandem RRM domains, the two RRM2–RRM3 domains in the Vt:raver1 asymmetric unit superimpose better onto each other (r.m.s.d. of 0.34 Å for 1301 atoms) [Fig. 1(D)] versus their superposition onto the apo RRM2–RRM3 structure (r.m.s.d. of 0.6 Å–0.7 Å for 1223 atoms). Indeed, residues 210–212 that connect RRM2 with RRM3 form a well-defined 310-helix in the apo structure while this region has a random coil structure in the Vt:RRM1–3 complex [Supporting Information Fig. S2(A)].23 Further, the C-terminal raver1 α-helix is about half a turn shorter in the apo structure, where it ends at Gln314 versus at Ala317 in its vinculin-bound state.
As seen in the Vt:RRM1–3 structure,23 the C-terminal hydrophobic α-helix (residues 304–316) is wedged into a hydrophobic cleft between the RRM2 and RRM3 domains [Fig. 1(A)]. The interdomain hydrophobic residues of the RRM2 and RRM3 domains and the C-terminal α-helix are strictly conserved among the raver1 protein family (human, mouse, rat, and bovine) and deletion of this α-helix rendered the protein insoluble (J.H.L., personal communication), suggesting that this α-helix is important for proper stabilization and/or folding of the RRM2 and RRM3 domains. Indeed, the C-terminal α-helix buries over 1170 Å2 of its solvent-accessible surface area on its interaction with the RRM2 and RRM3 domains.
A novel RRM1 domain orientation
The most pronounced difference between the RRM1–3 domains alone or in complex with Vt is the relative orientations of their respective RRM1 domains [Fig. 1(D)]. Indeed, analysis of the apo versus Vt-bound raver1 RRM1–3 structures by the DynDom server25 showed a 35° rotation of the RRM1 domain (residues 59–128) relative to the RRM2–RRM3 domain [residues 129–314; Fig. 1(D)]. The hinge region involves the Arg58-Arg59 and Leu127-Gln128-Pro129 psi and phi dihedral angles. In particular, the phi dihedral angle for the Arg58-Arg59 peptide bond differed by about 33° with a psi dihedral change of about 25° for Arg59 and corresponding consequences on RRM1–RRM2 interdomain interactions [Fig. 1(E)]. In the Vt:RRM1–3 complex, RRM2 residue Glu159 likely hinders the relative movement of RRM1 through its electrostatic interaction with Arg59 [Fig. 1(B)]. However, in the unbound RRM1–3 structure, the sulfate ion disrupts this interaction by directly binding to Arg59 and to Glu159 by water-mediated interactions. Thus, the phosphate group of RNA when bound to raver1 might cause similar RRM1–RRM2 interdomain movements. In contrast, as the vinculin binding interface resides over 20 Å distant from this hinge, vinculin binding does not appear to affect the orientation of the RRM1 domain [Fig. 1(D,E)].
The change in RRM1 orientation is accompanied by other local side-chain movements of vinculin–raver1 interface residues as seen for Tyr92 that binds to Vt residues Phe885, Pro886, Met899, and Arg935, where this residue is rotated by about 128° in the unbound raver1 structure [Fig. 1(D,E)]. Similarly, residues Tyr86 and Phe88 that are situated on β-strand β2 of the RRM1 domain are rotated by about 80° and 121°, respectively. Finally, the N-terminal α-helix engages in extensive interactions with the RRM2 domain [Fig. 1(A)] and might restrict the RRM1 domain flexibility. In support of this notion, recombinant RRM1–3 proteins lacking this amino-terminal α-helix would not crystallize (J.H.L., personal communication).
Comparison with the Prp24 three-tandem RRM domain structure
The only other three-tandem RRM structure reported is that of the splicing factor Prp24 at 2.7 Å resolution.26 The individual RRM domains of raver1 and Prp24 readily superimpose [Supporting Information Fig. S2(B,C)] even though superimposition of the RRM domains in our 2 Å raver1 structure is superior, with r.m.s.d. ranging from 1.2 Å to 2.3 Å for 113–381 atoms for the superposition of RRM1 onto RRM2 domains, of RRM1 onto RRM3, or of RRM2 onto RRM3 domains, respectively. However, the individual orientations of the three RRM domains of raver1 and Prp24 are distinct [Fig. 1(F)]. While the RRM2 domain in Prp24 forms extensive interdomain contacts with RRM1 and RRM3, they only bury a small solvent-accessible surface area (∼550 Å2), suggesting their intramolecular interactions might be dynamic.26 In contrast, the interdomain contacts of the raver1 RRM2 domain (residues 133–206) differ and the buried surface areas in the RRM1 (residues 60–127) versus RRM3 (residues 222–296) interfaces are about 400 Å2 and almost 1700 Å2, respectively.
Discussion
To our knowledge, this is only the second crystal structure of a protein having three tandem RRM domains, and in raver1 the constellation of three domains is distinct. Moreover, the β2–β3 loop in the raver1 RRM1 domain is shorter (by about three to five residues) compared to other RRM domain structures where it is involved in binding of RNA. In contrast, in the raver1 RRM1 domain this shorter loop is involved in vinculin binding suggesting that it is specific for protein–protein interactions.
In raver1, the RRM2 and RRM3 domains adopt virtually the same relative orientations in their vinculin-bound versus unbound states. Further, the structure of just the RRM2–RRM3 domains alone (Lee and Izard, unpublished data) shows the same conformation as seen for the vinculin-bound and apo RRM1–3 structures, even though the RRM2–RRM3 domains in all three structures have different crystal contacts. Thus, it seems that the RRM2–RRM3 domains are rigid with respect to each other probably due to the hydrophobic C-terminal α-helix that is wedged into a hydrophobic cleft between the RRM2 and RRM3 domains. In contrast, the RRM1 domain adopts distinct orientations in its vinculin-bound versus sulfate-bound states. Specifically, the electrostatic interactions between Arg58 and Gln128 seem to restrain the relative RRM1–RRM2 domain orientation given that it is the same for both molecules in the asymmetric unit in the Vt:RRM1–3 structure. Furthermore, the VBS of raver1 is located at a substantial distance from the RRM1–RRM2 hinge region, suggesting that vinculin binding does not affect the relative orientation of RRM1. Rather, the sulfate ion, an RNA phosphate mimic, engages in bifurcated interactions with Arg59 and water-mediated interactions with Glu159 to release the Arg59-Glu159 interaction seen in the sulfate-free raver1 structure. Finally, Glu159 of RRM2 participates in water-mediated binding to the sulfate ion and the adjacent Met181 likely has roles in orienting the base, similar to that of Met85 of human poly(A)-binding protein (PDB entry 1cvj), which suggests that the sulfate ion mimics the binding of RNA to raver1.
Materials and Methods
RRM1–3 crystallization and structure determination
The RRM1-3 domains (residues 39–321) were purified as described.23 The crystals of the RRM1–3 were grown at 20 °C by sitting-drop vapor diffusion from protein concentrated to 18.2 mg/mL against solution of 100 mM Tris–HCl buffer (pH 8.5), 24% polyethylene glycol 3350, and 200 mM lithium sulfate. Flash freezing was achieved by transferring the crystals directly to a reservoir solution containing 15% glycerol, and they were allowed to equilibrate for 2 min. Native X-ray diffraction data were collected at beamline 22ID of the Advanced Photon Source (SER-CAT) to 2 Å Bragg spacings and integrated and scaled using autoProc27 that uses XDS28 as the data-processing engine. The hexagonal raver1 RRM1–3 domain crystals belong to space group P65 with unit cell dimensions a = b = 104 Å, c = 50.4 Å, α = β = 90°, and γ = 120°. There is one RRM1–3 molecule in the asymmetric unit resulting in a solvent content of 51.1% (VM = 2.51 Å3/Da; Table I).
Table I.
X-ray Data Reduction and Crystallographic Refinement Statistics
| X-ray data reduction statistics | |
| Space group | P65 |
| Unit cell dimensions, a = b, c (Å) | 104, 50.4 |
| Resolution (last shell) | 90–1.99 Å (2.1–1.99 Å) |
| Total measurements | 142,114 |
| Number of unique reflections (last shell) | 21,054 (2749) |
| Wavelength (Å) | 1.0 |
| R mergea (last shell) | 0.034 (0.503) |
| I/σ(I) (last shell) | 29.4 (2.1) |
| Completeness (last shell) | 0.985 (0.9) |
| Redundancy (last shell) | 6.7 (4.1) |
| Crystallographic refinement statistics | |
| Resolution (last shell) | 20–1.99 Å (2.09–1.99 Å) |
| Number of reflections (working set) | 21,009 |
| Number of reflections (test set) | 1051 |
| R factorb (last shell) | 0.186 (0.222) |
| R free (last shell)c | 0.229 (0.24) |
| Number of amino acid residues | 279 |
| Number of protein atoms | 2190 |
| Number of solvent molecules | 235 |
| Number of sulfate atoms | 10 |
| Average B factor (Å2) | |
| Protein | 57.3 |
| Solvent | 57.1 |
| Sulfate | 82.2 |
| RMSD from ideal geometry | |
| Bond lengths (Å) | 0.01 |
| Bond angles (°) | 1.06 |
| Ramachandran plot statistics (%) | |
| Most favored | 93.8 |
| Additional allowed | 6.2 |
| Generously allowed | 0.0 |
| Disallowed | 0.0 |
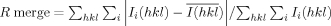
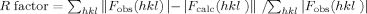 where 〈|Fcalc|〉 denotes the expectation of |Fcalc(hkl)| used in defining the likelihood refinement target.
where 〈|Fcalc|〉 denotes the expectation of |Fcalc(hkl)| used in defining the likelihood refinement target.
The free R factor is a cross-validation residual calculated by using about 5% reflections, which were randomly chosen and excluded from the refinement.
The RRM1–3 structure was determined by molecular replacement using the program MOLREP.29 Attempts to determine the structure using the Vt-bound RRM1–3 structure as a search model were unsuccessful. The refined RRM2–RRM3 domains of our Vt:RRM1–3 complex structure23 (PDB entry 3h2u) were used as the search model. Subsequently, more than 65% of the model was built automatically with ARP/wARP30 and REFMAC.31 The remaining model was then manually built into Fobs − Fcalc electron density maps using Coot32 in several iterative rounds. The RRM2–RRM3 model was fitted, which allowed the determination of the RRM1 structure by molecular replacement using the RRM1 model of the Vt:RRM1–3 structure23 (PDB entry 3h2u). Crystallographic refinement was performed using autoBUSTER33 applying translation, libration, and screw-motion to account for the anisotropic nature of the RRM3 domain and resulted in a final crystallographic R value of 0.19 (free R value = 0.22). The final refined model has an overall all-atom clash score of 3.93 as determined by MolProbity34 with a score of 1.63 and all residues were in favorable regions of the Ramachandran Plot. In the RRM1-3 structure, Cys239 and Cys251 are involved in disulfide bond formation and regions corresponding to amino acids residues 256–258 and 288–290 showed weak main chain electron density. The final crystallographic refinement statistics are shown in Table I.
Acknowledgments
We are indebted to our colleagues at Scripps Florida: John Cleveland for discussions and critical review of the manuscript, Zhen Wu and Philippe Bois for sequencing, and Philippe Bois for fruitful discussions. Finally, we are grateful to the staff at the Advanced Photon Source, SER-CAT, for their synchrotron support. This is publication no. 21181 from the Scripps Research Institute.
Glossary
Abbreviations:
- NES
nuclear export sequence
- NLS
nuclear localization signal
- PTB
polypyrimidine tract protein
- RNP
ribonucleoprotein
- RRM
RNA recognition motif
- TLS
translation, libration, and screw
- Vt
vinculin tail domain
Supplementary material
References
- 1.Huttelmaier S, Illenberger S, Grosheva I, Rudiger M, Singer RH, Jockusch BM. Raver1, a dual compartment protein, is a ligand for PTB/hnRNPI and microfilament attachment proteins. J Cell Biol. 2001;155:775–786. doi: 10.1083/jcb.200105044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gromak N, Rideau A, Southby J, Scadden AD, Gooding C, Huttelmaier S, Singer RH, Smith CW. The PTB interacting protein raver1 regulates alpha-tropomyosin alternative splicing. EMBO J. 2003;22:6356–6364. doi: 10.1093/emboj/cdg609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rideau AP, Gooding C, Simpson PJ, Monie TP, Lorenz M, Huttelmaier S, Singer RH, Matthews S, Curry S, Smith CW. A peptide motif in Raver1 mediates splicing repression by interaction with the PTB RRM2 domain. Nat Struct Mol Biol. 2006;13:839–848. doi: 10.1038/nsmb1137. [DOI] [PubMed] [Google Scholar]
- 4.Kenan DJ, Query CC, Keene JD. RNA recognition: towards identifying determinants of specificity. Trends Biochem Sci. 1991;16:214–220. doi: 10.1016/0968-0004(91)90088-d. [DOI] [PubMed] [Google Scholar]
- 5.Maris C, Dominguez C, Allain FH. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 2005;272:2118–2131. doi: 10.1111/j.1742-4658.2005.04653.x. [DOI] [PubMed] [Google Scholar]
- 6.Shamoo Y, Abdul-Manan N, Williams KR. Multiple RNA binding domains (RBDs) just don't add up. Nucleic Acids Res. 1995;23:725–728. doi: 10.1093/nar/23.5.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghetti A, Pinol-Roma S, Michael WM, Morandi C, Dreyfuss G. hnRNP I, the polypyrimidine tract-binding protedistinct nuclear localization and association with hnRNAs. Nucleic Acids Res. 1992;20:3671–3678. doi: 10.1093/nar/20.14.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Molony L, Burridge K. Molecular shape and self-association of vinculin and metavinculin. J Cell Biochem. 1985;29:31–36. doi: 10.1002/jcb.240290104. [DOI] [PubMed] [Google Scholar]
- 9.Geiger B, Tokuyasu KT, Dutton AH, Singer SJ. Vinculin, an intracellular protein localized at specialized sites where microfilament bundles terminate at cell membranes. Proc Natl Acad Sci USA. 1980;77:4127–4131. doi: 10.1073/pnas.77.7.4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilkins JA, Lin S. High-affinity interaction of vinculin with actin filaments in vitro. Cell. 1982;28:83–90. doi: 10.1016/0092-8674(82)90377-4. [DOI] [PubMed] [Google Scholar]
- 11.Jockusch BM, Isenberg G. Interaction of alpha-actinin and vinculin with actopposite effects on filament network formation. Proc Natl Acad Sci USA. 1981;78:3005–3009. doi: 10.1073/pnas.78.5.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burridge K, Mangeat P. An interaction between vinculin and talin. Nature. 1984;308:744–746. doi: 10.1038/308744a0. [DOI] [PubMed] [Google Scholar]
- 13.Jones P, Jackson P, Price GJ, Patel B, Ohanion V, Lear AL, Critchley DR. Identification of a talin binding site in the cytoskeletal protein vinculin. J Cell Biol. 1989;109:2917–2927. doi: 10.1083/jcb.109.6.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Izard T, Evans G, Borgon RA, Rush CL, Bricogne G, Bois PR. Vinculin activation by talin through helical bundle conversion. Nature. 2004;427:171–175. doi: 10.1038/nature02281. [DOI] [PubMed] [Google Scholar]
- 15.Rangarajan ES, Lee JH, Yogesha SD, Izard T. A helix replacement mechanism directs metavinculin functions. PLoS ONE. 2010;5:e10679. doi: 10.1371/journal.pone.0010679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pokutta S, Weis WI. Structure of the dimerization and beta-catenin-binding region of alpha-catenin. Mol Cell. 2000;5:533–543. doi: 10.1016/s1097-2765(00)80447-5. [DOI] [PubMed] [Google Scholar]
- 17.Johnson RP, Craig SW. An intramolecular association between the head and tail domains of vinculin modulates talin binding. J Biol Chem. 1994;269:12611–12619. [PubMed] [Google Scholar]
- 18.Izard T, Vonrhein C. Structural basis for amplifying vinculin activation by talin. J Biol Chem. 2004;279:27667–27678. doi: 10.1074/jbc.M403076200. [DOI] [PubMed] [Google Scholar]
- 19.Bois PR, Borgon RA, Vonrhein C, Izard T. Structural dynamics of alpha-actinin–vinculin interactions. Mol Cell Biol. 2005;25:6112–6122. doi: 10.1128/MCB.25.14.6112-6122.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 20.Bois PR, O'Hara BP, Nietlispach D, Kirkpatrick J, Izard T. The vinculin binding sites of talin and alpha-actinin are sufficient to activate vinculin. J Biol Chem. 2006;281:7228–7236. doi: 10.1074/jbc.M510397200. [DOI] [PubMed] [Google Scholar]
- 21.Izard T, Tran Van Nhieu G, Bois PR. Shigella applies molecular mimicry to subvert vinculin and invade host cells. J Cell Biol. 2006;175:465–475. doi: 10.1083/jcb.200605091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tran Van Nhieu G, Izard T. Vinculin binding in its closed conformation by a helix addition mechanism. EMBO J. 2007;26:4588–4596. doi: 10.1038/sj.emboj.7601863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JH, Rangarajan ES, Yogesha SD, Izard T. Raver1 interactions with vinculin and RNA suggest a feed-forward pathway in directing mRNA to focal adhesions. Structure. 2009;17:833–842. doi: 10.1016/j.str.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Madl T, Sattler M. Adhesion dance with raver. Structure. 2009;17:781–783. doi: 10.1016/j.str.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 25.Hayward S, Berendsen HJ. Systematic analysis of domain motions in proteins from conformational change: new results on citrate synthase and T4 lysozyme. Proteins. 1998;30:144–154. [PubMed] [Google Scholar]
- 26.Bae E, Reiter NJ, Bingman CA, Kwan SS, Lee D, Phillips GN, Jr, Butcher SE, Brow DA. Structure and interactions of the first three RNA recognition motifs of splicing factor prp24. J Mol Biol. 2007;367:1447–1458. doi: 10.1016/j.jmb.2007.01.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vonrhein C, Bricogne G. AutoPROC—a framework for automated data processing. Acta Crystallogr A. 2008;64:C78. [Google Scholar]
- 28.Kabsch W. XDS. Acta crystallographica. Section D, Biological crystallography. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 30.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 31.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 32.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 33.Smart OS, Brandl C, Flensburg P, Keller W, Paciorek C, Vonrhein C, Womack T, Bricogne G. Refinement with local structure similarity restraints (LSSR) enables exploitation of information from related structures and facilitates use of NCS. 2008. Abstr Annu Meet Am Crystallogr Assoc, Knoxville, TN abstract TP139.
- 34.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.