

Abstract
The growing practice of exploiting noninvasive fluorescence-based techniques to study G protein-coupled receptor pharmacology at the single cell and single molecule level demands the availability of high-quality fluorescent ligands. To this end, this study evaluated a new series of red-emitting ligands for the human β-adrenoceptor family. Upon the basis of the orthosteric ligands propranolol, alprenolol, and pindolol, the synthesized linker-modified congeners were coupled to the commercially available fluorophore BODIPY 630/650-X. This yielded high-affinity β-adrenoceptor fluorescent ligands for both the propranolol and alprenolol derivatives; however, the pindolol-based products displayed lower affinity. A fluorescent diethylene glycol linked propranolol derivative (18a) had the highest affinity (log KD of −9.53 and −8.46 as an antagonist of functional β2- and β1-mediated responses, respectively). Imaging studies with this compound further confirmed that it can be employed to selectively label the human β2-adrenoceptor in single living cells, with receptor-associated binding prevented by preincubation with the nonfluorescent β2-selective antagonist 3-(isopropylamino)-1-[(7-methyl-4-indanyl)oxy]butan-2-ol (ICI 118551) (J. Cardiovasc. Pharmacol.1983, 5, 430–437.)
Introduction
G-protein coupled receptors (GPCRs) remain a prime target for potential disease modulation, and therefore, research focused upon them persists as a significant component of the modern day drug discovery arena.(2) The β-adrenoceptors (β-AR) represent just one subset of this broad receptor family and are themselves further classified into three specific subtypes, β1-, β2-, and β3-AR.(3) For several decades these subtypes have been the molecular target for numerous drug discovery projects, and the drugs resulting from these endeavors have established themselves as key therapeutic agents in the treatment of numerous chronic cardiovascular and respiratory diseases.(4) More recently, their potential involvement in metastatic cancer progression has become apparent and rekindled further the therapeutic importance of this receptor class.5,6 Our current understanding of the molecular pharmacology of the β-AR has been derived primarily from the use of radioligand binding techniques for the elucidation of ligand–receptor interactions and the quantification of intracellular second messenger generation. As such, this type of experiment requires the use of large numbers of cells, and the data generated represents an average for the entire cell population. Fluorescent techniques, however, offer a wider spectrum of more detailed pharmacological investigations to be undertaken, especially the quantification and visualization of specific ligand–receptor complexes in intact cells.(7)
The recent discovery that GPCRs can regulate signaling pathways that are independent of G proteins has led to a realization that different signal transduction pathways can be selectively stimulated by agonists acting at a common cell surface receptor.8−11 GPCRs are not uniformly distributed at the cell surface but instead are organized within membrane compartments and microdomains,12,13 providing a mechanism by which intracellular signaling can be orchestrated in different areas of an individual cell and behave differently in specific cell types.(14) This new mechanistic information has focused attention on the need to develop fluorescent ligands to study the spatial and temporal aspects of ligand–receptor interactions.15,16 With their established clinical and therapeutic importance,(17) it is therefore not surprising that β-AR have been investigated within this area of pharmacological imaging.
To-date, however, only a handful of reports have been published detailing limited success in the synthesis of fluorescent β-AR ligands (Figure 1). The earliest studies detailed the use of fluorescent propranolol derivatives 9-aminoacridinylpropranolol (9-AAP) (1)18−22 and dansyl-propanolol (DAPN) (2)(23) to investigate receptor mapping and clustering. Following this, synthesis of an NBD–alprenolol derivative (3) was reported and utilized in further clustering studies.(24) However, it was later demonstrated that 3 and NBD–pindolol (4) were in fact both nonselective and noncompetitive.25,26 Additionally, two reports also detailed that during their use of 1 and 2 they were unable to distinguish between the binding of these ligands to cellular receptors and autofluorescent granules, concluding that the fluorescent output from these ligands was therefore of too low an intensity to be detected by fluorescence microscopy.27,28
Figure 1.

Previously reported fluorescent β-AR ligands.
A decade later, a significant advance was achieved through the synthesis of bordifluoropyrromethene- (BODIPY) (5) and fluorescein-labeled (6) β-AR ligands.(29) On the basis of the hydrophilic β-AR ligand 4-[3-[(1,1-dimethylethyl)amino]-2-hydroxypropoxy]-1,3-dihydro-2H-benzimidazol-2-one (CGP 12177),(30) it was demonstrated that the BODIPY conjugate (5) displayed similar binding characteristics when compared with the standard ligand. The fluorescein conjugate’s (6) receptor-specific signal was not sufficiently strong enough for subsequent pharmacological measurement and has indeed since been demonstrated to suffer from significant photobleaching during fluorescence microscopy experiments.(31) A variant of CGP 12177 (BODIPY TMR–CGP 12177) (7) has also been reported and shown to be a long-acting β2-AR partial agonist capable of labeling β2-AR in the plasma membrane of living cells.(32) More recently, the first description of a fluorescently labeled arterenol derivative (Alexa-NA) (8) and its use in fluorescence correlation spectroscopy (FCS) based studies to investigate β2-AR in living cells was published.(33) In addition, a europium-chelated fluorescent derivative of pindolol for use in time-resolved receptor–ligand binding assays has also been described, but its full structure was not revealed.(34)
Within this rapidly evolving arena of pharmacological imaging, we have previously reported the synthesis of red-shifted BODIPY-labeled fluorescent adenosine receptor agonists16,35,36,38 and antagonists37,38 and demonstrated their utility in measuring the diffusion of receptor species within defined membrane microdomains using the technique of fluorescence correlation spectroscopy (FCS).(39) Avoiding the issues of autofluorescence, highlighted as being severely problematic with many of the derivatives discussed above, was one of our primary objectives in designing pharmacologically useful fluorescent ligands. Clearly, fluorescent ligands that emit light in the red region of the electromagnetic spectrum meet this requirement and, in addition, often possess excellent photophysical properties suited for techniques such as LSCM and FCS. With the previously described fluorescent β-AR ligands either not meeting these requirements for fluorescence microscopy or, as in the case of BODIPY TMR–CGP 12177, no longer commercially available, we were keen to generate a new suite of red-shifted fluorescent β-AR ligands for use in LSCM- and FCS-based experiments. Three families of fluorescent ligands based on propranolol, alprenolol, and pindolol were synthesized, characterized, and pharmacologically assessed across the human forms of the β1-, β2-, and β3-ARs. Our choices of β-AR ligands were based primarily upon synthetic tractability, a desire to improve upon those ligands previously described, and to address three key questions surrounding ligand design: (i) Was the effect of linker length and composition similar to what we had observed for the corresponding adenosine ligands?(36) (ii) With structural extension of the pharmacophore to include both a linker and fluorophore, did the stereochemistry of the crucial carbinol center exert the same effect on affinity as that previously established for β-AR antagonists? (iii) Could the best fluorophore–linker combination from the initial “propranolol” library be transposed onto alternative orthosteric head-groups (alprenolol and pindolol) without deleterious effects on pharmacology.
Results and Discussion
Synthesis
We initially embarked upon the synthesis of a focused library of fluorophore-conjugated single enantiomer and racemic propranolol derivatives (Scheme 1). Commercially available 1-napthol (9) was therefore alkylated with either (2S)-glycidyl-3-nitrobenzenesulfonate or racemic epichlorohydrin as previously described to afford epoxides 10a and 10b, respectively.(40)
Scheme 1. Synthetic Route to BODIPY 630/650-X-Labeled Propranolol Derivatives.

Reagents and conditions: (i) for 10a, (2S)-glycidyl-3-nitrobenzenesulfonate, NaH, DMF, 56%; for 10b, epichlorohydrin, K2CO3, 2-butanone, Δ. (ii) 11a–c, DMF/H2O (9:1), 85 °C, 16 h, 35–70%. (iii) Pd/C, H2, MeOH, rt, 3 h, quantitative. (iv) BODIPY-X-630/650-OSu, DMF, rt, 4 h. (v) Benzyl 2-(2-(2-aminoethoxy)ethoxy)ethylcarbamate (15), DMF/H2O (9:1), 85 °C, 16 h, 35–67%.
Nucleophilic ring-opening of the epoxides was effected using a range of appropriately mono-Cbz-protected diamines (11a–c and 15) to afford carbinolamines 12a–f, 16a, and 16b. Subsequent hydrogenolysis of the Cbz protecting groups afforded the linker-modified congeners 13a–f, 17a, and 17b primed for chemoselective acylation of the terminal primary amine with commercially available 6-(((4,4-difluoro-5-(2-thienyl)-4-bora-3a,4a-diaza-s-indacene-3-yl)styryloxy)acetyl)aminohexanoic acid succinimidyl ester (BODIPY 630/650-X, SE; Invitrogen). Aminolysis of the fluorophore active ester proceeded smoothly and the fluorescently labeled ligands 14a–f, 18a, and 18b were isolated and purified using preparative thin-layer chromatography (PTLC); their purity was confirmed using RP-HPLC with photodiode array detection between 190 and 800 nm. Evaluation of these ligands highlighted that linker length exerted minimal overall influence on receptor affinity, and for expeditious reasons, the alprenolol and pindolol conjugates were therefore synthesized using only the ethylamino (short hydrocarbon) and ethoxyethoxyethylamino (long polyether) linker variants.
The S-enantiomer (23a and 26a) and racemic (23b and 26b) alprenolol-BODIPY fluorescent ligands were therefore synthesized using an identical strategy to that of the propranolol derivatives, but starting from commercially available 2-allylphenol and replacing the Cbz-protected diamines with their tert-butoxycarbonyl (Boc) equivalents (Scheme 2).
Scheme 2. Synthetic Route to BODIPY 630/650-X-Labeled Alprenolol Derivatives.

Reagents and conditions: (i) for 20a, (2S)-glycidyl-3-nitrobenzenesulfonate, NaH, DMF, 56%; for 20b, epichlorohydrin, K2CO3, 2-butanone, Δ, 54%. (ii) tert-Butyl 2-aminoethylcarbamate, DMF/H2O (9:1), 85 °C, 16 h, 35–70%. (iii) 2 M HCl in dioxane, 89–94%. (iv) BODIPY-X-630/650-OSu, DMF, rt, 4 h, 68–98%. (v) tert-Butyl 2-(2-(2-aminoethoxy)ethoxy)ethylcarbamate, DMF/H2O (9:1), 85 °C, 16 h, 77%.
Finally, having also established with these initial two series that the (S)-enantiomer exerted the expected shift in log KD values, we generated the final pindolol-based molecules in their inexpensive racemic form. Therefore, (±)-4-[(oxiran-2-yloxy)methyl]-1H-indole (28) was ring-opened using either 11a or 15 to afford the Cbz-protected carbinolamine congeners 29 and 32, respectively. Cbz hydrogenolysis and acylation of the relinquished amines with BODIPY 630/650-X, SE ultimately afforded the racemic fluorescent pindolol derivatives 31 and 33 (Scheme 3).
Scheme 3. Synthetic Route to BODIPY 630/650-X-Labeled Pindolol Derivatives.

Reagents and conditions: (i) epichlorohydrin, Cs2CO3, microwave (MW) 120 °C, 30 min, 97%. (ii) 11a, DIPEA, iPrOH/MeCN/H2O (7:2:1), MW 90 °C, 60 min, 48%. (iii) Pd/C, H2, MeOH, rt, 3 h, 88%. (iv) BODIPY-X-630/650-OSu, DMF, rt, 4 h. (v) Benzyl 2-(2-(2-aminoethoxy)ethoxy)ethylcarbamate (15), iPrOH/MeCN/H2O (7:2:1), MW 90 °C, 60 min, 40%.
Pharmacology
3H-CGP 12177 Whole Cell Binding Studies
Measurement of 3H-CGP 12177 binding to CHO cells expressing the human β1-, β2-, or β3-AR indicated that all compounds displayed the highest affinity for the human β2-AR (Table 1). The lowest affinity compounds were the pindolol derivatives 31 and 34. Increasing the linker chain length from C-2 to C-8 for the propranolol series caused a modest increase in affinity from C-2 (14a, 14d) to C4 (14b, 14e) but then a 10-fold reduction in affinity with a further doubling in linker chain length to C-8 (14c, 14f) (Table 1). It was notable that the C-8 derivative 14f did not completely displace the specific binding of 3H-CGP 12177 to the human β1-AR (maximal displacement of specific binding =65.9 ± 2.0%; Table 1; Figure 2a).
Table 1. Ligand-Binding Parameters for Fluorescent Antagonists Acting at the Human β-ARa.
| chirality | linkerb | β1 log KD | n | β2 log KD | n | β3 log KD | n | |
|---|---|---|---|---|---|---|---|---|
| propranolol | –8.22 ± 0.04 | 9 | –9.22 ± 0.03 | 10 | –6.67 ± 0.10 | 4 | ||
| 14a | S-enantiomer | C-2 | –7.44 ± 0.05 | 8 | –8.86 ± 0.03 | 8 | –7.35 ± 0.06 | 4 |
| 14b | S-enantiomer | C-4 | –7.48 ± 0.07 | 9 | –9.14 ± 0.06 | 9 | –6.91 ± 0.08 | 7 |
| 14c | S-enantiomer | C-8 | –6.73 ± 0.09c | 9 | –8.15 ± 0.06 | 9 | –7.17 ± 0.05 | 4 |
| 14d | racemate | C-2 | –7.05 ± 0.07 | 6 | –8.56 ± 0.04 | 6 | –7.03 ± 0.07 | 6 |
| 14e | racemate | C-4 | –7.25 ± 0.07 | 6 | –8.87 ± 0.04 | 6 | –6.98 ± 0.13 | 6 |
| 14f | racemate | C-8 | –6.72 ± 0.05d | 6 | –8.24 ± 0.05 | 6 | –7.11 ± 0.09 | 6 |
| 18a | S-enantiomer | PEG-8 | –7.76 ± 0.06 | 8 | –9.21 ± 0.06 | 8 | –7.09 ± 0.08 | 4 |
| 18b | racemate | PEG-8 | –7.14 ± 0.08 | 6 | –8.48 ± 0.06 | 6 | –6.53 ± 0.07 | 6 |
| alprenolol | –7.95 ± 0.03 | 6 | –9.30 ± 0.02 | 6 | –6.86 ± 0.08 | 6 | ||
| 23a | S-enantiomer | C-2 | –7.04 ± 0.05 | 7 | –8.54 ± 0.07 | 7 | –7.16 ± 0.09 | 7 |
| 23b | racemate | C-2 | –7.26 ± 0.07 | 6 | –8.63 ± 0.05 | 6 | –7.33 ± 0.12 | 6 |
| 26a | S-enantiomer | PEG-8 | –7.65 ± 0.07 | 6 | –9.03 ± 0.06 | 6 | –7.00 ± 0.06 | 5 |
| 26b | racemate | PEG-8 | –7.50 ± 0.05 | 12 | –8.91 ± 0.06 | 12 | –7.09 ± 0.04 | 10 |
| pindolol | –8.58 ± 0.04 | 7 | –9.27 ± 0.07 | 7 | –6.78 ± 0.10 | 7 | ||
| 31 | racemate | C2 | –6.06 ± 0.07 | 7 | –7.33 ± 0.09 | 7 | –6.48 ± 0.06 | 7 |
| 34 | racemate | PEG-8 | –7.01 ± 0.09 | 6 | –7.96 ± 0.10 | 6 | –6.42 ± 0.12 | 6 |
log KD values obtained from 3H-CGP 12177 binding in whole CHO cells expressing either the human β1-, β2-, or β3-AR. Values are mean ± SE from n separate experiments.
The linker composition describes the atoms between the amino group of the carbinolamine and the nearest amide nitrogen: C-2, ethyl; C-4, butyl, C-8, octyl; PEG-8, ethoxyethoxyethyl.
Apparent log KD value (85.3 ± 2.4% maximal inhibition of specific binding).
Apparent log KD value (65.9 ± 2.0% maximal inhibition of specific binding).
Figure 2.

Fluorescent ligand displacement of 3H-CGP 12177 specific binding at the human β-AR in whole cells. Inhibition of 3H-CGP 12177 specific binding to whole cells by compounds 18a, 18b, 14c, and 14f in (a) β1 cells, (b) β2 cells, and (c) β3 cells. Nonspecific binding was determined by (a) 10 μM CGP 20712A and (b and c) 10 μM ICI 118551.(1) The concentrations of 3H-CGP 12177 present in each case are (a and b) 1.3 nM and (c) 18 nM. Data points are mean ± SE of triplicate determinations. These single experiments are representative of (a and b) six and (c) four separate experiments.
This was seen to a lesser extent with the pure S-enantiomer 14c (85.3 ± 2.4% maximal inhibition; Figure 2a), suggesting that the R-enantiomer may be contributing an allosteric influence on the orthosteric binding site. It was also striking that 14f did not produce an effect to the same degree in cells expressing the human β2-AR (Figure 2b).
Substitution of a polyethylene glycol linker in place of the C-8 hydrocarbon chain in 14c provided the highest affinity ligand 18a at the human β2-AR (log KD = −9.21; Table 1). This compound also had lower but respectable affinities at the human β1-AR (log KD = −7.76) and β3-AR (log KD = −7.09). The equivalent molecule from the alprenolol series 26a also displayed high affinity for the β1-AR (log KD = −7.65) and β2-AR (log KD = −9.03; Figure 3). However, 34, from the pindolol series, was an order of magnitude less potent compared to its parent molecule (Table 1).
Figure 3.

Fluorescent ligand 26a displacement of 3H-CGP 12177 specific binding at the human β-adrenoceptors in whole cells. Inhibition of 3H-CGP 12177 specific binding to whole cells by compound 26a in (a) β1 and β2 cells and (b) β3 cells. Nonspecific binding was determined by (a) 10 μM CGP 20712A (β1) and (b) 10 μM ICI 118551 (β2 and β3). The concentrations of 3H-CGP 12177 present in each case are (a) 1.7 nM and (b) 6.4 nM. Data points are mean ± SE of triplicate determinations. These single experiments are representative of (a) six and (b) five separate experiments.
It was notable that the addition of fluorophore and linker did not markedly change the affinities of the parent compounds for the β3-AR. In the case of both propranolol and alprenolol, the C-2 linker derivatives had the highest affinity for this particular receptor (Table 1).
Functional Reporter Gene Studies
To gain some insight into the ability of these ligands to antagonize agonist-stimulated responses at each receptor, we evaluated their ability to attenuate functional activity in the CHO cells as they also expressed the target receptor and a six cyclic AMP response element (6 × CRE) reporter gene driving the expression of a human-secreted placental alkaline phosphatase reporter gene.41,42 Agonist responses were elicited by cimaterol (β1, site 1), salbutamol (β2), and fenoterol (β3). For the β1-AR site 2, agonist responses were elicited by CGP 12177. All of the propranolol derivatives were potent competitive antagonists of this response, with 18a once again displaying the highest affinity (Table 2; Figure 4).
Table 2. Fluorescent Ligand log KD Values from Antagonism of Agonist-Mediated Enhancement of CRE Reporter Gene Responses at the Human β-ARa.
| β1 (site 1) log KD | n | β1 (site 2) log KD | n | β2 log KD | n | β3 log KD | n | |
|---|---|---|---|---|---|---|---|---|
| propranolol | –8.74 ± 0.03 | 10 | –6.70 ± 0.10 | 21 | –9.62 ± 0.05 | 13 | –6.79 ± 0.02 | 6 |
| 14a | –8.08 ± 0.06 | 8 | –7.04 ± 0.09 | 9 | –9.37 ± 0.04 | 15 | –7.58 ± 0.07 | 13 |
| 14b | –7.87 ± 0.10 | 11 | –6.74 ± 0.09 | 7 | –9.56 ± 0.05 | 13 | –6.82 ± 0.09 | 6 |
| 14c | –7.28 ± 0.08 | 9 | –6.10 ± 0.10 | 13 | –8.76 ± 0.10 | 16 | –6.85 ± 0.05 | 7 |
| 14d | –7.12 ± 0.08 | 10 | –6.45 ± 0.10 | 10 | –8.59 ± 0.08 | 15 | –7.20 ± 0.09 | 12 |
| 14e | –7.51 ± 0.06 | 9 | –6.62 ± 0.08 | 8 | –9.13 ± 0.06 | 18 | –7.10 ± 0.05 | 10 |
| 14f | –6.66 ± 0.05 | 10 | >−6 | 5 | –8.60 ± 0.05 | 9 | –6.94 ± 0.09 | 12 |
| 18a | –8.46 ± 0.07 | 7 | –7.17 ± 0.06 | 7 | –9.53 ± 0.11 | 17 | –6.97 ± 0.08 | 6 |
| 18b | –7.38 ± 0.08 | 8 | –6.95 ± 0.10 | 5 | –9.36 ± 0.11 | 18 | –7.24 ± 0.09 | 9 |
| alprenolol | –8.37 ± 0.07 | 12 | –6.75 ± 0.12 | 7 | –9.53 ± 0.07 | 13 | –7.12 ± 0.08 | 12 |
| 23a | –7.33 ± 0.08 | 9 | –7.13 ± 0.11 | 9 | –8.74 ± 0.06 | 12 | –7.38 ± 0.05 | 8 |
| 23b | –7.36 ± 0.06 | 4 | –6.92 ± 0.09 | 4 | –8.80 ± 0.08 | 9 | –7.63 ± 0.19 | 9 |
| 26a | –8.12 ± 0.09 | 7 | –7.38 ± 0.08 | 3 | –9.37 ± 0.08 | 6 | –7.21 ± 0.07 | 7 |
| 26b | –7.76 ± 0.08 | 13 | –7.11 ± 0.11 | 11 | –9.21 ± 0.09 | 15 | –7.24 ± 0.10 | 17 |
| pindolol | –8.62 ± 0.07 | 9 | >−5 | 8 | –9.18 ± 0.04 | 6 | >−6 | 7 |
| 31 | –6.52 ± 0.06 | 4 | >−6 | 4 | –7.52 ± 0.05 | 8 | –6.75 ± 0.02 | 4 |
| 34 | –7.42 ± 0.05 | 8 | >−6 | 4 | –8.14 ± 0.07 | 12 | –6.54 ± 0.11 | 6 |
log KD values were obtained from inhibition of functional CRE-SPAP responses to cimaterol (β1, site 1), CGP 12177 (β1, site 2), salbutamol (β2), and fenoterol (β3) in CHO cells expressing either the human β1-, β2- or β3-AR. Values are the mean ± SE from n separate experiments.
Figure 4.

Fluorescent ligand 18a is a competitive antagonist at all three human β-AR as measured via a whole cell reporter gene assay. CRE-SPAP production in the absence and presence of 10, 100, and 1000 nM compound 18a in (a) β1 cells following stimulation by cimaterol, (b) β1 cells following stimulation by CGP 12177, (c) β2 cells following stimulation by salbutamol, and (d) β3 cells following stimulation by fenoterol. Bars represent basal CRE-SPAP production, that in response to 10 μM isoprenaline, and that in response to 10, 100, or 1000 nM compound 18a alone. Data points are mean ± SE of triplicate determinations. These single experiments are representative of (a) five, (b) seven, (c) 10, and (d) four separate experiments. The Schild slope for part (a) is 1.08.
The alprenolol and pindolol series showed partial agonist activity (Figure 5; Table 3) on all three β-AR subtypes. These observations are consistent with the agonist nature of the parent compounds on all three receptors (Table 3).41−43 Interestingly, the presence of the fluorophore/linker combination reduced the agonist efficacy of both alprenolol and pindolol at the β2-AR, particularly, but had a much smaller influence on agonist efficacy at the other two β-ARs (Table 3). This suggests that the presence of the BODIPY unit or the associated linker chemistry can modify agonist efficacy in a receptor subtype specific manner and implies that the design of ligands with receptor selectivity based on efficacy rather than ligand-binding affinity may be possible.(44) As a consequence of the partial agonist activity, the log KD values for alprenolol, pindolol, and their derivatives shown in Table 2 were calculated according to the partial agonist method of Stephenson.(45)
Figure 5.

Partial agonism effects of compound 26a are competitively antagonized at all three human β-AR. CRE-SPAP production in the absence and presence of 10, 100, and 1000 nM compound 26a in (a) β1 cells following stimulation by cimaterol, (b) β1 cells following stimulation by CGP 12177, (c) β2 cells following stimulation by salbutamol, and (d) β3 cells following stimulation by fenoterol. Bars represent basal CRE-SPAP production, that in response to 10 μM isoprenaline, and that in response to 10, 100, or 1000 nM compound 26a alone. Data points are mean ± SE of triplicate determinations. These single experiments are representative of (a) five, (b) eight, (c) four, and (d) five separate experiments.
Table 3. Partial Agonist Actions of Alprenolol and Pindolola Derivatives at the Three Human β-ARb.
| β1 %c | n | β2 %c | n | β3 %c | n | |
|---|---|---|---|---|---|---|
| alprenolol | 46.4 ± 3.1 | 13 | 52.8 ± 1.8 | 7 | 62.4 ± 3.2 | 12 |
| 23a | 40.8 ± 2.6 | 10 | 12.2 ± 1.4 | 4 | 56.6 ± 3.4 | 9 |
| 23b | 33.1 ± 2.5 | 4 | 11.7 ± 2.3 | 3 | 60.6 ± 5.0 | 5 |
| 26a | 28.9 ± 2.0 | 12 | 26.9 ± 2.1 | 5 | 63.5 ± 3.6 | 5 |
| 26b | 30.9 ± 1.8 | 14 | 22.9 ± 2.3 | 6 | 49.6 ± 2.3 | 13 |
| pindolol | 64.3 ± 2.6 | 8 | 52.3 ± 1.9 | 3 | 85.2 ± 3.2 | 7 |
| 31 | 31.1 ± 2.0 | 4 | 10.8 ± 1.0 | 4 | 49.4 ± 4.9 | 4 |
| 34 | 20.8 ± 1.8 | 8 | 11.0 ± 1.5 | 4 | 53.2 ± 3.5 | 8 |
The propranolol derivatives did not stimulate significant agonist responses.
Values are mean ± SE from n separate experiments.
Agonist action is expressed as a percentage of the CRE-SPAP responses to 10 μM isoprenaline in each experiment. As the top of the concentration response curve was not achieved by many fluorescent ligands at the maximum possible experimental concentration of 1 μM, the response to alprenolol, pindolol, and their derivatives at 1 μM is given.
Previous work on the human β1-AR has shown that this receptor can exist in two conformations that can both elicit functional responses. The first site has high affinity for the majority of β-antagonists and is the site at which isoprenaline and other catecholamines elicit their agonist responses. The second site can be activated by high concentrations of CGP 12177 and is generally relatively resistant to antagonism by standard β-blockers.(46) In order to evaluate the ability of fluorescent ligands to antagonize functional responses via these two sites, we used cimaterol as an agonist of site 1 and CGP 12177 as an agonist of site 2.47,10 All fluorescent ligands were effective antagonists of site 1 of the β1-AR with the PEG-8 derivatives 18a (Table 2), 26a, and 34 being the most potent. The inhibition of agonist responses to CGP 12177 provided the first indication of the affinity of these fluorescent ligands for site 2 of the β1-AR. All compounds tested had a lower affinity for site 2 than for site 1 (Table 2). However, it was striking that linker length and the presence of the fluorophore did not markedly change the affinity of the parent ligand for site 2 (Table 2). As noted from binding studies, this was also true of the β3-AR, and the functional studies confirmed the higher affinity of C2-linked propranolol and alprenolol derivatives for this receptor (Table 2).
Confocal Imaging
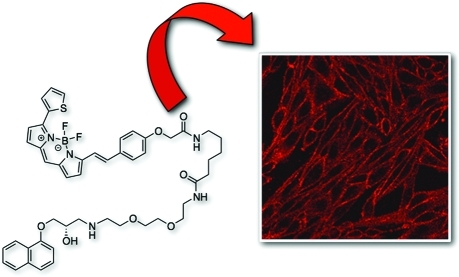
An essential requirement of a fluorescent ligand for any GPCR is that its binding to cell surface receptors can be visualized and that this binding displays the pharmacology of the target receptor. We have previously shown from our work on adenosine A1 and A3 receptors that the BODIPY 630/650 fluorophore is heavily quenched in aqueous environments but markedly increases its quantum yield when bound to cell surface receptors because of its location in a more lipid environment.(38) This property has proved highly beneficial in studying ligand-binding kinetics at the single cell level.(15) Figure 6 shows the binding of 3 nM 18a to CHO cells expressing the β2-AR. Bright labeling of cell surface β2-AR can be detected even in the continued presence of 18a.
Figure 6.

Confocal visualization of the effect of ICI 118551 or CGP 20712A on the binding of the BODIPY-630/650 derivative of propranolol 18a to CHO K1 cells expressing the β2-AR. Antagonists were incubated with cells for 30 min prior to addition of 3nM 18a for 10 min. Imaging of cells was undertaken in the continued presence of fluorescent ligand. The lower right-hand panel shows the binding of 18a to control CHO-K1 cells.
Quenching of the ligand present in the extracellular fluid is evident from the lack of fluorescence between the individual cells. Cell surface binding of 18a can be markedly inhibited by increasing concentrations (1–100 nM) of the β2-selective antagonist ICI 118551, consistent with its high affinity for the β2-AR. In contrast, the selective β1-AR antagonist 1-[2-((3-carbamoyl-4-hydroxy)phenoxy)ethylamino]-3-[4-(1-methyl-4-trifluoromethyl-2-imidazolyl)phenoxy]-2-propanol (CGP 20712A)(48) only began to displace at concentrations above 10 μM, which is consistent with its low affinity for this receptor.(49) Finally, 3nM 18a produced only a low level of binding to the cell surface of control CHO cells not expressing the β2-AR (Figure 6). This low level of binding was entirely consistent with the nonspecific binding observed in receptor-expressing cells incubated with 100 nM ICI 118551 (Figure 6).
Conclusions
A key feature of the current state of research into GPCRs is a growing awareness of the need to investigate their cellular location and involvement in protein–protein interactions. This requires the development of methodologies to investigate the allosteric regulation of the ligand–receptor binding by small molecules or associated signaling proteins. We have shown previously that fluorescent ligands can be used to provide insights into the membrane organization of GPCRs using fluorescence correlation spectroscopy37,16 and also to reveal novel allosteric interactions using kinetic analysis of ligand association and dissociation in single living cells.15,50 A key prerequisite for these approaches is the design and synthesis of novel fluorescent ligands with the correct pharmacological and biophysical properties. In the present study, we have designed, synthesized, and characterized 14 new red-emitting fluorescent ligands (14a–f, 18a,b, 23a,b, 26a,b, 31, and 34) for the human β-AR. Using alkyl- or polyether-based linker extension from the nitrogen atom of the conserved aryloxypropanolamine portion of propranolol, alprenolol, and pindolol generated a range of fully deprotected congeners, functionally equipped to undergo a high yielding chemoselective acylation with the commercially available fluorochrome BODIPY 630/650-X, SE (Molecular Probes). No protecting group manipulations were required following fluorophore attachment. This new range of fluorescent β-blockers displayed high affinity for the β2-AR, with some (18a and 26a) also possessing moderate affinity for the β1-AR. An evaluation of the pharmacology of the new fluorescent derivatives was undertaken. Compound 18a, in particular, displayed a log KD of −9.53 as an antagonist of functional responses mediated by the human β2-AR and a log KD of −8.46 as an antagonist of the high-affinity catecholamine site of the β1-AR.
Critically, we were further able to utilize these molecules to visualize ligand–receptor interactions using confocal microscopy. Confirmation of selective fluorescent labeling of, for example, the human β2-AR in single living cells was achieved by incubating CHO-β2 cells expressing the human β2-AR with a 3 nM concentration of 18a. This resulted in clear labeling of the cell membrane, with the receptor specific proportion of this binding blocked by preincubation with the nonfluorescent β2-selective antagonist ICI 118551. This fluorescent propranolol derivative should therefore be invaluable in future mechanistic studies aimed at unraveling the intricacies of cell signaling via this prototypical GPCR.
Experimental Section
General Chemistry Methods
Chemicals and solvents (HPLC grade) were purchased from the standard suppliers and were used without further purification. BODIPY fluorophores were obtained from Molecular Probes, Cambridge Bioscience, Cambridge, UK. Merck Kieselgel 60 (230–400 mesh) for flash chromatography was supplied by Merck KgaA (Darmstadt, Germany), and deuterated solvents were purchased from Goss International Limited. Unless otherwise stated, reactions were carried out at ambient temperature. Reactions were monitored by thin layer chromatography on commercially available precoated aluminum backed plates (Merck Kieselgel 60 F254). Visualization was by examination under UV light (254 and 366 nm). General staining employed KMnO4. A solution of ninhydrin (EtOH) was used for the visualization of primary amines. All organic extracts collected after aqueous workup procedures were dried over anhydrous magnesium sulfate, filtered under gravity, and evaporated to dryness. Organic solvents were evaporated in vacuo at a temperature of <35 °C (water bath). Purification using preparative layer chromatography (PLC) was carried out using Fluka silica gel GF254 on glass plates (200 mm × 200 mm × 1 mm).
Melting points were recorded on a Gallenkamp 3A 3790 apparatus and are uncorrected. FT-infrared spectra were recorded as thin films or KBr discs in the range of 4000–600 cm–1 using an Avatar 360 Nicolet FT-IR spectrophotometer. Optical rotation was measured on a Bellingham-Stanley ADP220 polarimeter. Mass spectra and HRMS TOF-ES mass spectra were recorded on a Waters 2795 Separation Module/Micromass LCT platform. Proton nuclear magnetic resonance spectra were recorded on a Bruker-AV 400 (400 MHz) spectrometer. Carbon nuclear magnetic resonance spectra were recorded at 100.6 MHz. Unless otherwise stated, spectra were recorded in CDCl3. Chemical shifts (δ) are recorded in ppm with reference to the chemical shift of the deuterated solvent/an internal tetramethylsilane standard. Coupling constants (J) are recorded in hertz and the signal multiplicities described by s, singlet; d, doublet; t, triplet; q, quartet; br, broad; m, multiplet; dd, doublet of doublets. Spectra were assigned using appropriate COSY, DEPT, and HMQC sequences. Analytical reverse-phase high-performance liquid chromatography (RP-HPLC) was performed on a Waters Millennium 995 LC system using columns, gradients, and flow rates as described. The eluent was monitored using photodiode array detection. Mobile phases were solvent A, water; solvent B, acetonitrile (both containing 0.06% v/v trifluoroacetic acid) degassed by helium bubble and sonication, respectively. The purities of all compounds tested in biological systems were therefore assessed as being ∼95%. Monoprotected diamines [benzyl 2-aminoethylcarbamate, tert-butyl 2-aminoethylcarbamate, tert-butyl 2-(2-(2-aminoethoxy)ethoxy)ethylcarbamate] were purchased (Sigma-Aldrich) or were synthesized [benzyl 4-aminobutylcarbamate, benzyl 8-aminooctylcarbamate, benzyl 2-(2-(2-aminoethoxy)ethoxy)ethylcarbamate] as previously described.36,51
(S)-2-((Naphthalen-1-yloxy)methyl)oxirane (10a)
A portion of 60% NaH in mineral oil (610 mg, equivalent to 366 mg of NaH, 15.26 mmol, 1.1 equiv) was dispersed in anhydrous DMF (2 mL) under an atmosphere of nitrogen at room temperature. To this vigorously stirred suspension was added a solution of 1-naphthol (2.00 g, 13.87 mmol) in anhydrous DMF (8 mL). The resulting pale green suspension was stirred at room temperature for 30 min. A solution of (S)-glycidyl nosylate (3.632 g, 14.01 mmol, 1.01 equiv) in anhydrous DMF (5 mL) was added dropwise to the reaction mixture. Once addition was complete, the mixture was heated at 60 °C overnight before cooling and quenching with aqueous saturated NH4Cl. The mixture was concentrated to dryness under reduced pressure and the resulting residue dispersed in water (100 mL). The aqueous slurry was extracted with Et2O (3 × 50 mL), and the combined organic extracts were washed with aqueous 1 M NaOH (50 mL) and brine (50 mL). After drying over anhydrous MgSO4, the organic extracts were concentrated under reduced pressure. The crude product was further purified by flash column chromatography (eluent EtOAc/petroleum ether 40–60 0:100 to 30:70 over 10 column volumes) to afford a colorless oil (2.44 g, 88%). [α]D26 = +16.7o (c 1.65, CHCl3); lit.(52) [α]D = +17.3o (c 2.0, CHCl3). 1H NMR: δ 2.86 (1H, dd, J = 4.9/2.7 Hz), 2.98 (1H, dd, J = 4.8/4.2 Hz), 3.48–3.54 (1H, m), 4.16 (1H, dd, J = 11.0/5.6 Hz), 4.41 (1H, dd, J = 11.0/3.2 Hz), 6.82 (1H, d, J = 7.0 Hz), 7.37 (1H, dd, J = 7.9 Hz), 7.43–7.54 (3H, m), 7.76–7.85 (1H, m), 8.27–8.35 (m, 1H). 13C NMR: δ 44.91, 50.39, 69.10, 105.12, 121.00, 122.16, 125.47, 125.73, 125.84, 126.65, 127.59, 134.66, 154.38. HRMS (TOF ES+): calcd for C13H13O2 201.0910, found 201.0904 (MH+).
(S)-2-((2-Allylphenoxy)methyl)oxirane (20a)
A portion of 60% NaH in mineral oil (656 mg, equivalent to 394 mg NaH, 16.40 mmol, 1.1 equiv) was dispersed in anhydrous DMF (2 mL) under an atmosphere of nitrogen at room temperature. To this vigorously stirred suspension was added a solution of 2-allylphenol (2.00 g, 14.91 mmol) in anhydrous DMF (8 mL). The resulting pale green suspension was stirred at room temperature for 30 min. A solution of (S)-glycidyl nosylate (3.903 g, 15.05 mmol, 1.01 equiv) in anhydrous DMF (5 mL) was added dropwise to the reaction mixture. Once addition was complete, the mixture was heated at 60 °C overnight before cooling and quenching with aqueous saturated NH4Cl. The mixture was concentrated to dryness under reduced pressure and the resulting residue dispersed in water (100 mL). The aqueous slurry was extracted with Et2O (3 × 50 mL), and the combined organic extracts were washed with aqueous 1 M NaOH (50 mL) and brine (50 mL). After drying over anhydrous MgSO4, the organic extracts were concentrated under reduced pressure. The crude product was further purified by flash column chromatography (eluent EtOAc/petroleum-ether 40–60 0:100 to 30:70 over 10 column volumes) to afford a clear yellow oil (2.97 g, quantitative). [α]D21 = +8.97° (c 4.18, DCM). 1H NMR: δ 2.78 (1H, dd, J = 5.0/2.7 Hz), 2.90 (1H, dd, J = 4.9/4.2 Hz), 3.33–3.39 (1H, m), 3.41 (2H, br d, J = 6.6 Hz), 3.99 (1H, m, J = 11.0/5.4 Hz), 4.23 (1H, dd, J = 11.0/3.1 Hz), 5.00–5.11 (2H, m), 5.93–6.06 (1H, m), 6.84 (1H, d, J = 8.2 Hz), 6.92 (1H, ddd, J = 7.4/7.4/1.1 Hz), 7.10–7.23 (2H, m). 13C NMR: δ 34.50, 44.80, 50.44, 68.88, 111.73, 115.62, 121.31, 127.46, 129.19, 130.11, 137.03, 156.28. HRMS (TOF ES+): calcd for C12H15O2 191.1067, found 191.1060 (MH+).
(±)-4-(Oxiran-2-ylmethoxy)-1H-indole (28)
4-Hydroxy-1H-indole (100 mg, 0.75 mmol) and Cs2CO3 (257 mg, 0.79 mmol, 1.05 equiv) were placed in a 10 mL MW vial, prior to the addition of (±)-epichlorohydrin (3.0 mL). The mixture was heated in the MW reactor at 120 °C (dynamic program, max. pressure 250 psi, max. power 300 W) for 30 min. The reaction mixture was diluted with water (20 mL) before extracting with DCM (3 × 10 mL). The combined organic extracts were dried over anhydrous Na2SO4 before concentration under reduced pressure. The crude product was further purified by flash column chromatography (eluent EtOAc/petroleum ether 40–60 0:100 to 30:70 over 16 column volumes), to afford a pale blue solid (0.138 g, 97%). Mp: 56–58 °C. 1H NMR: δ 2.83 (1H, dd, J = 4.9/2.7 Hz), 2.94 (1H, dd, J = 4.9/4.2 Hz), 3.44–3.52 (1H, m), 4.14 (1H, dd, J = 11.2/5.6 Hz), 4.38 (1H, dd, J = 11.2/3.2 Hz), 6.54 (1H, d, J = 7.5 Hz), 6.68–6.75 (1H, m), 7.03 (1H, d, J = 8.2 Hz), 7.08 (1H, dd, J = 2.8 Hz), 7.12 (1H, dd, J = 7.9 Hz), 8.30 (1H, br s). 13C NMR: δ 44.97, 50.48, 68.90, 99.90, 100.85, 105.11, 118.87, 122.63, 122.95, 137.50, 152.28. HRMS (TOF ES+): calcd for C11H12NO2 190.0863, found 190.0856 (MH+).
(±)-Benzyl 4-(2-Hydroxy-3-(naphthalen-1-yloxy)propylamino)butylcarbamate (12e)
A solution of 2-(napthalen-1-yloxymethyl)oxirane (10b) (0.20 g, 1 mmol) and benzyl 4-aminobutylcarbamate (11b) (0.56 g, 2.5 mmol) in DMF/H2O 9:1 (5 mL) was heated at 85 °C for 16 h. The solvent was evaporated under reduced pressure and the crude material was purified by flash column chromatography on silica employing a gradient from 0 to 10% MeOH in CH2Cl2 as eluent to afford the title compound as a yellow solid (0.245 g, 58%). Mp 90–91 °C. 1H NMR: 1.49–1.61 (4H, m), 2.45–2.74 (4H, m), 2.86 (1H, dd, J = 12.1, 7.5), 2.95 (1H, dd, J = 12.1, 3.7), 3.12–3.25 (2H, m), 4.09–4.25 (3H, m), 5.09 (2H, s), 5.19 (1H, br s), 6.81 (1H, d, J = 7.5), 7.27–7.40 (6H, m), 7.42–7.52 (3H, m), 7.76–7.84 (1H, m). 13C NMR: 27.5, 27.9, 41.0, 49.5, 52.2, 66.7, 68.5, 70.8, 105.1, 120.8, 121.9, 125.4, 125.7, 126.0, 126.6, 127.7, 128.2, 128.2, 128.6 (3 × CH), 134.6, 136.8, 154.4, 156.6. IR (NaCl, film): 2933, 1701, 1269, 1242, 1102, 772 cm-1. MS m/z (TOF ES+): 423 (MH+, 100%). HRMS (TOF ES+): calcd for C25H30N2O4 423.2284, found 423.2247.
(S)-tert-Butyl 2-(3-(2-Allylphenoxy)-2-hydroxypropylamino)ethylcarbamate (21a)
A solution of (S)-2-((2-allylphenoxy)methyl)oxirane (20a) (0.105 g, 0.553 mmol) and tert-butyl 2-aminoethylcarbamate (0.221 g, 1.38 mmol) in DMF/H2O 9:1 (3 mL) was heated at 85 °C for 16 h. The solvent was evaporated under reduced pressure and the target compound purified by flash column chromatography on silica (10% MeOH in CH2Cl2) to afford the title compound as a colorless oil (0.102 g, 53%). [α]D26 = +3.1° (c 0.8, 15:85 MeOH/CH2Cl2). 1H NMR: 1.44 (9H, s), 2.69–2.85 (2H, br s), 2.79 (2H, t, J = 5.8), 2.82 (1H, dd, J = 12.3, 7.7), 2.89 (1H, dd, J = 12.3, 4.1), 3.20–3.31 (2H, m), 3.38 (2H, d, J = 6.1), 3.98 (2H, d, J = 5.4), 4.06–4.14 (1H, m), 4.96–5.08 (3H, m), 5.97 (1H, ddt, J = 16.7, 10.4, 6.4), 6.83 (1H, d, J = 7.7), 6.91 (1H, t, J = 7.7), 7.13 (1H, dd, J = 7.7, 1.5), 7.17 (1H, td, J = 7.7, 1.5). 13C NMR: 28.5 (3 × CH3), 34.8, 40.3, 49.4, 51.8, 68.6, 70.5, 79.5, 111.5, 115.4, 121.1, 127.6, 128.6, 130.2, 137.4 156.4. HRMS (TOF ES+): calcd for C19H31N2O4 351.2284, found 351.2319 (MH+).
(±)-Benzyl 2-(2-(2-(3-(1H-Indol-4-yloxy)-2-hydroxypropylamino)ethoxy)ethoxy)ethylcarbamate (32)
(±)-4-(Oxiran-2-ylmethoxy)-1H-indole (28) (100 mg, 0.53 mmol) and benzyl 2-(2-(2-aminoethoxy)ethoxy)ethylcarbamate (15) (224 mg, 0.79 mmol, 1.5 equiv) were dissolved in propan-2-ol/acetonitrile/water (7:2:1, 5 mL) and heated in a MW reactor at 90 °C (dynamic program, max. pressure 250 psi, max. power 300 W) for 60 min. The reaction mixture was concentrated under reduced pressure and the residue purified by flash column chromatography (eluent 1 M methanolic NH3/DCM 0:100 to 10:90 over 12 column volumes), to give 100 mg of a pale yellow viscous oil (40%). 1H NMR: δ 8.72 (1H, br s), 7.23–7.41 (5H, m), 7.03–7.10 (2H, m), 7.00 (1H, d, J = 8.1 Hz), 6.61 (1H, br s), 6.46 (1H, d, J = 7.4 Hz), 5.67–5.88 (1H, br m), 5.07 (2H, s), 3.98–4.27 (3H, m), 3.53–3.65 (6H, m), 3.50 (2H, t, J = 4.9 Hz), 3.35 (2H, dt, J = 5.1/5.1 Hz), 2.72–2.95 (5H, m). 13C NMR: δ 156.73, 152.32, 137.46, 136.66, 128.55, 128.16, 128.12, 122.94, 122.60, 118.77, 104.97, 100.70, 99.62, 70.51, 70.20, 70.11, 68.46, 66.69, 51.97, 48.98, 40.91. IR (NaCl, film): 3319, 1707, 1253, 1090, 727 cm–1. HRMS (TOF ES–): calcd for C25H31N3O6 470.2297, found 470.2283 (MH–).
Method A. Cbz-Group Hydrogenolysis
To a solution of the Cbz-protected amine (100–130 μM) in MeOH/H2O (9:1) was added 10% Pd/C (1:1 w/w with protected amine). The solvent was briefly degassed and hydrogen was added via balloon; the mixture was then stirred at room temperature for 1 h. The reaction mixture was filtered through Celite and the pad was washed with MeOH (×2). The solvent was evaporated under reduced pressure to afford the deprotected amine, which was used without further purification.
(±)-1-(4-Aminobutylamino)-3-(napthalenyl-1-yloxy)propan-2-ol (13e)
The compound was prepared via method A to yield a colorless oil (quant.). 1H NMR (CD3OD): 1.63–1.79 (4H, m), 2.71–2.84 (2H, m), 2.89 (2H, t), 2.92 (1H, dd, J = 12.3, 8.7), 3.03 (1H, dd, J = 12.3, 3.6), 4.14 (1H, dd, J = 9.8, 5.3), 4.18 (1H, dd, J = 9.8, 5.3), 4.28 (1H, dtd, J = 8.7, 5.3, 3.6), 6.91 (1H, dd, J = 7.6, 0.9), 7.34–7.50 (4H, m), 7.76–7.82 (1H, m), 8.26–8.34 (1H, m). 13C NMR (CD3OD): 27.5, 27.7, 40.7, 49.7, 52.9, 69.2, 71.8, 106.0, 121.6, 122.9, 126.1, 126.9, 127.0, 127.4, 128.5, 136.0, 155.7. IR (NaCl, film): 3354, 2939, 1579, 1398, 1270, 1241, 1102, 795, 772 cm–1. HRMS (TOF ES+): calcd for C17H25N2O2 289.1916, found 289.1895 (MH+).
(S)-1-(2-Allylphenoxy)-3-(2-aminoethylamino)propan-2-ol (22a)
(S)-tert-Butyl 2-(3-(2-allylphenoxy)-2-hydroxypropylamino)ethylcarbamate (21a) (0.04 g, 0.114 mmol) was dissolved in a 2 M solution of HCl in dioxane (2 mL) and the reaction mixture was stirred at room temperature for 18 h. The solvent was evaporated under reduced pressure and the resultant residue was azeotroped sequentially with toluene and EtOAc to afford the dihydrochloride salt of the title compound as a white solid (0.036 g, 98%), which was used without further purification. Mp: 142–145 °C. [α]D26 = −2.2° (c 0.46, 15:85 MeOH/CH2Cl2). 1H NMR (CD3OD): 3.28 (1H, dd, J = 12.5, 9.8), 3.36–3.49 (7H, m), 4.01 (1H, dd, J = 10.0, 5.7), 4.09 (1H, dd, J = 10.0, 4.8), 4.32–4.40 (1H, m), 4.98–5.06 (2H, m), 5.94–6.05 (1H, m), 6.91 (1H, td, J = 7.7, 1.0), 6.95 (1H, d, J = 7.7), 7.13 (1H, dd, J = 7.7, 1.7), 7.18 (1H, td, J = 7.7, 1.7). 13C NMR (CD3OD): 35.3, 36.8, 45.9, 52.0, 66.8, 71.0, 112.7, 115.7, 122.3, 128.5, 129.8, 131.0, 138.5, 157.4. IR (KBr): 3299, 2956, 1600, 1491, 1455, 1258, 1214, 764 cm–1. HRMS (TOF ES+): calcd for C14H23N2O2 251.1760, found 251.1773 (MH+).
(±)-13-Amino-1-(1H-indol-4-yl)-1,8,11-trioxa-5-azatridecan-3-ol (33)
The compound was prepared via method A to yield a colorless oil (88%). 1H NMR (CD3OD): 2.86–3.02 (6H, m), 3.58–3.69 (8H, m), 4.12 (1H, dd, J = 9.9, 6.0), 4.15 (1H, dd, J = 9.9, 4.7), 4.16–4.24 (1H, m), 6.41 (1H, d, J = 3.0), 6.64 (1H, d, J = 7.6), 6.91 (1H, t, J = 7.8), 7.16 (1H, dd, J = 7.8, 0.7), 7.20 (1H, d, J = 3.3). 13C NMR (CD3OD): 41.1, 49.8, 52.9, 69.6, 69.6, 70.4, 71.3, 71.3, 71.7, 102.7, 103.6, 114.6, 120.4, 125.2, 128.2, 131.1, 146.7. IR (NaCl, film): 2919, 1576, 1253, 1091, 728 cm–1. HRMS (TOF ES+): calcd for C17H27N3O4 338.2080, found 338.2062 (MH+).
Method B. Coupling of Amines with BODIPY 630/650-X, SE
BODIPY 630/650-X, SE (1–2.5 mg, 1 equiv) and amine (2–4 equiv) were dissolved in anhydrous DMF (1 mL) [in the case of the alprenolol congener HCl salts, 2 equiv (with respect to the congener) of N,N-diisoproylethylamine (DIPEA) was also added (method B1)] and stirred under a nitrogen atmosphere, with the exclusion of light, for 2 h. The solvent was evaporated under reduced pressure and this crude mixture was purified by PTLC on silica using (10:90 MeOH/CH2Cl2) as eluent to give compounds 14a–f, 18a,b, 23a,b, 26a,b, 31, and 34 as blue amorphous solids. When submitted to analytical HPLC [YMC C8, 150 × 4.6 mm, 1 mL min–1, 35–100% B or 5–100% B, 30 min, monitored using photodiode array detection between 190 and 800 nm; mobile phases, solvent A, H2O; solvent B, MeCN (both A and B containing 0.06% TFA as additive)] all fluorescent conjugates were observed to elute as single and symmetrical peaks at the retention time tR given below for each compound. For the initial conjugates synthesized within the racemic propranolol series, we further analyzed compound purity by performing a second analytical HPLC [Jones C4, 150 × 4.6 mm, 1 mL min–1, 35–100% B, 30 min, monitored using photodiode array detection between 190 and 800 nm; solvent A, H2O; solvent B, MeCN (both A and B containing 0.06% TFA as additive)]. Again, all target compounds eluted as homogeneous single peaks (with the expected reduction in retention time). The identities of all the compounds were further analyzed by HRMS (TOF ES+). The purities of all compounds tested in biological systems were determined as being ∼95%.
(S)-N-[2-(2-Hydroxy-3-(naphthalen-1-yloxy)propylamino)ethyl]-6-(2-(2-(4,4-difluoro-4,4a-dihydro-5-(thiophen-2-yl)- 4-bora-3a,4a-diaza-s-indacene-3-yl)vinyl)phenoxy)acetamido)hexanamide (14a)
The compound was prepared via method B (78%). tR 9.61 min (YMC C8, 150 × 4.6 mm, 1 mL min–1, 35–100% B, 30 min; solvent A, H2O; solvent B, MeCN, both A and B contain 0.06% TFA as additive). HRMS (TOF ES+): calcd for C44H47BF2N5O5S 806.3359, found 806.3425 (MH+).
(S)-N-[2-(2-Hydroxy-3-(naphthalen-1-yloxy)propylamino)butyl]-6-(2-(2-(4,4-difluoro-4,4a-dihydro-5-(thiophen-2-yl)-4-bora-3a,4a-diaza-s-indacene-3-yl)vinyl)phenoxy)acetamido)hexanamide (14b)
The compound was prepared via method B (80%). tR 15.68 min (YMC C8, 150 × 4.6 mm, 1 mL min–1, 5–100% B, 30 min; solvent A, H2O; solvent B, MeCN, both A and B contain 0.06% TFA as additive). HRMS (TOF ES+): calcd for C46H51BF2N5O5S 834.3672, found 834.3690 (MH+).
(S)-N-[2-(2-Hydroxy-3-(naphthalen-1-yloxy)propylamino)octyl]-6-(2-(2-(4,4-difluoro-4,4a-dihydro-5-(thiophen-2-yl)-4-bora-3a,4a-diaza-s-indacene-3-yl)vinyl)phenoxy)acetamido)hexanamide (14c)
The compound was prepared via method B (71%). tR 10.22 min (YMC C8, 150 × 4.6 mm, 1 mL min–1, 35–100% B, 30 min; solvent A, H2O; solvent B, MeCN, both A and B contain 0.06% TFA as additive). HRMS (TOF ES+): calcd for C50H59BF2N5O5S 890.4298, found 890.4278 (MH+).
(±)-N-[2-(2-Hydroxy-3-(naphthalen-1-yloxy)propylamino)ethyl]-6-(2-(2-(4,4-difluoro-4,4a-dihydro-5-(thiophen-2-yl)-4-bora-3a,4a-diaza-s-indacene-3-yl)vinyl)phenoxy)acetamido)hexanamide (14d)
The compound was prepared via method B (98%). tR1 10.37 min (YMC C8, 150 × 4.6 mm, 1 mL min–1, 35–100% B, 30 min). tR2 7.45 min (Jones C4, 150 × 4.6 mm, 1 mL min–1, 35–100% B, 30 min; solvent A, H2O; solvent B, MeCN, both A and B contain 0.06% TFA as additive). HRMS (TOF ES+): calcd for C44H47BF2N5O5S 806.3359, found 806.3279 (MH+).
(±)-N-[4-(2-Hydroxy-3-(naphthalen-1-yloxy)propylamino)butyl]-6-(2-(2-(4,4-difluoro-4,4a-dihydro-5-(thiophen-2-yl)-4-bora-3a,4a-diaza-s-indacene-3-yl)vinyl)phenoxy)acetamido)hexanamide (14e)
The compound was prepared via method B (77%). tR1 9.48 min (YMC C8, 150 × 4.6 mm, 1 mL min–1, 35–100% B, 30 min). tR2 7.54 min (Jones C4, 150 × 4.6 mm, 1 mL min–1, 35–100% B, 30 min). 1H NMR (CD3OD): 1.22–1.33 (2H, m), 1.47–1.64 (6H, m), 1.66–1.76 (2H, m), 2.16 (2H, t, J = 7.3), 3.06 (2H, t, J = 8.0), 3.16–3.33 (6H, m), 4.13 (1H, dd, J = 9.9, 5.7), 4.20 (1H, dd, J = 9.9, 5.0), 4.34–4.41 (1H, m), 4.55 (2H, s), 6.83 (1H, d, J = 4.2), 6.90 (1H, d, J = 7.1), 7.02 (2H, app d, J = 8.8), 7.11 (1H, d, J = 4.2), 7.11 (1H, d, J = 4.2), 7.18 (1H, d, J = 4.4), 7.19 (1H, dd, J = 5.0, 3.8), 7.33 (1H, s), 7.37 (1H, t, J = 7.9), 7.43–7.56 (4H, m), 7.57–7.63 (3H, m), 7.81 (1H, dd, J = 7.1, 1.5), 8.12 (1H, dd, J = 3.9, 1.0), 8.22 (1H, br t, J = 6.0), 8.24–8.28 (1H, m). HRMS (TOF ES+): calcd for C46H51BF2N5O5S 834.3672, found 834.3622 (MH+).
(±)-N2-(2-Hydroxy-3-(naphthalen-1-yloxy)propylamino)octyl]-6-(2-(2-(4,4-difluoro-4,4a-dihydro-5-(thiophen-2-yl)-4-bora-3a,4a-diaza-s-indacene-3-yl)vinyl)phenoxy)acetamido)hexanamide (14f)
The compound was prepared via method B (69%). tR 14.21 min (YMC C8, 150 × 4.6 mm, 1 mL min–1, 5–100% B, 30 min; solvent A, H2O; solvent B, MeCN, both A and B contain 0.06% TFA as additive). HRMS (TOF ES+): calcd for C50H59BF2N5O5S 890.4298, found 890.4321 (MH+).
(S)-N-[3-Hydroxy-1-(naphthalen-1-yl)-1,8,11-trioxa-5-azatridecan-13-yl]-6-(2-(2-(4,4-difluoro-4,4a-dihydro-5-(thiophen-2-yl)-4-bora-3a,4a-diaza-s-indacene-3-yl)vinyl)phenoxy)acetamido)hexanamide (18a)
The compound was prepared via method B (87%). tR 10.75 min (YMC C8, 150 × 4.6 mm, 1 mL min–1, 35–100% B, 30 min; solvent A, H2O; solvent B, MeCN, both A and B contain 0.06% TFA as additive). HRMS (TOF ES+): calcd for C48H55BF2N5O7S 894.3883, found 894.3849 (MH+).
(±)-N-[3-Hydroxy-1-(naphthalen-1-yl)-1,8,11-trioxa-5-azatridecan-13-yl]-6-(2-(2-(4,4-difluoro-4,4a-dihydro-5-(thiophen-2-yl)-4-bora-3a,4a-diaza-s-indacene-3-yl)vinyl)phenoxy)acetamido)hexanamide (18b)
The compound was prepared via method B (81%). tR 9.52 min (YMC C8, 150 × 4.6 mm, 1 mL min–1, 35–100% B, 30 min; solvent A, H2O; solvent B, MeCN, both A and B contain 0.06% TFA as additive). HRMS (TOF ES+): calcd for C48H55BF2N5O7S 894.3883, found 894.3926 (MH+).
(S)-N-[2-(3-(2-Allylphenoxy)-2-hydroxypropylamino)ethyl]-6-(2-(2-(4,4-difluoro-4,4a-dihydro-5-(thiophen-2-yl)-4-bora-3a,4a-diaza-s-indacene-3-yl)vinyl)phenoxy)acetamido)hexanamide (23a)
The compound was prepared via method B1 (79%). tR 9.33 min (YMC C8, 150 × 4.6 mm, 1 mL min–1, 35–100% B, 30 min). 1H NMR (CD3OD): 1.24–1.35 (2H, m), 1.54 (2H, tt, J = 7.6, 7.6), 1.60 (2H, tt, J = 7.6, 7.6), 2.17 (2H, t, J = 7.5), 2.82–2.94 (3H, m), 3.00 (1H, dd, J = 12.5, 3.5), 3.23–3.31 (2H, m), 3.33–3.40 (4H, m), 3.92 (1H, dd, J = 9.6, 5.4), 3.97 (1H, dd, J = 9.6, 5.1), 4.07–4.16 (1H, m), 4.57 (2H, s), 4.96–5.04 (2H, m), 5.97 (1H, ddt, J = 16.9, 10.3, 6.5), 6.82–6.92 (3H, m), 7.05 (2H, app d, J = 9.0), 7.08–7.19 (4H, m), 7.20 (1H, d, J = 3.8), 7.21 (1H, d, J = 3.8), 7.35 (1H, s), 7.55 (2H, br d, J = 7.7), 7.59–7.65 (3H, m), 8.12 (1H, dd, J = 3.8, 1.0). HRMS (TOF ES+): calcd for C43H49BF2N5O5S 796.3516, found 796.3474 (MH+).
(±)-N-[2-(3-(2-Allylphenoxy)-2-hydroxypropylamino)ethyl]-6-(2-(2-(4,4-difluoro-4,4a-dihydro-5-(thiophen-2-yl)-4-bora-3a,4a-diaza-s-indacene-3-yl)vinyl)phenoxy)acetamido)hexanamide (23b)
The compound was prepared via method B1 (29%). tR 13.33 min (YMC C8, 150 × 4.6 mm, 1 mL min–1, 5–100% B, 30 min; solvent A, H2O; solvent B, MeCN, both A and B contain 0.06% TFA as additive). HRMS (TOF ES+): calcd for C43H49BF2N5O5S 796.3516, found 795.3345 (MH+).
(S)-N-[3-Hydroxy-1-(2-(prop-2-en-1-yl)phenyl)-1,8,11-trioxa-5-azatridecan-13-yl]-6-(2-(2-(4,4-difluoro-4,4a-dihydro-5-(thiophen-2-yl)-4-bora-3a,4a-diaza-s-indacene-3-yl)vinyl)phenoxy)acetamido)hexanamide (26a)
The compound was prepared via method B1 (53%). tR 13.35 min (YMC C8, 150 × 4.6 mm, 1 mL min–1, 5–100% B, 30 min; solvent A, H2O; solvent B, MeCN, both A and B contain 0.06% TFA as additive). HRMS (TOF ES+): calcd for C47H57BF2N5O7S 884.4040, found 884.4086 (MH+).
(±)-N-[3-Hydroxy-1-(2-(prop-2-en-1-yl)phenyl)-1,8,11-trioxa-5-azatridecan-13-yl]-6-(2-(2-(4,4-difluoro-4,4a-dihydro-5-(thiophen-2-yl)-4-bora-3a,4a-diaza-s-indacene-3-yl)vinyl)phenoxy)acetamido)hexanamide (26b)
The compound was prepared via method B1 (68%). tR 9.57 min (YMC C8, 150 × 4.6 mm, 1 mL min–1, 35–100% B, 30 min; solvent A, H2O; solvent B, MeCN, both A and B contain 0.06% TFA as additive). HRMS (TOF ES+): calcd for C47H57BF2N5O7S 884.4040, found 884.4045 (MH+).
(±)-N-[2-(2-Hydroxy-3-(1H-indol-4-yloxy)propylamino)ethyl]-6-(2-(2-(4,4-difluoro-4,4a-dihydro-5-(thiophen-2-yl)-4-bora-3a,4a-diaza-s-indacene-3-yl)vinyl)phenoxy)acetamido)hexanamide (31)
The compound was prepared via method B (39%). tR 9.19 min (YMC C8, 150 × 4.6 mm, 1 mL min–1, 35–100% B, 30 min; solvent A, H2O; solvent B, MeCN, both A and B contain 0.06% TFA as additive). HRMS (TOF ES+): calcd for C42H46BF2N6O5S 795.3312, found 795.3345 (MH+).
(±)-N-[3-Hydroxy-1-(1H-indol-4-yl)-1,8,11-trioxa-5-azatridecan-13-yl]-6-(2-(2-(4,4-difluoro-4,4a-dihydro-5-(thiophen-2-yl)-4-bora-3a,4a-diaza-s-indacene-3-yl)vinyl)phenoxy)acetamido)hexanamide (34)
The compound was prepared via method B (68%). tR 9.40 min (YMC C8, 150 × 4.6 mm, 1 mL min–1, 35–100% B, 30 min). 1H NMR (CD3OD): 1.21–1.32 (2H, m), 1.51 (2H, tt, J = 7.6, 7.6), 1.57 (2H, tt, J = 7.6, 7.6), 2.14 (2H, t, J = 7.4), 3.11–3.22 (3H, m), 3.25 (2H, t, J = 6.9), 3.28–3.37 (2H, m), 3.44–3.50 (1H, m), 3.47 (2H, t, J = 5.6), 3.53–3.58 (2H, m), 3.58–3.63 (2H, m), 3.72 (2H, t, J = 4.8), 4.13 (1H, dd, J = 9.8, 5.4), 4.18 (1H, dd, J = 9.8, 5.0), 4.26–4.35 (1H, m), 4.55 (2H, s), 6.43 (1H, d, J = 3.2), 6.63 (1H, d, J = 7.7), 6.85 (1H, d, J = 4.5), 6.92 (1H, t, J = 7.8), 7.03 (2H, app d, J = 9.0), 7.11–7.15 (2H, m), 7.17–7.22 (4H, m), 7.35 (1H, s), 7.55 (2H, br d, J = 8.3), 7.58–7.63 (3H, m), 8.12 (1H, dd, J = 3.8, 1.0). HRMS (TOF ES+): calcd for C46H54BF2N6O7S 883.3836, found 883.3886 (MH+).
General Pharmacology Methods: Cell Lines and Cell Culture
CHO-K1 stably expressing a six cyclic AMP response (CRE) secreted placental alkaline phosphatase (SPAP) reporter gene and either human β1 (1146 fmol/mg of protein), human β2 (466 fmol/mg of protein), or human β3-AR (790 fmol/mg of protein) were used throughout this study.(44) Cells were grown in Dulbecco’s modified Eagle’s medium nutrient mix F12 (DMEM/F12) containing 10% fetal calf serum and 2 mM l-glutamine in a 37 °C humidified 5% CO2:95% air atmosphere.
3H-CGP 12177 Whole Cell Binding
Cells were grown to confluence in white-sided tissue culture treated 96-well view plates. 3H-CGP 12177 whole cell competition binding was performed as previously described,(45) using 3H-CGP 12177 in the ranges of 0.71–3.14 nM (for β1 and β2) and 4.64–63.0 nM (β3) (total volume 200 μL per well).
CRE-SPAP Production
Cells were grown to confluence in clear plastic tissue culture treated 96-well plates, and CRE-SPAP secretion into the media was measured between 5 and 6 h after the addition of agonist as previously described.(36)
Confocal Imaging
Cells were grown in Labtek eight-well plates (Nunc Nalgene, Rochester, NY) for at least 18 h and grown to 80–100% confluence before imaging. Following washing of the cells in HEPES-buffered saline solution (HBSS; 25 mM HEPES, 10 mM glucose, 146 mM NaCl, 5 mM KCl, 1 mM MgSO4, 2 mM sodium pyruvate, 1.3 mM CaCl2), live cell imaging was performed at room temperature using a Zeiss LSM 710 laser scanning confocal microscope and a Zeiss Plan-Neofluar 40 × 1.3 NA oil-immersion objective. A 633 nm HeNe laser was used for the excitation of the BODIPY630/650–18a conjugate with emission being detected using a 650 nm long-pass filter. The pinhole diameter (1 Airy Unit; 1.1 μm optical slice), laser power, and gain remained constant between experiments.
Data Analysis: Whole Cell Binding
All data points on each competition binding curve were performed in triplicate and each 96-well plate also contained six determinations of total and nonspecific binding. In all cases where a KD value is stated, the competing ligand completely inhibited the specific binding of 3H-CGP 12177. For compounds 14c and 14f, where inhibition of binding was only 66% and 85%, respectively, an apparent KD value is stated. A one-site sigmoidal response curve was then fitted to the data using Graphpad Prism 2.01 and the IC50 was then determined as the concentration required to inhibit 50% of the specific binding.
where A in the concentration of the competing ligand, IC50 is the concentration at which half of the specific binding of 3H-CGP 12177 has been inhibited, and NS is the nonspecific binding.
From the IC50 value and the known concentration of 3H-CGP 12177, a KD value was calculated using the equation
Inhibition of Agonist-Stimulated CRE-SPAP Production
Agonist responses were best described by a one-site sigmoidal concentration response curve (eq 3)
where Emax is the maximum response, [A] is the agonist concentration, and EC50 is the concentration of agonist that produces 50% of the maximal response
The affinities of antagonist drugs were calculated (KD values) from the shift of the agonist concentration responses in the presence of a fixed concentration of antagonist using eq 4:
where DR (dose ratio) is the ratio of the agonist concentration required to stimulate an identical response in the presence and absence of a fixed concentration of antagonist, [B].
Where clear partial agonism was observed (e.g., Figure 5), the affinity was calculated according to the method(45) of Stephenson using eq 5:
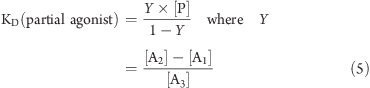 |
where [P] in the concentration of the partial agonist, [A1] in the concentration of the agonist at the point where the partial agonist alone agonist causes the same response, [A2] is the concentration of agonist causing a given response above that achieved by the partial agonist, and [A3] the concentration of the agonist, in the presence partial agonist, causing the same stimulation as [A2].
Acknowledgments
J.G.B. is a Wellcome Trust Clinician Scientist Fellow (073377/Z/03/Z). This work was supported by The BBSRC (Grant Number BB/0521581/1) and The University of Nottingham. The molecules described in this paper are the subject of an international patent application, WO 2004/088312.
Glossary
Abbreviations Used
- β-AR
β-adrenoceptor
- BODIPY 630/650 or BY630
6-(((4,4-difluoro-5-(2-thienyl)-4-bora-3a,4a-diaza-s-indacene-3-yl)styryloxy)acetyl)aminohexanoic acid
- BODIPY 630/650-X, SE
6-(((4,4-difluoro-5-(2-thienyl)-4-bora-3a,4a-diaza-s-indacene-3-yl)styryloxy)acetyl)aminohexanoic acid succinimidyl ester
- Cbz
benzyloxycarbonyl
- CHO
chinese hamster ovary cell
- CGP 20712A
1-[2-((3-carbamoyl-4-hydroxy)phenoxy)ethylamino]-3-[4-(1-methyl-4-trifluoromethyl-2-imidazolyl)phenoxy]-2-propanol
- CGP 12177
4-[3-[(1,1-dimethylethyl)amino]-2-hydroxypropoxy]-1,3-dihydro-2H-benzimidazol-2-one
- FCS
fluorescence correlation spectroscopy
- GPCR
G-protein coupled receptor
- ICI 118551
3-(isopropylamino)-1-[(7-methyl-4-indanyl)oxy]butan-2-ol
- LSCM
laser scanning confocal microscopy
- MW, microwave; NBD
7-nitrobenzo-2-oxa-1,3-diazole.
Supporting Information Available
Full experimental procedures for compounds 12a–d, 12f, 13a–d, 13f, 14a–d, 14f, 16a,b, 17a,b, 18a,b, 21b, 22b, 23b, 24a,b, 25a,b, 26a,b, 29–31. This material is available free of charge via the Internet at http://pubs.acs.org
S.J.H. and B.K. are founding directors of the University of Nottingham spin-off company CellAura Technologies Ltd. R.M. is currently employed by CellAura Technologies Ltd.
Author Contributions
∥ These authors contributed equally to this work
Supplementary Material
References
- Bilski A. J.; Halliday S. E.; Fitzgerald J. D.; Wale J. L. The pharmacology of a beta 2-selective adrenoceptor antagonist (ICI 118,551). J. Cardiovasc. Pharmacol. 1983, 5, 430–437. [DOI] [PubMed] [Google Scholar]
- Jacoby E.; Bouhelal R.; Gerspacher M.; Seuwen K. The 7 TM G-protein coupled receptor target family. ChemMedChem 2006, 1, 760–782. [DOI] [PubMed] [Google Scholar]
- Bylund D. B.; Bond R. A.; Eikenburg D. C.; J. Hieble P. J. Hills R.; Minneman K. P.; Parra S.. Beta-adrenoceptors. http://www.iuphar-db.org/DATABASE/FamilyMenuForward?familyId=4.
- Michel M. C.; Insel P. A.. Adrenergic receptors in clinical medicine. In The Receptors: The Adrenergic Receptors: In the 21st Century; Perez D. M., Ed.; Humana Press: New York, 2007; pp 129–147. [Google Scholar]
- Powe D. G.; Voss M. J.; Zänker K. S.; Habashy H. O.; Green A. R.; Ellis I. O.; Entschladen F. Beta-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget 2010, 1, 628–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan E. K.; Priceman S. J.; Cox B. F.; Yu S.; Pimente M. A.; Tangkanangnukul V.; Arevalo J. M. C.; Morizono K.; Karanikolas B. D. W.; Wu L.; Sood A. K.; Cole S. W. The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res. 2010, 70, 7042–7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leopoldo M.; Lacivita E.; Berardi F.; Perrone R. Developments in fluorescent probes for receptor research. Drug Discovery Today 2009, 14, 706–712. [DOI] [PubMed] [Google Scholar]
- Rajagopal S.; Rajagopal K.; Leftkowitz R. J. Teaching old receptors new tricks: Biasing seven-transmembrane receptors. Nat. Rev. Drug Discov. 2010, 9, 373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T.; Miller L. J. Seven transmembrane receptors as shapeshifting proteins: The impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol. Rev. 2010, 62, 265–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker J. G.; Hill S. J. Multiple GPCR conformations and signalling pathways: Implications for antagonist affinity estimates. Trends Pharmacol. Sci. 2007, 28, 374–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C.; Hill S. J. GPCR signalling: Understanding the pathway to successful drug discovery. Methods Mol. Biol. 2009, 552, 39–50. [DOI] [PubMed] [Google Scholar]
- Hill S. J. G-protein-coupled receptors: Past, present and future. Br. J. Pharmacol. 2006, 147, S27–S37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel H. H.; Murray F.; Insel P. A. Caveolae as organizers of pharmacologically relevant signal transduction molecules. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 359–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin A. B.; Butcher A. J.; Kong K. C. Location, location, location. Site-specific GPCR phosphorylation offers a mechanism for cell-type specific signalling. Trends Pharmacol. Sci. 2008, 29, 413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May L. T.; Self T. J.; Briddon S. J.; Hill S. J. The effect of allosteric modulators on the kinetics of agonist-G protein-coupled receptor interactions in single living cells. Mol. Pharmacol. 2010, 78, 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeaux Y.; Briddon S. J.; Alexander S. P. H.; Kellam B.; Hill S. J. Agonist-occupied A3 adenosine receptors exist within heterogeneous complexes in membrane microdomains of individual living cells. FASEB J. 2008, 22, 850–860. [DOI] [PubMed] [Google Scholar]
- Baker J. G.; Hill S. J.; Summers R. J. Evolution of β-blockers: From anti-anginal drugs to ligand-directed signalling. Trends Pharmacol. Sci. 2011, 32, 227–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahav M.; Melamed E.; Dafna Z.; Atlas D. Localization of beta receptors in the anterior segment of the rat eye by a fluorescent analogue of propranolol. Invest. Ophthalmol. Vis. Sci. 1978, 17, 645–651. [PubMed] [Google Scholar]
- Melamed E.; Lahav M.; Atlas D. Histochemical evidence for beta-adrenergic receptors in the rat spinal cord. Brain Res. 1976, 116, 511–515. [DOI] [PubMed] [Google Scholar]
- Melamed E.; Lahav M.; Atlas D. Beta-adrenergic receptors in rat myocardium: Direct detection by a new fluorescent beta-blocker. Experientia 1976, 32, 1387–1389. [DOI] [PubMed] [Google Scholar]
- Melamed E.; Lahav M.; Atlas D. Direct localisation of beta-adrenoceptor sites in rat cerebellum by a new fluorescent analogue of propranolol. Nature 1976, 261, 420–422. [DOI] [PubMed] [Google Scholar]
- Melamed E.; Lahav M.; Atlas D. Beta-adrenergic receptors in rat cerebral cortex: Histochemical localization by a fluorescent beta-blocker. Brain Res. 1977, 128, 379–384. [DOI] [PubMed] [Google Scholar]
- Atlas D.; Melamed E. Direct mapping of beta-adrenergic receptors in the rat central nervous system by a novel fluorescent beta-blocker. Brain Res. 1978, 150, 377–385. [DOI] [PubMed] [Google Scholar]
- Henis Y. I.; Hekman M.; Elson E. L.; Helmreich E. J. Lateral motion of beta receptors in membranes of cultured liver cells. Proc. Natl. Acad. Sci. U. S. A. 1982, 79, 2907–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademaker B.; Kramer K.; van Ingen H.; Kranendonk M.; Timmerman H. Non-specific binding of the fluorescent beta-adrenergic receptor probe alprenolol–NBD. J. Receptor Res. 1985, 5, 121–131. [DOI] [PubMed] [Google Scholar]
- Rademaker B.; Kramer K.; Bast A.; Timmerman H. Irreversible binding of the fluorescent beta-adrenoceptor probes alprenolol–NBD and pindolol–NBD to specific and non-specific binding sites. Res. Commun. Chem. Pathol. Pharmacol. 1988, 60, 147–159. [PubMed] [Google Scholar]
- Hess A. Visualization of beta-adrenergic receptor sites with fluorescent beta-adrenergic blocker probes, or autofluorescent granules?. Brain Res. 1979, 160, 533–538. [DOI] [PubMed] [Google Scholar]
- Barnes P.; Koppel H.; Lewis P.; Hutson C.; Blair I.; Dollery C. A fluorescent analogue of propranolol does not label beta-adrenoceptor sites. Brain Res. 1980, 181, 209–213. [DOI] [PubMed] [Google Scholar]
- Heithier H.; Hallmann D.; Boege F.; Reiländer H.; Dees C.; Jaeggi K. A.; Arndt-Jovin D.; Jovin T. M.; Helmreich E. J. Synthesis and properties of fluorescent beta-adrenoceptor ligands. Biochemistry 1994, 33, 9126–9134. [DOI] [PubMed] [Google Scholar]
- Staehelin M.; Simons P.; Jaeggi K.; Wigger N. CGP-12177—A hydrophilic beta-adrenergic receptor radioligand reveals high-affinity binding of agonists to intact-cells. J. Biol. Chem. 1983, 258, 3496–3502. [PubMed] [Google Scholar]
- McGrath J. C., Daly C. J.Use of fluorescent ligands and receptors to visualize adrenergic receptors. In The Receptors: The Adrenergic Receptors: In the 21st Century; Perez D. M., Ed.; Humana Press: New York, 2007; pp 151–172. [Google Scholar]
- Baker J. G.; Hall I. P.; Hill S. J. Pharmacology and direct visualisation of BODIPY-TMR–CGP: A long-acting fluorescent β2-adrenoceptor agonist. Br. J. Pharmacol. 2003, 139, 232–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegener O.; Prenner L.; Runkel F.; Baader S. L.; Kappler J.; Häberlein H. Dynamics of beta2-adrenergic receptor-ligand complexes on living cells. Biochemistry 2004, 43, 6190–6199. [DOI] [PubMed] [Google Scholar]
- Martikkala E.; Lehmusto M.; Lilja M.; Rozwandowicz-Jansen A.; Lunden J.; Tomohiro T.; Hanninen P.; Petaja-Repo U.; Harma H. Cell-based beta-2-adrenergic receptor-ligand binding assay using synthesized europium-labeled ligands and time-resolved fluorescence. Anal. Biochem. 2009, 392, 103–109. [DOI] [PubMed] [Google Scholar]
- Briddon S. J.; Middleton R. J.; Yates A. S.; George M. W.; Kellam B.; Hill S. J. Application of fluorescence correlation spectroscopy to the measurement of agonist binding to a G-protein coupled receptor at the single cell level. Faraday Discuss 2004, 126, 197–207. [DOI] [PubMed] [Google Scholar]; Discussion 245–254.
- Middleton R. J.; Briddon S. J.; Cordeaux Y.; Yates A. S.; Dale C. L.; George M. W.; Baker J. G.; Hill S. J.; Kellam B. New fluorescent adenosine A1-receptor agonists that allow quantification of ligand–receptor interactions in microdomains of single living cells. J. Med. Chem. 2007, 50, 782–793. [DOI] [PubMed] [Google Scholar]
- Briddon S. J.; Middleton R. J.; Cordeaux Y.; Flavin F. M.; Weinstein J. A.; George M. W.; Kellam B.; Hill S. J. Quantitative analysis of the formation and diffusion of A1-adenosine receptor-antagonist complexes in single living cells. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 4673–4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker J. G.; Middleton R.; Adams L.; May L. T.; Briddon S. J.; Kellam B.; Hill S. J. Influence of fluorophore and linker composition on the pharmacology of fluorescent adenosine A1 receptor ligands. Br. J. Pharmacol. 2010, 159, 772–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briddon S. J.; Hill S. J. Pharmacology under the microscope: The use of fluorescence correlation spectroscopy to determine the properties of ligand–receptor complexes. Trends Pharmacol. Sci. 2007, 28, 637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunder J. M.; Onami T.; Sharpless K. B. Arenesulfonate derivatives of homochiral glycidol: Versatile chiral building blocks for organic synthesis. J. Org. Chem. 1989, 54, 1295–1304. [Google Scholar]
- Baker J. G.; Hall I. P.; Hill S. J. Agonist actions of ’β-blockers’ provide evidence for two agonist activation sites on the human β1-adrenoceptor. Mol. Pharmacol. 2003, 63, 1312–1321. [DOI] [PubMed] [Google Scholar]
- Baker J. G.; Hall I. P.; Hill S. J. Agonist and inverse agonist actions of “β-blockers” at the human β2-adrenoceptor provide evidence for agonist-directed signalling. Mol. Pharmacol. 2003, 64, 1357–1369. [DOI] [PubMed] [Google Scholar]
- Baker J. G. Evidence for a secondary state of the human β3-adrenoceptor. Mol. Pharmacol. 2005, 68, 1645–1655. [DOI] [PubMed] [Google Scholar]
- Baker J. G. The selectivity of beta-adrenoceptor agonists at human beta1-, beta2- and beta3-adrenoceptors. Br. J. Pharmacol. 2010, 160, 1048–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson R. P. A modification of receptor theory. Br. J. Pharmacol. Chemother. 1956, 11, 379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Granneman J. G. (2001). The putative beta4-adrenergic receptor is a novel state of the beta1-adrenergic receptor. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E199–E202. [DOI] [PubMed] [Google Scholar]; b Kaumann A. J.; Engelhardt S.; Hein L.; Molenaar P.; Lohse M. Abolition of (−)-CGP 12177-evoked cardiostimulation in double beta1/beta2-adrenoceptor knockout mice. Obligatory role of beta1-adrenoceptors for putative beta4-adrenoceptor pharmacology. Naunyn Schmiedebergs Arch. Pharmacol. 2001, 363, 87–93. [DOI] [PubMed] [Google Scholar]; c Kaumann A. J.; Molenaar P. (2008). The low-affinity site of the beta1-adrenoceptor and its relevance to cardiovascular pharmacology. Pharmacol Ther. 2008, 118, 303–336. [DOI] [PubMed] [Google Scholar]; d Arch J. R. Do low-affinity states of beta-adrenoceptors have roles in physiology and medicine?. Br. J. Pharmacol. 2004, 143, 517–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker J. G. Site of action of beta-ligands at the human beta1-adrenoceptor. J. Pharmacol. Exp. Ther. 2005, 313, 1163–1171. [DOI] [PubMed] [Google Scholar]
- Dooley D. J.; Bittiger H.; Reymann N. C. CGP 20712A: A useful tool for quantitating beta 1- and beta 2-adrenoceptors. Eur. J. Pharmacol. 1986, 130, 137–139. [DOI] [PubMed] [Google Scholar]
- Baker J. G. The selectivity of beta-adrenoceptor antagonists at the human β1, β2 and β3 adrenoceptors. Br. J. Pharmacol. 2005, 144, 317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May L. T.; Briddon S. J.; Hill S. J. Antagonist selective modulation of adenosine A1- and A3- receptor pharmacology by the food dye Brilliant Black BN: Evidence for allosteric interactions. Mol. Pharmacol. 2010, 77, 678–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collet M.; Lenger J.; Jenssen K.; Hannes P. P.; Sewald N. Molecular tools for metalloprotease sub-proteome generation. J. Biotechnol. 2007, 129, 316–328. [DOI] [PubMed] [Google Scholar]
- Chen R.; Nguyen P.; You Z.; Sinsheimer J. E. Enantioselective detoxication of optical isomers of glycidyl ethers. Chirality 1993, 5, 501–504. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.