

Abstract
The characterization of a novel yeast-splicing factor, Luc7p, is presented. The LUC7 gene was identified by a mutation that causes lethality in a yeast strain lacking the nuclear cap-binding complex (CBC). Luc7p is similar in sequence to metazoan proteins that have arginine–serine and arginine–glutamic acid repeat sequences characteristic of a family of splicing factors. We show that Luc7p is a component of yeast U1 snRNP and is essential for vegetative growth. The composition of yeast U1 snRNP is altered in luc7 mutant strains. Extracts of these strains are unable to support any of the defined steps of splicing unless recombinant Luc7p is added. Although the in vivo defect in splicing wild-type reporter introns in a luc7 mutant strain is comparatively mild, splicing of introns with nonconsensus 5′ splice site or branchpoint sequences is more defective in the mutant strain than in wild-type strains. By use of reporters that have two competing 5′ splice sites, a loss of efficient splicing to the cap proximal splice site is observed in luc7 cells, analogous to the defect seen in strains lacking CBC. CBC can be coprecipitated with U1 snRNP from wild-type, but not from luc7, yeast strains. These data suggest that the loss of Luc7p disrupts U1 snRNP–CBC interaction, and that this interaction contributes to normal 5′ splice site recognition.
Keywords: Pre-mRNA processing, U1 snRNP, 5′ splice site recognition, cap-binding complex, alternative splicing
The formation of mRNAs in the nuclei of eukaryotic cells involves several co- and post-transcriptional processing events. These include 5′ end capping, 3′ end formation, usually by cleavage and polyadenylation, and frequently the removal of intervening sequences by splicing. Pre-mRNA splicing can be conceptually divided into distinct stages. The initial step is recognition of conserved intronic sequences near the 5′ splice site and branchpoint region by a subset of splicing factors. This is followed by assembly of multiple additional splicing factors to form the spliceosome. Rearrangements within the spliceosome then occur, accompanying the two chemical steps of intron removal. Spliced mRNA is released for export to the cytoplasm while intronic RNA is degraded and splicing factors are recycled (Moore et al. 1993).
The first defined step of splicing consists of the formation of commitment complexes in yeast (Séraphin and Rosbash 1989a) and E complex in mammals (Michaud and Reed 1991). In yeast, two forms of commitment complex are experimentally separable, CC1 and CC2 (Séraphin and Rosbash 1989a). It is likely, though not definitively proven, that CC1 is a precursor of CC2. Both contain U1 snRNP, which interacts with the 5′ splice site. CC2 additionally contains at least two proteins, BBP and Mud2p, that bind to the branchpoint sequence and an adjacent pyrimidine-rich tract, respectively (Séraphin and Rosbash 1991; Abovich et al. 1994; Abovich and Rosbash 1997; Berglund et al. 1998). mBBP/SF1 and U2AF65, the mammalian homologs of these proteins, are present in E complex (Ruskin et al. 1988; Michaud and Reed 1991, 1993; Krämer 1992; Rain et al. 1998).
These facts about early steps in spliceosome formation point to a critical role for U1 snRNP in 5′ splice site definition and choice, and lead to the question of how the choice between two alternative 5′ splice sites that can both be spliced to a common 3′ splice site is made. Examination of alternative splicing in vertebrates suggests that factors that are not components of U1 snRNP can influence the selection of splice sites (for examples, see Chabot and Steitz 1987; Eperon et al. 1993; Kohtz et al. 1994). Recent work in yeast has shown, however, that at least one U1 snRNP protein can also influence 5′ splice site choice (Puig et al. 1999).
Yeast U1 snRNA is significantly larger than vertebrate U1 snRNA (Kretzner et al. 1987; Siliciano et al. 1987). The yeast-specific regions of the RNA are not absolutely essential for survival, but nevertheless they play a role in splicing (Liao et al. 1990; Siliciano et al. 1991). Yeast U1 snRNP, as biochemically purified, is considerably more complex than vertebrate U1 snRNP. Both contain the Sm core proteins and three U1-specific proteins, U1 70K/Snp1p, U1A/Mud1p, and U1C/yU1-C. In addition, the yeast U1 snRNP contains at least six specific proteins (Snu71p, Snu65p, Snu56p, Prp39p, Prp40p, and Nam8p) that have no currently characterized vertebrate homologs (Neubauer et al. 1997; Gottschalk et al. 1998). U1 snRNP interacts with the 5′ splice site via base-pairing through U1 snRNA (Zhuang and Weiner 1986; Séraphin et al. 1988; Siliciano and Guthrie 1988; Séraphin and Rosbash 1989b). Recent data indicate that the yeast U1 snRNP proteins also make extensive contact with the pre-mRNA both upstream and downstream of the 5′ splice site (Puig et al. 1999; Zhang and Rosbash 1999). These interactions are likely to increase the stability of U1 snRNP-5′ splice site binding. In addition, at least one U1 snRNP protein–pre-mRNA interaction, involving Nam8p, is affected by the sequence of the pre-mRNA to which the protein binds. The sequence specificity of this interaction can affect 5′ splice site choice (Puig et al. 1999).
Other signals on a pre-mRNA can also influence binding of U1 snRNP to a 5′ splice site or other steps that affect the efficiency of intron recognition and removal. Examples include the effects of adjacent introns or 3′ end formation signals (for review, see Berget 1995), exon enhancer sequences (for review, see Hertel et al. 1997), and, in the case of the cap-proximal intron, the cap structure (Konarska et al. 1984; Krainer et al. 1984; Ohno et al. 1987).
The effect of the cap structure is mediated by the nuclear cap-binding complex (CBC), a conserved heterodimeric complex composed of CBP80 and CBP20 (Izaurralde et al. 1994, 1995; Colot et al. 1996; Görlich et al. 1996; Lewis et al. 1996a,b). In both yeast and mammals, CBC appears to act by increasing the efficiency of recognition of the cap-proximal 5′ splice site by U1 snRNP during commitment complex/E complex assembly (Colot et al. 1996; Lewis et al. 1996a,b). Much of the initial evidence for this mechanism came from biochemical experiments (Izaurralde et al. 1994; Lewis et al. 1996b) but in yeast a considerable body of genetic data indicates that CBC plays an important role in commitment complex assembly. The gene encoding yCBP20, MUD13, was identified by a mutation that caused synthetic lethality in combination with a nonlethal deletion of part of U1 snRNA (Colot et al. 1996). A more extensive search for genes whose mutation led to synthetic lethality in the absence of CBC (Fortes et al. 1999) led to the identification of LUC genes (lethal unless CBC is produced). The LUC collection includes genes that encode several components of the commitment complex, including both Mud2p/Luc2p and several protein components of yeast U1 snRNP. Some of these genes encode proteins conserved between yeast and vertebrates, like SmD3/Luc6p or Mud1p/Luc1p, the yeast homolog of the human U1A protein, and others encode several of the recently identified yeast-specific U1 snRNP proteins, Nam8p/Luc3p, Snu56p/Luc4p, and Snu71p/Luc5p (Neubauer et al. 1997; Gottschalk et al. 1998; Fortes et al. 1999).
One functionally uncharacterized gene identified in the screen was named LUC7. Here we demonstrate that Luc7p is an additional component of the yeast U1 snRNP. LUC7 is an essential gene, and Luc7p is required for commitment complex formation in vitro. In the presence of a temperature-sensitive form of Luc7p, the protein composition of U1 snRNP is altered. Although the defective U1 snRNP still appears to be partially active in vivo, splicing efficiency is reduced and 5′ splice site selection is altered. The change in 5′ splice site recognition is similar to that seen in the absence of CBC, suggesting that CBC–U1 snRNP interaction is affected by the absence of Luc7p. Biochemical data that support this hypothesis are presented.
Results
LUC7 was originally identified by complementation of a mutation that caused lethality in yeast strains that did not produce CBC, but was not further characterized (Fortes et al. 1999; see also below). As a first step in the analysis of LUC7, the phenotype caused by deletion of the gene was determined. One allele of LUC7 was disrupted in a diploid yeast strain by replacement of the entire ORF with sequences encoding the wild-type URA3 gene. After sporulation and dissection, maximally two spores from each tetrad gave rise to a growing colony (Fig. 1A). The growing cells contained the nondisrupted LUC7 allele, indicating that LUC7 is an essential gene. While this work was in progress, LUC7 was disrupted as part of a systematic analysis of Saccharomyces cerevisiae chromosome IV, and reported to be essential for growth (Lopez et al. 1998). Separately, in a screen for temperature-sensitive mutants blocked in the cell cycle, a further allele of LUC7 was identified, luc7-1 (see Materials and Methods). On shift to 37°C, haploid strains carrying luc7-1 exhibited a growth defect after ∼2 hr, and they stopped growing altogether after ∼5 hr (Fig. 1B).
Figure 1.

LUC7 is essential in yeast. In a diploid strain, one chromosomal copy of LUC7 was substituted by a URA3 marker with homologous recombination. As shown in A, after sporulation and tetrad dissection, only two of the four spores gave rise to colonies. None of the surviving progeny carried the URA+ marker and all retained the LUC7 gene. This indicates that LUC7 is an essential gene. (B) A temperature-sensitive mutant of LUC7 (luc7-1) was isolated. The growth of the strain carrying luc7-1 after shift to the restrictive temperature (37°C) is compared with the growth of a wild-type strain (WT). Note that after 2 hr at 37°C, the growth rate of the culture carrying luc7-1 decreases and growth stops after 5 hr at 37°C (arrows). (C) The luc7-2 mutant can grow on FOA plates when transformed with a vector that expresses wild-type LUC7 (+pLEU–LUC7) but not when transformed with an empty vector (+pLEU) or a vector expressing the mutant forms of LUC7 (+pLEU–luc7-1 or +pLEU–luc7-2). Because growth on FOA selects against the plasmid from which yCBP80 and yCBP20 are expressed, this indicates that LUC7 can rescue the synthetic lethality in strains lacking CBC and LUC7, but that neither luc7-1 nor luc7-2 can rescue this defect. (D) The WR244 temperature-sensitive strain (luc7-1) can grow at 37°C when transformed with a vector expressing wild-type LUC7 (+pLEU–LUC7) but not when transformed with an empty vector (+pLEU) or vectors expressing the mutant forms of LUC7 (+pLEU–luc7-1 and +pLEU–luc7-2). Growth of WR244 at 30°C in the presence of these plasmids is shown at left.
The PF433 strain was isolated in a synthetic lethal screen with yCBC. This strain carries the luc7-2 allele and, in addition, lacks chromosomal copies of both GCR3 and MUD13, the genes encoding the two subunits of yeast CBC. This genetic combination causes synthetic lethality (Fortes et al. 1999). PF433 can grow when carrying a plasmid expressing GCR3 and MUD13, as well as the URA3 marker. When transferred to FOA-containing plates to select for loss of this plasmid, luc7-2 cells could not grow (Fig. 1C, left). To analyze the ability of the two mutant forms of LUC7 to complement the synthetic lethal phenotype, complementation tests were carried out. The growth defect on FOA was complemented by an expression plasmid carrying wild-type LUC7 but not by plasmids from which either the luc7-1 or luc7-2 alleles were expressed (Fig. 1C). Similarly, the temperature-sensitive defect of a strain carrying luc7-1 was complemented by a plasmid expressing LUC7, but not by plasmids expressing either luc7-1 or luc7-2 (Fig. 1D).
Luc7p, the conceptual protein product of LUC7 (Fig. 2), includes sequences that encode two putative zinc finger motifs, structures known to bind either nucleic acids or proteins. The first zinc finger is of the C3H (cysteine 3, histidine) type, whereas the second is a typical C2H2 zinc finger of the class often found in RNA-binding proteins (Matsushima et al. 1997). Sequencing and further analysis of the mutant alleles revealed that the luc7-1 phenotype was due to a point mutation that lies between the two zinc fingers (glutamate to lysine at amino acid position 149), whereas the luc7-2 phenotype was due to a frameshift at position 195 (Fig. 2B), just upstream of the second zinc finger.
Figure 2.


Schematic representation of LUC7 and its relatives. (A) The LUC7 gene appears to have been duplicated several times during evolution (see phylogenetic tree at left) to generate a family of related proteins in C. elegans (ceLuc7A1, ceLuc7A2, and ceLuc7B) and Homo sapiens (hLuc7A, hLuc7B1, and hLuc7B2). Luc7 proteins contain zinc fingers of the CH3 and C2H2 type (hatched boxes). The homologs in higher eukaryotes are extended at their carboxyl termini with domains that contain RE (highlighted with black lines) and RS (gray lines) repeats. These repeats are characteristic of splicing factors. A vertical line represents two repeats. (B) Sequence alignment of yeast Luc7p and two human homologs. The zinc fingers are boxed and the position of the conserved C and H residues is indicated. The positions of the mutations found in luc7-1 and luc7-2 are indicated. The extended RE- and RS-rich carboxyl termini of the human proteins are not shown.
Examination of protein sequence databases revealed the existence of metazoan relatives of Luc7p, including three in human and Caenorhabditis elegans (Fig. 2A) and others in Arabidopsis thaliana, Drosophila melanogaster, and other eukaryotes. The regions encoding the zinc finger motifs are particularly highly conserved (57% similarity conserved across the whole family) (Fig. 2). A phylogenetic tree was constructed with the sequence information. LUC7 appears to have been duplicated early in evolution, leading to the Luc7A and Luc7B subfamilies in higher eukaryotes (Fig. 2A). Interestingly, all metazoan LUC7 family members contain carboxy-terminal extensions with multiple arginine–serine (RS) or arginine–glutamate (RE) repeats, characteristic of a large number of metazoan splicing factors (Neugebauer et al. 1995; Staknis and Reed 1995) (Fig. 2A). This, together with the synthetic lethality data, suggested that Luc7p might be involved in pre-mRNA splicing.
Luc7p mutation affects splicing
The effect of mutation of LUC7 on pre-mRNA splicing was tested in vivo. Initially, a series of reporter constructs derived from the ribosomal protein 51A (RP51A) gene were utilized. They contained either the wild-type RP51A intron or derivatives whose splicing efficiency was reduced by mutation at the 5′ splice site or branchpoint (Teem and Rosbash 1983; Jacquier et al. 1985; Pascolo and Séraphin 1997). Splicing of these three pre-mRNAs was tested with RNA extracted from luc7-1 cells grown at either permissive (30°C) or nonpermissive (37°C) temperature either in the presence (+) or absence (−) of a plasmid from which wild-type Luc7p was expressed (Fig. 3). As demonstrated previously, expression of Luc7p complements the luc7-1 phenotype (Fig. 1).
Figure 3.
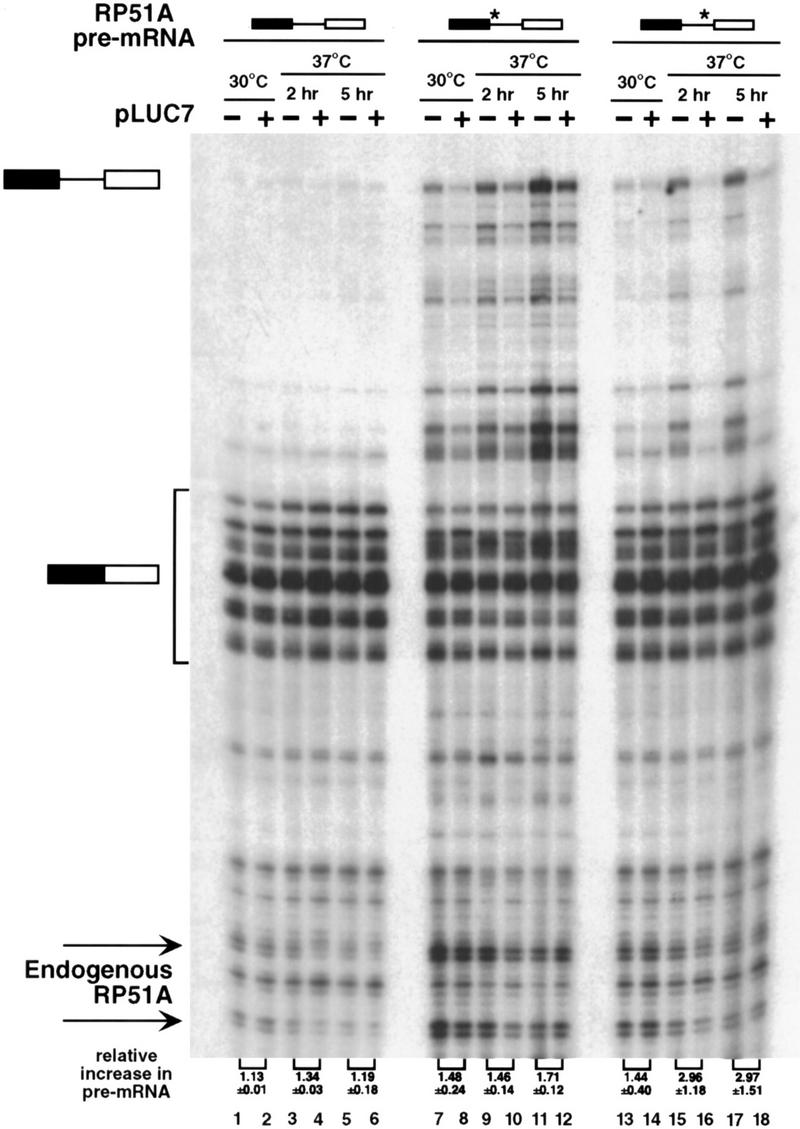
In vivo splicing is less efficient in the luc7-1 cells. WR244 cells (temperature-sensitive luc7-1) untransformed (−) or transformed with a plasmid that expresses LUC7 (+) were transformed with plasmids that express RP51A pre-mRNA with consensus splicing sequences (lanes 1–6), with a weak 5′ splice site (GUAUaU, lanes 7–12), or with a weak branchpoint sequence (UAuUAAC, lanes 13–18), as schematized at top where a star represents the mutant sequence. Total RNA was isolated from these strains grown at 30°C or after shift to 37°C for 2 or 5 hr and RP51A mRNAs (indicated schematically at left) were visualized by primer extension. The multiple species of spliced mRNA are due to the existence of several transcriptional start sites. Signals were quantified with a fluorimager (Fujifilm). The numbers at the bottom represent the increase in the ratio of pre-mRNA to mRNA in the matched pairs of Luc7p expressing and nonexpressing strains.
Splicing of the wild-type RP51A intron was affected to only a minor extent by the luc7-1 mutation, as seen by the small increase in pre-mRNA relative to mRNA in the strain at 37°C (Fig. 3, lanes 1–6, cf. + and − lanes). Splicing of the chromosomally encoded RP51A and actin pre-mRNAs was also not detectably reduced (data not shown). However, pre-mRNA accumulation was readily visible in cells carrying luc7-1 when the splicing efficiency of the reporter was compromised by mutation of either the 5′ splice site from GUAUGU to GUAUaU or the branchpoint from UACUAAC to UAuUAAC (Fig. 3, lanes 7–18). The defect in splicing was already detected at permissive temperature by the reporter with the mutant 5′ splice site (lanes 7,8), although it was exacerbated further by raising the temperature to 37°C (lanes 9–12).
Luc7p is a component of U1 snRNP
As a first step to examining the role of Luc7p in splicing in more detail, its association with U snRNAs was examined. To this end, a yeast strain carrying a protein A-tagged version of Luc7p was constructed. Extracts were made from this strain, a control strain lacking a protein A tag, and, as a positive control, a strain carrying a protein A-tagged version of the U1 snRNP component Nam8p (Gottschalk et al. 1998; Puig et al. 1999). The extracts were fractionated on IgG–agarose and RNA extracted from input, supernatant, and pellet fractions was analyzed by primer extension. U snRNAs immunoprecipitated with anti-trimethyl guanosine cap antibodies were used as size markers (Fig. 4A, lane 11). Only minor background quantities of U snRNAs were found in the control pellet fraction (Fig. 4A, lanes 1–3). However, U1 snRNA was specifically and efficiently precipitated from protein A-tagged Nam8p (Nam8p–ProtA) and Luc7p–ProtA strains (lanes 4–9), suggesting that Luc7p, like Nam8p, is a U1 snRNP component.
Figure 4.

Luc7p is a U1 snRNP protein. (A) Extracts prepared from a control strain (wild-type), a strain with protein A-tagged Nam8p (NAM8–ProtA), or a strain with protein A-tagged Luc7p (LUC7–ProtA) were incubated with beads containing anti-rabbit antibody. After binding, supernatant (S) and pellet (P) fractions were recovered. Primer extension of 50% of the input (I), unbound (S), and bound (P) RNAs was carried out with oligonucleotides specific for U4, U2, U6, U5, and U1 snRNAs. The extended products were separated by electrophoresis, as indicated at left. U1 RNA is highlighted with an arrow. Wild-type extracts precipitated with an α-trimethyl guanosine cap antibody (α-TMG) served as a positive control (lanes 10 and 11). (B,C) Extracts prepared from a control strain (wild-type) or strains expressing a NAM8–ProtA, MUD10–ProtA, or LUC7–ProtA were incubated with beads containing rabbit IgG. After extensive washing with buffer containing 0.15 m NaCl, proteins were sequentially eluted with buffers containing 0.2, 0.5, and 2 m NaCl. Extensive washing was carried out with each elution buffer before changing the salt concentration in the elution. Eluted proteins were analyzed by Western blotting with anti-Snu71p, anti-Nam8p, anti-yCBP80, and anti-Npl3p antibodies as shown in B or schematically in C, in which + indicates detection of the protein by Western blotting. Lane 1 represents 25% of wild-type input.
To confirm this conclusion, similar experiments were carried out, but instead of analyzing U snRNAs, proteins that coprecipitated with Luc7p–ProtA were examined. In this case, proteins precipitated via association with Nam8p–ProtA and with a protein A-tagged version of Mud10p/Snu56p were also examined. Mud10p/Snu56p is a U1 snRNP protein (Gottschalk et al. 1998) that, like Nam8p and Luc7p, can be mutated to a form that causes synthetic lethality in the absence of CBC (Fortes et al. 1999).
Two U1 snRNP proteins, Snu71p and Nam8p (Gottschalk et al. 1998), coprecipitated with Luc7p–ProtA even after washing in buffer containing up to two molar NaCl, indicating that Luc7p is, like these two proteins, a stable U1 snRNP component (Fig. 4B,C). This was confirmed in an independent biochemical study of U1 snRNP composition (Rigaut et al. 1999). Proteins that exhibit a weaker association with U1 snRNP were examined next. A minor amount of yCBP80 and Npl3p (see Gottschalk et al. 1998) also specifically coprecipitated with all three protein A-tagged U1 snRNP proteins (Fig. 4B,C). However, these latter associations were disrupted by moderate and low salt washes, respectively (Fig. 4B,C). There seemed to be no preferential association between any of the U1 snRNP proteins tested and yCBP80 under these conditions, suggesting that the precipitation seen reflects association of a fraction of CBC with the U1 snRNP.
Mutation of Luc7p destabilizes U1 snRNP
To further examine the basis for the splicing defect, U1 snRNP was examined in luc7-1 cells grown at 30°C or 37°C . Cells containing either only the luc7-1 allele (−) or also carrying LUC7 on a plasmid (+) were compared. No difference in the accumulation of U1 snRNA was seen in noncomplemented luc7-1 cells, even 5 hr after transfer to 37°C (Fig. 5A). The luc7-1 mutation did, however, affect the association of Luc7p with U1 snRNA. When U1 snRNA association to protein A-tagged wild-type Luc7p (Luc7p–ProtA) and Luc7-1p–ProtA was compared, U1 could only be coprecipitated with the wild-type protein. Luc7-1p association with U1 snRNA was not detectable above background even when U1 snRNP was prepared under mild conditions (0.15 m NaCl) from cells grown at permissive temperature (Fig. 5B, cf. lanes 1–6 with 7–10). By Western blotting, no change in the amount of Luc7-1p–ProtA was seen after temperature shift (data not shown), demonstrating that the loss of U1 snRNP association was not due to degradation of the protein.
Figure 5.



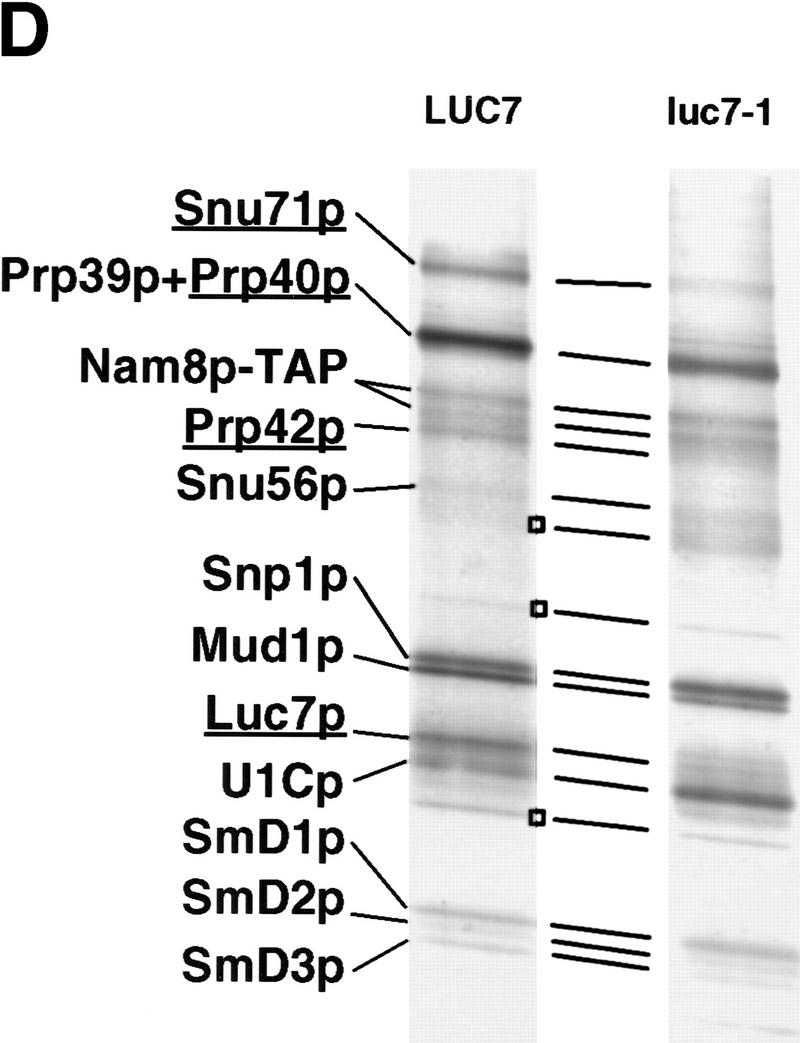
U1 snRNP composition is altered in the luc7-1 temperature-sensitive cells. (A) U1 snRNA accumulation is not affected by the luc7-1 mutation. Untransformed WR244 (luc7-1) cells (−) or cells transformed (+) with a plasmid that expresses LUC7 (pLUC7) were grown at 30°C or shifted for 2 or 5 hr to 37°C. U1 snRNA and U2 snRNA levels were analyzed by primer extension. (B) Extracts were prepared from a control strain (wild-type), LUC7–ProtA or luc7-1–ProtA, grown at 30°C or at 37°C for 2 hr. Protein A-containing complexes were selected from these extracts with IgG beads. After binding, supernatant (S) and pellet (P) fractions were recovered and U1 snRNA (right) was visualized by primer extension. (C) Extracts prepared as described in A were immunoprecipitated with preimmune (αPI), anti-Snu71p (α-Snu71p), anti-Nam8p (α-Nam8p), and anti-trimethyl guanosine cap (α TMG) antibodies. One-fourth of the input and the bound fractions were analyzed by primer extension with a U1-specific oligonucleotide or by Western blotting with anti-Snu71p and anti-Nam8p antibodies (right of the corresponding panels). (D) U1 snRNP proteins were purified and separated by electrophoresis from LUC7 wild-type yeast and from luc7-1 mutant yeast via TAP-tagged Nam8p. Proteins from the fractions were separated by SDS-PAGE. (Left) U1 snRNP proteins; (□) major contaminants. Proteins that are known to be absent (Snu71p, Luc7p) or that may be absent (Prp40p, Prp42p) from U1 snRNP prepared from luc7-1 cells are underlined. Nam8p–TAP runs as a doublet, as determined by mass spectrometry.
To examine U1 snRNP composition in the mutant strain extracts prepared from either luc7-1 cells or luc7-1 cells complemented with LUC7 were immunoprecipitated with three different antibodies. The first was directed against the trimethyl cap structure and the other two against the U1 snRNP proteins Snu71p and Nam8p (Gottschalk et al. 1998). U1 snRNA was efficiently precipitated with anti-trimethyl guanosine cap antibodies (Fig. 5C, cf. lanes 1–2 with 15–20). Because, at least in vertebrates, trimethyl capping requires assembly of U snRNAs with the U snRNP core proteins (Mattaj 1986), this suggested that U1 snRNP assembly had occurred to some extent in luc7-1 cells. U1 snRNA immunoprecipitation by anti-Nam8p antibodies was also efficient (lanes 9–14). In contrast, only background levels of U1 snRNA were precipitated with anti-Snu71p antibodies in the absence of LUC7, even when immunoprecipitation was carried out in 0.15 m NaCl on extracts of cells grown at permissive temperature (lanes 3–8). Anti-Snu71p antibodies immunoprecipitated Snu71p, but not Nam8p, from luc7-1 cells (lanes 3–8, bottom). Conversely, anti-Nam8p antibodies precipitated Nam8p but not Snu71p from the mutant cells (lanes 9–14, middle). Similarly, anti-trimethyl cap antibodies only precipitated Snu71p in the presence of wild-type Luc7p (lanes 15–20). These results demonstrate the lack of association of Snu71p with Nam8p or with U1 snRNA in extracts of the luc7-1 strain. This defect in association is not due to a decrease in the amount of either Snu71p or U1 snRNA present in the mutant cells (Fig. 5C).
A more complete analysis of U1 snRNP composition in luc7-1 cells was undertaken, with the recently developed TAP (tandem affinity purification) method (Rigaut et al. 1999). Because the results in Figure 5C indicated that Nam8p remained in the U1 snRNP in the mutant cells, tagged Nam8p was chosen for the analysis. Because of problems in extract preparation from luc7-1 cells at nonpermissive temperature, U1 snRNP from wild-type cells was compared with U1 snRNP from luc7-1 cells grown at 30°C, in which Luc7p–U1 snRNA interaction is already defective (Fig. 5B).
The protein profile of purified U1 snRNP (Fig. 5D, left) was compared with previous purifications (Neubauer et al. 1997; Gottschalk et al. 1998; Rigaut et al. 1999). Many U1 snRNP proteins were detected in U1 snRNP purified from luc7-1 cells (Fig. 5D, right). The SmD proteins and several yeast U1-specific proteins were present in both cases. As expected from the results in Figure 5, B and C, both Luc7p and Snu71p were absent from the U1 snRNP extracted from mutant cells (Fig. 5D), although both proteins were present at normal levels in the extracts as determined by Western blotting (Fig. 5C; data not shown). This indicates that the stable association of these proteins with the snRNP depends on the integrity of Luc7p. Other changes in U1 snRNP composition in luc7 cells are possible. The relative amount of protein in the Prp39p/Prp40p doublet was reduced in luc7-1 extract (Fig. 5D). Only peptides from Prp39p were detected when this band was subjected to mass spectrometric analysis (data not shown). Similarly, no peptides corresponding to Prp42p were detected in the samples prepared from luc7-1 cells. Mud10p/Snu56p was, however, found to be present in U1 snRNP in the luc7-1 extract (data not shown). The negative results with Prp40p and Prp42p suggest that these proteins, like Snu71p, may be less stably associated with U1 snRNP in luc7-1 cells, but this possibility needs to be tested more definitively. The apparent increase in recovery of a protein that migrated just below U1Cp was not reproduced in other experiments, whereas all the other changes were reproducibly seen (data not shown).
Luc7p is required for commitment complex assembly
U1 snRNP is a component of both yeast commitment complexes, CC1 and CC2 (Séraphin and Rosbash 1989a, 1991). Commitment complexes formed using extracts of a protein A-tagged Luc7p strain could be supershifted with IgG (data not shown). Thus, Luc7p is found in both commitment complexes. To determine whether luc7-1 extracts could form commitment complexes, native gel electrophoresis was carried out under conditions in which commitment complex formation is favored with either a wild-type pre-mRNA, able to form CC2, or a mutant pre-mRNA in which the branchpoint sequence has been deleted and that is therefore only able to form CC1 (Séraphin and Rosbash 1989a). Extracts of luc7-1 cells, whether grown at 30°C or 37°C, exhibited strong defects in CC1 and CC2 formation (Fig. 6, lanes 3,9,17,22,29,37). As expected, these defects were reversed when luc7-1 cells were complemented with a LUC7-expressing plasmid (Fig. 6, lanes 5–8, 13–16, 25–28, 33–36). Commitment complex assembly could also be partly restored to luc7-1 extracts by the addition of recombinant Luc7p produced in Escherichia coli (lanes 9–12, 17–20, 29–32, 37–40). Thus, the biochemical defect in luc7-1 extracts, a failure to assemble stable commitment complexes, could be rescued by provision of Luc7p alone. Note that the extracts were prepared in such a way that some prespliceosomes or spliceosomes were formed on the wild-type pre-mRNA substrate (Fig. 6, lanes 25–40). Complementation with Luc7p rescued the defect in both spliceosome assembly (lanes 25–40) and in pre-mRNA splicing (data not shown) of the luc7-1 extracts.
Figure 6.

Recombinant Luc7p can restore commitment complex formation in luc7-1 extracts. Splicing extracts were isolated from control cells (wild-type), or from untransformed luc7-1 cells (−) or cells transformed with a plasmid that expresses LUC7 (+pLUC7). The cells were either grown continuously at 30°C or grown for 5 hr after shift to 37°C as indicated. The extracts were incubated with a wild-type (CC2 probe) or mutant (CC1 probe) pre-mRNA probe in the presence or absence of increasing amounts of recombinant Luc-7p (His-Luc7p). Complexes were separated by native gel electrophoresis. CC1, CC2, and pre-spliceosomes plus spliceosomes (SPL) are indicated. The mobility of the probes incubated without extracts (CC1 probe and CC2 probe) and the mobility of probe CC1 incubated in the presence of the highest concentration of recombinant His–Luc7p are also shown.
Mutation of LUC7 affects 5′ splice site choice
The results presented in Figure 5 suggest that U1 snRNP composition may be altered in luc7 mutant cells even if splicing of at least the RP51A reporter was still reasonably efficient (Fig. 3). Therefore, we assayed more stringently for a functional defect in the mutant U1 snRNP. Because LUC7 interacts genetically with CBC, it was reasoned that mutation of Luc7p might affect the choice of a 5′ splice site in relation to its proximity to the 5′ cap. Pre-mRNAs based on the RP51A pre-mRNA, but containing duplicated 5′ splice sites and flanking sequences (Séraphin and Kandels-Lewis 1993), were used as reporters in these experiments to examine whether mutation of LUC7 would affect the two competing 5′ splice sites differentially. We assayed constructs in which both 5′ splice sites were wild type, or where either the cap distal, the cap proximal, or both 5′ splice sites were mutant. In all cases, the mutated 5′ splice sites carried UC instead of AG at the last two positions of the 5′ exon (Séraphin and Kandels-Lewis 1993).
Splicing of these constructs was examined by primer extension, and the products of splicing from the cap-distal and cap-proximal 5′ splice sites were detected (Fig. 7A, the top and bottom groups of products, respectively). With three of the reporters, a clear reduction in the usage of the cap-proximal 5′ splice site was seen in luc7-1 cells grown at 37°C (Fig. 7A, − lanes 1,3,9,11,13,15). These effects were reversed when the luc7-1 cells were complemented with plasmid-borne LUC7 (Fig. 7A, + lanes 2,4,10,12,14,16). Only when the cap-distal 5′ splice site was mutant and the cap-proximal wild type (lanes 5–8) was the change in relative usage of the two splice sites in the absence of Luc7p not observed. Thus, where two identical 5′ splice sites are in competition, the presence of Luc7p favors splicing to the cap-proximal of the two. These results are summarized in Figure 7C. Here the ratio of usage of the proximal and distal splice sites in the mutant strain are presented with reference to the wild type, whose ratio has been set to 1. Thus, a value lower than 1 represents a relative preference for the distal 5′ splice site. Similar effects on relative usage of the two splice sites were obtained when a gcr3 deletion strain, which lacks yCBP80, was compared with a strain producing CBC (Fig. 7B, lanes 1–8). For reasons that are not understood, the defect in this case does not appear evenly distributed among the transcripts arising from different transcription initiaton points. When quantified, however, the overall proximal:distal ratio changes similarly in luc7 and gcr3 cells. Similar results were observed in strains lacking yCBP20 (data not shown) or lacking both components of CBC (Fig. 7C).
Figure 7.



5′ splice site choice is altered in the WR244 (luc7-1) strain. (A) 5′ splice site competition assay in luc7-1 cells. The untransformed WR244 (luc7-1) strain (−) or the strain transformed with a plasmid that expresses LUC7 (+) were transformed with different plasmids that express RP51A pre-mRNA with an intron in which the 5′ splice site and surrounding sequences have been duplicated such that cap-proximal and cap-distal 5′ splice sequences bear strong or weak consensus sequences in all possible combinations. The weak consensus 5′ splice sites have UC instead of AG at the last two positions of the 5′ exon. The splice sites with the nonconsensus exon sequence are indicated by an asterisk. Selection between tandem 5′ splice sites was assayed in extracts isolated from these strains grown continuously at 30°C or after shift to 37°C for 2 hr. RP51A mRNAs (indicated schematically at left) were visualized by primer extension. (B) 5′ splice site competition in the absence of CBP80. The assay is identical to that described in A, and was carried out either in a wild-type strain (wild-type) or in a gcr3 (cbp80-Δ) strain. (C) 5′ splice site competition in strains lacking commitment complex proteins. The same competition assay as in A and B was carried out in strains lacking Luc7p, CBC, Mud1p, or Mud2p or in matching wild-type strains with the same genetic background. The usage of the proximal and distal 5′ splice sites was determined in each case by primer extension analysis and the results were quantified with a fluorimager (Fujifilm). Average proximal-to-distal splice site usage ratios from several experiments are shown. In each case, the ratio of usage of the two splice sites in the wild-type strain was defined as 1 and the effect of gene deletion or inactivation calculated with respect to the wild type.
To test whether this phenotype was unique for luc7 and cbc mutants, yeast strains mutant in two other commitment complex proteins were analyzed. Mud1p and Mud2p were chosen as, like Luc7p, they can both cause synthetic lethality in the absence of CBC (Fortes et al. 1999). Mud1p is a U1 snRNP protein, whereas Mud2p binds to the branchpoint and has been shown to physically interact with CBC (Fortes et al. 1999). In vivo splicing of reporters containing duplicated 5′ splice site regions was assayed in mud1 or mud2 deletion strains. The effect of the lack of these proteins on splice site choice was less pronounced than the lack of Luc7p or CBC (Fig. 7C). Lack of Mud1p caused a preference for the cap proximal site, whereas lack of Mud2p caused the cap distal site to be slightly favored (Fig. 7C). We also tested the effect of NAM8 deletion on splice site choice using these reporter constructs. As predicted by a previous study (Puig et al. 1999; O. Puig and B. Séraphin, pers. comm.) the absence of Nam8p favored use of the cap proximal 5′ splice site, although this effect was minor with the reporter used here (data not shown).
The concordance of the results in the luc7 and cbc strains suggested that the defective U1 snRNP present in luc7 mutant cells might have a reduced ability to interact with CBC. To test this directly, we used the assay introduced in Figure 4. A luc7-1 strain containing a protein A-tagged version of Nam8p was constructed. The Nam8p–ProtA fusion was used as it allows quantitative precipitation of U1 snRNPs from both wild-type and luc7-1 strains (Figs. 4 and 5). A fraction of yCBP80 was specifically coprecipitated on IgG beads from extracts of a strain containing Nam8p–ProtA but not wild-type Nam8p (Fig. 4B and Fig. 8, lanes 2,3,5,6). The amount of bound yCBP80 was drastically reduced in extracts of luc7-1 cells containing Nam8p–ProtA and grown at permissive temperature (Fig. 8, lanes 4,7), in which U1 snRNP composition is already altered. The amount of bound yCBC was more drastically affected when extracts were made from luc7-1 cells grown at the nonpermissive temperature (data not shown). This reduction in coprecipitation was not due to instability of yCBP80, because similar amounts of the protein were present in the flowthrough from the column (Fig. 8, lanes 2–4). Thus, interaction between U1 snRNP and yCBC is disrupted in luc7-1 cell extracts.
Figure 8.

Luc7p affects U1 snRNP–CBC interaction. Extracts were isolated from three yeast strains, a LUC7 control strain (WT), a LUC7 strain carrying Nam8p–ProtA (Nam8–ProtA), or a luc7-1 strain carrying Nam8–ProtA (Nam8–ProtA/ts) grown at permissive temperature. After fractionation on IgG agarose, both bound fractions and unbound fractions (5% of the latter) were separated by SDS-PAGE and analyzed by Western blotting with anti-CBP80 antibodies. Lane 1 is 5% of the input wild-type extract.
Discussion
Luc7p, a newly identified component of U1 snRNP, is described. Whereas mammalian U1 snRNP has a relatively small number of associated components, the Sm core proteins, U1 snRNA, and three U1-specific proteins, U1-70K, U1A, and U1C (Lührmann et al. 1990), yeast U1 snRNP is more complex. U1 snRNA in yeast is considerably larger than in mammals (Kretzner et al. 1987; Siliciano et al. 1987). In addition to homologs of the mammalian core and U1-specific proteins, yeast U1 snRNP was shown recently to contain six additional proteins, several encoded by essential genes (Neubauer et al. 1997; Gottschalk et al. 1998). The identification of Luc7p as an additional U1 snRNP-specific protein (Rigaut et al. 1999; this work) brings the total number of protein components in the yeast snRNP to 17. Given the extensive genetic and biochemical analysis applied to yeast U1 snRNP, it is probable that all stably associated proteins have now been described.
Luc7p affects U1 snRNP composition and 5′ splice site recognition
Luc7p is encoded by an essential gene, and both in vivo and in vitro data show that mutation of LUC7 causes a defect in pre-mRNA splicing. The complete defect in all steps of splicing in extracts from luc7-1 cells (Fig. 6; data not shown) strongly suggests that the reason why Luc7p is essential is because of its participation in splicing. The lack of a major effect of mutation of Luc7p on efficiently spliced reporter genes in vivo is similar to previous observations with other commitment complex components (Liao et al. 1993; Abovich et al. 1994; Colot et al. 1996; Puig et al. 1999). However, it is possible that Luc7p has an additional function that is not related to pre-mRNA splicing and is essential for yeast growth.
When extracts were prepared from a temperature-sensitive luc7 strain grown at either permissive or nonpermissive temperature, the mutant Luc7p was not detectably associated with either U1 snRNA or other U1 snRNP proteins. In addition, another U1 snRNP component, Snu71p, was also no longer stably bound to the snRNP. Other changes in U1 snRNP composition were observed. However, the in vitro splicing defect in extracts of these mutant cells could be complemented by the simple addition of recombinant Luc7p, suggesting that U1 snRNP could be reassembled from the disassembled components and Luc7p. Although these data are suggestive of a Luc7p-containing U1 snRNP subcomplex, they do not prove its existence. An alternative explanation for the data would be that Luc7p would bind to the partially assembled U1 snRNP and cause a change in the conformation of either its RNA or protein components that allows the formation of a U1 snRNP to proceed to completion. We note that it is difficult to extrapolate directly from these in vitro experiments to the state of assembly of U1 snRNP in vivo, although the in vivo splicing data do indicate that U1 snRNP activity is affected in luc7-1 cells.
The fact that splicing of at least some reporter introns was affected to only a very minor extent in the temperature-sensitive luc7 strain grown at nonpermissive temperature raises the possibility that neither Luc7p nor the other components whose stable association with U1 snRNA depend on Luc7p, notably the essential Snu71p (Gottschalk et al. 1998), are absolutely required for the splicing of every intron. For example, the mutant U1 snRNP subcomplex was sufficient to allow efficient splicing of the RP51A pre-mRNA. If the essential function of LUC7, as argued above, is pre-mRNA splicing, there must be at least one intron in another essential gene whose splicing is defective enough in luc7 strains to cause inviability. This in turn suggests that at least Luc7p and Snu71p, and possibly other U1 snRNP-specific proteins, may only be required for splicing of certain specific introns.
Evidence for this was obtained by analysis of a reporter construct containing an intron with a duplicated 5′ splice site. In the absence of Luc7p, splicing to the cap-proximal 5′ splice site was greatly reduced, whereas splicing to the cap-distal site was little affected. Note that because the reporter in question had a duplication of 25 nucleotides upstream of the 5′ splice site and 48 nucleotides downstream (Séraphin and Kandels-Lewis 1993), this was unlikely to be due to an effect on an intrinsic sequence-specific recognition of the 5′ splice sites by U1 snRNP. These sequences all lie within the region that can be cross-linked to U1 snRNP proteins (Zhang and Rosbash 1999; Puig et al. 1999). Rather, it was suggestive of loss of interactions with factors that contacted the RNA outside of the duplicated region, either upstream or downstream of the tandem 5′ splice sites, and affected their recognition by U1 snRNP. Analysis of strains lacking the commitment complex components Mud1p, Mud2p, or Nam8p showed that removal of these proteins also affected splice site choice in the competition assay. The effects of depletion of individual commitment complex components on splice site choice were, however, both quantitatively and qualitatively diverse.
U1 snRNP–CBC interaction
LUC7 was identified by a mutation that caused lethality in the absence of CBC, and a defect in recognition of a cap proximal intron, similar to that of luc7-1 cells, was seen in strains lacking yCBC. Biochemically, this defect was observed as a lack of coimmunoprecipitation of yCBC with U1 snRNP, supporting previous data from both yeast and human systems that suggested a direct or indirect role for CBC in U1 snRNP interaction with cap-proximal 5′ splice sites (Colot et al. 1996; Lewis et al. 1996a,b). We have failed in attempts to detect a direct physical interaction between Luc7p and yCBC (Fortes et al. 1999). It is therefore likely that the absence of Luc7p from the U1 snRNP changes the snRNP conformation such that interaction between yCBC and another U1 snRNP component is affected. The only U1 snRNP component thus far shown to interact directly with yeast CBC is Mud10p/Snu56p (Fortes et al. 1999). This protein is present in U1 snRNPs in luc7 mutant cells (Fig. 5D; data not shown). The reduction in CBC interaction with U1 snRNP in these cells may therefore either be due to the lack of an as yet uncharacterized interaction with CBC, or to a change in U1 snRNP that prevents CBC–Snu56p interaction. Note that although yCBC is not essential, whereas Luc7p is, mediating interaction with CBC cannot be the sole function of Luc7p.
The data, together with recent work on Nam8p (Puig et al. 1999), strongly suggest that not all 5′ splice site–U1 snRNP interactions are identical, and that different U1 snRNP components will differentially affect the splicing of different introns. This may result from interaction between U1 snRNP components and other splicing factors, like yCBC in the case of Luc7p, or with the pre-mRNA, in the case of Nam8p and several other U1 snRNP proteins (Nakagawa and Ogawa 1997; Puig et al. 1999; Zhang and Rosbash 1999). This suggestion of substrate-specific functions for proteins that are generally considered to belong to the basal splicing machinery is very analogous to the recent observation that some components of basal RNA polymerase II transcription complexes are only required for transcription from certain specific promoters (Holstege et al. 1998).
Yeast and vertebrate U1 snRNPs
Relatives of several of the yeast U1-specific proteins that have no characterized vertebrate counterparts are present in the DNA databases and are therefore probably present in vertebrate cells (Gottschalk et al. 1998; Puig et al. 1999; this paper). Good examples are the three human relatives of Luc7p described here. These proteins contain not only the zinc finger region of Luc7p but also, unlike Luc7p, carry multiple RS and RE repeats. These sequences are characteristic of a large family of metazoan splicing factors (Neugebauer et al. 1995; Staknis and Reed 1995), increasing the likelihood that the vertebrate Luc7p relatives are involved in pre-mRNA splicing. However, in contrast to the situation in yeast, these proteins do not appear to be stable components of the U1 snRNP (Lührmann et al. 1990). This leads us to propose the following possible rationale for the fact that yeast U1 snRNP contains many more stable components than does vertebrate U1 snRNP.
Vertebrate splicing is subject to a much greater level of regulation than yeast splicing (Black 1995; Fu 1995; Manley and Tacke 1996). In addition, even some nonregulated introns may require interaction with exon-bound proteins, like the SR proteins, to be spliced efficiently (Schaal and Maniatis 1999). We therefore suggest that for splicing of any specific vertebrate intron some, but probably not all, of the homologs of the yeast-specific U1 snRNP proteins will have to be assembled in situ with the core vertebrate U1 snRNP to form the functional holo vertebrate U1 snRNP. This will involve binding of these proteins to either the pre-mRNA directly in a sequence-dependent manner, analogous to that of Nam8p (Puig et al. 1999), or to other proteins that bind to the pre-mRNA. The result would effectively increase the available functional complexity of the U1 snRNP.
Examples of other proteins that could influence U1 snRNP composition include mammalian CBC, which might recruit a mammalian Luc7p homolog to the cap-proximal 5′ splice site via direct or indirect interactions. An alternative way to bring in human Luc7p, perhaps a different homolog of Luc7p, to a 5′ splice site might be through proteins bound to an exon enhancer. The composition of U1 snRNP could therefore be different on different 5′ splice sites in a context-dependent manner. In this way, many different combinations of factors could potentially give rise to an active spliceosome, increasing the potential for splicing regulation. This hypothesis could also help to explain why vertebrate splicing is so dependent on the SR protein family that helps assemble spliceosomes through networks of weak protein–protein interactions, whereas yeast has either no or very few SR proteins. Much further work will be required to test this hypothesis, but there is preliminary evidence that even the much more stable holo yeast U1 snRNP may not be of uniform composition. It was reported that after deletion of the NAM8 gene another yeast U1 snRNP protein, Snu65p, replaced Nam8p and became a much more abundant component of the yeast U1 snRNP population (Gottschalk et al. 1998).
Materials and methods
Media, strains, and biological materials
Standard media and techniques were used for yeast (Sherman 1991) and E. coli manipulation (Sambrook et al. 1989). Growth curves were assayed as described (Fortes et al. 1999) and yeast was transformed by the lithium acetate method (Ito et al. 1983).
The wild-type strains used were the diploid BSY320 (MATa/MATα; leu2-3, 112/leu2-3, 112; arg4/arg4; ade2/ade2; trp1-289/trp1-289; ura3-52/ura3-52) and the haploid MGD425-13D (Séraphin et al. 1988). Mutant strains were PF433 (MATa; ade2; ade3; his3; leu2-3,112; trp1; ura3; ycbp80/gcr3::TRP1; ycbp20/mud13::HIS3; [pHT8020; YCBP80/GCR3; YCBP20/MUD13; URA3; ADE3]; luc7-2) isolated in a synthetic lethal screen with yCBC (Fortes et al. 1999) and WR244 (MATa, ura3, leu2, can1; cyh2; luc7-1), a temperature-sensitive strain that was selected as follows. Strain WR140 (MATa, cdc15-7, ura3, trp1, ade1, can1, cyh2) was mutagenized with ethyl methane sulfonate to 40% viability at 23°C. Replica-plated colonies were screened for loss of viability after a 3-hr incubation at 36°C, followed by downshifting to 23°C. Of 49,582 colonies screened, 49 candidates lost viability after incubation at 36°C. A subset of these candidates were secondarily screened for those that accumulated as microcolonies of at least two to four cells following the shift to 36°C and that in unsonicated, fixed samples exhibited two well-separated DAPI-staining bodies. By these secondary screens, 25 candidates were interpreted as being able to reach or pass late-nuclear division. One of these candidates, strain 15A286, was shown through a series of crosses and complementation experiments to contain a temperature-sensitive allele of LUC7, luc7-1. Strain WR244 was derived from 15A286 through standard crosses.
To tag Nam8p, Mud10p, and Luc7p, a URA3 marker fused to the sequence coding for two IgG-binding domains of protein A (Puig et al. 1998) was introduced in frame with the NAM8, MUD10, and LUC7 coding regions. To obtain the NAM8–ProtA strain, BSY593 was used (Gottschalk et al. 1998), MUD10–ProtA (O. Puig, unpubl.), and LUC7–ProtA strains were obtained from MGD425-13D and luc7-1–ProtA was similarly constructed from WR244. Western blot analysis and PCR were used to verify the structure of the tagged genes. The same ProtA–URA3 construct was introduced in the genome of BSY320 to disrupt the LUC7 gene completely. For the diploid strain, successful gene disruption was confirmed by PCR.
NAM8 was also fused to the TAP tag (Rigaut et al. 1999). This fragment was introduced in frame with the NAM8 gene into the genome of MGD425-13D strain to obtain the strain NAM8–TAP (O. Puig, unpubl.) or into the genome of the WR244 strain. Western blot analysis and PCR were used to verify the structure of the tagged genes.
DNA constructs
Oligonucleotides corresponding to position −400 and position +205 relative to the LUC7 ORF were used to PCR amplify the wild-type LUC7 gene, the luc7-1 temperature-sensitive allele, and the luc7-2 synthetic lethal allele. These DNA fragments were introduced into the BamHI site of pRS315 (pLEU), a single copy plasmid with a LEU2 marker (Sikorski and Hieter 1989) to generate pLEU–LUC7, pLEU–luc7-1, and pLEU–luc7-2, respectively.
pET–Luc7 contains LUC7 ligated between the NdeI and BamHI sites of pET21a. His-tagged Luc7p can be expressed from this plasmid in E. coli under the control of T7 promoter and purified with Ni–NTA column chromatography (Clontech).
Reporters used to analyze commitment complexes are transcribed from pBS195, encoding wild-type RP51 sequences and pBS199, derived from a pBS195 in which the UACUAAC branchpoint region has been deleted (Séraphin and Rosbash 1989a).
Reporters used to analyze in vivo splicing efficiency have been described previously. All express RP51A pre-mRNA under the control of a GAL inducible promoter; wild-type intron (pHZ18; Teem and Rosbash 1983); 5′ splice mutant GUAUaU (pHZ12; Jacquier et al. 1985), and branchpoint mutant UAuUAAC (pBS64; Pascolo and Séraphin 1997). The reporters with wild-type and mutant-duplicated 5′ splice sites, pBS450 (AG/AG), pBS452 (AG/UC), pBS456 (UC/AG), and pBS458 (UC/UC) have been described (Séraphin and Kandels-Lewis 1993).
Sequence analysis
Sequences were compiled and analyzed with computer software from the Wisconsin Package version 9.1, Genetics Computer Group (GCG), Madison, WI, BLAST (Altschul et al. 1997), and Clustal_X (Thompson et al. 1998). ceLuc7A1 is predicted from C. elegans cosmid C50D2 (GenBank accession no. AF040642) and is similar to the predicted gene product C50D2.8 (accession no. 2746789), except that the last two exons are encoded by C50D2 120606–12659 and 12074–12247, respectively. ceLuc7A2 is identical to the predicted C. elegans protein Y119D3_450.D. This sequence was produced by the C. elegans Sequencing Group at the Sanger Centre and can be obtained from ftp://ftp.sanger.ac.uk/pub/databases/wormpep/excluded. ceLuc7B is identical to the C. elegans predicted gene product B0495.8 (accession no. Q09217). hLuc7A was derived by clustering of human ESTs with accession numbers AA535263, AA311424, AA451779, AA331496, AA332577, R94890, AA587998, N55709, AA493284, AA737596, and AA628673. hLuc7B1 was derived by clustering of human ESTs with accession numbers AA143197, AA352322, AA730774, N29757, AA143213, AA613804, C21369, T91335, AA317465, AA621718, H24668, W95695, and human genomic sequences with accession numbers Z69706 (nucleotides 16971–17147) and Z69890 (nucleotides 849–992 and 2923–3033). hLuc7B2 was derived from human ESTs AA009903, N91986, Z44294, AA081610, H17407, R73628, AA213413, AA496763, H17408, T62184, AA307963, N25025, and Z44289. Luc7 proteins are predicted in D. melanogaster from ESTs AA942407, AA439479, and AI107196 (dmLuc7A) and from ESTs AI258257, AI292994, AA978748, AA951418, AA802732, AA390380, AA949554, and AA201483 (dmLuc7B).
Protein and RNA analysis in vitro
Splicing mini-extracts (Séraphin and Rosbash 1989a) of control or protein A-tagged wild-type strains (NAM8–ProtA, MUD10–ProtA, and LUC7–ProtA) were used to characterize binding partners of Luc7p. Luc7-1p interactions were studied in WR244 and a strain derived from WR244 carrying luc7-1p–ProtA and either Nam8p–ProtA or Mud10p–ProtA. As a control, the same strains transformed with the pLEU–LUC7 wild-type plasmid were used. Cells were grown at 30°C to an optical density at 600 nm of 1 and shifted to 37°C for 2 or 5 hr. Total RNA (Pikielny and Rosbash 1985) or splicing mini-extracts (Séraphin and Rosbash 1989a) were isolated from these cells at permissive or restrictive temperatures. U1 and U2 snRNA levels were analyzed by primer extension as described below.
Splicing mini-extracts were incubated in IPP buffer (10 mm Tris-HCl at pH 8.0, 150 mm NaCl, and 0.1% NP40) with rabbit IgG–agarose (Sigma), or with protein A agarose linked to anti-2,2,7-trimethylguanosine (Ab-1, Calbiochem), anti-Snu71p, anti-Nam8p (Gottschalk et al. 1998), or preimmune antibodies. The beads were washed extensively with the same buffer and bound RNA or proteins were analyzed. For RNA analysis, the input, flow-through, and bound fractions were incubated with a proteinase K-SDS buffer (2 mg/ml proteinase K, 1 mg/ml tRNA, 0.6% SDS, 25 mm EDTA, and 25 mm Tris-HCl at pH 8) for 20 min at 65°C. After phenol extraction and ethanol precipitation, RNAs were analyzed by primer extension as described (Séraphin 1995).
For protein analysis, proteins were eluted from the column either with 2 m NaCl containing IPP buffer or stepwise with the same buffer containing 0.2, 0.5, and 2 m NaCl. After each elution, the column was washed extensively with the same buffer before the salt concentration was changed. Proteins were analyzed by Western blotting with anti-Snu71p, anti-Nam8p, anti-Npl3p, and anti-yCBP80 as described (Görlich et al. 1996; Gottschalk et al. 1998). U1 snRNP purification with the TAP (Tandem Affinity Purification) system was as described (Rigaut et al. 1999).
Splicing assays
For commitment complex assays, splicing mini-extracts were incubated with CC1 or CC2 probes as described (Séraphin and Rosbash 1989a, 1991). For supershift experiments, after commitment complex formation extracts were incubated with buffer D or buffer D containing 0.1 or 1 mg of rabbit IgG. When His–Luc7p was used, extracts were mixed, before commitment complex formation, with buffer D or 0.1, 0.2, or 0.5 mg of purified His–Luc7p in buffer D.
Splicing efficiency in vivo was assayed in either the WR244 (luc7-1) strain or WR244 transformed with a pLEU–LUC7 wild-type plasmid as a control. These strains were also transformed with a collection of plasmids that express RP51A pre-mRNA under the control of a GAL-inducible promoter. Strains transformed with these plasmids were grown at 30°C to an optical density at 600 nm of 0.8 in minimal medium containing 2% lactate–2% glycerol as carbon sources. Cells were then maintained at 30°C or shifted to 37°C for 20 min. 2% galactose was added to the cells to induce RP51A pre-mRNA expression for 2 or 5 hr. Total RNA was isolated from the cells (Pikielny and Rosbash 1985) and RP51A RNAs were detected by primer extension from the second exon with an oligonucleotide primer of sequence CACGCTTGACGGTCTTGGT. The same protocol was used to analyze the in vivo splicing efficiency in wild-type cells or cells genetically depleted of CBC, Mud1p, Mud2p or Nam8p, but shift to 37°C was omitted.
Acknowledgments
We thank Oscar Puig for providing yeast strains, splicing reporter and other plasmid constructs, and stimulating discussions. We also acknowledge Alexander Gottschalk and Reinhard Lührmann for antibodies against Snu71p, Npl3p, and Nam8p, Dirk Görlich for antibody against yCBP80, Michael Rosbash and Nadja Abovich for mud1 and mud2 strains, and Angela Bachi and Matthias Wilm for mass spectrometric analysis of U1 snRNP fractions. Gert-Jan Arts, Scott Kuersten, Mutsuhito Ohno, Juan Valcárcel, Giulia Guarguaglini, Alexandra Segref, Martin Hetzer, Tobias Walther, Reinhard Lührmann, and David Tollervey provided valuable comments on the manuscript. P.F. was a recipient of fellowships from the Human Frontier Science Program Organization (HFSPO) and the European Union Training and Mobility Research (TMR) program.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL mattaj@embl-heidelberg.de; FAX +49 6221 387 518.
References
- Abovich N, Rosbash M. Cross-intron bridging interactions in the yeast commitment complex are conserved in mammals. Cell. 1997;89:403–412. doi: 10.1016/s0092-8674(00)80221-4. [DOI] [PubMed] [Google Scholar]
- Abovich N, Liao XC, Rosbash M. The yeast MUD2 protein: An interaction with PRP11 defines a bridge between commitment complexes and U2 snRNP addition. Genes & Dev. 1994;8:843–854. doi: 10.1101/gad.8.7.843. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berget SM. Exon recognition in vertebrate splicing. J Biol Chem. 1995;270:2411–2414. doi: 10.1074/jbc.270.6.2411. [DOI] [PubMed] [Google Scholar]
- Berglund JA, Fleming ML, Rosbash M. The KH domain of the branchpoint sequence binding protein determines specificity for the pre-mRNA branchpoint sequence. RNA. 1998;4:998–1006. doi: 10.1017/s1355838298980499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black DL. Finding splice sites within a wilderness of RNA. RNA. 1995;1:763–771. [PMC free article] [PubMed] [Google Scholar]
- Chabot B, Steitz JA. Multiple interactions between the splicing substrate and small nuclear ribonucleoproteins in spliceosomes. Mol Cell Biol. 1987;7:281–293. doi: 10.1128/mcb.7.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colot HV, Stutz F, Rosbash M. The yeast splicing factor Mud13p is a commitment complex component and corresponds to CBP20, the small subunit of the nuclear cap-binding complex. Genes & Dev. 1996;10:1699–1708. doi: 10.1101/gad.10.13.1699. [DOI] [PubMed] [Google Scholar]
- Eperon IC, Ireland DC, Smith RA, Mayeda A, Krainer AR. Pathways for selection of 5′ splice sites by U1 snRNPs and SF2/ASF. EMBO J. 1993;12:3607–3617. doi: 10.1002/j.1460-2075.1993.tb06034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortes, P., J. Kufel, M. Fornerod, M. Polycarpou-Schwarz, D. Lafontaine, D. Tollervey, and I.W. Mattaj. 1999. Genetic and physical interactions involving the yeast nuclear cap-binding complex. Mol. Cell. Biol. (in press). [DOI] [PMC free article] [PubMed]
- Fu XD. The superfamily of arginine/serine-rich splicing factors. RNA. 1995;1:663–680. [PMC free article] [PubMed] [Google Scholar]
- Görlich D, Kraft R, Kostka S, Vogel F, Hartmann E, Laskey RA, Mattaj IW, Izaurralde E. Importin provides a link between nuclear protein import and U snRNA export. Cell. 1996;87:21–32. doi: 10.1016/s0092-8674(00)81319-7. [DOI] [PubMed] [Google Scholar]
- Gottschalk A, Tang J, Puig O, Salgado J, Neubauer G, Colot HV, Mann M, Séraphin B, Rosbash M, Lührmann R, Fabrizio P. A comprehensive biochemical and genetic analysis of the yeast U1 snRNP reveals five novel proteins. RNA. 1998;4:374–393. [PMC free article] [PubMed] [Google Scholar]
- Hertel KJ, Lynch KW, Maniatis T. Common themes in the function of transcription and splicing enhancers. Curr Opin Cell Biol. 1997;9:350–357. doi: 10.1016/s0955-0674(97)80007-5. [DOI] [PubMed] [Google Scholar]
- Holstege FC, Jennings EG, Wyrick JJ, Lee TI, Hengartner CJ, Green MR, Golub TR, Lander ES, Young RA. Dissecting the regulatory circuitry of a eukaryotic genome. Cell. 1998;95:717–728. doi: 10.1016/s0092-8674(00)81641-4. [DOI] [PubMed] [Google Scholar]
- Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izaurralde E, Lewis J, McGuigan C, Jankowska M, Darzynkiewicz E, Mattaj IW. A nuclear cap binding protein complex involved in pre-mRNA splicing. Cell. 1994;78:657–668. doi: 10.1016/0092-8674(94)90530-4. [DOI] [PubMed] [Google Scholar]
- Izaurralde E, Lewis J, Gamberi C, Jarmolowski A, McGuigan C, Mattaj IW. A cap-binding protein complex mediating U snRNA export. Nature. 1995;376:709–712. doi: 10.1038/376709a0. [DOI] [PubMed] [Google Scholar]
- Jacquier A, Rodriguez JR, Rosbash M. A quantitative analysis of the effects of 5′ junction and TACTAAC box mutants and mutant combinations on yeast mRNA splicing. Cell. 1985;43:423–430. doi: 10.1016/0092-8674(85)90172-2. [DOI] [PubMed] [Google Scholar]
- Kohtz JD, Jamison SF, Will CL, Zuo P, Lührmann R, Garcia-Blanco MA, Manley JL. Protein-protein interactions and 5′-splice-site recognition in mammalian mRNA precursors. Nature. 1994;368:119–124. doi: 10.1038/368119a0. [DOI] [PubMed] [Google Scholar]
- Konarska MM, Padgett RA, Sharp PA. Recognition of cap structure in splicing in vitro of mRNA precursors. Cell. 1984;38:731–736. doi: 10.1016/0092-8674(84)90268-x. [DOI] [PubMed] [Google Scholar]
- Krainer AR, Maniatis T, Ruskin B, Green MR. Normal and mutant human beta-globin pre-mRNAs are faithfully and efficiently spliced in vitro. Cell. 1984;36:993–1005. doi: 10.1016/0092-8674(84)90049-7. [DOI] [PubMed] [Google Scholar]
- Krämer A. Purification of splicing factor SF1, a heat-stable protein that functions in the assembly of a presplicing complex. Mol Cell Biol. 1992;12:4545–4552. doi: 10.1128/mcb.12.10.4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzner L, Rymond BC, Rosbash M. S. cerevisiae U1 RNA is large and has limited primary sequence homology to metazoan U1 snRNA. Cell. 1987;50:593–602. doi: 10.1016/0092-8674(87)90032-8. [DOI] [PubMed] [Google Scholar]
- Lewis JD, Gorlich D, Mattaj IW. A yeast cap binding protein complex (yCBC) acts at an early step in pre- mRNA splicing. Nucleic Acids Res. 1996a;24:3332–3336. doi: 10.1093/nar/24.17.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JD, Izaurralde E, Jarmolowski A, McGuigan C, Mattaj IW. A nuclear cap-binding complex facilitates association of U1 snRNP with the cap-proximal 5′ splice site. Genes & Dev. 1996b;10:1683–1698. doi: 10.1101/gad.10.13.1683. [DOI] [PubMed] [Google Scholar]
- Liao XC, Kretzner L, Séraphin B, Rosbash M. Universally conserved and yeast-specific U1 snRNA sequences are important but not essential for U1 snRNP function. Genes & Dev. 1990;4:1766–1774. doi: 10.1101/gad.4.10.1766. [DOI] [PubMed] [Google Scholar]
- Liao XC, Tang J, Rosbash M. An enhancer screen identifies a gene that encodes the yeast U1 snRNP A protein: Implications for snRNP protein function in pre-mRNA splicing. Genes & Dev. 1993;7:419–428. doi: 10.1101/gad.7.3.419. [DOI] [PubMed] [Google Scholar]
- Lopez MC, Sanchez M, Ferminan E, Dominguez A. Disruption of six Saccharomyces cerevisiae genes from chromosome IV and basic phenotypic analysis of deletion mutants. Yeast. 1998;14:1199–1208. doi: 10.1002/(SICI)1097-0061(19980930)14:13<1199::AID-YEA309>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Lührmann R, Kastner B, Bach M. Structure of spliceosomal snRNPs and their role in pre-mRNA splicing. Biochim Biophys Acta. 1990;1087:265–292. doi: 10.1016/0167-4781(90)90001-i. [DOI] [PubMed] [Google Scholar]
- Manley JL, Tacke R. SR proteins and splicing control. Genes & Dev. 1996;10:1569–1579. doi: 10.1101/gad.10.13.1569. [DOI] [PubMed] [Google Scholar]
- Matsushima Y, Matsumura K, Kitagawa Y. Zinc finger-like motif conserved in a family of RNA binding proteins. Biosci Biotechnol Biochem. 1997;61:905–906. doi: 10.1271/bbb.61.905. [DOI] [PubMed] [Google Scholar]
- Mattaj IW. Cap trimethylation of U snRNA is cytoplasmic and dependent on U snRNP protein binding. Cell. 1986;46:905–911. doi: 10.1016/0092-8674(86)90072-3. [DOI] [PubMed] [Google Scholar]
- Michaud S, Reed R. An ATP-independent complex commits pre-mRNA to the mammalian spliceosome assembly pathway. Genes & Dev. 1991;5:2534–2546. doi: 10.1101/gad.5.12b.2534. [DOI] [PubMed] [Google Scholar]
- ————— A functional association between the 5′ and 3′ splice site is established in the earliest prespliceosome complex (E) in mammals. Genes & Dev. 1993;7:1008–1020. doi: 10.1101/gad.7.6.1008. [DOI] [PubMed] [Google Scholar]
- Moore M, Query C, Sharp P. Splicing of precursors to messenger RNAs by the spliceosome. In: Gesteland RF, Atkins JF, editors. The RNA world. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1993. pp. 303–357. [Google Scholar]
- Nakagawa T, Owaga H. Involvement of the MRE2 gene of yeast in formation of meiosis-specific double-strand breaks and crossover recombination through RNA splicing. Genes Cells. 1997;2:65–79. doi: 10.1046/j.1365-2443.1997.d01-283.x. [DOI] [PubMed] [Google Scholar]
- Neubauer G, Gottschalk A, Fabrizio P, Séraphin B, Lührmann R, Mann M. Identification of the proteins of the yeast U1 small nuclear ribonucleoprotein complex by mass spectrometry. Proc Natl Acad Sci. 1997;94:385–390. doi: 10.1073/pnas.94.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer KM, Stolk JA, Roth MB. A conserved epitope on a subset of SR proteins defines a larger family of Pre-mRNA splicing factors. J Cell Biol. 1995;129:899–908. doi: 10.1083/jcb.129.4.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M, Sakamoto H, Shimura Y. Preferential excision of the 5′ proximal intron from mRNA precursors with two introns as mediated by the cap structure. Proc Natl Acad Sci. 1987;84:5187–5191. doi: 10.1073/pnas.84.15.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascolo E, Séraphin B. The branchpoint residue is recognized during commitment complex formation before being bulged out of the U2 snRNA-pre-mRNA duplex. Mol Cell Biol. 1997;17:3469–3476. doi: 10.1128/mcb.17.7.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikielny CW, Rosbash M. mRNA splicing efficiency in yeast and the contribution of nonconserved sequences. Cell. 1985;41:119–126. doi: 10.1016/0092-8674(85)90066-2. [DOI] [PubMed] [Google Scholar]
- Puig O, Rutz B, Luukkonen BG, Kandels-Lewis S, Bragado-Nilsson E, Séraphin B. New constructs and strategies for efficient PCR-based gene manipulations in yeast. Yeast. 1998;14:1139–1146. doi: 10.1002/(SICI)1097-0061(19980915)14:12<1139::AID-YEA306>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Puig O, Gottschalk A, Fabrizio P, Séraphin B. Interaction of the U1 snRNP with non-conserved intronic sequences affects 5′ splice site selection. Genes & Dev. 1999;13:569–580. doi: 10.1101/gad.13.5.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rain JC, Rafi Z, Rhani Z, Legrain P, Krämer A. Conservation of functional domains involved in RNA binding and protein-protein interactions in human and Saccharomyces cerevisiae pre-mRNA splicing factor SF1. RNA. 1998;4:551–565. doi: 10.1017/s1355838298980335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigaut, G., A. Shevchenko, B. Rutz, M. Wilm, M. Mann, and B. Séraphin. 1999. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. (in press). [DOI] [PubMed]
- Ruskin B, Zamore PD, Green MR. A factor, U2AF, is required for U2 snRNP binding and splicing complex assembly. Cell. 1988;52:207–219. doi: 10.1016/0092-8674(88)90509-0. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning. A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Schaal TD, Maniatis T. Multiple distinct splicing enhancers in the protein-coding sequences of a constitutively spliced pre-mRNA. Mol Cell Biol. 1999;19:261–273. doi: 10.1128/mcb.19.1.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Séraphin B. Sm and Sm-like proteins belong to a large family: Identification of proteins of the U6 as well as the U1, U2, U4 and U5 snRNPs. EMBO J. 1995;14:2089–2098. doi: 10.1002/j.1460-2075.1995.tb07200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Séraphin B, Rosbash M. Identification of functional U1 snRNA-pre-mRNA complexes committed to spliceosome assembly and splicing. Cell. 1989a;59:349–358. doi: 10.1016/0092-8674(89)90296-1. [DOI] [PubMed] [Google Scholar]
- ————— Mutational analysis of the interactions between U1 small nuclear RNA and pre-mRNA of yeast. Gene. 1989b;82:145–151. doi: 10.1016/0378-1119(89)90039-5. [DOI] [PubMed] [Google Scholar]
- ————— The yeast branchpoint sequence is not required for the formation of a stable U1 snRNA-pre-mRNA complex and is recognized in the absence of U2 snRNA. EMBO J. 1991;10:1209–1216. doi: 10.1002/j.1460-2075.1991.tb08062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Séraphin B, Kandels-Lewis S. 3′ splice site recognition in S. cerevisiae does not require base pairing with U1 snRNA. Cell. 1993;73:803–812. doi: 10.1016/0092-8674(93)90258-r. [DOI] [PubMed] [Google Scholar]
- Séraphin B, Kretzner L, Rosbash M. A U1 snRNA: pre-mRNA base pairing interaction is required early in yeast spliceosome assembly but does not uniquely define the 5′ cleavage site. EMBO J. 1988;7:2533–2538. doi: 10.1002/j.1460-2075.1988.tb03101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F. Getting started with yeast. Methods Enzymol. 1991;194:3–11. doi: 10.1016/0076-6879(91)94004-v. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siliciano PG, Guthrie C. 5′ splice site selection in yeast: Genetic alterations in base-pairing with U1 reveal additional requirements. Genes & Dev. 1988;2:1258–1267. doi: 10.1101/gad.2.10.1258. [DOI] [PubMed] [Google Scholar]
- Siliciano PG, Jones MH, Guthrie C. Saccharomyces cerevisiae has a U1-like small nuclear RNA with unexpected properties. Science. 1987;237:1484–1487. doi: 10.1126/science.3306922. [DOI] [PubMed] [Google Scholar]
- Siliciano PG, Kivens WJ, Guthrie C. More than half of yeast U1 snRNA is dispensable for growth. Nucleic Acids Res. 1991;19:6367–6372. doi: 10.1093/nar/19.23.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staknis D, Reed R. Members of a family of proteins (the RD family) detected by a U1 70K monoclonal antibody are present in spliceosomal complexes. Nucleic Acids Res. 1995;23:4081–4086. doi: 10.1093/nar/23.20.4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teem JL, Rosbash M. Expression of a beta-galactosidase gene containing the ribosomal protein 51 intron is sensitive to the rna2 mutation of yeast. Proc Natl Acad Sci. 1983;80:4403–4407. doi: 10.1073/pnas.80.14.4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1998;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Rosbash M. Identification of eight proteins that cross-link to pre-mRNA in the yeast commitment complex. Genes & Dev. 1999;13:581–592. doi: 10.1101/gad.13.5.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang Y, Weiner AM. A compensatory base change in U1 snRNA suppresses a 5′ splice site mutation. Cell. 1986;46:827–835. doi: 10.1016/0092-8674(86)90064-4. [DOI] [PubMed] [Google Scholar]