

Summary
Pseudomonas
exotoxin A (PE) is a highly toxic protein secreted by the opportunistic pathogen Pseudomonas aeruginosa. The modular structure and corresponding mechanism of action of PE make it amenable to extensive modifications that can redirect its potent cytotoxicity from disease to a therapeutic function. In combination with a variety of artificial targeting elements, such as receptor ligands and antibody fragments, PE becomes a selective agent for the elimination of specific cell populations. This review summarizes our current understanding of PE, its intoxication pathway, and ongoing efforts to convert this toxin into a treatment for cancer.
Keywords: Pseudomonas exotoxin A, recombinant immunotoxins, cancer therapy, intracellular trafficking, antibody conjugates, moxetumomab pasudotox
Introduction
The natural world abounds with an enormous variety of toxins, poisonous substances that are naturally produced by living organisms [1]. Typically, only small quantities of toxins are necessary to damage cells, although the specific target and the toxic dose may vary extensively. Microorganisms secrete toxins as virulence factors during pathogenic infection, and as secondary metabolites that can contaminate local environments. Well known examples include diphtheria toxin and ergot alkaloids. Higher organisms use toxins as components in complex venoms and accumulate them as defense factors to deter predators. Overall, toxins can take many forms, appear in sizes ranging from small molecules to large proteins, and have diverse mechanisms of action, but they normally serve similar functions related to predation and/or defense.
Although frequently hazardous and occasionally lethal, many toxins have the potential for therapeutic application by removing the molecule from its natural context. Strategies such as altering the route of delivery, changing the dose, eliminating supporting or synergizing molecules (such as from a complex mixture like a venom), or even modifying the structure of the molecule may convert a dangerous toxin into a valuable therapeutic resource. One recent example is the botulinum toxins, which are potent paralytic neurotoxins produced by the microbes of the Clostridium genus, most notably Clostridium botulinum. Botulinum toxin type A has been approved as the drug on a botulinum toxin A (Botox® and Botox Cosmetic®; Allergan, Inc., Irvine, CA), for both therapeutic and cosmetic purposes. Although the toxin has an estimated human LD50 of ~ 1 ng per kg of body weight [2], the extremely low dose employed clinically and its delivery via a site-specific injection make the agent safe for widespread use.
Other toxins must be more heavily modified for therapeutic purposes. Diphtheria toxin (DT) is an extremely potent cytotoxic protein that is the primary virulence factor secreted by the bacterium Corynebacterium diphtheriae, the pathogen that causes the disease diphtheria [3]. The LD50 of diphtheria toxin in humans has been reported as ≤ 100 ng per kg of body weight [2], yet the toxin was converted into the first recombinant toxin to be FDA approved for the intravenous therapy of cutaneous T-cell lymphoma. Denileukin diftitox (Ontak®; Esai, Inc., Woodcliff Lake, NJ) is a recombinant form of DT that has been engineered by replacing the native receptor-binding domain of DT with interleukin-2 (IL-2). This substitution alters the toxin’s target from the membrane-associated heparin-binding epidermal-growth-factor-like growth factor [4] to the IL-2 receptor, redirecting its potent cytotoxicity toward a therapeutic purpose (for recent reviews of denileukin diftitox see [5,6]).
A comparable strategy to alter the target of an intracellular toxin has been employed for Pseudomonas exotoxin A (PE), a protein toxin with many similarities to DT. PE and DT are only distantly related, but they both belong to a class of cytotoxic proteins, the A–B toxins, that require cellular uptake through receptor-mediated endocytosis for activity. The overall structure of these proteins consists of a receptor-binding domain (B subunit) linked to a domain with cytotoxic activity (A subunit) that is delivered to the cytosol. Although their B subunits have very different targets, the A subunit of both PE and DT is a NAD+-diphthamide ADP-ribosyltransferase (EC 2.4.2.36) that targets and inactivates eukaryotic translation elongation factor 2 (eEF2). This halts protein synthesis and eventually leads to cell death. A recently identified third member of the NAD+-diphthamide ADP-ribosyltransferase toxin subfamily, cholera exotoxin (CE, also known as cholix toxin) from Vibrio cholerae, has extensive sequence (36% identity, 50% similarity) and structural (2.04 Ǻ Cα RMSD) resemblance to PE and presumably utilizes a similar intoxication pathway [7,8]. PE, CE, DT, and other toxins that utilize receptor-meditated endocytosis can potentially be redirected for therapeutic purposes by replacing their native receptor-binding domains with other targeting elements. This review will discuss our current understanding of PE intoxication and efforts to convert PE into a viable therapeutic agent.
Pseudomonas exotoxin A
Pseudomonas aeruginosa is a ubiquitous, Gram-negative, aerobic bacillus that is often encountered as an opportunistic human pathogen, although infections in healthy individuals are rare. Roughly ten percent of hospital-acquired infections are caused by P. aeruginosa, and certain patient populations, such as individuals with cystic fibrosis or burn wounds, are especially prone to this infection (for a review see [9]). The bacterium is known to possess a number of virulence determinants, the most toxic of which is the protein PE [10]. Studies in mice have identified the median lethal dose of PE as approximately 200 ng, and evidence suggests that PE may play a major role in the virulence of P. aeruginosa. Strains of P. aeruginosa deficient in PE production are less virulent than strains producing PE, and patients who survive infection from PE-producing strains typically have high antibody titers against PE [3,11].
PE (see GenBank accession number AAB59097) is synthesized as a single 638-residue (69-kDa) polypeptide that is processed by the removal of a 25-residue N-terminal sequence before secretion as the 613-residue (66-kDa) native toxin. All sequence numbering in this review is based on the 613-residue native toxin. The initial X-ray crystallographic structure of native PE revealed three major structural domains [12]. The N-terminal domain I is divided into nonsequential but structurally adjacent domains Ia (residues 1-252) and Ib (365–404). The residues between domains Ia and Ib comprise domain II (253–364), and the remaining C-terminal residues make up domain III (405–613). Native PE contains 8 cysteines that form 4 disulfide bonds in sequential order; two lie in domain Ia (C11-C15 & C197-C214), one in domain II (C265-C287), and one in domain Ib (C372-C379). Figure 1 illustrates the domain structure of native PE.
Fig. 1.
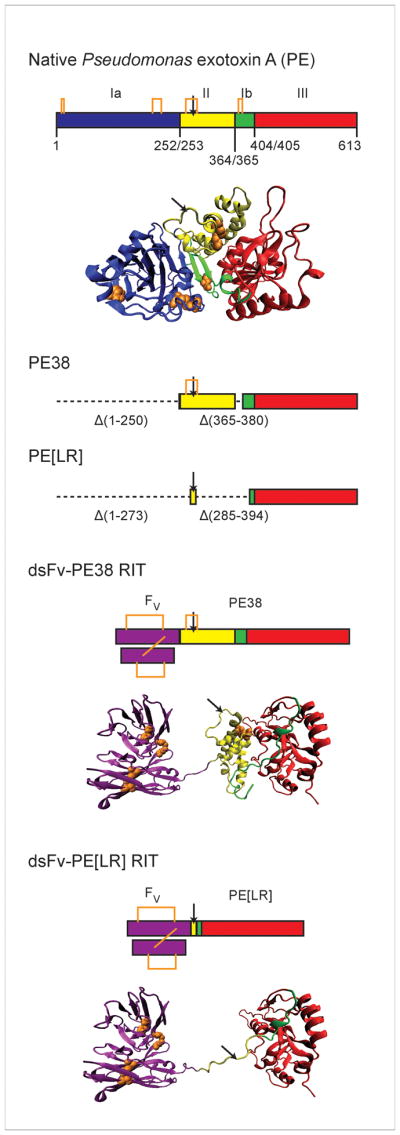
PE and PE-based RITs. Native PE consists of three structural domains organized from a single polypeptide sequence. Domain I is separated into the structurally adjacent but discontinuous Domain Ia (blue; residues 1-252) and Domain Ib (green; 365–404) by Domain II (yellow; 253–364). Domain III (red; 405–613) lies at the C-terminus. A cartoon model (created using VMD [13]) based on the X-ray crystal structure of PE (PDB ID 1IKQ) is shown, excluding those residues absent from the electron density map (607–613). RITs based on PE are chimeric molecules that fuse antibodies to fragments of PE, most frequently a 38-kDa truncation known as PE38 that contains extensive deletions in Domain Ia (Δ1–250) and Ib (Δ365–380). Recently, a smaller fragment, PE[LR] (Δ1–273 and Δ285–394), has been developed for use in RITs. Structural models of RITs using a dsFv joined to PE38 or PE[LR] are presented. The Fv is shown in purple. Models are hypothetical only and do not represent actual structural determinations. The dsFv-PE38 RIT contains a gap in the structure that corresponds to the deletion of residues 365–380 in Domain Ib. Disulfide bonds in PE and the Fv are shown in orange. The site of furin cleavage is indicated with a black arrow.
Functionally, domain I of PE is the receptor-binding domain, and is the major component of the B subunit. It targets the low density lipoprotein receptor related protein (LRP1; also known as CD91 or the α2-macroglobulin receptor) or the closely related variant LRP1B for subsequent cellular internalization by receptor-mediated endocytosis [14,15]. Domain III is the catalytically active domain, and is the primary constituent of the A subunit. It catalyzes the inactivation of eEF2 by transferring an ADP-ribosyl group from NAD+ to the diphthamide residue, a highly conserved, post-translationally modified histidine that is unique to eEF2. Although domain III is structurally defined by residues 405-613 of the native toxin, full catalytic activity requires a portion of domain Ib [16,17]. We have defined the catalytically functional domain III as consisting of residues 395-613 [18]. Domain II was proposed to be involved in toxin translocation and intracellular trafficking, but supporting evidence for this function is not consistent.
PE-based therapeutics
PE can be converted into an agent that selectively eliminates cells by changing its target to a different cell surface receptor. The new target is typically specified by attaching either an anti-receptor antibody or a receptor ligand to PE through chemical conjugation or recombinant protein engineering. Our laboratory has focused efforts over many years on the generation of PE-based recombinant immunotoxins (RITs), recombinant proteins that combine antibodies with protein toxins. Initial studies in which full-length PE was chemically conjugated to whole mAbs or receptor ligands [19,20] gradually gave way to the more efficient production of recombinant molecules in which domain Ia of PE was replaced by a ligand [21] or the variable fragment (Fv) of a mAb [22]. Single-chain Fv (scFv) molecules, which utilize the heavy chain (VH) and light chain (VL) fragments of the Fv covalently connected with a flexible polypeptide linker sequence [23,24], were recombinantly inserted at the N-terminus of a cytotoxic fragment of PE. To enhance the stability of recombinant immunotoxins, disulfide-stabilized Fv (dsFv) molecules were subsequently developed. The dsFv divides the VH and VL into separate polypeptides that are covalently connected through a disulfide bond engineered into the framework region of the Fv [25–27]. A cytotoxic fragment of PE can be inserted at the C-terminus of one of the two Fv polypeptide chains (see Fig. 1). The generation and production of PE-based RITs has been described [28].
The most commonly employed cytotoxic fragment of PE in RITs is a 38-kDa version known as PE38 [29] (Fig 1). PE38 contains a deletion of the majority of domain Ia (Δ1–250) and a portion of domain Ib (Δ365–380) from native PE. Several RITs incorporating a 38-kDa fragment of PE are in preclinical evaluation or have already reached clinical trials (see Table I). PE38 RITs undergoing preclinical testing include an anti-glycoprotein NMB (scFv) for the treatment of malignant gliomas and melanomas [30], an anti-HIV-1 gp120 (scFv) for the treatment of HIV [31,32], and a RIT targeted to osteosarcomas using a dsFv from the TP-3 mAb [33,34].
Table 1.
Several PE-based recombinant toxins currently in development for the treatment of cancers
| Agent | Alternative Names | Target | Stage of Development | Cancer |
|---|---|---|---|---|
| BL22 | RFB4(dsFv)-PE38 CAT-3888 |
CD22 | clinical trials completed; superseded by moxetumomab pasudotox | B cell malignancies |
| Moxetumomab pasudotox | RFB4[GTHW](dsFv)-PE38 HA22 CAT-8015 |
CD22 | clinical trials | B cell malignancies |
| LMB-2 | anti-TAC(scFv)-PE38 | CD25 (IL-2R α chain) | clinical trials | T and B cell malignancies |
| SS1P | SS1(dsFv)-PE38 | mesothelin | clinical trials | mesothelioma, lung cancer |
| MR1-1 | MR1-1KDEL MR1(scFv)-PE38KDEL |
epidermal growth factor receptor vIII | clinical trials | brain tumors |
| Cervene | TP-38 TGFα-PE38 |
epidermal growth factor receptor | clinical trials | brain and CNS tumors |
| Cintredekin besudotox | IL13-PE38QQR | interleukin-13 receptor | clinical trials | glioblastoma multiforme |
| G49[F6V](scFv)-PE38 | - | Glycoprotein NMB | preclinical | glioblastoma multiforme |
RITs that have progressed to clinical trials include the anti-CD22 RIT RFB4(dsFv)PE38, also known as BL22 or CAT-3888, for the treatment of B-cell malignancies [35–37]. The RFB4 Fv was subsequently affinity-optimized by phage display selection to create the second generation molecule RFB4[GTHW](dsFv)-PE38 [38], known variously as HA22 or CAT-8015, and now called moxetumomab pasudotox. Moxetumomab pasudotox is currently undergoing extensive clinical testing for the treatment of hematologic malignancies ([39,40]; ongoing studies can be found under ClinicalTrial.gov identifiers NCT00462189, NCT00457860, NCT00515892, NCT01086644, NCT00659425, and NCT00586924). Other RITs from our laboratory in clinical trials include the anti-mesothelin SS1(dsFv)PE38, called SS1P, for the treatment of lung cancer and mesothelioma ([41,42]; ongoing studies can be found under ClinicalTrial.gov identifiers NCT01041118, NCT00575770, and NCT01051934), and the anti-TAC(scFv)PE38, called LMB-2, which targets the IL-2 receptor (IL-2R) for the treatment of hematologic malignancies ([43]; ongoing studies can be found under ClinicalTrial.gov identifiers NCT00924170, NCT00077922, NCT00080535, and NCT00321555). Extensive lists of PE-based therapeutics at both the pre-clinical and clinical stages have been published [45,46], and additional agents continue to be developed. We have recently generated a new variant of PE, PE[LR] (Fig. 1), that shows decreased immunogenicity and nonspecific toxicity in mice while retaining cytotoxicity against malignant cells [46].
The strategy of re-routing A–B toxins, like DT and PE, through a different cellular target works well for several reasons. The cytotoxic A domain is stable and fully active independent of the receptor-binding B domain, which can be replaced by a component that confers alternate specificity, like a ligand or an antibody. Additionally, the available tools for recombinant DNA manipulation and protein expression allow us to easily generate these chimeric molecules, and protein engineering techniques give us powerful ways to develop and select improved variants. Furthermore, we can differentiate between normal and malignant cells using tumor-associated cell-surface receptors as markers. By specifically targeting these receptors with PE we can eliminate cancers while avoiding toxicities to normal tissue that are frequently associated with general chemotherapeutic strategies. Lastly, these proteins are extremely potent toxins that have been naturally selected for their ability to kill eukaryotic cells. Their activities typically require no major enhancement to function at a therapeutic level.
The PE intoxication pathway
A basic outline of the PE intoxication pathway is well understood. The secreted toxin binds to an LRP1 or LRP1B cell surface receptor, is internalized by receptor-mediated endocytosis, and undergoes intracellular trafficking to reach the cytosol. In the cytosol PE encounters eEF2 and transfers an ADP-ribosyl group from NAD+ to the diphthamide residue. This irreversibly inactivates eEF2, halts protein synthesis, and ultimately leads to cell death. A general description of the pathway is deceptively simple, and many of the specifics are not clear. Figure 2 attempts to presents a comprehensive description of PE intoxication, the details of which are discussed below. The pathway described in Figure 2 is not necessarily complete, but represents our current understanding of PE intoxication.
Fig. 2.
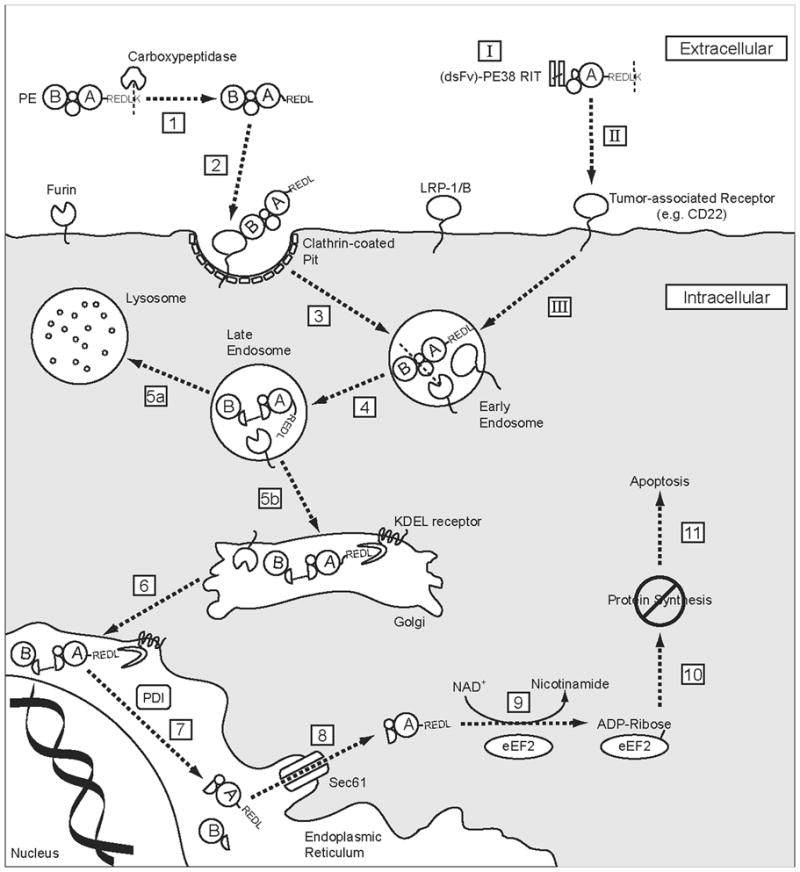
PE intoxication pathway. Native PE can be divided into two fragments with functions of receptor binding (B) and catalytic activity (A). After secretion into the extracellular environment, PE is cleaved by a carboxypeptidase (1) to remove the C-terminal lysine residue and expose the ER localization signal (REDL). The B fragment subsequently recognizes its cell-surface receptor, LRP1 or LRP1B (2), and is internalized via receptor-mediated endocytosis in clathrin-coated pits (3). Within the endocytic pathway, PE encounters the endoprotease furin, which cleaves at a site in domain II and separates the polypeptide backbone between the A and B fragments (4). A disulfide bond preserves a covalent linkage between the two fragments. While in the endocytic pathway, PE can either follow a productive trafficking route to the Golgi (5b) or continue to the lysosome for terminal degradation (5a). In the Golgi, PE encounters KDEL receptors that recognize the REDL C-terminal signal and transport PE to the ER in a retrograde manner (6). At an undetermined point in the pathway, possibly by PDI in the ER, the disulfide bond connecting the A and B fragments is reduced and the two fragments separate (7). The A fragment is subsequently transported into the cytosol (8), possibly by exploiting the ERAD pathway through the Sec61 translocon. In the cytosol PE transfers an ADP-ribosyl (ADPr) group from NAD+ to the diphthamide residue of eEF2 (9). This halts protein synthesis (10) and ultimately leads to apoptotic cell death (11). RITs based on PE (I) target tumor-associated cell surface receptors for internalization (II), and are generally thought to undergo an intoxication pathway similar to that of PE (III).
PE in the endocytic pathway
Similar to DT, native PE is a secreted as a proenzyme that must be activated before it displays catalytic activity [47]. Full activation can be accomplished under reducing and denaturing conditions and proteolysis, and appears to involve structural rearrangements that reveal the previously obscured NAD+ binding cleft in domain III [48]. RITs using versions of PE without domain Ia do not require a structural arrangement to expose the NAD+ binding site. This difference is unlikely to affect PE intoxication in RITs, although it does eliminate the requirement for catalytic activation.
After endocytosis, PE undergoes an essential proteolytic processing step at a cleavage site between residues R279 and G280 of domain II [49,50]. Using SDS-PAGE, two bands corresponding to the A and B subunits of PE were initially observed: a 28-kDa N-terminal fragment (B subunit) and a cytotoxic 37-kDa C-terminal fragment (A subunit), which was enriched in the cytosolic fraction of treated cells. PE mutated so that it did not undergo this processing step failed to kill cells. Subsequent research implicated the intracellular protease furin (EC 3.4.21.75) in this process [51–53], and supporting evidence has accumulated [54–59]. PE that is treated with furin prior to intoxication is more active than untreated PE. In addition, PE is less active on cell lines that are furin deficient, or on cells treated with furin inhibitors.
Furin is a ubiquitous, Ca2+-dependent, transmembrane serine endoprotease that is a member of the subtilisin-like family of proprotein convertases [60]. It plays an active role in the maturation of many cellular proteins, and its prevalence is frequently exploited by bacterial toxins and viruses during intoxication and infection. Furin contains a luminal catalytic domain and a cytoplasmic domain that controls its cycling between the trans-Golgi network and the plasma membrane. PE could potentially encounter furin at either of these sites, or in the endosomal network during intracellular trafficking between them.
In addition to furin cleavage of the PE polypeptide backbone, separation of the A and B fragments must be preceded by the reduction of a disulfide bond between residues C265 and C287, which provides a second covalent linkage. Thus, both a reduction and a proteolysis step are necessary for PE intoxication [61]. The C265-C287 disulfide bond is buried in the crystal structure of native PE [12] and must be exposed by unfolding before it can be reduced [61]. This observation suggests that furin cleavage precedes reduction, although the order of events in vivo has not been established experimentally.
The subcellular location of the reduction event is difficult to pinpoint. The general redox state of the extracellular environment is normally more oxidizing, while the intracellular environment is more reducing (for a review see [62]), but numerous factors can influence the redox balance and different subcellular compartments can have very different redox potentials. One suggestion has been that the reduction of PE is accomplished by protein disulfide-isomerases (PDIs; EC 5.3.4.1), because in vitro experimental evidence suggests that PE can be reduced by PDIs [61]. PDIs are a family of enzymes that catalyze the formation and breakage of disulfide bonds in proteins (for a review see [63]). They are abundant in the endoplasmic reticulum (ER) and Golgi, but also found in other intracellular locations and on the cell surface [64,65]. PE could potentially encounter PDIs at every stage of the intoxication pathway. The relative abundance of PDIs in the ER, however, suggests that PE would be more likely to encounter PDIs there.
Indirect support for the involvement of PDIs in PE intoxication comes from the pathways of other protein toxins. The protein toxins ricin and cholera toxin (CT) both follow routes through the ER and into the cytosol following receptor-mediated endocytosis. In vivo and in vitro evidence supports the involvement of PDIs in a reductive separation event essential to ricin and CT [66–70]. The PDI family of proteins has additionally been associated with retrograde transport of polypeptides from the ER in the process of ER-associated degradation (ERAD), a mechanism that may be exploited by PE to reach the cytosol and is discussed later in this review.
The precise role played by intracellular processing of PE in its intoxication pathway is not entirely clear. Separation of the A and B subunits serves to activate the PE proenzyme, but RITs that do not require activation for catalytic activity still need a cleavable furin site for full activity (JW unpublished observations). Separation of the catalytic and binding domains may therefore serve an additional function, perhaps by exposing sequences in domain II necessary for intracellular trafficking. PE38 RITs retain all of domain II, including the furin cleavage site and C265-C287 disulfide bond (Fig. 1). Unlike native PE, however, separation of the catalytic and binding fragments is not always essential for cytotoxicity. The RIT HA22 (anti-CD22/PE38) remains active on CD22-positive cells even with an R279G mutation that prevents furin cleavage, although it is 3-fold less active than wild-type HA22 (JW unpublished data). The same R279G mutation in the RIT SS1-LR/GGS (anti-mesothelin/PE[LR]) is completely inactive on mesothelin-positive cells. Current research is exploring these differences. Both the cell line and the target receptor appear to play major roles in determining the outcome of intoxication.
PE in the endoplasmic reticulum
The intoxication pathways of DT and PE are remarkably similar in several respects (see [71] for a review). Both are secreted as proenzymes, internalized by receptor-mediated endocytosis, processed by furin, and reduced to separate the catalytic (A) from the binding (B) fragments. Following these steps, however, their respective pathways diverge dramatically. While DT pursues a route directly from acidified endocytic vesicles into the cytosol [72], PE follows a path through the ER.
The evidence for an ER-dependent PE intoxication pathway is extensive. It was initially observed that the R609EDL612 sequence immediately adjacent to the C-terminal residue of PE was essential for cytotoxicity [73]. Deletions in the REDL sequence of PE eliminate its cytotoxicity, but replacement with a similar sequence, KDEL, restores activity. The KDEL sequence is a well-defined ER retention and retrieval signal in mammalian cells [74] that is recognized by integral membrane proteins known as KDEL receptors (KDEL-R [75]; see [76] for a review). The subcellular localization of KDEL-R appears to be a dynamic cycle between the Golgi and the ER [77,78]. This is consistent with the proposed function of KDEL-Rs in returning to the ER proteins that have escaped into the Golgi.
The REDL C-terminal sequence of PE, which also occurs on several ER-resident proteins, is a variant of the canonical KDEL sequence and is recognized and retained in the ER by KDEL-R [79]. As anticipated, the over expression of KDEL-R1 (hERD2) sensitizes cells to PE. Conversely, cells become resistant to PE when KDEL transport is restricted by microinjected antibodies to KDEL-R1 or by expression of lysozyme-KDEL, which competes for binding to free receptor [80]. Before KDEL-R can recognize PE, however, the C-terminal residue, K613, must be removed in order to expose the REDL signal sequence. Binding to KDEL-R is seriously impaired if the terminal lysine residue is not removed [81]. The removal of K613 appears to occur early in the intoxication process, possibly by plasma carboxypeptidase(s) in the bloodstream [82].
Analysis of KDEL-R binding to oligopeptides ending with various sequences showed that the REDL native sequence of PE had nearly a 100-fold weaker affinity than the canonical KDEL sequence [81]. This result suggests that replacing the native REDL sequence with KDEL might enhance the cytotoxicity of PE-based RITs by increasing the efficiency of Golgi to ER transport, and multiple studies have supported this hypothesis [81,83]. Unfortunately, the therapeutic benefit of enhanced cytotoxicity is offset by an accompanying increase in nonspecific toxicity in laboratory animals (Robert J. Kreitman, JW and IP, unpublished results).
Based on the perturbation of different trafficking pathways, it has been suggested that PE can exploit routes to the ER other than through KDEL-R [84]. While alternative pathways to the ER certainly exist and are used by other toxins, most notably a KDEL-R-independent lipid transport route used by Shiga toxin [85,86], the evidence indicates that the vast majority of PE reaches the ER through KDEL-R. Deletion of the ER localization signal at the C-terminus of PE reduces its activity by 1000-fold or more [73]. Our experience with PE-based RITs has shown that the C-terminal ER localization sequence of PE is essential for cytotoxicity (unpublished observations). An additional mechanism has been suggested in which PE can translocate directly from acidified endocytic vesicles into the cytosol, using an approach similar to DT [87]. This proposal also conflicts with the observation that the C-terminal ER localization signal of PE is essential. It is possible that differences between cell lines may account for the conflicting experimental observations, and more work needs to be done to clarify the matter.
An exit pathway from the ER to the cytosol is suggested by the evidence for an association between PE and the Sec61p ER translocation pore [88,89]. This suggests that PE may be exported from the ER into the cytosol through the Sec61p membrane channel in a manner similar to the retrotranslocation (a.k.a. dislocation) of polypeptides destined for proteasomal degradation by luminal ER-associated degradation (ERAD-L) [90]. Presumably, this would entail a chaperone-assisted unfolding step in the ER followed by translocation and refolding in the cytosol. It is possible that processed PE and other protein toxins like CT and Shiga toxin mimic the presence of a misfolded protein in the ER in order to exploit the ERAD system for transport across the ER membrane to the cytosol [91,92]. To date, we are unaware of direct evidence for transport of PE through the Sec61p translocon.
Additional support for the hypothesis that PE exploits the ERAD system is the amino acid bias against lysine residues in its catalytic fragment [93]. Sequence analyses of the catalytic (A) fragments of PE and other protein toxins show that arginine residues are much more highly preferred over lysine when examining the occurrence of basic amino acids. Interestingly, this paradigm does not hold true for the B fragments, in which lysine residue occur with normal frequency. There are 15 total lysine residues in native PE, but only three lysines in its A fragment (residues 280-613): K590, K606, and K613. All three of these residues are located near the C-terminus of PE, and K613 must be removed to expose the C-terminal REDL ER localization signal. This suggests a selective pressure against the inclusion of lysine residues in the protein sequence of the A fragment, but not the B fragment of PE. Since only the A fragment must traffic to the cytosol for activity, the lack of lysine residues may protect it from the ubiquitin/proteasome system, the terminal step of ERAD in which proteins are targeted for degradation by polyubiquitination of lysine ε-amino groups [94]. Both ricin and abrin toxins engineered to contain additional lysine residues have shown enhanced ubiquitin-mediated proteasomal degradation [95]. PE may similarly lack lysine residues to avoid degradation in the cytosol while exploiting an ERAD transport pathway.
PE in the Cytosol
Once PE reaches the cytosol it exerts its catalytic activity on EF2. The translation factor EF2 (reviewed by [96]) is an essential component of protein synthesis, during which it catalyzes the coordinated movement of the growing polypeptide chain along the ribosome. In eukaryotes (eEF2) and archaea (aEF2), but not bacteria (EF2, formerly EF-G), the protein contains a unique and rigidly conserved post-translationally modified histidine, known as a diphthamide residue. The purpose of the diphthamide residue is unclear, but it is strictly conserved among eukaryotes and archaea. Gene knockout studies in mice have shown that enzymes in the diphthamide biosynthesis pathway are essential for normal development [97,98], but it is not clear if the diphthamide residue itself is essential. Lack of a diphthamide did not have a significant impact on the activity of aEF2 in vitro [99]. In addition, mammalian and yeast cultured cells lacking the diphthamide modification on EF2 are viable and resistant to NAD+-diphthamide ADP-ribosyltransferases, but may show effects such as temperature sensitivity and decreased growth rate [100–107]. Several hypotheses for the necessity of the diphthamide have been proposed, including its involvement in protection from ribosome-inactivating proteins like ricin [108] or preservation of translational fidelity [109], but no consensus has been reached. The existence of bacterial NAD+-diphthamide ADP-ribosyltransferases (PE, DT, and CE), however, demonstrates that bacteria have found the diphthamide residue an appealing target to differentiate themselves from archaea and eukaryotes.
Since the initial determination that PE halts protein synthesis in a manner identical to DT [110], the catalytic mechanism of PE has been extensively studied [111–117]. Several residues in domain III of PE have been identified as playing important roles in catalysis, including Glu553, His440, Tyr481, and Tyr470. Studies of the reaction itself indicate that an ADP-ribosyl group derived from NAD+ is transferred to the N3 atom of the diphthamide imidazole using a random third-order SN1 mechanism. NAD+ is cleaved to produce nicotinamide, which is released, and an ADP-ribosyl oxacarbenium ion intermediate, which contains a positively charged ribosyl group that reacts with the diphthamide imidazole N3 atom. The molecular mechanism by which the ADP-ribosylation of eEF2 halts protein synthesis remains unclear, but it seems possible that the ADP-ribose moiety interferes with an interaction between eEF2 and RNA at the diphthamide site [118].
We also do not know precisely how ADP-ribosylation of eEF2 leads to cell death, although halting translation almost certainly leads to growth inhibition and arrest. Studies that have examined cell death following treatment with PE or PE-based RITs have reported results consistent with apoptotic cell death [119–122], but little is known concerning the intermediate steps after ADP-ribosylation of eEF2 and prior to caspase activation. Recently, it was reported that apoptosis induced in mouse embryonic fibroblasts by PE or other protein synthesis inhibitors was dependent on the degradation of Mcl-1 and release of Bak [123]. The anti-apoptotic protein Mcl-1 is rapidly turned over in the cell, and inhibition of its synthesis may shift the balance of apoptotic signals towards cell death [124]. It seems possible that this mechanism could be common among different cell types and protein synthesis inhibitors.
Unanswered questions
At this point it should be clear that our understanding of PE intoxication is incomplete. One important missing element is an understanding of the role of domain II in PE intoxication. It has been suggested that domain II assists in the translocation of the toxin into the cytosol [16,87] and that it plays a role in proper folding, stability, and secretion by P. aeruginosa [125–127], but there is no consensus. Domains Ia and III have independent, experimentally verified functions that can be directly assessed, but speculation concerning the function of domain II has been made primarily by inference. Domain Ib also has no independent function, but it is structurally contiguous with domain Ia and a portion of domain Ib is functionally essential to the catalytic activity domain III. At least a portion of domain II is devoted to maintaining the covalent attachments between the A and B toxin fragments; it contains the furin protease cleavage site and flanking cysteines (Cys265-Cys287) that form a disulfide bond. It seems unlikely, however, that the entirety of domain II exists simply to provide a site for the separation of the A and B fragments.
Work on PE-based RITs has shown that the majority of domain II is not essential for activity, although it can have a large influence on cytotoxicity [46]. Depending on the cells examined and the receptor targeted, mutations that eliminate all of domain II except for the furin cleavage site can enhance, reduce, or have no impact on cytotoxicity. Eliminating the furin cleavage site by deletion or preventing cleavage with a point mutation in the site has either reduced the cytotoxicity of the RIT or completely abolished it. An explanation for these differing effects is unknown and currently under study, but it raises the issue that our understanding of the PE intoxication pathway can be complicated by the use of recombinant immunotoxins. Much of the information accumulated over years of study concerns PE-based RITs rather than native PE. Not only is the protein heavily modified from its native form, but the target receptor is also changed. This could potentially influence cytotoxicity in a variety of ways, from changing the number of receptor sites per cell to altering the rate of internalization of the receptor or influencing the intracellular trafficking. The proteome of the target cell also influences the pathway. We have observed large differences in the cytotoxicity of PE and PE-based RITs on different cell lines. The assumption that the route of trafficking is conserved following internalization in different cell lines and through different receptors is not necessarily accurate, but our understanding of PE trafficking is currently insufficient to make such distinctions.
Another unanswered question concerns the fraction of the internalized PE that productively traffics to the cytosol. Based on work with DT [128] and unpublished data from our laboratory using PE, it has been proposed that as few as one molecule of PE in the cytosol may be sufficient to kill a cell. Typically, cells in culture require treatment with concentrations of PE greater than 1000 molecules/cell (~10−16 g/cell) in order to ensure cell death. This number is close to an estimate of the toxin load/cell in a mouse xenograft tumor model. Tumor-bearing mice treated with a PE-based RIT required 400–750 molecules/cell to ensure tumor remission [129]. Together, these studies suggest that less than 1% of the internalized toxin may successfully traffic into the cytosol. The remainder appears to follow an unproductive path into lysosomes. This estimate agrees with observations of cells treated with labeled PE ([130,131] and JW, unpublished observations). The stability of the A fragment of PE in the cytosol has also not been examined, although its relative lack of lysine residues may hamper ubiquitination-dependent proteasomal degradation and enhance cytosolic stability.
Clinical trials of PE-based RITs
Although no PE-based therapies have been approved by the FDA, several have reached the point of advanced clinical trials in their development (Table 1). The examples provided in this review do not constitute an exhaustive list. At the time of this review, a search for “immunotoxin” on the NIH clinical trials database (www.clinicaltrials.gov) revealed at least 16 active studies involving PE that has been redirected to selectively eliminate cells. The majority of these trials involve PE-based RITs developed in our laboratory, and they are discussed below.
The RIT BL22 (anti-CD22/PE38) has undergone several early-phase clinical trials for the treatment of B cell malignancies [35–37]. These trials have validated the use of CD22 as a target and highlighted several potential problems with this treatment. BL22 was most effective in patients with drug-resistant hairy cell leukemia (HCL), whose response rates were 81% (25/31) in a phase I trial [35] and 69% (25/36) in a phase II trial [36]. Dose-limiting toxicity was related to a completely reversible hemolytic uremic syndrome (HUS) resulting from the destruction of red blood cells. High levels of neutralizing antibodies developed in 24% (11/46) of patients in the phase I trial and 11% (4/36) of patients in the phase II trial.
Clinical trials of BL22 have been superseded by moxetumomab pasudotox, a modified RIT whose Fv has undergone selection for enhanced CD22 affinity by phage display [38]. As previously discussed, there are at least 6 active clinical trials of moxetumomab pasudotox. Preliminary results from a phase I study in patients with relapsed or refractory HCL (trial identifier NCT00462189) show a response rate of 81% (26/32), even though neutralizing antibodies eventually developed in 44% (14/32) of patients [132]. There is a notable lack of dose-limiting toxicity due to HUS with moxetumomab pasudotox, and a maximum tolerated dose has not yet been established. An additional phase I clinical trial in pediatric patients with acute lymphoblastic leukemia (ALL) or non-Hodgkin’s lymphoma (trial identifier NCT00659425) shows activity in patients with ALL [133]. Of the ALL patients evaluated, 25% (3/12) had complete responses, 50% (6/12) had partial responses (hematologic activity), 17% (2/12) had stable disease, and 8% (1/12) had progressive disease. Two patients eventually developed high levels of neutralizing antibodies, and two patients developed a dose-limiting capillary leak syndrome.
In addition to CD22, CD25 (IL-2R α chain) has been targeted for the treatment of various leukemias and lymphomas. The anti-CD25 RIT LMB-2 has undergone a phase I clinical trial [43] showing an overall response rate of 23% (8/35), and there are at least 4 active clinical trials of LMB-2 (listed above). Immunogenicity and nonspecific toxicities continue to be problematic. Of the patients evaluated in the phase I study, 29% (10/34) showed high levels of neutralizing antibodies to PE38. Toxicities were reversible and most commonly low level transaminase elevations and mild fever. LMB-2 has also been used clinically in a partially successful effort to deplete patients of CD25+ regulatory T lymphocytes and thereby enhance the immune response to vaccination with tumor-specific antigens [134].
Another PE-based RIT that has reached clinical trials is the anti-mesothelin SS1P. Two phase I trials treating patients with mesothelioma, pancreatic cancer, or ovarian cancer have been completed [41,42], and at least 2 studies are currently active. Patient responses to SS1P were modest, with a few minor responses. Toxicities associated with treatment were typically mild. Immunogenicity appears to constitute the major obstacle to SS1P treatment. In the two studies 88% (30/34) and 75% (18/24) of patients developed high levels of neutralizing antibodies to SS1P after a single cycle of treatment. These rates were significantly higher than the immunogenicity observed when treating hematologic malignancies, possibly due to patients with blood cancers having an immune system that is compromised as a result of disease and/or prior chemotherapy. Primarily as a result of the immunogenicity, very few patients qualified to receive more than a single cycle of treatment, which might account for the low efficacy of SS1P. Preliminary results from a phase I clinical trial combining SS1P with chemotherapy to treat patients newly diagnosed with advanced-stage pleural mesothelioma (trial identifier NCT00575770) show good results [135]. SS1P is well tolerated when combined with pemetrexed and cisplatin, and 50% (7/14) of patients showed a partial response to treatment.
The future of PE-based RITs
Many obstacles have been overcome in the development of RITs for the treatment of cancer, and striking responses have been observed in many patients with HCL, but several properties of RITs still need improvement. One of the most significant problems we have encountered in the clinical trials is immune response leading to the generation of neutralizing antibodies. Immunogenicity can be a major difficulty for protein therapeutics, particularly those derived from nonhuman sources [136]. For PE-based RITs, neutralizing antibodies are a common occurrence that is a major limitation in patients with solid tumors who have an intact immune system. Antibody formation is much less of a barrier to treating patients with hematologic malignancies, whose immune systems are typically suppressed, and multiple treatment cycles can usually be given. Mouse studies show that PE38 RITs are no more immunogenic than most foreign proteins. Antibody responses typically do not occur until several weeks after the initial treatment [137–139]. Nevertheless, it is clear that lower immunogenicity would benefit PE-based RITs. This is especially apparent with SS1P; in approximately 80% of patients only a single cycle (3 doses) can be administered before the development of neutralizing antibodies.
Several strategies have been attempted to overcome the issue of immunogenicity in PE-based RITs. PEGylation is a common strategy to reduce the immunogenicity and alter the pharmacokinetics of proteins (reviewed by [140] and many others). We have PEGylated various PE RITs [141–143], but found their efficacy was greatly diminished. An alternate strategy is to treat patients with general immunosuppressive drugs concurrent with RIT therapy in order to prevent, delay, or otherwise limit the production of neutralizing antibodies. This strategy is currently being assessed clinically using LMB-2 in conjunction with fludarabine and cyclophosphamide ([40]; ClinicalTrials.gov study identifier NCT00924170), but previous attempts to reduce immunogenicity in this manner have been unsuccessful. Clinical trials using cyclophosphamide [144] or cyclosporine A [145] in combination with a ricin-based immunotoxin failed to decrease the antibody response. An attempt to treat patients with rituximab (anti-CD20 mAb) prior to treatment with a PE-based RIT also failed to suppress the antibody response [146].
A third strategy is the elimination of immunogenic epitopes in PE by mutation. The targeted removal of B cell (antibody) epitopes (reviewed by [147,148]) in PE38 has progressed the furthest [137–139,149]. This strategy has been effective at dramatically decreasing immunogenicity in mice, but much work remains to be done to ensure low immunogenicity in humans. The identification and elimination of T cell epitopes is a complementary approach [150]. We propose that the strategy of epitope elimination offers the best promise for the deimmunization of PE-based RITs.
Other potential strategies to avoid immunogenicity have a more uncertain forecast. One option is to utilize human proteins that can act as cytotoxins, since fully human proteins should theoretically not elicit an immune response in patients. Examples include proapoptotic proteins and RNase. While these “humanized” RITs [151] are generally beyond the purview of this review, the potential existence of an endogenous eEF-2 ADP-ribosyltransferase in mammals [152–155] suggests that a human equivalent to PE may exist. A RIT engineered out of such a protein might be non-immunogenic, but it must first be identified and characterized.
Strategies to enhance the efficacy of RITs might also be practical. If only a single cycle of treatment is required to achieve a complete tumor remission, the appearance of neutralizing antibodies following that cycle of treatment might be irrelevant to the outcome. One strategy is to combine PE with other chemotherapeutic drugs to achieve a synergistic effect without nonspecific toxicity. Combination therapy with standard chemotherapy drugs has shown promise in preclinical studies of SS1P [156–158], and clinical trials are currently underway to test SS1P within a combination therapy regimen (ClinicalTrials.gov study identifiers NCT01041118 and NCT00575770). Research into the observed synergy suggests that it may be a result of both chemotherapy-induced tumor cell depletion and lower levels of free mesothelin shed into the extracellular space [158]. Free mesothelin competes with cell surface mesothelin for SS1P, acting as an unproductive sink for the immunotoxin [159]. Other potential combination drugs are those that could act at specific stages in the PE intoxication pathway to enhance productive intoxication, or those that can enhance the initiation of apoptotic cell death in PE-treated cells. For example, a pro-apoptotic BH3 domain mimetic has recently been shown to synergistically enhance the cytotoxicity of PE-based recombinant immunotoxins [160].
Further engineering of PE to enhance productive intoxication might also be possible. As previously described, the initial development of immunotoxins utilized full-length native PE chemically conjugated to mAbs. Improved protein engineering techniques permitted the combination of PE and mAb fragments (PE38 and Fv) into RITs, which were more efficient to produce and highly active with fewer side effects. Variant RITs have subsequently been developed to enhance ER trafficking by replacing the C-terminal residues with a KDEL sequence [161] and to limit endolysosomal proteolytic degradation by deleting protease-sensitive regions [46]. Other potential engineering targets include the furin cleavage site of PE, which is remarkably inefficiently cleaved by furin [162] and may benefit from enhanced cleavage efficiency.
Additional obstacles exist outside of PE and its intoxication pathway. Target selection and the targeting element are at least as important as the toxin portion of RITs. A RIT’s ability to discriminate between normal and malignant cells is fundamental to its success, making the identification and validation of a target the most important stage in their early development. In addition to selectivity, factors such as receptor site density, internalization rate, and internalization route can influence RIT efficacy. For example, both CD19 and CD22 represent excellent targets for RITs in the selective elimination of mature B cells and associated malignancies. Although CD19 is more heavily expressed than CD22, CD22 internalizes much more rapidly and is a much better target for PE-based RITs [163]. The stability of the targeting element is also essential. The dsFv immunotoxin variants are typically much more stable than the scFv molecules originally developed for RITs, and are thus more clinically useful [25]. A detailed discussion of receptor targeting is beyond the scope of this review, but it plays an essential role in the therapeutic efficacy of RITs.
Concluding remarks
Substantial progress has been made in the development of PE-based therapeutics over the past 30 years. Initial tentative steps to transform a potent bacterial toxin into a selective agent for the elimination of cells have become purposeful strides to generate the immunotoxins of today and, we anticipate, the medicines of tomorrow. Advances in our understanding of PE and its intoxication pathway have fueled the translation of basic research into clinical therapies that have the opportunity to make a large positive impact on human health. High expectations should be tempered by the realization that obstacles remain to be overcome for these RITs to achieve their maximum potential. Many of the details of the PE intoxication process remain uncertain, and must be addressed before we can claim success. Future advances in PE therapeutics will be dependent on a clear and comprehensive grasp of PE and its mechanism.
Acknowledgments
The authors would like to thank David Fitzgerald and Dawn Walker for their helpful comments during the preparation of this manuscript. This work is supported in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. Work on BL22 and moxetumomab pasudotox is supported in part by MedImmune, LLC under a Cooperative Research and Development Agreement.
Abbreviations
- ALL
acute lymphoblastic leukemia
- CE
cholera exotoxin
- CT
cholera toxin
- dsFv
disulfide-stabilized variable fragment
- DT
diphtheria toxin
- aEF2
archaeal translation elongation factor 2
- eEF2
eukaryotic translation elongation factor 2
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- ERAD-L
luminal ERAD
- Fv
variable fragment
- HCL
hairy cell leukemia
- HUS
hemolytic uremic syndrome
- IL-2
interleukin-2
- IL-2R
IL-2 receptor
- KDEL-R
KDEL receptor
- LRP1
low density lipoprotein receptor related protein 1
- LRP1B
low density lipoprotein receptor related protein 1B
- PDIs
protein disulfide-isomerases
- PE
Pseudomonas exotoxin A
- RITs
recombinant immunotoxins
- scFv
single-chain Fv
References
- 1.Pugh MB, et al., editors. Stedman’s Medical Dictionary. 28. Lippincott Williams & Wilkins; Baltimore, MD: 2006. [Google Scholar]
- 2.Gill DM. Bacterial toxins: a table of lethal amounts. Microbiol Rev. 1982;46:86–94. doi: 10.1128/mr.46.1.86-94.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murray PR, Rosenthal KS, Pfaller MA, editors. Medical Microbiology. 6. Elsevier, Inc; Philadelphia, PA: 2009. [Google Scholar]
- 4.Naglich JG, Metherall JE, Russell DW, Eidels L. Expression cloning of a diphtheria toxin receptor: identity with a heparin-binding EGF-like growth factor precursor. Cell. 1992;69:1051–1061. doi: 10.1016/0092-8674(92)90623-k. [DOI] [PubMed] [Google Scholar]
- 5.Manoukian G, Hagemeister F. Denileukin diftitox: a novel immunotoxin. Expert Opin Biol Ther. 2009;9:1445–1451. doi: 10.1517/14712590903348135. [DOI] [PubMed] [Google Scholar]
- 6.Kadin ME, Vonderheid EC. Targeted therapies: Denileukin diftitox--a step towards a ‘magic bullet’ for CTCL. Nat Rev Clin Oncol. 2010;7:430–432. doi: 10.1038/nrclinonc.2010.105. [DOI] [PubMed] [Google Scholar]
- 7.Jørgensen R, Purdy AE, Fieldhouse RJ, Kimber MS, Bartlett DH, Merrill AR. Cholix toxin, a novel ADP-ribosylating factor from Vibrio cholerae. J Biol Chem. 2008;283:10671–10678. doi: 10.1074/jbc.M710008200. [DOI] [PubMed] [Google Scholar]
- 8.Sarnovsky R, Tendler T, Makowski M, Kiley M, Antignani A, Traini R, Zhang J, Hassan R, FitzGerald DJ. Initial characterization of an immunotoxin constructed from domains II and III of cholera exotoxin. Cancer Immunol Immunother. 2010;59:737–746. doi: 10.1007/s00262-009-0794-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Driscoll JA, Brody SL, Kollef MH. The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs. 2007;67:351–368. doi: 10.2165/00003495-200767030-00003. [DOI] [PubMed] [Google Scholar]
- 10.Liu PV. The roles of various fractions of Pseudomonas aeruginosa in its pathogenesis. 3. Identity of the lethal toxins produced in vitro and in vivo. J Infect Dis. 1966;116:481–489. doi: 10.1093/infdis/116.4.481. [DOI] [PubMed] [Google Scholar]
- 11.Iglewski BH. In: Pseudomonas in Medical Microbiology. 4. Barson S, editor. University of Texas Medical Branch at Galveston; Galveston, TX: 1966. [Google Scholar]
- 12.Allured VS, Collier RJ, Carroll SF, McKay DB. Structure of exotoxin A of Pseudomonas aeruginosa at 3.0-Angstrom resolution. Proc Natl Acad Sci USA. 1986;83:1320–1324. doi: 10.1073/pnas.83.5.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Humphrey W, Dalke A, Schulten K. VMD - Visual Molecular Dynamics. J Molec Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 14.Kounnas MZ, Morris RE, Thompson MR, FitzGerald DJ, Strickland DK, Saelinger CB. The alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein binds and internalizes Pseudomonas exotoxin A. J Biol Chem. 1992;267:12420–12423. [PubMed] [Google Scholar]
- 15.Pastrana DV, Hanson AJ, Knisely J, Bu G, Fitzgerald DJ. LRP 1 B functions as a receptor for Pseudomonas exotoxin. Biochim Biophys Acta. 2005;1741:234–239. doi: 10.1016/j.bbadis.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 16.Hwang J, Fitzgerald DJ, Adhya S, Pastan I. Functional domains of Pseudomonas exotoxin identified by deletion analysis of the gene expressed in E. coli. Cell. 1987;48:129–136. doi: 10.1016/0092-8674(87)90363-1. [DOI] [PubMed] [Google Scholar]
- 17.Siegall CB, Chaudhary VK, FitzGerald DJ, Pastan I. Functional analysis of domains II, Ib, and III of Pseudomonas exotoxin. J Biol Chem. 1989;264:14256–14261. [PubMed] [Google Scholar]
- 18.Kihara A, Pastan I. Analysis of sequences required for the cytotoxic action of a chimeric toxin composed of Pseudomonas exotoxin and transforming growth factor alpha. Bioconjug Chem. 1994;5:532–538. doi: 10.1021/bc00030a008. [DOI] [PubMed] [Google Scholar]
- 19.FitzGerald DJ, Padmanabhan R, Pastan I, Willingham MC. Adenovirus-induced release of epidermal growth factor and Pseudomonas toxin into the cytosol of KB cells during receptor-mediated endocytosis. Cell. 1983;32:607–617. doi: 10.1016/0092-8674(83)90480-4. [DOI] [PubMed] [Google Scholar]
- 20.FitzGerald DJ, Trowbridge IS, Pastan I, Willingham MC. Enhancement of toxicity of antitransferrin receptor antibody-Pseudomonas exotoxin conjugates by adenovirus. Proc Natl Acad Sci USA. 1983;80:4134–4138. doi: 10.1073/pnas.80.13.4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chaudhary VK, FitzGerald DJ, Adhya S, Pastan I. Activity of a recombinant fusion protein between transforming growth factor type alpha and Pseudomonas toxin. Proc Natl Acad Sci USA. 1987;84:4538–4542. doi: 10.1073/pnas.84.13.4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chaudhary VK, Queen C, Junghans RP, Waldmann TA, FitzGerald DJ, Pastan I. A recombinant immunotoxin consisting of two antibody variable domains fused to Pseudomonas exotoxin. Nature. 1989;339:394–397. doi: 10.1038/339394a0. [DOI] [PubMed] [Google Scholar]
- 23.Bird RE, Hardman KD, Jacobson JW, Johnson S, Kaufman BM, Lee SM, Lee T, Pope SH, Riordan GS, Whitlow M. Single-chain antigen-binding proteins. Science. 1988;242:423–426. doi: 10.1126/science.3140379. (Erratum: 1989, Science 244, 409) [DOI] [PubMed] [Google Scholar]
- 24.Huston JS, Levinson D, Mudgett-Hunter M, Tai MS, Novotný J, Margolies MN, Ridge RJ, Bruccoleri RE, Haber E, Crea R, Oppermann H. Protein engineering of antibody binding sites: recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc Natl Acad Sci USA. 1988;85:5879–5883. doi: 10.1073/pnas.85.16.5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brinkmann U, Reiter Y, Jung SH, Lee B, Pastan I. A recombinant immunotoxin containing a disulfide-stabilized Fv fragment. Proc Natl Acad Sci USA. 1993;90:7538–7542. doi: 10.1073/pnas.90.16.7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reiter Y, Brinkmann U, Kreitman RJ, Jung SH, Lee B, Pastan I. Stabilization of the Fv fragments in recombinant immunotoxins by disulfide bonds engineered into conserved framework regions. Biochemistry. 1994;33:5451–5459. doi: 10.1021/bi00184a014. [DOI] [PubMed] [Google Scholar]
- 27.Reiter Y, Brinkmann U, Webber KO, Jung SH, Lee B, Pastan I. Engineering interchain disulfide bonds into conserved framework regions of Fv fragments: improved biochemical characteristics of recombinant immunotoxins containing disulfide-stabilized Fv. Protein Eng. 1994;7:697–704. doi: 10.1093/protein/7.5.697. [DOI] [PubMed] [Google Scholar]
- 28.Pastan I, Beers R, Bera TK. Recombinant immunotoxins in the treatment of cancer. Methods Mol Biol. 2004;248:503–518. doi: 10.1385/1-59259-666-5:503. [DOI] [PubMed] [Google Scholar]
- 29.Kreitman RJ, Siegall CB, Chaudhary VK, FitzGerald DJ, Pastan I. Properties of chimeric toxins with two recognition domains: interleukin 6 and transforming growth factor alpha at different locations in Pseudomonas exotoxin. Bioconjug Chem. 1992;3:63–68. doi: 10.1021/bc00013a010. [DOI] [PubMed] [Google Scholar]
- 30.Kuan CT, Wakiya K, Keir ST, Li J, Herndon JE, 2nd, Pastan I, Bigner DD. Affinity-matured anti-glycoprotein NMB recombinant immunotoxins targeting malignant gliomas and melanomas. Int J Cancer. 2010 doi: 10.1002/ijc.25645. [Epub ahead of print-PMID: 20824708] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berger EA, Pastan I. Immunotoxin complementation of HAART to deplete persisting HIV-infected cell reservoirs. PLoS Pathog. 2010;6:e1000803. doi: 10.1371/journal.ppat.1000803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kennedy PE, Bera TK, Wang QC, Gallo M, Wagner W, Lewis MG, Berger EA, Pastan I. Anti-HIV-1 immunotoxin 3B3(Fv)-PE38: enhanced potency against clinical isolates in human PBMCs and macrophages, and negligible hepatotoxicity in macaques. J Leukoc Biol. 2006;80:1175–1182. doi: 10.1189/jlb.0306139. [DOI] [PubMed] [Google Scholar]
- 33.Onda M, Olafsen T, Tsutsumi Y, Bruland OS, Pastan I. Cytotoxicity of antiosteosarcoma recombinant immunotoxins composed of TP-3 Fv fragments and a truncated Pseudomonas exotoxin A. J Immunother. 2001;24:144–150. [PubMed] [Google Scholar]
- 34.Onda M, Bruland ØS, Pastan I. TP-3 immunotoxins improve antitumor activity in mice with osteosarcoma. Clin Orthop Relat Res. 2005;430:142–148. doi: 10.1097/01.blo.0000137544.30200.b6. [DOI] [PubMed] [Google Scholar]
- 35.Kreitman RJ, Squires DR, Stetler-Stevenson M, Noel P, FitzGerald DJ, Wilson WH, Pastan I. Phase I trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with B-cell malignancies. J Clin Oncol. 2005;23:6719–6729. doi: 10.1200/JCO.2005.11.437. [DOI] [PubMed] [Google Scholar]
- 36.Kreitman RJ, Stetler-Stevenson M, Margulies I, Noel P, Fitzgerald DJ, Wilson WH, Pastan I. Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. J Clin Oncol. 2009;27:2983–2990. doi: 10.1200/JCO.2008.20.2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wayne AS, Kreitman RJ, Findley HW, Lew G, Delbrook C, Steinberg SM, Stetler-Stevenson M, Fitzgerald DJ, Pastan I. Anti-CD22 immunotoxin RFB4(dsFv)-PE38 (BL22) for CD22-positive hematologic malignancies of childhood: preclinical studies and phase I clinical trial. Clin Cancer Res. 2010;16:1894–1903. doi: 10.1158/1078-0432.CCR-09-2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salvatore G, Beers R, Margulies I, Kreitman RJ, Pastan I. Improved cytotoxic activity toward cell lines and fresh leukemia cells of a mutant anti-CD22 immunotoxin obtained by antibody phage display. Clin Cancer Res. 2002;8:995–1002. [PubMed] [Google Scholar]
- 39.Alderson RF, Kreitman RJ, Chen T, Yeung P, Herbst R, Fox JA, Pastan I. CAT-8015: a second-generation Pseudomonas exotoxin A-based immunotherapy targeting CD22-expressing hematologic malignancies. Clin Cancer Res. 2009;15:832–839. doi: 10.1158/1078-0432.CCR-08-1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kreitman RJ. Recombinant immunotoxins for the treatment of chemoresistant hematologic malignancies. Curr Pharm Des. 2009;15:2652–2664. doi: 10.2174/138161209788923949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hassan R, Bullock S, Premkumar A, Kreitman RJ, Kindler H, Willingham MC, Pastan I. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin Cancer Res. 13:5144–5149. doi: 10.1158/1078-0432.CCR-07-0869. [DOI] [PubMed] [Google Scholar]
- 42.Kreitman RJ, Hassan R, Fitzgerald DJ, Pastan I. Phase I trial of continuous infusion anti-mesothelin recombinant immunotoxin SS1P. Clin Cancer Res. 2009;15:5274–5279. doi: 10.1158/1078-0432.CCR-09-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kreitman RJ, Wilson WH, White JD, Stetler-Stevenson M, Jaffe ES, Giardina S, Waldmann TA, Pastan I. Phase I trial of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) in patients with hematologic malignancies. J Clin Oncol. 2000;18:1622–1636. doi: 10.1200/JCO.2000.18.8.1622. [DOI] [PubMed] [Google Scholar]
- 44.Wolf P, Elsässer-Beile U. Pseudomonas exotoxin A: from virulence factor to anti-cancer agent. Int J Med Microbiol. 2009;299:161–176. doi: 10.1016/j.ijmm.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 45.Shapira A, Benhar I. Toxin-based therapeutic approaches. Toxins. 2010;2:2519–2583. doi: 10.3390/toxins2112519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weldon JE, Xiang L, Chertov O, Margulies I, Kreitman RJ, Fitzgerald DJ, Pastan I. A protease-resistant immunotoxin against CD22 with greatly increased activity against CLL and diminished animal toxicity. Blood. 2009;113:3792–3800. doi: 10.1182/blood-2008-08-173195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leppla SH, Martin OC, Muehl LA. The exotoxin P. aeruginosa: a proenzyme having an unusual mode of activation. Biochem Biophys Res Commun. 1978;81:532–538. doi: 10.1016/0006-291x(78)91567-x. [DOI] [PubMed] [Google Scholar]
- 48.Wedekind JE, Trame CB, Dorywalska M, Koehl P, Raschke TM, McKee M, FitzGerald D, Collier RJ, McKay DB. Refined crystallographic structure of Pseudomonas aeruginosa exotoxin A and its implications for the molecular mechanism of toxicity. J Mol Biol. 2001;314:823–837. doi: 10.1006/jmbi.2001.5195. [DOI] [PubMed] [Google Scholar]
- 49.Ogata M, Chaudhary VK, Pastan I, FitzGerald DJ. Processing of Pseudomonas exotoxin by a cellular protease results in the generation of a 37,000-Da toxin fragment that is translocated to the cytosol. J Biol Chem. 1990;265:20678–20685. [PubMed] [Google Scholar]
- 50.Ogata M, Fryling CM, Pastan I, FitzGerald DJ. Cell-mediated cleavage of Pseudomonas exotoxin between Arg279 and Gly280 generates the enzymatically active fragment which translocates to the cytosol. J Biol Chem. 1992;267:25396–25401. [PubMed] [Google Scholar]
- 51.Fryling C, Ogata M, FitzGerald D. Characterization of a cellular protease that cleaves Pseudomonas exotoxin. Infect Immun. 1992;60:497–502. doi: 10.1128/iai.60.2.497-502.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moehring JM, Inocencio NM, Robertson BJ, Moehring TJ. Expression of mouse furin in a Chinese hamster cell resistant to Pseudomonas exotoxin A and viruses complements the genetic lesion. J Biol Chem. 1993;268:2590–2594. [PubMed] [Google Scholar]
- 53.Chiron MF, Fryling CM, FitzGerald DJ. Cleavage of pseudomonas exotoxin and diphtheria toxin by a furin-like enzyme prepared from beef liver. J Biol Chem. 1994;269:18167–18176. [PubMed] [Google Scholar]
- 54.Inocencio NM, Moehring JM, Moehring TJ. Furin activates Pseudomonas exotoxin A by specific cleavage in vivo and in vitro. J Biol Chem. 1994;269:31831–31835. [PubMed] [Google Scholar]
- 55.Gordon VM, Klimpel KR, Arora N, Henderson MA, Leppla SH. Proteolytic activation of bacterial toxins by eukaryotic cells is performed by furin and by additional cellular proteases. Infect Immun. 1995;63:82–87. doi: 10.1128/iai.63.1.82-87.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gu M, Gordon VM, Fitzgerald DJ, Leppla SH. Furin regulates both the activation of Pseudomonas exotoxin A and the Quantity of the toxin receptor expressed on target cells. Infect Immun. 1996;64:524–527. doi: 10.1128/iai.64.2.524-527.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sarac MS, Cameron A, Lindberg I. The furin inhibitor hexa-D-arginine blocks the activation of Pseudomonas aeruginosa exotoxin A in vivo. Infect Immun. 2002;70:7136–7139. doi: 10.1128/IAI.70.12.7136-7139.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shiryaev SA, Remacle AG, Ratnikov BI, Nelson NA, Savinov AY, Wei G, Bottini M, Rega MF, Parent A, Desjardins R, Fugere M, Day R, Sabet M, Pellecchia M, Liddington RC, Smith JW, Mustelin T, Guiney DG, Lebl M, Strongin AY. Targeting host cell furin proprotein convertases as a therapeutic strategy against bacterial toxins and viral pathogens. J Biol Chem. 2007;282:20847–20853. doi: 10.1074/jbc.M703847200. [DOI] [PubMed] [Google Scholar]
- 59.Ornatowski W, Poschet JF, Perkett E, Taylor-Cousar JL, Deretic V. Elevated furin levels in human cystic fibrosis cells result in hypersusceptibility to exotoxin A-induced cytotoxicity. J Clin Invest. 2007;117:3489–3497. doi: 10.1172/JCI31499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thomas G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol. 2002;3:753–766. doi: 10.1038/nrm934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McKee ML, FitzGerald DJ. Reduction of furin-nicked Pseudomonas exotoxin A: an unfolding story. Biochemistry. 1999;38:16507–16513. doi: 10.1021/bi991308+. [DOI] [PubMed] [Google Scholar]
- 62.Chaiswing L, Oberley TD. Extracellular/microenvironmental redox state. Antioxid Redox Signal. 2010;13:449–465. doi: 10.1089/ars.2009.3020. [DOI] [PubMed] [Google Scholar]
- 63.Appenzeller-Herzog C, Ellgaard L. The human PDI family: versatility packed into a single fold. Biochim Biophys Acta. 2008;1783:535–548. doi: 10.1016/j.bbamcr.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 64.Turano C, Coppari S, Altieri F, Ferraro A. Proteins of the PDI family: unpredicted non-ER locations and functions. J Cell Physiol. 2002;193:154–163. doi: 10.1002/jcp.10172. [DOI] [PubMed] [Google Scholar]
- 65.Kimura T, Horibe T, Sakamoto C, Shitara Y, Fujiwara F, Komiya T, Yamamoto A, Hayano T, Takahashi N, Kikuchi M. Evidence for mitochondrial localization of P5, a member of the protein disulphide isomerase family. J Biochem. 2008;144:187–196. doi: 10.1093/jb/mvn057. [DOI] [PubMed] [Google Scholar]
- 66.Orlandi PA. Protein-disulfide isomerase-mediated reduction of the A subunit of cholera toxin in a human intestinal cell line. J Biol Chem. 1997;272:4591–4599. [PubMed] [Google Scholar]
- 67.Majoul I, Ferrari D, Söling HD. Reduction of protein disulfide bonds in an oxidizing environment. The disulfide bridge of cholera toxin A-subunit is reduced in the endoplasmic reticulum. FEBS Lett. 1997;401:104–108. doi: 10.1016/s0014-5793(96)01447-0. [DOI] [PubMed] [Google Scholar]
- 68.Tsai B, Rodighiero C, Lencer WI, Rapoport TA. Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell. 2001;104:937–948. doi: 10.1016/s0092-8674(01)00289-6. [DOI] [PubMed] [Google Scholar]
- 69.Spooner RA, Watson PD, Marsden CJ, Smith DC, Moore KA, Cook JP, Lord JM, Roberts LM. Protein disulphide-isomerase reduces ricin to its A and B chains in the endoplasmic reticulum. Biochem J. 2004;383:285–293. doi: 10.1042/BJ20040742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moore P, Bernardi KM, Tsai B. The Ero1alpha-PDI redox cycle regulates retro-translocation of cholera toxin. Mol Biol Cell. 2010;21:1305–1313. doi: 10.1091/mbc.E09-09-0826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Watson P, Spooner RA. Toxin entry and trafficking in mammalian cells. Adv Drug Deliv Rev. 2010;58:1581–1596. doi: 10.1016/j.addr.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 72.Beaumelle B, Bensammar L, Bienvenüe A. Selective translocation of the A chain of diphtheria toxin across the membrane of purified endosomes. J Biol Chem. 1992;267:11525–11531. [PubMed] [Google Scholar]
- 73.Chaudhary VK, Jinno Y, FitzGerald D, Pastan I. Pseudomonas exotoxin contains a specific sequence at the carboxyl terminus that is required for cytotoxicity. Proc Natl Acad Sci USA. 1990;87:308–312. doi: 10.1073/pnas.87.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Munro S, Pelham HR. A C-terminal signal prevents secretion of luminal ER proteins. Cell. 1987;48:899–907. doi: 10.1016/0092-8674(87)90086-9. [DOI] [PubMed] [Google Scholar]
- 75.Lewis MJ, Pelham HR. A human homologue of the yeast HDEL receptor. Nature. 1990;348:162–163. doi: 10.1038/348162a0. [DOI] [PubMed] [Google Scholar]
- 76.Capitani M, Sallese M. The KDEL receptor: new functions for an old protein. FEBS Lett. 2009;583:3863–3871. doi: 10.1016/j.febslet.2009.10.053. [DOI] [PubMed] [Google Scholar]
- 77.Tang BL, Wong SH, Qi XL, Low SH, Hong W. Molecular cloning, characterization, subcellular localization and dynamics of p23, the mammalian KDEL receptor. J Cell Biol. 1993;120:325–338. doi: 10.1083/jcb.120.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Griffiths G, Ericsson M, Krijnse-Locker J, Nilsson T, Goud B, Soling HD, Tang BL, Wong SH, Hong W. Localization of the Lys, Asp, Glu, Leu tetrapeptide receptor to the Golgi complex and the intermediate compartment in mammalian cells. J Cell Biol. 1994;127:1557–1574. doi: 10.1083/jcb.127.6.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Raykhel I, Alanen H, Salo K, Jurvansuu J, Nguyen VD, Latva-Ranta M, Ruddock L. A molecular specificity code for the three mammalian KDEL receptors. J Cell Biol. 2007;179:1193–1204. doi: 10.1083/jcb.200705180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jackson ME, Simpson JC, Girod A, Pepperkok R, Roberts LM, Lord JM. The KDEL retrieval system is exploited by Pseudomonas exotoxin A, but not by Shiga-like toxin-1, during retrograde transport from the Golgi complex to the endoplasmic reticulum. J Cell Sci. 1999;112:467–475. doi: 10.1242/jcs.112.4.467. [DOI] [PubMed] [Google Scholar]
- 81.Kreitman RJ, Pastan I. Importance of the glutamate residue of KDEL in increasing the cytotoxicity of Pseudomonas exotoxin derivatives and for increased binding to the KDEL receptor. Biochem J. 1995;307:29–37. doi: 10.1042/bj3070029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hessler JL, Kreitman RJ. An early step in Pseudomonas exotoxin action is removal of the terminal lysine residue, which allows binding to the KDEL receptor. Biochemistry. 1997;36:14577–14582. doi: 10.1021/bi971447w. [DOI] [PubMed] [Google Scholar]
- 83.Seetharam S, Chaudhary VK, FitzGerald D, Pastan I. Increased cytotoxic activity of Pseudomonas exotoxin and two chimeric toxins ending in KDEL. J Biol Chem. 1991;266:17376–17381. [PubMed] [Google Scholar]
- 84.Smith DC, Spooner RA, Watson PD, Murray JL, Hodge TW, Amessou M, Johannes L, Lord JM, Roberts LM. Internalized Pseudomonas exotoxin A can exploit multiple pathways to reach the endoplasmic reticulum. Traffic. 2006;7:379–393. doi: 10.1111/j.1600-0854.2006.00391.x. [DOI] [PubMed] [Google Scholar]
- 85.Mallard F, Antony C, Tenza D, Salamero J, Goud B, Johannes L. Direct pathway from early/recycling endosomes to the Golgi apparatus revealed through the study of shiga toxin B-fragment transport. J Cell Biol. 1998;143:973–990. doi: 10.1083/jcb.143.4.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Girod A, Storrie B, Simpson JC, Johannes L, Goud B, Roberts LM, Lord JM, Nilsson T, Pepperkok R. Evidence for a COP-I-independent transport route from the Golgi complex to the endoplasmic reticulum. Nat Cell Biol. 1999;1:423–430. doi: 10.1038/15658. [DOI] [PubMed] [Google Scholar]
- 87.Méré J, Morlon-Guyot J, Bonhoure A, Chiche L, Beaumelle B. Acid-triggered membrane insertion of Pseudomonas exotoxin A involves an original mechanism based on pH-regulated tryptophan exposure. J Biol Chem. 2005;280:21194–21201. doi: 10.1074/jbc.M412656200. [DOI] [PubMed] [Google Scholar]
- 88.Koopmann JO, Albring J, Hüter E, Bulbuc N, Spee P, Neefjes J, Hämmerling GJ, Momburg F. Export of antigenic peptides from the endoplasmic reticulum intersects with retrograde protein translocation through the Sec61p channel. Immunity. 2000;13:117–127. doi: 10.1016/s1074-7613(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 89.Wirth A, Jung M, Bies C, Frien M, Tyedmers J, Zimmermann R, Wagner R. The Sec61p complex is a dynamic precursor activated channel. Mol Cell. 2003;12:261–268. doi: 10.1016/s1097-2765(03)00283-1. [DOI] [PubMed] [Google Scholar]
- 90.Willer M, Forte GM, Stirling CJ. Sec61p is required for ERAD-L: genetic dissection of the translocation and ERAD-L functions of Sec61p using novel derivatives of CPY. J Biol Chem. 2008;283:33883–33888. doi: 10.1074/jbc.M803054200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hazes B, Read RJ. Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry. 1997;36:11051–11054. doi: 10.1021/bi971383p. [DOI] [PubMed] [Google Scholar]
- 92.Lord JM, Roberts LM, Lencer WI. Entry of protein toxins into mammalian cells by crossing the endoplasmic reticulum membrane: co-opting basic mechanisms of endoplasmic reticulum-associated degradation. Curr Top Microbiol Immunol. 2005;300:149–168. doi: 10.1007/3-540-28007-3_7. [DOI] [PubMed] [Google Scholar]
- 93.London E, Luongo CL. Domain-specific bias in arginine/lysine usage by protein toxins. Biochem Biophys Res Commun. 1989;160:333–339. doi: 10.1016/0006-291x(89)91660-4. [DOI] [PubMed] [Google Scholar]
- 94.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 95.Deeks ED, Cook JP, Day PJ, Smith DC, Roberts LM, Lord JM. The low lysine content of ricin A chain reduces the risk of proteolytic degradation after translocation from the endoplasmic reticulum to the cytosol. Biochemistry. 2002;41:3405–3413. doi: 10.1021/bi011580v. [DOI] [PubMed] [Google Scholar]
- 96.Merrick WC, Nyborg J. Protein Bioynthesis Elongation Cycle. In: Sonenberg N, Hershey JWB, Mathews MB, editors. Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2000. pp. 89–126. [Google Scholar]
- 97.Liu S, Wiggins JF, Sreenath T, Kulkarni AB, Ward JM, Leppla SH. Dph3, a small protein required for diphthamide biosynthesis, is essential in mouse development. Mol Cell Biol. 2006;26:3835–3841. doi: 10.1128/MCB.26.10.3835-3841.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Webb TR, Cross SH, McKie L, Edgar R, Vizor L, Harrison J, Peters J, Jackson IJ. Diphthamide modification of eEF2 requires a J-domain protein and is essential for normal development. J Cell Sci. 2008;121:3140–3145. doi: 10.1242/jcs.035550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.De Vendittis E, Amatruda MR, Raimo G, Bocchini V. Heterologous expression in Escherichia coli of the gene encoding an archaeal thermoacidophilic elongation factor 2. Properties of the recombinant protein. Biochimie. 1997;79:303–308. doi: 10.1016/s0300-9084(97)83518-3. [DOI] [PubMed] [Google Scholar]
- 100.Foley BT, Moehring JM, Moehring TJ. A mutation in codon 717 of the CHO-K1 elongation factor 2 gene prevents the first step in the biosynthesis of diphthamide. Somat Cell Mol Genet. 1992;18:227–231. doi: 10.1007/BF01233859. [DOI] [PubMed] [Google Scholar]
- 101.Foley BT, Moehring JM, Moehring TJ. Mutations in the elongation factor 2 gene which confer resistance to diphtheria toxin and Pseudomonas exotoxin A. Genetic and biochemical analyses. J Biol Chem. 1995;270:23218–23225. doi: 10.1074/jbc.270.39.23218. [DOI] [PubMed] [Google Scholar]
- 102.Kimata Y, Harashima S, Kohno K. Expression of non-ADP-ribosylatable, diphtheria toxin-resistant elongation factor 2 in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 1993;191:1145–1151. doi: 10.1006/bbrc.1993.1336. [DOI] [PubMed] [Google Scholar]
- 103.Phan LD, Perentesis JP, Bodley JW. Saccharomyces cerevisiae elongation factor 2. Mutagenesis of the histidine precursor of diphthamide yields a functional protein that is resistant to diphtheria toxin. J Biol Chem. 1993;268:8665–8668. [PubMed] [Google Scholar]
- 104.Kimata Y, Kohno K. Elongation factor 2 mutants deficient in diphthamide formation show temperature-sensitive cell growth. J Biol Chem. 1994;269:13497–13501. [PubMed] [Google Scholar]
- 105.Liu S, Milne GT, Kuremsky JG, Fink GR, Leppla SH. Identification of the proteins required for biosynthesis of diphthamide, the target of bacterial ADP-ribosylating toxins on translation elongation factor 2. Mol Cell Biol. 2004;24:9487–9497. doi: 10.1128/MCB.24.21.9487-9497.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ivankovic M, Rubelj I, Matulic M, Reich E, Brdar B. Site-specific mutagenesis of the histidine precursor of diphthamide in the human elongation factor-2 gene confers resistance to diphtheria toxin. Mutat Res. 2006;609:34–42. doi: 10.1016/j.mrgentox.2006.06.027. [DOI] [PubMed] [Google Scholar]
- 107.Roy V, Ghani K, Caruso M. A dominant-negative approach that prevents diphthamide formation confers resistance to Pseudomonas exotoxin A and diphtheria toxin. PLoS One. 2010;5:e15753. doi: 10.1371/journal.pone.0015753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gupta PK, Liu S, Batavia MP, Leppla SH. The diphthamide modification on elongation factor-2 renders mammalian cells resistant to ricin. Cell Microbiol. 2008;10:1687–1694. doi: 10.1111/j.1462-5822.2008.01159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ortiz PA, Ulloque R, Kihara GK, Zheng H, Kinzy TG. Translation elongation factor 2 anticodon mimicry domain mutants affect fidelity and diphtheria toxin resistance. J Biol Chem. 2006;281:32639–32648. doi: 10.1074/jbc.M607076200. [DOI] [PubMed] [Google Scholar]
- 110.Iglewski BH, Kabat D. NAD-dependent inhibition of protein synthesis by Pseudomonas aeruginosa toxin. Proc Natl Acad Sci USA. 1975;72:2284–2288. doi: 10.1073/pnas.72.6.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Van Ness BG, Howard JB, Bodley JW. ADP-ribosylation of elongation factor 2 by diphtheria toxin. NMR spectra and proposed structures of ribosyl-diphthamide and its hydrolysis products. J Biol Chem. 1980;255:10710–10716. [PubMed] [Google Scholar]
- 112.Wilson BA, Collier RJ. Diphtheria toxin and Pseudomonas aeruginosa exotoxin A: active-site structure and enzymic mechanism. Curr Top Microbiol Immunol. 1992;175:27–41. doi: 10.1007/978-3-642-76966-5_2. [DOI] [PubMed] [Google Scholar]
- 113.Beattie BK, Prentice GA, Merrill AR. Investigation into the catalytic role for the tryptophan residues within domain III of Pseudomonas aeruginosa exotoxin A. Biochemistry. 1996;35:15134–15142. doi: 10.1021/bi961985t. [DOI] [PubMed] [Google Scholar]
- 114.Armstrong S, Yates SP, Merrill AR. Insight into the catalytic mechanism of Pseudomonas aeruginosa exotoxin A. Studies of toxin interaction with eukaryotic elongation factor-2. J Biol Chem. 2002;277:46669–46675. doi: 10.1074/jbc.M206916200. [DOI] [PubMed] [Google Scholar]
- 115.Armstrong S, Merrill AR. Toward the elucidation of the catalytic mechanism of the mono-ADP-ribosyltransferase activity of Pseudomonas aeruginosa exotoxin A. Biochemistry. 2004;43:183–194. doi: 10.1021/bi034772u. [DOI] [PubMed] [Google Scholar]
- 116.Jørgensen R, Merrill AR, Yates SP, Marquez VE, Schwan AL, Boesen T, Andersen GR. Exotoxin A-eEF2 complex structure indicates ADP ribosylation by ribosome mimicry. Nature. 2005;436:979–984. doi: 10.1038/nature03871. [DOI] [PubMed] [Google Scholar]
- 117.Jørgensen R, Wang Y, Visschedyk D, Merrill AR. The nature and character of the transition state for the ADP-ribosyltransferase reaction. EMBO Rep. 2008;9:802–809. doi: 10.1038/embor.2008.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jørgensen R, Yates SP, Teal DJ, Nilsson J, Prentice GA, Merrill AR, Andersen GR. Crystal structure of ADP-ribosylated ribosomal translocase from Saccharomyces cerevisiae. J Biol Chem. 2004;279:45919–45925. doi: 10.1074/jbc.M406218200. [DOI] [PubMed] [Google Scholar]
- 119.Decker T, Oelsner M, Kreitman RJ, Salvatore G, Wang QC, Pastan I, Peschel C, Licht T. Induction of caspase-dependent programmed cell death in B-cell chronic lymphocytic leukemia by anti-CD22 immunotoxins. Blood. 2004;103:2718–2726. doi: 10.1182/blood-2003-04-1317. [DOI] [PubMed] [Google Scholar]
- 120.Sharma AK, FitzGerald D. Pseudomonas exotoxin kills Drosophila S2 cells via apoptosis. Toxicon. 2010;56:1025–1034. doi: 10.1016/j.toxicon.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bogner C, Dechow T, Ringshausen I, Wagner M, Oelsner M, Lutzny G, Licht T, Peschel C, Pastan I, Kreitman RJ, Decker T. Immunotoxin BL22 induces apoptosis in mantle cell lymphoma (MCL) cells dependent on Bcl-2 expression. Br J Haematol. 2010;148:99–109. doi: 10.1111/j.1365-2141.2009.07939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mussai F, Campana D, Bhojwani D, Stetler-Stevenson M, Steinberg SM, Wayne AS, Pastan I. Cytotoxicity of the anti-CD22 immunotoxin HA22 (CAT-8015) against paediatric acute lymphoblastic leukaemia. Br J Haematol. 2010;150:352–358. doi: 10.1111/j.1365-2141.2010.08251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Du X, Youle RJ, FitzGerald DJ, Pastan I. Pseudomonas exotoxin A-mediated apoptosis is Bak dependent and preceded by the degradation of Mcl-1. Mol Cell Biol. 2010;30:3444–3452. doi: 10.1128/MCB.00813-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Adams KW, Cooper GM. Rapid turnover of mcl-1 couples translation to cell survival and apoptosis. J Biol Chem. 2007;282:6192–6200. doi: 10.1074/jbc.M610643200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chaudhary VK, Xu YH, FitzGerald D, Adhya S, Pastan I. Role of domain II of Pseudomonas exotoxin in the secretion of proteins into the periplasm and medium by Escherichia coli. Proc Natl Acad Sci USA. 1988;85:2939–2943. doi: 10.1073/pnas.85.9.2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kasturi S, Kihara A, FitzGerald D, Pastan I. Alanine scanning mutagenesis identifies surface amino acids on domain II of Pseudomonas exotoxin required for cytotoxicity, proper folding, and secretion into periplasm. J Biol Chem. 1992;267:23427–23433. [PubMed] [Google Scholar]
- 127.Voulhoux R, Taupiac MP, Czjzek M, Beaumelle B, Filloux A. Influence of deletions within domain II of exotoxin A on its extracellular secretion from Pseudomonas aeruginosa. J Bacteriol. 2000;182:4051–4058. doi: 10.1128/jb.182.14.4051-4058.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yamaizumi M, Mekada E, Uchida T, Okada Y. One molecule of diphtheria toxin fragment A introduced into a cell can kill the cell. Cell. 1978;15:245–250. doi: 10.1016/0092-8674(78)90099-5. [DOI] [PubMed] [Google Scholar]
- 129.Kreitman RJ, Pastan I. Accumulation of a recombinant immunotoxin in a tumor in vivo: fewer than 1000 molecules per cell are sufficient for complete responses. Cancer Res. 1998;58:968–975. [PubMed] [Google Scholar]
- 130.Fitzgerald D. Why toxins! Semin Cancer Biol. 1996;7:87–95. doi: 10.1006/scbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 131.Bard F, Mazelin L, Péchoux-Longin C, Malhotra V, Jurdic P. Src regulates Golgi structure and KDEL receptor-dependent retrograde transport to the endoplasmic reticulum. J Biol Chem. 2003;278:46601–46606. doi: 10.1074/jbc.M302221200. [DOI] [PubMed] [Google Scholar]
- 132.Kreitman RJ, Tallman MS, Coutre S, Robak T, Wilson WH, Stetler-Stevenson M, Noel P, Fitzgerald DJ, Buzoinu M, Lechleider RJ, Pastan I. A phase I study of Moxetumomab pasudotox, an anti-CD22 recombinant immunotoxin, in relapsed/refractory hairy cell leukemia (HCL): updated results. American Society of Hematology (ASH) annual meeting abstract; December 4–7, 2010; Orlando, FL. 2010. [Google Scholar]
- 133.Wayne AS, Bhojwani D, Richards K, Stetler-Stevenson M, Silverman LB, Jeha S, Pui C, McDevitt J, Fitzgerald DJ, Kreitman RJ, Lechleider RJ, Pastan I. A phase I study of Moxetumomab pasudotox, an anti-CD22 recombinant immunotoxin, in patients with pediatric acute lymphoblastic leukemia (ALL): updated results. American Society of Pediatric Hematology/Oncology (ASPHO) annual meeting abstract; April 13–16, 2011; Baltimore MD. 2011. [Google Scholar]
- 134.Powell DJ, Jr, Felipe-Silva A, Merino MJ, Ahmadzadeh M, Allen T, Levy C, White DE, Mavroukakis S, Kreitman RJ, Rosenberg SA, Pastan I. Administration of a CD25-directed immunotoxin, LMB-2, to patients with metastatic melanoma induces a selective partial reduction in regulatory T cells in vivo. J Immunol. 2007;179:4919–4928. doi: 10.4049/jimmunol.179.7.4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hassan R, Sharon E, Schuler B, Mallory Y, Zhang J, Ling A, Pastan I. Anti-tumor activity of SS1P with pemetrexed and cisplatin for newly diagnosed patients with advanced pleural mesothelioma and utility of serum mesothelin as a marker of tumor response. American Society of Clinical Oncology (ASCO) annual meeting abstract; June 3–7, 2011; Chicago, IL. 2011. [Google Scholar]
- 136.De Groot AS, Scott DW. Immunogenicity of protein therapeutics. Trends Immunol. 2007;28:482–490. doi: 10.1016/j.it.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 137.Onda M, Beers R, Xiang L, Nagata S, Wang QC, Pastan I. An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. Proc Natl Acad Sci USA. 2008;105:11311–11316. doi: 10.1073/pnas.0804851105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hansen JK, Weldon JE, Xiang L, Beers R, Onda M, Pastan I. A recombinant immunotoxin targeting CD22 with low immunogenicity, low nonspecific toxicity, and high antitumor activity in mice. J Immunother. 2010;33:297–304. doi: 10.1097/CJI.0b013e3181cd1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Onda M, Beers R, Xiang L, Lee B, Weldon JE, Kreitman RJ, Pastan I. A recombinant immunotoxin against B cell malignancies with no immunogenicity in mice by removal of B cell epitopes. Proc Natl Acad Sci USA. 2011 doi: 10.1073/pnas.1102746108. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jevsevar S, Kunstelj M, Porekar VG. PEGylation of therapeutic proteins. Biotechnol J. 2010;5:113–128. doi: 10.1002/biot.200900218. [DOI] [PubMed] [Google Scholar]
- 141.Benhar I, Wang QC, FitzGerald D, Pastan I. Pseudomonas exotoxin A mutants. Replacement of surface-exposed residues in domain III with cysteine residues that can be modified with polyethylene glycol in a site-specific manner. J Biol Chem. 1994;269:13398–13404. [PubMed] [Google Scholar]
- 142.Tsutsumi Y, Onda M, Nagata S, Lee B, Kreitman RJ, Pastan I. Site-specific chemical modification with polyethylene glycol of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) improves antitumor activity and reduces animal toxicity and immunogenicity. Proc Natl Acad Sci USA. 2000;97:8548–8553. doi: 10.1073/pnas.140210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Filpula D, Yang K, Basu A, Hassan R, Xiang L, Zhang Z, Wang M, Wang QC, Ho M, Beers R, Zhao H, Peng P, Zhou J, Li X, Petti G, Janjua A, Liu J, Wu D, Yu D, Zhang Z, Longley C, FitzGerald D, Kreitman RJ, Pastan I. Releasable PEGylation of mesothelin targeted immunotoxin SS1P achieves single dosage complete regression of a human carcinoma in mice. Bioconjug Chem. 2007;18:773–784. doi: 10.1021/bc060314x. [DOI] [PubMed] [Google Scholar]
- 144.Oratz R, Speyer JL, Wernz JC, Hochster H, Meyers M, Mischak R, Spitler LE. Antimelanoma monoclonal antibody-ricin A chain immunoconjugate (XMMME-001-RTA) plus cyclophosphamide in the treatment of metastatic malignant melanoma: results of a phase II trial. J Biol Response Mod. 1990;9:345–354. [PubMed] [Google Scholar]
- 145.Selvaggi K, Saria EA, Schwartz R, Vlock DR, Ackerman S, Wedel N, Kirkwood JM, Jones H, Ernstoff MS. Phase I/II study of murine monoclonal antibody-ricin A chain (XOMAZYME-Mel) immunoconjugate plus cyclosporine A in patients with metastatic melanoma. J Immunother Emphasis Tumor Immunol. 1993;13:201–207. doi: 10.1097/00002371-199304000-00007. [DOI] [PubMed] [Google Scholar]
- 146.Hassan R, Williams-Gould J, Watson T, Pai-Scherf L, Pastan I. Pretreatment with rituximab does not inhibit the human immune response against the immunogenic protein LMB-1. Clin Cancer Res. 2004;10:16–18. doi: 10.1158/1078-0432.ccr-1160-3. [DOI] [PubMed] [Google Scholar]
- 147.Onda M. Reducing the immunogenicity of protein therapeutics. Curr Drug Targets. 2009;10:131–139. doi: 10.2174/138945009787354511. [DOI] [PubMed] [Google Scholar]
- 148.Nagata S, Pastan I. Removal of B cell epitopes as a practical approach for reducing the immunogenicity of foreign protein-based therapeutics. Adv Drug Deliv Rev. 2009;61:977–985. doi: 10.1016/j.addr.2009.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Onda M, Nagata S, FitzGerald DJ, Beers R, Fisher RJ, Vincent JJ, Lee B, Nakamura M, Hwang J, Kreitman RJ, Hassan R, Pastan I. Characterization of the B cell epitopes associated with a truncated form of Pseudomonas exotoxin (PE38) used to make immunotoxins for the treatment of cancer patients. J Immunol. 2006;177:8822–8834. doi: 10.4049/jimmunol.177.12.8822. [DOI] [PubMed] [Google Scholar]
- 150.Yeung VP, Chang J, Miller J, Barnett C, Stickler M, Harding FA. Elimination of an immunodominant CD4+ T cell epitope in human IFN-beta does not result in an in vivo response directed at the subdominant epitope. J Immunol. 2004;172:6658–6665. doi: 10.4049/jimmunol.172.11.6658. [DOI] [PubMed] [Google Scholar]
- 151.Mathew M, Verma RS. Humanized immunotoxins: a new generation of immunotoxins for targeted cancer therapy. Cancer Sci. 2009;100:1359–1365. doi: 10.1111/j.1349-7006.2009.01192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sitikov AS, Davydova EK, Ovchinnikov LP. Endogenous ADP-ribosylation of elongation factor 2 in polyribosome fraction of rabbit reticulocytes. FEBS Lett. 1984;176:261–263. doi: 10.1016/0014-5793(84)80953-9. [DOI] [PubMed] [Google Scholar]
- 153.Fendrick JL, Iglewski WJ, Moehring JM, Moehring TJ. Characterization of the endogenous ADP-ribosylation of wild-type and mutant elongation factor 2 in eukaryotic cells. Eur J Biochem. 1992;205:25–31. doi: 10.1111/j.1432-1033.1992.tb16748.x. [DOI] [PubMed] [Google Scholar]
- 154.Bektaş M, Akçakaya H, Aroymak A, Nurten R, Bermek E. Effect of oxidative stress on in vivo ADP-ribosylation of eukaryotic elongation factor 2. Int J Biochem Cell Biol. 2005;37:91–99. doi: 10.1016/j.biocel.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 155.Jäger D, Werdan K, Müller-Werdan U. Endogenous ADP-ribosylation of elongation factor-2 by interleukin-1β. Mol Cell Biochem. 2011;348:125–128. doi: 10.1007/s11010-010-0646-8. [DOI] [PubMed] [Google Scholar]
- 156.Hassan R, Broaddus VC, Wilson S, Liewehr DJ, Zhang J. Anti-mesothelin immunotoxin SS1P in combination with gemcitabine results in increased activity against mesothelin-expressing tumor xenografts. Clin Cancer Res. 2007;13:7166–7171. doi: 10.1158/1078-0432.CCR-07-1592. [DOI] [PubMed] [Google Scholar]
- 157.Zhang Y, Xiang L, Hassan R, Paik CH, Carrasquillo JA, Jang BS, Le N, Ho M, Pastan I. Synergistic antitumor activity of taxol and immunotoxin SS1P in tumor-bearing mice. Clin Cancer Res. 2006;12:4695–4701. doi: 10.1158/1078-0432.CCR-06-0346. [DOI] [PubMed] [Google Scholar]
- 158.Zhang Y, Hansen JK, Xiang L, Kawa S, Onda M, Ho M, Hassan R, Pastan I. A flow cytometry method to quantitate internalized immunotoxins shows that taxol synergistically increases cellular immunotoxins uptake. Cancer Res. 2010;70:1082–1089. doi: 10.1158/0008-5472.CAN-09-2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhang Y, Pastan I. High shed antigen levels within tumors: an additional barrier to immunoconjugate therapy. Clin Cancer Res. 2008;14:7981–7986. doi: 10.1158/1078-0432.CCR-08-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Traini R, Ben-Josef G, Pastrana DV, Moskatel E, Sharma AK, Antignani A, Fitzgerald DJ. ABT-737 overcomes resistance to immunotoxin-mediated apoptosis and enhances the delivery of Pseudomonas exotoxin-based proteins to the cell cytosol. Mol Cancer Ther. 2010;9:2007–2015. doi: 10.1158/1535-7163.MCT-10-0257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kreitman RJ, Schneider WP, Queen C, Tsudo M, Fitzgerald DJ, Waldmann TA, Pastan I. Mik-beta 1(Fv)-PE40, a recombinant immunotoxin cytotoxic toward cells bearing the beta-chain of the IL-2 receptor. J Immunol. 1992;149:2810–2815. [PubMed] [Google Scholar]
- 162.Izidoro MA, Gouvea IE, Santos JA, Assis DM, Oliveira V, Judice WA, Juliano MA, Lindberg I, Juliano L. A study of human furin specificity using synthetic peptides derived from natural substrates, and effects of potassium ions. Arch Biochem Biophys. 2009;487:105–114. doi: 10.1016/j.abb.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Du X, Beers R, Fitzgerald DJ, Pastan I. Differential cellular internalization of anti-CD19 and -CD22 immunotoxins results in different cytotoxic activity. Cancer Res. 2008;68:6300–6305. doi: 10.1158/0008-5472.CAN-08-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]