

Abstract
Zn2+ is coreleased with glutamate from mossy fiber terminals and can influence synaptic function. Here, we demonstrate that synaptically released Zn2+ activates a selective postsynaptic Zn2+-sensing receptor (ZnR) in the CA3 region of the hippocampus. ZnR activation induced intracellular release of Ca2+, as well as phosphorylation of extracellular-regulated kinase and Ca2+/calmodulin kinase II. Blockade of synaptic transmission by tetrodotoxin or CdCl inhibited the ZnR-mediated Ca2+ rises. The responses mediated by ZnR were largely attenuated by the extracellular Zn2+ chelator, CaEDTA, and in slices from mice lacking vesicular Zn2+, suggesting that synaptically released Zn2+ triggers the metabotropic activity. Knockdown of the expression of the orphan G-protein-coupled receptor 39 (GPR39) attenuated ZnR activity in a neuronal cell line. Importantly, we observed widespread GPR39 labeling in CA3 neurons, suggesting a role for this receptor in mediating ZnR signaling in the hippocampus. Our results describe a unique role for synaptic Zn2+ acting as the physiological ligand of a metabotropic receptor and provide a novel pathway by which synaptic Zn2+ can regulate neuronal function.
Introduction
Histochemically reactive or “free” Zn2+ in the brain is mostly localized within glutamatergic synaptic vesicles in the hippocampus and in a variety of forebrain areas (Frederickson and Moncrieff, 1994). Uptake of Zn2+ ions into synaptic vesicles is mediated by the Zn2+ transporter, zinc transporter 3 (ZnT3), and deletion of the gene encoding it results in the elimination of the synaptic Zn2+ pool (Palmiter et al., 1996; Cole et al., 1999). At mossy fiber synapses, Zn2+ is coreleased with glutamate in an activity-dependent manner (Vogt et al., 2000; Qian and Noebels, 2005). The hippocampal mossy fibers, by virtue of their high concentration of synaptic Zn2+ and their well defined circuitry, have been the focus of many of the studies on the role of Zn2+ in the CNS.
While increases in intracellular free Zn2+ (Sensi et al., 1999) have been associated with neuronal death (Aizenman et al., 2000; McLaughlin et al., 2001; Sensi and Jeng, 2004; Zhang et al., 2004), a physiological role for synaptically released Zn2+ is still largely unknown. Prior work has focused primarily on the function of synaptic Zn2+ as a modulator of ionotropic receptors. For example, it has been shown that Zn2+ modifies the activity of postsynaptic NMDA receptors through interactions with specific low and high affinity binding sites (Paoletti et al., 1997; Rachline et al., 2005; Izumi et al., 2006). Additionally, zinc ions have been shown to block the activity of GABAA receptors (Hosie et al., 2003; Ruiz et al., 2004). Synaptic plasticity has thus been reported to be affected by Zn2+ by shaping NMDA (Vogt et al., 2000) or GABA synaptic potentials (Ruiz et al., 2004), by modulating assembly of the postsynaptic density (Gundelfinger et al., 2006), or by regulating long-term potentiation (LTP) (Li et al., 2001; Huang et al., 2008). Despite these findings, Zn2+-dependent allosteric regulation of ionotropic pathways may not fully explain how endogenous Zn2+ is linked to all these physiological processes (Paoletti et al., 2009).
Metabotropic receptors play a critical role in synaptic transmission (Nicholls et al., 2006). These receptors mediate slow synaptic potentials, activate intracellular signaling pathways, and indirectly modulate ion channels and transporters. Metabotropic receptors (mGluRs) are also involved in changes in synaptic plasticity via activation of the mitogen-activated protein (MAP) kinase pathways and in the regulation of gene expression (Volk et al., 2006). Activation of the mossy fibers has been shown to induce intracellular Ca2+ release from postsynaptic CA3 neurons via a metabotropic pathway (Kapur et al., 2001). Although synaptic Zn2+ is released in this region, a role for this ion in metabotropic signaling has heretofore not been demonstrated.
We previously characterized a Zn2+-sensing receptor, ZnR, in epithelial cells, which induces release of intracellular Ca2+ mediated by a Gαq-protein through the IP3 pathway (Hershfinkel et al., 2001). In the present study, we demonstrate that synaptic Zn2+ specifically triggers metabotropic activity in the CA3 hippocampal region through activation of a ZnR. Our results also suggest that the orphan G-protein-coupled receptor 39 (GPR39) mediates neuronal ZnR activity.
Materials and Methods
Slice preparation.
Experimental procedures were performed in accordance with protocols approved by the committee for the Ethical Care and Use of Animals in Research at Ben Gurion University. After decapitation, brains were quickly removed and placed into ice-cold (4°C) artificial CSF (ACSF). Coronal slices, 400 μm thick, from mice postnatal day 8 (P8) to P16, were produced on a vibrating microtome (Campden Instruments) as described previously (Qian and Noebels, 2005; Frederickson et al., 2006). Slices were maintained at room temperature in ACSF for 1 h, containing the following (in mm): 124 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 10 dextrose, 2 MgSO4, and 2 CaCl2, saturated with 95% O2 and 5% CO2.
Cell culture and GPR39 silencing.
Human neuroblastoma cells, SH-SY5Y, were cultured in DMEM as described previously (Kan et al., 2007). Cells were seeded on 10 mm glass coverslips to 80% confluency and imaged after 24 h as described below. For gene silencing experiments, cells were cotransfected with the silencing plasmids, 3 μg of shGPR39 or shT1R3, and 1 μg YFP plasmid in 35 mm plates, using Lipofectamine 2000 as described by the manufacturer (Invitrogen), and imaged 48 h after transfection. The target sequence of the GPR39 for shRNA was CCATGGAGTTCTACAGCATtt and that of T1R3 was CUUAGGAUGAAGGGGGACUtt.
Fluorescence imaging.
Slices were loaded with the Ca2+ indicator Fura-2 A.M. (25 μm, TefLabs) for imaging experiments, or Fluo-4 A.M. (25 μm, Invitrogen), for confocal analysis, in the presence of 0.02% pluronic acid for 20 min, as described previously (Beierlein et al., 2002). The imaging system consisted of an upright fluorescence microscope (Olympus BX51; Olympus Optical), Polychrome II monochromator (TILL Photonics) and a SensiCam cooled charge-coupled device (PCO). Fluorescence imaging measurements were acquired with Imaging Workbench 2 (Axon Instruments) as described previously (Hershfinkel et al., 2001). The ratio of the Fura-2 fluorescence response excited at 340 and 380 nm was determined. Representative traces from each experiment, averaged over ∼60 cells from at least 3 slices ± SEM are shown. In some experiments, the intracellular Ca2+ concentration was determined by calibration of intracellular Ca2+ in slices superfused with the calcium ionophore, Bromo-A23187, in the presence of 5 mm Ca2+ (Rmax) or 5 mm EGTA (Rmin), as described previously (Grynkiewicz et al., 1985; Hershfinkel et al., 2001), using a Kd of 220 nm (for Fig. 1, Rmax = 1.56 and Rmin = 0.84, and for Fig. 6, Rmax = 1.62 and Rmin = 0.89).
Figure 1.

A metabotropic Zn2+-dependent Ca2+ response is observed in the hippocampus. A, Fluorescence image before (left), and after (right) application of 200 μm zinc (×10). Inset shows high magnification images (×40). B, Intracellular Ca2+ signal in the pyramidal cell layer, CA3, after application of extracellular Zn2+ (200 μm) in the presence or absence of 2 mm Ca2+ in the ACSF perfusate. The response after application of ATP (300 μm) is shown in the inset. C, ER Ca2+ stores were depleted using TG (1 μm), an ER Ca2+ pump inhibitor, and the purinergic agonist, ATP (300 μm). Subsequently, Zn2+ (200 μm) was applied and Fura-2 fluorescence was monitored. D, A summary of the averaged Zn2+-dependent Ca2+ response in the presence or absence of extracellular or intracellular Ca2+, is presented (**p < 0.01, n = 10 for each treatment). E, Zn2+ was applied at the indicated concentrations, and the calibrated intracellular Ca2+ level as a function of increasing Zn2+ concentration is shown (n = 4 for each concentration). The data were fitted to a Michaelis–Menten equation and yielded an apparent Km of 146 ± 26 μm for Zn2+ and a Hill coefficient of 1 ± 0.8.
Figure 6.
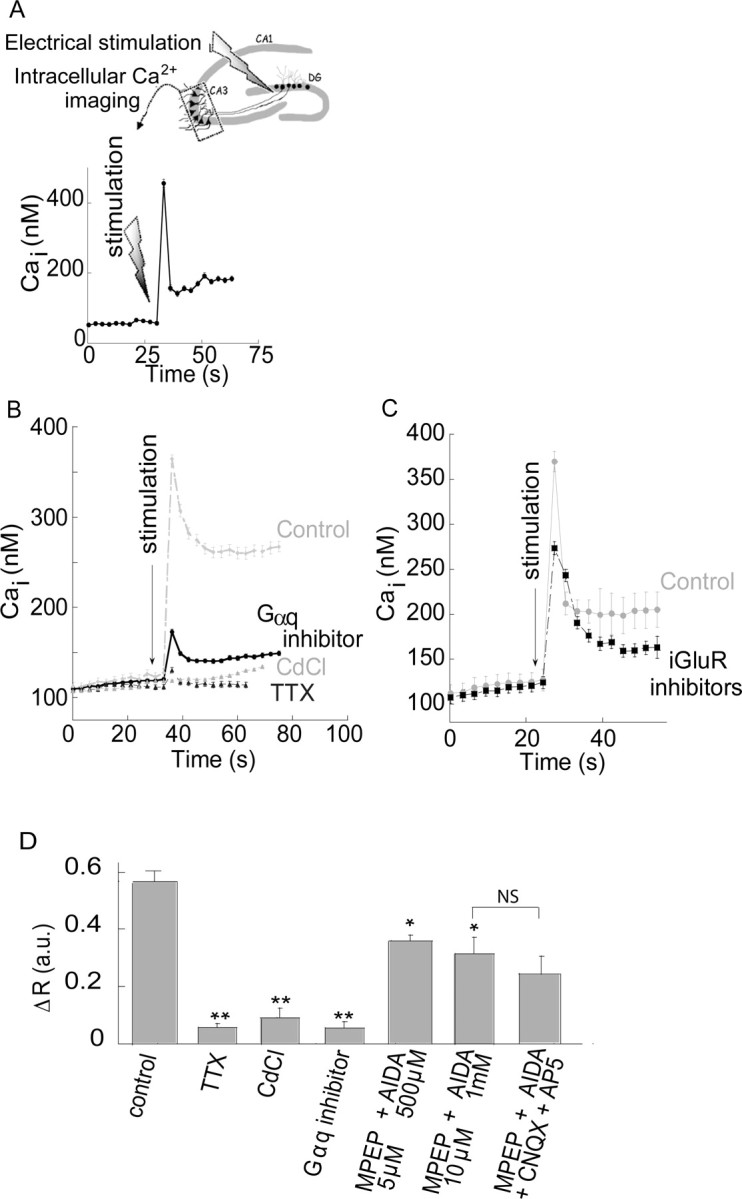
Mossy fiber stimulation triggers a metabotropic Ca2+ rise that is partially independent of the mGluRs. A, Schematic representation of the experimental setup for electrical stimulation. A stimulating electrode was placed at the mossy fiber axons, near the dentate gyrus (DG) and the Fura-2 signal was monitored in the CA3 pyramidal cell layer. Shown is a representative time lapse Ca2+ signal acquired from a single cell, after electrical stimulation (66 Hz, 100 μA for 150 ms, total of 10 pulses) of the mossy fibers at the marked time. B, The Ca2+ response in the CA3 region after electrical stimulation of the mossy fibers in the presence or absence of the Gαq inhibitor (1 μm, YM-254890), with the voltage-gated Na+-channel blocker TTX (1 μm), or with the voltage-gated Ca2+ channel blocker CdCl (200 μm). Representative responses averaged over 25 cells in 1 slice. C, Slices were treated with the iGluR inhibitors (CNQX, 20 μm and AP5, 50 μm) subsequently the mossy fibers were stimulated and the Ca2+ response is shown. Representative responses averaged over 24 cells in 1 slice. D, The averaged responses of the Ca2+ rise triggered after electrical stimulation of the mossy fibers in control slices (n = 23) or in slices treated with TTX (n = 6), CdCl (n = 6), the Gαq inhibitor (n = 6), the mGluR1 and 5 inhibitors (AIDA and MPEP, respectively, n = 6) or the mGluRs inhibitors together with the iGluRs inhibitors (1 mm AIDA, 10 μm MPEP, 20 μm CNQX and 50 μm AP5, n = 4) (*p < 0.05, **p < 0.01; NS, nonsignificant).
Because calcium stores are often rapidly depleted, and store refilling may take several seconds, and because ZnR activity may undergo rapid desensitization, repetitive application of Zn2+ or electrical stimuli were avoided. We therefore monitored Ca2+ responses triggered by only a single stimulation per slice. If required, cells were preincubated in ACSF containing Zn2+ (for the desensitization protocol) or inhibitors before imaging. Slices were imaged within 15–30 min after loading with the intracellular dye, except for the desensitization experiment which required extended time for the recovery of the intracellular Ca2+. In some of the experiments, the fluorescence response after stimulation remained at an elevated plateau or was followed by a slow rise, an effect that was more apparent in the presence of inhibitors, since the signal was attenuated but this nonrelated, elevated plateau persisted. This elevated fluorescence may arise from several factors, including activation of the store operated channels, continuous release of Ca2+ from the IP3 sensitive store (Bianchi et al., 1999; Nakamura et al., 2000), or slow permeation of Zn2+, or Cd2+ (see Fig. 6A) via voltage sensitive Ca2+ channels, leading to Fura-2 fluorescence (Hinkle et al., 1987).
The change in the fluorescence response, “ΔR,” was determined by subtracting the maximal signal after stimulation from the baseline signal. The ΔR (the number of slices for each experiment is indicated in the figure legend) was averaged and is presented in the bar graphs. Statistical analyses were performed using a student's t test, compared with control or ANOVA with post hoc comparisons as relevant.
For studying the metal selectivity of the receptor, the ΔR was obtained after application of each heavy metal in the presence or absence of the Gαq inhibitor. The ΔR obtained in the presence of the inhibitor was subtracted from the ΔR measured in its absence, to eliminate the fluorescence response triggered via Gq-independent pathways. Thus, we determined the residual Gαq-dependent fluorescent response triggered by the ions, which was marked as ΔRGq inhibitor.
Confocal imaging analysis was performed using slices loaded with the Ca2+ indicator, Fluo-4 (5 μm). Images were acquired by a Zeiss LSM 510 system using a 20× objective (488 nm excitation). Changes in Fluo-4 signal were monitored after the application of Zn2+ and subsequently slices were washed and stained on stage with Sulforhodamine 101, SR101 (Sigma, 1 μm in ACSF) (Nimmerjahn et al., 2004) for 2 min. The slices were subsequently rinsed in ACSF and imaged at an excitation wavelength of 540 nm.
Fluorescence imaging of cultured cells was performed as described previously (Hershfinkel et al., 2001). Cells were loaded with Fura-2 (3 μm) for 20 min and allowed to recover for 30 min. Transfected cells were identified by monitoring YFP fluorescence (excitation at 480 nm) and Ca2+ signals were monitored and averaged only on the transfected cells.
Mossy fiber stimulation.
Transverse slices were prepared from mice at P8–P16 as described above, and imaged within 60–90 min to avoid synaptic Zn2+ depletion (Yin et al., 2002; Frederickson et al., 2006). Slices were loaded with Fura-2 as described above and intracellular Ca2+ imaging was performed by digitally acquiring the fluorescent images every 3 s. A bipolar tungsten electrode placed near the hilus of the dentate gyrus was used to stimulate mossy fibers. Electrical stimuli (66 Hz, 100 μA for 150 ms, total of 10 pulses) were delivered to the mossy fibers using Master-8 stimulator unit (A.M.P.I.), and intracellular Ca2+ was monitored in the CA3 hippocampal region.
Desensitization protocol.
Slices were pretreated with 75 μm Zn2+ for 15 min, washed, and allowed to recover in ACSF for 60–100 min. Control slices were treated with ACSF for the same time period. Slices were then loaded with Fura-2 and imaged using the same protocol as above. Finally, Zn2+ (300 μm) was reapplied and the Ca2+ response compared between control and Zn2+-pretreated slices.
Immunohistochemical analysis.
For GPR39 labeling, mice were perfused intracardially with 1% sodium sulfide in PBS (50 ml) followed by 4% paraformaldehyde in PBS (50 ml) and 5 μm paraffin-embedded sections were prepared. Sections were then deparaffinized in xylene and rehydrated in decreasing concentrations of ethanol. Antigen retrieval was achieved by boiling the slide-mounted sections in 0.01 m citric acid buffer, pH 6.0, in a microwave for 10 min. The sections were then rinsed in PBS, incubated in blocking solution for 30 min in room temperature, and incubated overnight in primary antibody. The following dilutions were used: rabbit anti-GPR39 (Novus Biological, 1:1000); NeuN (NeuN Alexa 488, Millipore, 1:500) and MAP2 (Santa Cruz Biotechnology, 1:200), at room temperature. After rinsing in PBS, the sections were incubated for 1 h with affinity-purified cy2 or cy3-conjugated goat anti-rabbit IgG (The Jackson Laboratory), rinsed in PBS, and mounted with anti-fade solution (Kierrkegaard). The tissue was imaged in a confocal microscope (Zeiss LSM 510) with 488 nm or 543 nm excitation wavelength for cy2 and cy3 staining, respectively.
For kinase phosphorylation analysis, 400 μm slices were incubated with Zn2+ (100 μm for 90 or 180 s). Some slices were pretreated with the Gαq inhibitor (1 μm, YM-254890, 10 min) or the ionotropic glutamate receptor (iGluR) inhibitors, CNQX (20 μm) and AP5 (50 μm), and the Ca2+ channel blocker, nimodipine (1 μm). Immunohistochemical analysis was performed as described previously (Sekler et al., 2002). Briefly, slices were frozen in OCT embedding media (Tissue-TEK), sectioned on a cryostat (10 μm) and fixed in 4% paraformaldehyde at room temperature (20 min). Sections were rinsed in PBS, incubated in blocking solution for 30 min at room temperature, and then incubated for 3 h with anti-phosphorylated extracellular regulated kinase (pERK1/2) (Sigma-Aldrich) at 1:100 or overnight with anti-phosphorylated Ca2+/calmodulin kinase (pCaMKII) (Santa Cruz Biotechnology, 1:200). This was followed by 1 h incubation in Cy3-conjugated secondary antibody (The Jackson Laboratory). Immunolabeled sections were mounted with DAPI-containing mounting medium (Immunomount) and viewed in a Zeiss Axiomat microscope equipped with appropriate fluorescence barrier and excitation filters or in a Zeiss LSM 510 confocal microscope. Images were analyzed and digitally captured into the ImageJ analysis software to quantify pERK1/2 or pCaMKII phosphorylation, and normalized to the respective DAPI staining to control for the number of cells in the region of interest. The images were assigned false color scales and superimposed with Adobe Photoshop 7.0 software (Adobe Systems). Graphs represent mean of the normalized fluorescence determined from at least 3 regions obtained from three independent experiments ± SEM.
Results
A metabotropic response is triggered by Zn2+
To determine if Zn2+ activates metabotropic signaling in the brain, we used coronal hippocampal slices. Cells were loaded with Fura-2 and treated with Zn2+ by direct delivery into the ACSF perfusate. Application of 200 μm Zn2+ triggered a rapid increase in Fura-2 fluorescence in the pyramidal cell layer of the CA3 hippocampal region (Fig. 1A,B) in the presence or absence of extracellular Ca2+. This response was similar to that induced by ATP (300 μm, see inset), shown previously to trigger a metabotropic purinergic response (Yamazaki et al., 2002). After application of Zn2+, the decline to resting Ca2+ levels was somewhat slower in the presence of extracellular Ca2+, possibly due to Ca2+ influx via the store-operated Ca2+ channel, activated after store depletion (Wu et al., 2004). Because Zn2+ can permeate into neurons and because Fura-2 has a high affinity for this ion (Kd = 2 nm), it was important to establish that the fluorescence signal observed was related to changes in intracellular Ca2+ and not Zn2+. Endoplasmic reticulum (ER) Ca2+ stores were depleted, using the ER Ca2+ pump inhibitor, thapsigargin (TG, 1 μm), applied with the purinergic agonist ATP (300 μm) to enhance intracellular Ca2+ release. Subsequent applications of Zn2+ (200 μm) (Fig. 1C,D) failed to elicit an increase in Fura-2 signal (10 ± 2% of response in control), indicating that the fluorescence response is not mediated by Zn2+ permeation, but rather by release of Ca2+ from TG-sensitive stores. We then assessed the Zn2+ concentration dependence of the observed Ca2+ rise (Fig. 1E). The Zn2+-dependent Ca2+ response was observed over a wide range of Zn2+ concentrations (i.e., from tens to hundreds micromolar) with an apparent Km of 146 ± 26 μm. Together, these results indicate that Zn2+ triggers an intracellular Ca2+ signal in the CA3 region, which is extracellular Ca2+ independent, and mediated by Ca2+ release from TG-sensitive stores (Fig. 1D).
We next asked if the Zn2+-induced Ca2+ signal is mediated through a Gq-coupled receptor and via the IP3 pathway, both characteristic of the described previously ZnR-mediated responses in non-neuronal cells (Hershfinkel et al., 2001). Application of 200 μm Zn2+ after treatment with the phospholipase C (PLC) inhibitor U73122 (1 μm) resulted in a Ca2+ rise that was inhibited by 77 ± 3% compared with control. In the presence of the inactive analog of this inhibitor U73343 (1 μm) the Zn2+-dependent response was similar (92 ± 11%) to control (Fig. 2A,B). The Gαq inhibitor YM-254890 (1 μm) (Sharir and Hershfinkel, 2005) inhibited the Zn2+-dependent Ca2+ response by 68 ± 4% compared with control (Fig. 2B). Since mGluRs mediate metabotropic signaling in this region, we compared the Zn2+-dependent response in the presence or absence of mGluR inhibitors. To ascertain that the mGluRs were blocked, saturating concentrations of the inhibitors were used (Nakamura et al., 2000), and their efficacy was also verified by monitoring their ability to block the Ca2+ response triggered by the group I mGluR agonist DHPG ((S)-3,5-dihydroxyphenylglycine) (Bianchi et al., 1999). Application of the mGluR5 inhibitor MPEP (2-methyl-6-(phenylethynyl)-pyridine; 5 μm), with the mGluR1 inhibitor AIDA (1-aminoindan-1,5-dicarboxylic acid; 500 μm), attenuated the Ca2+ signal induced by 100 μm DHPG by 62 ± 6% (n = 8), while 10 μm MPEP in the presence of 1 mm AIDA reduced the DHPG-dependent response by 69 ± 5% (n = 8). In contrast, the Zn2+-dependent Ca2+ signal was not reduced at either concentration of the mGluR inhibitors (p > 0.3) (Fig. 2B), suggesting that the Zn2+-triggered response is not related to mGluRs, but is mediated via a distinct metabotropic pathway. The metal selectivity of the metabotropic response was then assessed, focusing on Cd2+ and Pb2+. Cd2+ is a group IIB metal, like Zn2+, which binds with high affinity to Zn2+ binding sites on proteins such as metallothionein (Henkel and Krebs, 2004). Pb2+ is a divalent metal that has been shown to alter synaptic function (White et al., 2007). To avoid background effects mediated by permeation of the ions, and to resolve if these ions trigger the metabotropic Ca2+ response, we determined the difference between the maximal Fura-2 signal, triggered by Cd2+, Pb2+ or Zn2+, in the presence or absence of the Gαq inhibitor (ΔRGq inhibitor, see Materials and Methods). As shown in Figure 2C, the Zn2+-dependent Fura-2 response was largely blocked by the Gαq inhibitor (1 μm), yielding a ΔRGq inhibitor = 0.65 ± 0.05. In contrast, the inhibitor did not significantly reduce Cd2+ or Pb2+-dependent fluorescence (ΔRGq inhibitor = 0.05 ± 0.09 or 0.1 ± 0.05, respectively). This indicates that Cd2+ and Pb2+ do not induce a metabotropic Ca2+ rise, and that the ZnR response is selective for Zn2+.
Figure 2.

The Ca2+ rise triggered by Zn2+ is mediated by a Gq- and PLC-dependent pathway. A, Slices were pretreated (as marked) with the PLC inhibitor U73122 (1 μm, active form) or its inactive derivative U73343 (1 μm, control) and Zn2+ (200 μm) was applied. As control, ATP (300 μm) was subsequently applied. The Ca2+ signal (Fura-2) is shown. B, The averaged Ca2+ rise triggered by Zn2+ in the presence of the inhibitors of PLC (U73122, 1 μm) or Gαq (YM-254890, 1 μm) or the mGluR1 and 5 inhibitors (AIDA and MPEP, respectively) at the indicated concentrations (**p < 0.01, n = 20 for control and n = 9 for all other treatments). C, The fluorescent response observed after application of Cd2+ and Pb2+ were monitored in the absence or presence of the Gαq inhibitor (1 μm, YM-254890). The difference (ΔRGq inhibitor) between the response in the presence or absence of the inhibitor is shown (**p < 0.01, n = 5). D, Slices were pretreated with 75 μm Zn2+ or ACSF for 15 min, washed, and allowed to recover in ACSF for 60–100 min. Zn2+ (300 μm) was then reapplied while monitoring the Ca2+ response. As control, ATP (300 μm) was subsequently applied (**p < 0.01, n = 7).
Previous work in our lab has shown that the epithelial ZnR undergoes profound functional desensitization after prolonged exposure to Zn2+ at concentrations which can activate the ZnR (Azriel-Tamir et al., 2004; Sharir and Hershfinkel, 2005). Such a regulatory mechanism would be especially relevant in the CNS, considering the pathophysiological implications of sustained activation of the IP3 pathway (Tang et al., 2003), and the potential excessive release of synaptic Zn2+ during intense excitatory activity (Weiss et al., 2000). To determine if Zn2+-dependent signaling in neurons undergoes desensitization, slices were pretreated with 75 μm Zn2+ for 15 min and then washed in ACSF (see Materials and Methods). We subsequently applied 300 μm Zn2+ while monitoring the Ca2+ response. The Zn2+ and ATP-dependent Ca2+ response observed in the control slices (pretreated with ACSF) is smaller than that shown in Figure 2B, and may result from the prolonged interval before imaging. The Zn2+-dependent Ca2+ response in slices that were pre-exposed to Zn2+ was only 15 ± 2% compared with control (Fig. 2D) (ΔR = 0.33 ± 0.02 in control compared with 0.05 ± 0.01 in the Zn2+ pretreated slices), while application of ATP triggered a robust Ca2+ rise (Fig. 2D). As shown, ATP-induced Ca2+ responses were somewhat larger in Zn2+-desensitized preparations, likely due to the fact that intracellular stores had not been depleted by a ZnR-mediated response. Together, these results indicate that the Zn2+-dependent metabotropic response triggered in the CA3 region is mediated through a Gq-coupled receptor via the IP3 pathway, and undergoes a Zn2+-dependent desensitization similar to that described previously for the epithelial ZnR.
To determine whether neurons or glia exhibit metabotropic Zn2+-dependent Ca2+ release, Zn2+ was applied as before and the Ca2+ response monitored in a confocal microscope (Fig. 3A). Slices were subsequently stained with the astroglial marker SR101 (Sulforhodamine 101) (Nimmerjahn et al., 2004). As shown in Figure 3B, cells which exhibited a Zn2+-dependent Ca2+ rise (white arrows) were not labeled with the astrocyte marker, SR101 suggesting that ZnR activity is not found in astroglia. To further address this issue, we asked if ZnR activity is observed in an established neuronal cell line, SH-SY5Y, a subclone of the SK-N-SH cell line. SH-SY5Y cells were previously used to study neurite outgrowth, cell growth and glutamate toxicity (Canals et al., 2005; Smith et al., 2009). As shown in Figure 3C, application of 200 μm Zn2+, a concentration which triggered robust ZnR activity in the slices (Fig. 1), was also followed by an intracellular Ca2+ rise in the SH-SY5Y cells. When SH-SY5Y cells were treated with the Gαq inhibitor, YM-254890 (1 μm), the Zn2+-induced Ca2+ response was inhibited by 95 ± 3% compared with control (Fig. 3C) (ΔR = 0.61 ± 0.15 in control and 0.03 ± 0.01 in the presence of the inhibitor), indicating that the Zn2+-dependent Ca2+ response is mediated via a Gq-dependent pathway.
Figure 3.

Zn2+-dependent Ca2+ rise is monitored in CA3 neurons but not in astrocytes. A, Confocal microscope analysis of slices that were loaded with the Ca2+ indicator, Fluo-4 (5 μm). The Ca2+ rise after application of Zn2+ in two representative cells is shown. B, Cells that showed Zn2+-dependent Ca2+ rise were indicated (arrows) and subsequently SR101 (1 μm) was added to mark astroglia cells (red). C, Zn2+ (200 μm) was applied to SH-SY5Y neuronal cells loaded with Fura-2 in the absence or presence of the Gαq inhibitor (1 μm YM-254890) (**p < 0.01, n = 9).
MAP and CAM kinase pathways are activated by metabotropic Zn2+-dependent signaling
Activation of extracellular regulated kinase (ERK1/2) and Ca2+/calmodulin-dependent protein kinase (CaMKII) play fundamental roles in hippocampal synaptic plasticity and neuronal survival (Hansen et al., 2003; Giovannini, 2006; Luo and DeFranco, 2006; Cohen-Matsliah et al., 2007). Because intracellular Ca2+ rise can activate these pathways (Berkeley and Levey, 2003), we sought to determine if the Zn2+-dependent metabotropic response could trigger ERK1/2 or CaMKII phosphorylation in neurons. As shown in Figure 4, A and B, application of Zn2+ (100 μm, 90 s) led to a fourfold increase in pERK1/2 in the CA3 region compared with control. Colocalization with DAPI using confocal microscopy indicated that pERK1/2 staining was abundant in the nuclear and perinuclear regions (Fig. 4A, bottom). Application of blockers of the iGluRs (20 μm CNQX and 50 μm AP5) (Fig. 4B), and the Ca2+ channel blocker, nimodipine (1 μm), did not attenuate the Zn2+-dependent phosphorylation of ERK1/2, indicating that it is mediated by extracellular Zn2+ and not by its potential permeation through these pathways. As illustrated in Figure 4, C and D, application of Zn2+ also resulted in an increase of ∼50% in phosphorylated CaMKII. Finally, to assess the role of the metabotropic pathway in mediating Zn2+-dependent kinase phosphorylation, slices were treated with the Gαq inhibitor YM-254890 (1 μm) before application of Zn2+. As shown in Figure 4, application of the Gαq inhibitor reduced the Zn2+-dependent phosphorylation of both ERK1/2 and CaMKII to baseline levels. Together, our results indicate that a Gαq-coupled pathway mediates Zn2+-dependent ERK1/2 and CaMKII phosphorylation in the CA3 region of the hippocampus.
Figure 4.
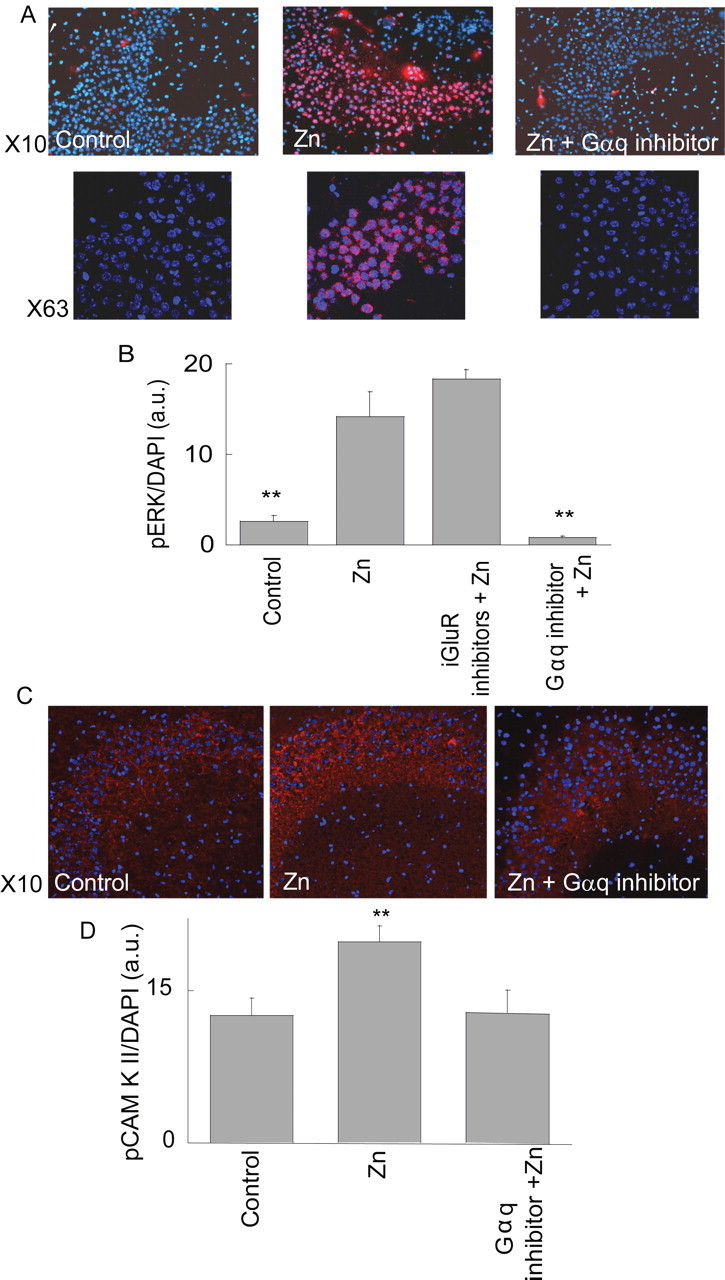
The metabotropic Zn2+-dependent response is followed by phosphorylation of ERK1/2 and CaMKII in the CA3 region. A, Zn2+ (100 μm, 90 s) was applied to slices in the presence or absence of the Gαq inhibitor (1 μm, YM-254890). Slices were then reacted with pERK1/2 antibody, shown in red, and DAPI (blue). Images acquired at ×10 (top) and ×63 (confocal, bottom) are shown. B, Quantitative analysis of ERK1/2 phosphorylation normalized to DAPI staining in the CA3 region. Shown is the phosphorylation after application of Zn2+ in the absence or presence of the ionotropic glutamate inhibitors (CNQX, 20 μm and AP5, 50 μm) and Ca2+ channel blocker (nimodipine 1 μm), or the Gαq inhibitor (1 μm YM-254890) (**p < 0.01, n = 6). C, Slices treated with Zn2+ (100 μm, 3 min), in the presence or absence of the Gαq inhibitor, were then reacted with pCaMKII antibodies (red) and DAPI (blue). D, Quantitative analysis of pCaMKII staining normalized to DAPI staining (**p < 0.01, n = 6).
GPR39 mediates neuronal Zn2+-dependent signaling
It was recently suggested that a member of the ghrelin receptor family of GPCRs, namely GPR39, can be activated by serum Zn2+ (Yasuda et al., 2007). The physiological significance of the Zn2+-dependent signal mediated by GPR39 activation remains unclear, however (Holst et al., 2007; Yasuda et al., 2007). We hypothesized that GPR39 might be the ZnR itself and thus sought to determine if it mediates neuronal Zn2+-dependent signaling. The endogenous expression of GPR39 was initially determined by immunoblot analysis of the neuronal SH-SY5Y cells (Fig. 5A). This cell line is particularly useful for ectopic expression of genes and RNA constructs because of the high transfection efficiency that is typically achieved. A short hairpin RNA (shGPR39) construct targeted to silence GPR39 efficiently lowered GPR39 expression in these cells, as determined by immunoblot analysis (Fig. 5A). As a control, a hairpin construct targeted against T1R3, a closely related GPCR, did not alter GPR39 expression in SH-SY5Y cells (Fig. 5A). The Ca2+ response to application of Zn2+ was then monitored in SH-SY5Y cells transfected with shGPR39 or in cells transfected with either shT1R3 or with an empty vector. As shown in Figure 5, B and C, the Zn2+-dependent Ca2+ response was similar in vector and shT1R3-expressing cells, but significantly attenuated (by 73 ± 7%) in cells transfected with shGPR39. We then asked whether GPR39 was endogenously expressed in the hippocampus. Immunofluorescent labeling of GPR39 (Fig. 5D) was observed as punctuate staining at the periphery of cells expressing the neuronal nuclear marker (NeuN) in CA3. Some punctuate staining was also observed near dendrites in the CA3 region (Fig. 5D, inset). This suggests that GPR39 is present on the cells that exhibit Zn2+-dependent metabotropic activity. These results are consistent with the hypothesis that GPR39 may mediate ZnR signaling in CA3 neurons.
Figure 5.
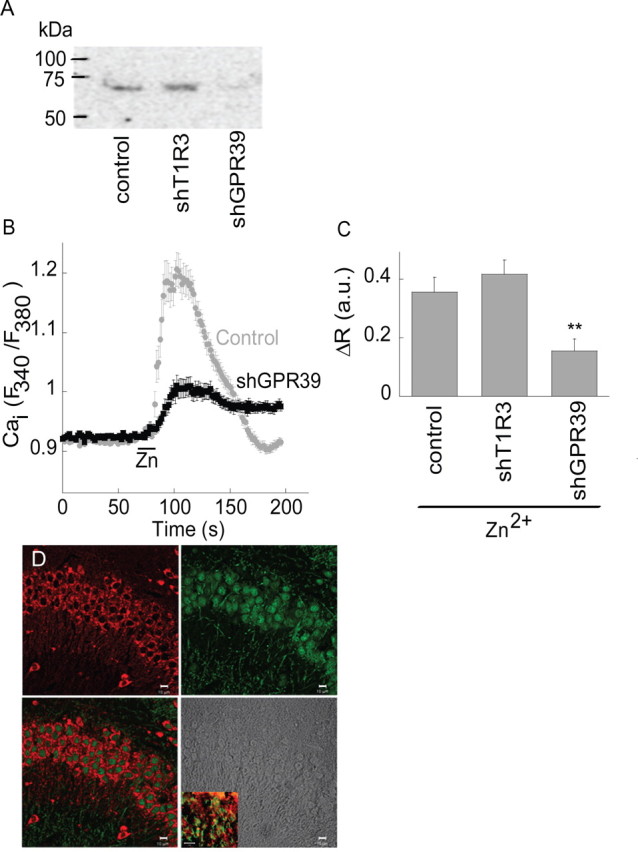
ZnR signaling is mediated by GPR39. A, Western blot analysis of GPR39 expression in SH-SY5Y cells transfected with control vector, shGPR39 or shT1R3 plasmids. B, The Ca2+ response after application of Zn2+ (100 μm) in cells transfected with the shGPR39 or vector (control). C, Quantitative analysis of the Zn2+-dependent Ca2+ rise in cells transfected with the shGPR39, shT1R3 or control (**p < 0.01, n = 9). D, Confocal images of GPR39 labeling (red) and the neuronal marker NeuN (green) in the CA3. A combined image of both indicates that GPR39 is expressed in neuronal cells (bottom left). Bright field image is also shown (bottom right). Insert shows staining of the dendritic marker MAP2 (green) combined with GPR39 labeling (red) in the CA3 neurons (scale bar, 10 μm).
Synaptic Zn2+ triggers ZnR-dependent response in the CA3 region
While the experiments described above strongly support the notion that a metabotropic receptor is activated in neurons in the CA3 region by exogenous Zn2+, induction of the Ca2+ response by synaptically released Zn2+ is required to demonstrate the physiological significance of this phenomenon. To address this issue, synaptic Zn2+ release was induced by electrical stimulation of the mossy fibers using a protocol consisting of 10 pulses at 66 Hz (Fig. 6A) while monitoring intracellular Ca2+ changes in CA3 neurons. As shown in Figure 6B, a robust increase in intracellular Ca2+ concentration was triggered after the electrical stimulation. This response was blocked by application of the voltage-gated Na+-channel blocker tetrodotoxin (1 μm, 91 ± 3% inhibition compared with control) (Fig. 6B,D). We also applied CdCl (200 μm) at a concentration that did not attenuate a metabotropic signal triggered by ATP (data not shown) yet is known to effectively inhibit the Ca2+-channels that govern synaptic release (Hinkle et al., 1987; Rosenmund and Stevens, 1996). Similar to the effect of tetrodoxin (TTX), application of CdCl inhibited the fluorescence response in the CA3 cells after stimulation of the mossy fibers (Fig. 6B,D) (86 ± 5% inhibition compared with control), suggesting that the metabotropic Ca2+ response in the CA3 region is triggered by synaptic transmission. Application of the Gαq inhibitor YM-254890 (1 μm) also resulted in a 92 ± 4% inhibition of the responses, compared with control slices (Fig. 6B,D). The inhibitory action of YM-254890 indicates that a Gq-coupled receptor triggers the Ca2+ response in the CA3 neurons after mossy fiber stimulation. The observed Ca2+ signals occurred over a range of several seconds, characteristic of metabotropic receptor-mediated physiological responses (Fig. 1) (Kapur et al., 2001). Inhibitors of iGluRs, namely CNQX (20 μm) and AP5 (50 μm), failed to attenuate the Ca2+ rise (Fig. 6C). Finally, in slices that were treated with AIDA and MPEP, inhibitors, respectively, of mGluR1 and mGluR5, the Ca2+ response triggered by the electrical stimulation was reduced by ∼50%, compared with the response in control slices (Fig. 6D). A similar inhibitory effect was observed in slices treated with both the mGluR and iGluR inhibitors (1 mm AIDA, 10 μm MPEP, 20 μm CNQX and 50 μm AP5). Thus, a significant, residual metabotropic Ca2+ response persisted in the presence of mGluR inhibitors, while the Gαq inhibitor completely inhibited this response (Fig. 6D). This suggests that after synaptic release a receptor distinct from the mGluRs is responsible for a substantial metabotropic Ca2+ response in CA3.
To assess the role of Zn2+ in triggering the synaptically mediated metabotropic Ca2+ response, electrical stimulation was performed in the presence of the cell-impermeable Zn2+ chelator, CaEDTA, which does not significantly affect extracellular Ca2+ concentration (Qian and Noebels, 2005). As shown in Figure 7, A and B, in the presence of 10 μΜ CaEDTA, the Ca2+ response within the CA3 layer was only slightly lower than that in control. However, an inhibitory effect was observed in the presence of either 150 μm or 1 mm CaEDTA (Fig. 7B) (inhibition of 51 ± 5% or 54 ± 7%, respectively, compared with control). This indicates that synaptically released Zn2+ mediates a major part of the metabotropic Ca2+ response. The relatively short time required to induce the metabotropic effect indicates that CaEDTA is unlikely to affect this signaling by regulating the protein expression pattern, as was previously demonstrated for GluR2 activation (Calderone et al., 2004). Finally, we asked if the mGluR inhibitors together with the chelator have an additive effect in blocking the metabotropic Ca2+ response. As shown in Figure 7B, the metabotropic signal after electrical stimulation of the mossy fibers in the presence of the mGluR inhibitors (1 mm AIDA and 10 μm MPEP) and CaEDTA (150 μm) was attenuated by 84 ± 6%. Thus, the coapplication of the chelator together with the mGluR inhibitors yielded a significantly smaller response than that monitored in the presence of either CaEDTA (p < 0.05) or the inhibitors alone (p < 0.05).
Figure 7.

The metabotropic Ca2+ response is attenuated in the absence of synaptic Zn2+. A, The Ca2+ response in cells in the CA3 region after electrical stimulation of the mossy fibers in the presence or absence of the extracellular Zn2+ chelator CaEDTA at the indicated concentrations. Representative responses averaged over 28 cells in 1 slice. B, The averaged Ca2+ response in the presence or absence of CaEDTA (n = 9) at the indicated concentration, or 150 μm CaEDTA in the presence of the mGluR inhibitors (n = 6), or CaEDTA with the mGluR and iGluR inhibitors (n = 4) (*p < 0.05 compared with control and #p < 0.05 compared with CaEDTA alone; NS, nonsignificant). The control is the same as in Figure 6D. C, The response of cells in the CA3 region after electrical stimulation of the mossy fibers in slices obtained from ZnT3 KO versus WT, control, mice. Representative responses averaged over 23 cells in 1 slice. D, Summary of the Ca2+ responses triggered in slices from the ZnT3 KO and WT mice after electrical stimulation or the application of exogenous Zn2+ using the paradigm described in Figure 1 (**p < 0.01, n = 11 for each treatment, the WT control is the same as in Fig. 6D for the stimulation induced response and to Fig. 2B for the exogenous Zn2+ application). E, The Ca2+ rise triggered in slices from the ZnT3 KO mice in the presence or absence of CaEDTA. Representative response averaged over 26 cells in 1 slice. F, Averaged response after electrical stimulation of the mossy fibers in slices from ZnT3 KO mice in the presence of CaEDTA (n = 7) or the mGluR inhibitors (500 μm AIDA, 5 μm MPEP, n = 7) (**p < 0.01).
Mice deficient in ZnT3 lack synaptic Zn2+ while maintaining apparently normal synaptic activity, including release of glutamate and GABA (Cole et al., 2000, 2001; Lopantsev et al., 2003). In agreement with previous studies (Qian and Noebels, 2005), we observed that stimulation of the mossy fibers was followed by a significantly lower fluorescence signal by the extracellular Zn2+-sensitive dye, Newport green (1 μm), in slices from ZnT3 knock-out (KO) mice compared with wild-type controls (data not shown). These mice can thus serve as a useful model to determine the role of synaptic Zn2+ in activating the ZnR and subsequent metabotropic Ca2+ signaling. The Ca2+ response after stimulation of the mossy fibers in slices from ZnT3 KO mice was 56 ± 7% of the response in slices from wild-type (WT) mice (Fig. 7C). Thus, in slices from mice lacking synaptic Zn2+, we observed ∼45% inhibition of the metabotropic signaling in CA3 (Fig. 7D), similar to the reduction of the metabotropic signal by CaEDTA in WT mice (Fig. 7B). Application of exogenous Zn2+ to slices from the ZnT3 KO mice, however, was followed by a Ca2+ rise (108 ± 7%) similar to WT controls (Fig. 7D). This Ca2+ response triggered by exogenous Zn2+ in slices from ZnT3 KO mice indicates that the lack of synaptic Zn2+, rather than a deficiency of ZnR, is responsible for the attenuated metabotropic response. Application of CaEDTA (150 μm) to slices from ZnT3 KO mice yielded a Ca2+ response that did not significantly differ from the response obtained in nontreated ZnT3 KO slices (Fig. 7E,F). Finally, application of the mGluR inhibitors, using the same experimental paradigm described in Figure 6D, was followed by a Ca2+ response that was much smaller than the response in the nontreated, ZnT3 KO slices (Fig. 7F) (73 ± 3% inhibition). Thus, the mGluR inhibitors exhibit significantly (p < 0.01) greater inhibition of the response in ZnT3 KO slices than their effect in the WT slices. The inhibitory effect of the extracellular Zn2+ chelator in slices from the WT, but not the ZnT3 KO mice, together with the substantially attenuated metabotropic Zn2+ response in the ZnT3 KO mice, support the conclusion that synaptic Zn2+, released from mossy fiber terminals, activates a ZnR and triggers Ca2+ signaling in CA3 neurons of the hippocampus.
Discussion
Previous studies have suggested that synaptic Zn2+ is a neuromodulator, acting allosterically at ionotropic synaptic receptors (Vogt et al., 2000; Smart et al., 2004; Paoletti et al., 2009). The present work demonstrates, for the first time, that Zn2+ specifically activates metabotropic signaling in neurons, and does so via a Zn2+-sensing receptor. This finding is supported by the attenuation of the Zn2+-dependent Ca2+ rise after depletion of Ca2+ stores in the ER, and after inhibition of the IP3 pathway using a Gαq inhibitor or PLC blocker. A role for the IP3 pathway in ZnR signaling is consistent with a previous study suggesting that metabotropic signaling in the CA3 region is IP3-dependent rather than rayonidine-receptor dependent (Kapur et al., 2001). The lack of a Zn2+-dependent Ca2+ rise in cells labeled by the astroglial marker, SR101, suggests that although glial cells express metabotropic receptors, the Zn2+-dependent metabotropic response is mediated primarily by neurons.
Our results indicate that the transient metabotropic Ca2+ response in CA3 neurons is triggered by synaptically released Zn2+ after mossy fiber stimulation. The Ca2+ response triggered by electrical stimulation was inhibited by blockade of synaptic transmission. Furthermore, the response is attenuated in the presence of an extracellular Zn2+ chelator, CaEDTA, or in slices from mice lacking synaptic Zn2+. The synaptic Zn2+-dependent Ca2+ response was also mediated via a Gq-coupled mechanism. We conclude, therefore, that in addition to glutamate, metabotropic signaling in the CA3 cells is mediated by synaptically released Zn2+.
We used CaEDTA at concentrations sufficient to remove Zn2+ from the extracellular milieu, based on steady state conditions (http://www.stanford.edu/∼cpatton/webmaxc/webmaxcS.htm) and the expected concentration of released-Zn2+ (Vogt et al., 2000; Qian and Noebels, 2005). At low concentration of the chelator (10 μm), no significant effect on ZnR activity was apparent. This is consistent with the fact that ZnR is not activated by baseline concentrations of extracellular Zn2+, because of its relative low affinity (Km ∼150 μm) (Fig. 1), unlike the high affinity Zn2+ binding sites on the NMDA receptor (Qian and Noebels, 2005). In the presence of 150 μm or 1 mm CaEDTA, however, the ZnR response was reduced. In previous studies, CaEDTA, at similar concentrations, did not effectively reduce a transient rise in extracellular Zn2+ because of its slow chelation activity (Qian and Noebels, 2005). The effective inhibition of the ZnR response is probably related to the relatively low affinity of the ZnR to Zn2+, suggesting such that even partial reduction of extracellular Zn2+ will suffice to lower its concentration below the threshold of ZnR. Such reduction in extracellular Zn2+ may not be apparent using high affinity Zn2+-sensitive dyes. In addition, metabotropic receptors are often distantly located from the release site of neurotransmitters (Knöpfel and Uusisaari, 2008), as we also show for the GPR39 (Fig. 5). Thus, prolonged diffusion time of Zn2+ toward its receptor may enable more effective chelation of Zn2+ by CaEDTA.
While some studies have suggested that synaptic Zn2+ does not affect activity-dependent plasticity, but rather, reshapes the NMDA response (Vogt et al., 2000), others support an effect of synaptic Zn2+ on modulation of long-term synaptic changes in the hippocampus (Xie and Smart, 1994; Li et al., 2001; Izumi et al., 2006). Furthermore, synaptic Zn2+ was shown previously to facilitate LTP in fear conditioning pathways by attenuating GABAergic inhibition (Kodirov et al., 2006). Recently, synaptic Zn2+ was suggested to enhance mossy fiber LTP by activating intracellular signaling of TrkB receptors (Huang et al., 2008; Nagappan et al., 2008). Interestingly, TrkB signaling is strongly regulated by GPCRs, via signaling pathway similar to that activated by ZnR (Hwang et al., 2005; Chen et al., 2007). It has also been reported that TrkB is activated by extracellular Zn2+ through regulation of metalloproteases (Hwang et al., 2005). An intriguing question remains whether ZnR links synaptic Zn2+ to regulation of LTP.
Our results indicate that extracellular Zn2+, via the ZnR, activates mitogen-activated protein kinase (MAPK) and CaMKII pathways. Activation of the MAPK pathway by intracellular Zn2+ rise (Seo et al., 2001; Harris et al., 2004), or after oxidative or nitrosative stress (McLaughlin et al., 2001; Du et al., 2003; Zhang et al., 2004, 2007), is associated with neuronal death. In contrast, activation of MAPK by extracellular Zn2+, in neurons, has been shown to be anti-apoptotic (An et al., 2005). Activation of CaMKII has also been linked to neuronal survival, and inactivation of CaMKII during seizure increases neuronal cell death (Hansen et al., 2003). Zinc has been shown to activate CaMKII (Lengyel et al., 2000), while an extracellular Zn2+ chelator suppressed CaMKII activity in the hippocampus (Tan and Chen, 1997). In the absence of synaptic Zn2+, hippocampal CA3 neurons are more vulnerable to seizure-induced cell death (Domínguez et al., 2003; Côté et al., 2005), consistent with a prosurvival role for ZnR-dependent activation of MAPK and CaMKII. Finally, both the MAPK and CaMKII pathways are critical players in synaptic plasticity, involved in hippocampal learning and memory (Rosenblum et al., 2002; Miyamoto, 2006). Our results show that Zn2+, applied for a shortduration (i.e., 90 s), induces translocation of pERK1/2 to the nucleus.
Changes in expression and function of metabotropic glutamate receptors appear to be critical factors in the etiology of epileptic seizures (Pitsch et al., 2007). Interestingly, chelation of synaptic Zn2+, as well as dietary Zn2+ deficiency, are associated with increased susceptibility to seizures (Fukahori and Itoh, 1990; Domínguez et al., 2003). These effects complement the phenotype of the ZnT3 knock-out mice, which are more susceptible to kainate-induced seizures (Cole et al., 2000). At present, it is unclear how synaptic Zn2+ is related to seizure activity. The involvement of Zn2+ in modulating GABA and NMDA receptors does not dictate either an inhibitory or excitatory role for this metal, and thus, provides no clear basis for assigning it a role in seizure generation. As such, ZnR function in the hippocampus may represent a novel contributor to the overall regulation of neuronal excitability by Zn2+.
Effects of Zn2+ on synaptic plasticity and neuronal excitability, and effects of Zn2+ deficiency on learning and memory in mice and humans are well documented (Cole et al., 2000; Lopantsev et al., 2003; Smart et al., 2004; Kodirov et al., 2006). Since ZnT3 is considered the principal transport system for the loading of Zn2+ ions into synaptic vesicles, and knockout of this transporter completely eliminates Zn2+ from these vesicles (Frederickson et al., 2006; Linkous et al., 2008), the relatively mild phenotype characterizing ZnT3 KO mice has been somewhat unexpected. Our finding that the metabotropic Ca2+ response in slices from ZnT3 KO mice was unaffected by CaEDTA, suggests that it is not mediated via the ZnR.
The metabotropic Zn2+-dependent activity we observed in CA3 neurons is remarkably similar to the extracellular Zn2+-dependent response we previously characterized in epithelial cells (Hershfinkel et al., 2001; Sharir and Hershfinkel, 2005; Dubi et al., 2008). The molecular identity of the receptor mediating this response had been unknown. The orphan GPCR GPR39 is a member of the ghrelin receptor family (McKee et al., 1997). Obestatin was initially proposed as the putative ligand for GPR39 (Zhang et al., 2005), yet, subsequent studies failed to confirm this (Lauwers et al., 2006). It was subsequently suggested that Zn2+ may directly interact with this receptor and enhance its signaling (Holst et al., 2004). The results presented here suggest that GPR39 may indeed directly mediate Zn2+-dependent signaling. The metal selectivity and dose dependence of the neuronal ZnR present in the hippocampus (Fig. 2), and those described for GPR39 (Yasuda et al., 2007) are strikingly similar. It has been reported, moreover, that GPR39 mRNA is expressed in the CNS, and specifically in the hippocampus (Jackson et al., 2006). Consistent with these findings, our immunofluorescence analysis indicates that GPR39 expression is highly enriched in the CA3 neurons that are postsynaptic to the Zn2+-rich mossy fibers, the same cells that mediate synaptically released Zn2+-dependent metabotropic activity. Thus, we propose that GPR39 is a critical component of ZnR metabotropic signaling, and, most likely, that GPR39 and ZnR are one and the same receptor.
In conclusion, we have identified Zn2+-dependent metabotropic activity in hippocampal CA3 neurons. This activity in brain areas rich in synaptic Zn2+ may represent the long-sought link between dynamic changes in extracellular Zn2+ and neuronal signaling mediated by this metal. Our data suggest that specific Zn2+-sensing receptor activity, putatively mediated by GPR39, triggers synaptic Zn2+-dependent metabotropic signaling in the CA3 region. This is the first evidence that Zn2+ is not only a modulatory ion, but acts via a specific postsynaptic receptor to trigger neuronal metabotropic signaling.
Footnotes
This work was supported by United States–Israel Binational Science Foundation Grant 2003201 (M.H., J.T.R.), a Rich Foundation grant, and Israel Science Foundation Grant 585/05 (M.H.). We thank Dr. Richard Palmiter for the ZnT3 KO mice, Dr. Meredin Stoltenberg for advice on the ZnT3 KO experiments, and Drs. Elias Aizenman and Edi Barkai for valuable discussions and critical reading of this manuscript. We thank Astellas Pharma Inc. for generously providing the Gαq inhibitor YM-254890.
References
- Aizenman E, Stout AK, Hartnett KA, Dineley KE, McLaughlin B, Reynolds IJ. Induction of neuronal apoptosis by thiol oxidation: putative role of intracellular zinc release. J Neurochem. 2000;75:1878–1888. doi: 10.1046/j.1471-4159.2000.0751878.x. [DOI] [PubMed] [Google Scholar]
- An WL, Pei JJ, Nishimura T, Winblad B, Cowburn RF. Zinc-induced anti-apoptotic effects in SH-SY5Y neuroblastoma cells via the extracellular signal-regulated kinase 1/2. Brain Res Mol Brain Res. 2005;135:40–47. doi: 10.1016/j.molbrainres.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Azriel-Tamir H, Sharir H, Schwartz B, Hershfinkel M. Extracellular zinc triggers ERK-dependent activation of Na+/H+ exchange in colonocytes mediated by the zinc-sensing receptor. J Biol Chem. 2004;279:51804–51816. doi: 10.1074/jbc.M406581200. [DOI] [PubMed] [Google Scholar]
- Beierlein M, Fall CP, Rinzel J, Yuste R. Thalamocortical bursts trigger recurrent activity in neocortical networks: layer 4 as a frequency-dependent gate. J Neurosci. 2002;22:9885–9894. doi: 10.1523/JNEUROSCI.22-22-09885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkeley JL, Levey AI. Cell-specific extracellular signal-regulated kinase activation by multiple G protein-coupled receptor families in hippocampus. Mol Pharmacol. 2003;63:128–135. doi: 10.1124/mol.63.1.128. [DOI] [PubMed] [Google Scholar]
- Bianchi R, Young SR, Wong RK. Group I mGluR activation causes voltage-dependent and -independent Ca2+ rises in hippocampal pyramidal cells. J Neurophysiol. 1999;81:2903–2913. doi: 10.1152/jn.1999.81.6.2903. [DOI] [PubMed] [Google Scholar]
- Calderone A, Jover T, Mashiko T, Noh KM, Tanaka H, Bennett MV, Zukin RS. Late calcium EDTA rescues hippocampal CA1 neurons from global ischemia-induced death. J Neurosci. 2004;24:9903–9913. doi: 10.1523/JNEUROSCI.1713-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canals M, Angulo E, Casadó V, Canela EI, Mallol J, Viñals F, Staines W, Tinner B, Hillion J, Agnati L, Fuxe K, Ferré S, Lluis C, Franco R. Molecular mechanisms involved in the adenosine A and A receptor-induced neuronal differentiation in neuroblastoma cells and striatal primary cultures. J Neurochem. 2005;92:337–348. doi: 10.1111/j.1471-4159.2004.02856.x. [DOI] [PubMed] [Google Scholar]
- Chen MJ, Nguyen TV, Pike CJ, Russo-Neustadt AA. Norepinephrine induces BDNF and activates the PI-3K and MAPK cascades in embryonic hippocampal neurons. Cell Signal. 2007;19:114–128. doi: 10.1016/j.cellsig.2006.05.028. [DOI] [PubMed] [Google Scholar]
- Cohen-Matsliah SI, Brosh I, Rosenblum K, Barkai E. A novel role for extracellular signal-regulated kinase in maintaining long-term memory-relevant excitability changes. J Neurosci. 2007;27:12584–12589. doi: 10.1523/JNEUROSCI.3728-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole TB, Wenzel HJ, Kafer KE, Schwartzkroin PA, Palmiter RD. Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc Natl Acad Sci U S A. 1999;96:1716–1721. doi: 10.1073/pnas.96.4.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole TB, Robbins CA, Wenzel HJ, Schwartzkroin PA, Palmiter RD. Seizures and neuronal damage in mice lacking vesicular zinc. Epilepsy Res. 2000;39:153–169. doi: 10.1016/s0920-1211(99)00121-7. [DOI] [PubMed] [Google Scholar]
- Cole TB, Martyanova A, Palmiter RD. Removing zinc from synaptic vesicles does not impair spatial learning, memory, or sensorimotor functions in the mouse. Brain Res. 2001;891:253–265. doi: 10.1016/s0006-8993(00)03220-0. [DOI] [PubMed] [Google Scholar]
- Côté A, Chiasson M, Peralta MR, 3rd, Lafortune K, Pellegrini L, Tóth K. Cell type-specific action of seizure-induced intracellular zinc accumulation in the rat hippocampus. J Physiol. 2005;566:821–837. doi: 10.1113/jphysiol.2005.089458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domínguez MI, Blasco-Ibáñez JM, Crespo C, Marqués-Marí AI, Martínez-Guijarro FJ. Zinc chelation during non-lesioning overexcitation results in neuronal death in the mouse hippocampus. Neuroscience. 2003;116:791–806. doi: 10.1016/s0306-4522(02)00731-5. [DOI] [PubMed] [Google Scholar]
- Du L, Zhang X, Han YY, Burke NA, Kochanek PM, Watkins SC, Graham SH, Carcillo JA, Szabó C, Clark RS. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J Biol Chem. 2003;278:18426–18433. doi: 10.1074/jbc.M301295200. [DOI] [PubMed] [Google Scholar]
- Dubi N, Gheber L, Fishman D, Sekler I, Hershfinkel M. Extracellular zinc and zinc-citrate, acting through a putative zinc sensing receptor, regulate growth and survival of prostate cancer cells. Carcinogenesis. 2008;29:1692–1700. doi: 10.1093/carcin/bgn027. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Moncrieff DW. Zinc-containing neurons. Biol Signals. 1994;3:127–139. doi: 10.1159/000109536. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Giblin LJ, 3rd, Balaji RV, Rengarajan B, Masalha R, Frederickson CJ, Zeng Y, Lopez EV, Koh JY, Chorin U, Besser L, Hershfinkel M, Li Y, Thompson RB, Krezel A. Synaptic release of zinc from brain slices: factors governing release, imaging, and accurate calculation of concentration. J Neurosci Methods. 2006;154:19–29. doi: 10.1016/j.jneumeth.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Fukahori M, Itoh M. Effects of dietary zinc status on seizure susceptibility and hippocampal zinc content in the El (epilepsy) mouse. Brain Res. 1990;529:16–22. doi: 10.1016/0006-8993(90)90806-m. [DOI] [PubMed] [Google Scholar]
- Giovannini MG. The role of the extracellular signal-regulated kinase pathway in memory encoding. Rev Neurosci. 2006;17:619–634. doi: 10.1515/revneuro.2006.17.6.619. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Gundelfinger ED, Boeckers TM, Baron MK, Bowie JU. A role for zinc in postsynaptic density asSAMbly and plasticity? Trends Biochem Sci. 2006;31:366–373. doi: 10.1016/j.tibs.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Hansen MR, Bok J, Devaiah AK, Zha XM, Green SH. Ca2+/calmodulin-dependent protein kinases II and IV both promote survival but differ in their effects on axon growth in spiral ganglion neurons. J Neurosci Res. 2003;72:169–184. doi: 10.1002/jnr.10551. [DOI] [PubMed] [Google Scholar]
- Harris FM, Brecht WJ, Xu Q, Mahley RW, Huang Y. Increased tau phosphorylation in apolipoprotein E4 transgenic mice is associated with activation of extracellular signal-regulated kinase: modulation by zinc. J Biol Chem. 2004;279:44795–44801. doi: 10.1074/jbc.M408127200. [DOI] [PubMed] [Google Scholar]
- Henkel G, Krebs B. Metallothioneins: zinc, cadmium, mercury, and copper thiolates and selenolates mimicking protein active site features–structural aspects and biological implications. Chem Rev. 2004;104:801–824. doi: 10.1021/cr020620d. [DOI] [PubMed] [Google Scholar]
- Hershfinkel M, Moran A, Grossman N, Sekler I. A zinc-sensing receptor triggers the release of intracellular Ca2+ and regulates ion transport. Proc Natl Acad Sci U S A. 2001;98:11749–11754. doi: 10.1073/pnas.201193398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinkle PM, Kinsella PA, Osterhoudt KC. Cadmium uptake and toxicity via voltage-sensitive calcium channels. J Biol Chem. 1987;262:16333–16337. [PubMed] [Google Scholar]
- Holst B, Holliday ND, Bach A, Elling CE, Cox HM, Schwartz TW. Common structural basis for constitutive activity of the ghrelin receptor family. J Biol Chem. 2004;279:53806–53817. doi: 10.1074/jbc.M407676200. [DOI] [PubMed] [Google Scholar]
- Holst B, Egerod KL, Schild E, Vickers SP, Cheetham S, Gerlach LO, Storjohann L, Stidsen CE, Jones R, Beck-Sickinger AG, Schwartz TW. GPR39 signaling is stimulated by zinc ions but not by obestatin. Endocrinology. 2007;148:13–20. doi: 10.1210/en.2006-0933. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Dunne EL, Harvey RJ, Smart TG. Zinc-mediated inhibition of GABA(A) receptors: discrete binding sites underlie subtype specificity. Nat Neurosci. 2003;6:362–369. doi: 10.1038/nn1030. [DOI] [PubMed] [Google Scholar]
- Huang YZ, Pan E, Xiong ZQ, McNamara JO. Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy fiber-CA3 pyramid synapse. Neuron. 2008;57:546–558. doi: 10.1016/j.neuron.2007.11.026. [DOI] [PubMed] [Google Scholar]
- Hwang JJ, Park MH, Choi SY, Koh JY. Activation of the Trk signaling pathway by extracellular zinc: role of metalloproteinases. J Biol Chem. 2005;280:11995–12001. doi: 10.1074/jbc.M403172200. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Auberson YP, Zorumski CF. Zinc modulates bidirectional hippocampal plasticity by effects on NMDA receptors. J Neurosci. 2006;26:7181–7188. doi: 10.1523/JNEUROSCI.1258-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson VR, Nothacker HP, Civelli O. GPR39 receptor expression in the mouse brain. Neuroreport. 2006;17:813–816. doi: 10.1097/01.wnr.0000215779.76602.93. [DOI] [PubMed] [Google Scholar]
- Kan I, Ben-Zur T, Barhum Y, Levy YS, Burstein A, Charlow T, Bulvik S, Melamed E, Offen D. Dopaminergic differentiation of human mesenchymal stem cells–utilization of bioassay for tyrosine hydroxylase expression. Neurosci Lett. 2007;419:28–33. doi: 10.1016/j.neulet.2007.03.070. [DOI] [PubMed] [Google Scholar]
- Kapur A, Yeckel M, Johnston D. Hippocampal mossy fiber activity evokes Ca2+ release in CA3 pyramidal neurons via a metabotropic glutamate receptor pathway. Neuroscience. 2001;107:59–69. doi: 10.1016/s0306-4522(01)00293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knöpfel T, Uusisaari M. Modulation of excitation by metabotropic glutamate receptors. Results Probl Cell Differ. 2008;44:163–175. doi: 10.1007/400_2007_035. [DOI] [PubMed] [Google Scholar]
- Kodirov SA, Takizawa S, Joseph J, Kandel ER, Shumyatsky GP, Bolshakov VY. Synaptically released zinc gates long-term potentiation in fear conditioning pathways. Proc Natl Acad Sci U S A. 2006;103:15218–15223. doi: 10.1073/pnas.0607131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauwers E, Landuyt B, Arckens L, Schoofs L, Luyten W. Obestatin does not activate orphan G protein-coupled receptor GPR39. Biochem Biophys Res Commun. 2006;351:21–25. doi: 10.1016/j.bbrc.2006.09.141. [DOI] [PubMed] [Google Scholar]
- Lengyel I, Fieuw-Makaroff S, Hall AL, Sim AT, Rostas JA, Dunkley PR. Modulation of the phosphorylation and activity of calcium/calmodulin-dependent protein kinase II by zinc. J Neurochem. 2000;75:594–605. doi: 10.1046/j.1471-4159.2000.0750594.x. [DOI] [PubMed] [Google Scholar]
- Li Y, Hough CJ, Frederickson CJ, Sarvey JM. Induction of mossy fiber→CA3 long-term potentiation requires translocation of synaptically released Zn2+ J Neurosci. 2001;21:8015–8025. doi: 10.1523/JNEUROSCI.21-20-08015.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkous DH, Flinn JM, Koh JY, Lanzirotti A, Bertsch PM, Jones BF, Giblin LJ, Frederickson CJ. Evidence that the ZNT3 protein controls the total amount of elemental zinc in synaptic vesicles. J Histochem Cytochem. 2008;56:3–6. doi: 10.1369/jhc.6A7035.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopantsev V, Wenzel HJ, Cole TB, Palmiter RD, Schwartzkroin PA. Lack of vesicular zinc in mossy fibers does not affect synaptic excitability of CA3 pyramidal cells in zinc transporter 3 knockout mice. Neuroscience. 2003;116:237–248. doi: 10.1016/s0306-4522(02)00570-5. [DOI] [PubMed] [Google Scholar]
- Luo Y, DeFranco DB. Opposing roles for ERK1/2 in neuronal oxidative toxicity: distinct mechanisms of ERK1/2 action at early versus late phases of oxidative stress. J Biol Chem. 2006;281:16436–16442. doi: 10.1074/jbc.M512430200. [DOI] [PubMed] [Google Scholar]
- McKee KK, Tan CP, Palyha OC, Liu J, Feighner SD, Hreniuk DL, Smith RG, Howard AD, Van der Ploeg LH. Cloning and characterization of two human G protein-coupled receptor genes (GPR38 and GPR39) related to the growth hormone secretagogue and neurotensin receptors. Genomics. 1997;46:426–434. doi: 10.1006/geno.1997.5069. [DOI] [PubMed] [Google Scholar]
- McLaughlin B, Pal S, Tran MP, Parsons AA, Barone FC, Erhardt JA, Aizenman E. p38 activation is required upstream of potassium current enhancement and caspase cleavage in thiol oxidant-induced neuronal apoptosis. J Neurosci. 2001;21:3303–3311. doi: 10.1523/JNEUROSCI.21-10-03303.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto E. Molecular mechanism of neuronal plasticity: induction and maintenance of long-term potentiation in the hippocampus. J Pharmacol Sci. 2006;100:433–442. doi: 10.1254/jphs.cpj06007x. [DOI] [PubMed] [Google Scholar]
- Nagappan G, Woo NH, Lu B. Ama “zinc” link between TrkB transactivation and synaptic plasticity. Neuron. 2008;57:477–479. doi: 10.1016/j.neuron.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Nakamura K, Lasser-Ross N, Barbara JG, Sandler VM, Ross WN. Inositol 1,4,5-trisphosphate (IP3)-mediated Ca2+ release evoked by metabotropic agonists and backpropagating action potentials in hippocampal CA1 pyramidal neurons. J Neurosci. 2000;20:8365–8376. doi: 10.1523/JNEUROSCI.20-22-08365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls RE, Zhang XL, Bailey CP, Conklin BR, Kandel ER, Stanton PK. mGluR2 acts through inhibitory Galpha subunits to regulate transmission and long-term plasticity at hippocampal mossy fiber-CA3 synapses. Proc Natl Acad Sci U S A. 2006;103:6380–6385. doi: 10.1073/pnas.0601267103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Kerr JN, Helmchen F. Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat Methods. 2004;1:31–37. doi: 10.1038/nmeth706. [DOI] [PubMed] [Google Scholar]
- Palmiter RD, Cole TB, Quaife CJ, Findley SD. ZnT-3, a putative transporter of zinc into synaptic vesicles. Proc Natl Acad Sci U S A. 1996;93:14934–14939. doi: 10.1073/pnas.93.25.14934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Ascher P, Neyton J. High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J Neurosci. 1997;17:5711–5725. doi: 10.1523/JNEUROSCI.17-15-05711.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Vergnano AM, Barbour B, Casado M. Zinc at glutamatergic synapses. Neuroscience. 2009;158:126–136. doi: 10.1016/j.neuroscience.2008.01.061. [DOI] [PubMed] [Google Scholar]
- Pitsch J, Schoch S, Gueler N, Flor PJ, van der Putten H, Becker AJ. Functional role of mGluR1 and mGluR4 in pilocarpine-induced temporal lobe epilepsy. Neurobiol Dis. 2007;26:623–633. doi: 10.1016/j.nbd.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Qian J, Noebels JL. Visualization of transmitter release with zinc fluorescence detection at the mouse hippocampal mossy fibre synapse. J Physiol. 2005;566:747–758. doi: 10.1113/jphysiol.2005.089276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachline J, Perin-Dureau F, Le Goff A, Neyton J, Paoletti P. The micromolar zinc-binding domain on the NMDA receptor subunit NR2B. J Neurosci. 2005;25:308–317. doi: 10.1523/JNEUROSCI.3967-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum K, Futter M, Voss K, Erent M, Skehel PA, French P, Obosi L, Jones MW, Bliss TV. The role of extracellular regulated kinases I/II in late-phase long-term potentiation. J Neurosci. 2002;22:5432–5441. doi: 10.1523/JNEUROSCI.22-13-05432.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Ruiz A, Walker MC, Fabian-Fine R, Kullmann DM. Endogenous zinc inhibits GABA(A) receptors in a hippocampal pathway. J Neurophysiol. 2004;91:1091–1096. doi: 10.1152/jn.00755.2003. [DOI] [PubMed] [Google Scholar]
- Sekler I, Moran A, Hershfinkel M, Dori A, Margulis A, Birenzweig N, Nitzan Y, Silverman WF. Distribution of the zinc transporter ZnT-1 in comparison with chelatable zinc in the mouse brain. J Comp Neurol. 2002;447:201–209. doi: 10.1002/cne.10224. [DOI] [PubMed] [Google Scholar]
- Sensi SL, Jeng JM. Rethinking the excitotoxic ionic milieu: the emerging role of Zn(2+) in ischemic neuronal injury. Curr Mol Med. 2004;4:87–111. doi: 10.2174/1566524043479211. [DOI] [PubMed] [Google Scholar]
- Sensi SL, Yin HZ, Carriedo SG, Rao SS, Weiss JH. Preferential Zn2+ influx through Ca2+-permeable AMPA/kainate channels triggers prolonged mitochondrial superoxide production. Proc Natl Acad Sci U S A. 1999;96:2414–2419. doi: 10.1073/pnas.96.5.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo SR, Chong SA, Lee SI, Sung JY, Ahn YS, Chung KC, Seo JT. Zn2+-induced ERK activation mediated by reactive oxygen species causes cell death in differentiated PC12 cells. J Neurochem. 2001;78:600–610. doi: 10.1046/j.1471-4159.2001.00438.x. [DOI] [PubMed] [Google Scholar]
- Sharir H, Hershfinkel M. The extracellular zinc-sensing receptor mediates intercellular communication by inducing ATP release. Biochem Biophys Res Commun. 2005;332:845–852. doi: 10.1016/j.bbrc.2005.05.036. [DOI] [PubMed] [Google Scholar]
- Smart TG, Hosie AM, Miller PS. Zn2+ ions: modulators of excitatory and inhibitory synaptic activity. Neuroscientist. 2004;10:432–442. doi: 10.1177/1073858404263463. [DOI] [PubMed] [Google Scholar]
- Smith IF, Wiltgen SM, Parker I. Localization of puff sites adjacent to the plasma membrane: functional and spatial characterization of Ca(2+) signaling in SH-SY5Y cells utilizing membrane-permeant caged IP(3) Cell Calcium. 2009;45:65–76. doi: 10.1016/j.ceca.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan SE, Chen SS. The activation of calcium/calmodulin-dependent protein kinase II after glutamate or potassium stimulation in hippocampal slices. Brain Res Bull. 1997;43:269–273. doi: 10.1016/s0361-9230(97)00004-x. [DOI] [PubMed] [Google Scholar]
- Tang TS, Tu H, Chan EY, Maximov A, Wang Z, Wellington CL, Hayden MR, Bezprozvanny I. Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron. 2003;39:227–239. doi: 10.1016/s0896-6273(03)00366-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt K, Mellor J, Tong G, Nicoll R. The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron. 2000;26:187–196. doi: 10.1016/s0896-6273(00)81149-6. [DOI] [PubMed] [Google Scholar]
- Volk LJ, Daly CA, Huber KM. Differential roles for group 1 mGluR subtypes in induction and expression of chemically induced hippocampal long-term depression. J Neurophysiol. 2006;95:2427–2438. doi: 10.1152/jn.00383.2005. [DOI] [PubMed] [Google Scholar]
- Weiss JH, Sensi SL, Koh JY. Zn(2+): a novel ionic mediator of neural injury in brain disease. Trends Pharmacol Sci. 2000;21:395–401. doi: 10.1016/s0165-6147(00)01541-8. [DOI] [PubMed] [Google Scholar]
- White LD, Cory-Slechta DA, Gilbert ME, Tiffany-Castiglioni E, Zawia NH, Virgolini M, Rossi-George A, Lasley SM, Qian YC, Basha MR. New and evolving concepts in the neurotoxicology of lead. Toxicol Appl Pharmacol. 2007;225:1–27. doi: 10.1016/j.taap.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Wu X, Zagranichnaya TK, Gurda GT, Eves EM, Villereal ML. A TRPC1/TRPC3-mediated increase in store-operated calcium entry is required for differentiation of H19–7 hippocampal neuronal cells. J Biol Chem. 2004;279:43392–43402. doi: 10.1074/jbc.M408959200. [DOI] [PubMed] [Google Scholar]
- Xie X, Smart TG. Modulation of long-term potentiation in rat hippocampal pyramidal neurons by zinc. Pflugers Arch. 1994;427:481–486. doi: 10.1007/BF00374264. [DOI] [PubMed] [Google Scholar]
- Yamazaki Y, Fujii S, Nakamura T, Miyakawa H, Kudo Y, Kato H, Ito K. Changes in [Ca2+]. (i) during adenosine triphosphate-induced synaptic plasticity in hippocampal CA1 neurons of the guinea pig. Neurosci Lett. 2002;324:65–68. doi: 10.1016/s0304-3940(02)00175-1. [DOI] [PubMed] [Google Scholar]
- Yasuda S, Miyazaki T, Munechika K, Yamashita M, Ikeda Y, Kamizono A. Isolation of Zn2+ as an endogenous agonist of GPR39 from fetal bovine serum. J Recept Signal Transduct Res. 2007;27:235–246. doi: 10.1080/10799890701506147. [DOI] [PubMed] [Google Scholar]
- Yin HZ, Sensi SL, Ogoshi F, Weiss JH. Blockade of Ca2+-permeable AMPA/kainate channels decreases oxygen-glucose deprivation-induced Zn2+ accumulation and neuronal loss in hippocampal pyramidal neurons. J Neurosci. 2002;22:1273–1279. doi: 10.1523/JNEUROSCI.22-04-01273.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JV, Ren PG, Avsian-Kretchmer O, Luo CW, Rauch R, Klein C, Hsueh AJ. Obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin's effects on food intake. Science. 2005;310:996–999. doi: 10.1126/science.1117255. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang H, Li J, Jimenez DA, Levitan ES, Aizenman E, Rosenberg PA. Peroxynitrite-induced neuronal apoptosis is mediated by intracellular zinc release and 12-lipoxygenase activation. J Neurosci. 2004;24:10616–10627. doi: 10.1523/JNEUROSCI.2469-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Aizenman E, DeFranco DB, Rosenberg PA. Intracellular zinc release, 12-lipoxygenase activation and MAPK dependent neuronal and oligodendroglial death. Mol Med. 2007;13:350–355. doi: 10.2119/2007-00042.Zhang. [DOI] [PMC free article] [PubMed] [Google Scholar]