

Abstract
Nuclear receptor-mediated activation of transcription involves coactivation by cofactors collectively denoted the steroid receptor coactivators (SRCs). The process also involves the subsequent recruitment of p300/CBP and PCAF to a complex that synergistically regulates transcription and remodels the chromatin. PCAF and p300 have also been demonstrated to function as critical coactivators for the muscle-specific basic helix–loop–helix (bHLH) protein MyoD during myogenic commitment. Skeletal muscle differentiation and the activation of muscle-specific gene expression is dependent on the concerted action of another bHLH factor, myogenin, and the MADS protein, MEF-2, which function in a cooperative manner. We examined the functional role of one SRC, GRIP-1, in muscle differentiation, an ideal paradigm for the analysis of the determinative events that govern the cell's decision to divide or differentiate. We observed that the mRNA encoding GRIP-1 is expressed in proliferating myoblasts and post-mitotic differentiated myotubes, and that protein levels increase during differentiation. Exogenous/ectopic expression studies with GRIP-1 sense and antisense vectors in myogenic C2C12 cells demonstrated that this SRC is necessary for (1) induction/activation of myogenin, MEF-2, and the crucial cell cycle regulator, p21, and (2) contractile protein expression and myotube formation. Furthermore, we demonstrate that the SRC GRIP-1 coactivates MEF-2C-mediated transcription. GRIP-1 also coactivates the synergistic transactivation of E box-dependent transcription by myogenin and MEF-2C. GST-pulldowns, mammalian two-hybrid analysis, and immunoprecipitation demonstrate that the mechanism involves direct interactions between MEF-2C and GRIP-1 and is associated with the ability of the SRC to interact with the MADS domain of MEF-2C. The HLH region of myogenin mediates the direct interaction of myogenin and GRIP-1. Interestingly, interaction with myogenic factors is mediated by two regions of GRIP-1, an amino-terminal bHLH–PAS region and the carboxy-terminal region between amino acids 1158 and 1423 (which encodes an activation domain, has HAT activity, and interacts with the coactivator-associated arginine methyltransferase). This work demonstrates that GRIP-1 potentiates skeletal muscle differentiation by acting as a critical coactivator for MEF-2C-mediated transactivation and is the first study to ascribe a function to the amino-terminal bHLH–PAS region of SRCs.
Keywords: GRIP-1, gene expression, skeletal muscle differentiation, myogenic cells
The process of myogenesis involves two events, determination and differentiation. Determination is the process by which embryonic precursors become committed to the muscle lineage. Differentiation is the mechanism by which these committed cells (myoblasts) acquire the contractile phenotype and involves the fusion of dividing myoblasts to form post-mitotic multinucleated cells. Insight into the molecular mechanisms that control these events was provided by the characterization of the myoD gene family (myoD, myf-5, myogenin, and MRF-4/myf-6/herculin) whose members encode basic helix–loop– helix (bHLH) proteins. Members of the skeletal muscle-specific MyoD family of proteins inhibit cell proliferation and directly/indirectly regulate a cascade of muscle-specific gene expression. The four members of the myoD gene family have the capacity to both auto- and cross-regulate their own and each others' expression (Ludolph and Konieczny 1995, and references therein; Molkentin and Olson 1996; Yun and Wold 1996).
Gene-targeting studies indicated that myoD and myf-5 are required for commitment/determination (Rudnicki et al. 1993), whereas myogenin (Hasty et al. 1993; for review, see Olson et al. 1996) is specifically required for differentiation. In cell culture, myoD/myf-5 are expressed in proliferating myoblasts and are markers for the committed myoblast state; in contrast, myogenin expression strictly coincides with the terminal differentiation.
The bHLH proteins contain a 68 amino acid-conserved basic/myc-like region, which is necessary and sufficient for myogenic conversion. The basic region and adjacent residues that form an HLH motif mediate DNA binding and dimerization, respectively (Lassar et al. 1991; Tapscott and Weintraub 1991; Weintraub et al. 1991; Olson 1992; Olson and Klein 1994). MyoD forms heterodimers with ubiquitously expressed members of the E2A HLH protein family, (alternatively spliced products of the E2A gene—E2–5, E12, E47; Henthorn et al. 1990; Murre et al. 1991; Sun and Baltimore 1991; Kadesh 1993). The formation of these functional heterodimers is negatively regulated by another ubiquitously expressed HLH protein, designated Id. The MyoD–E2A heterodimers bind to the E box motif (CANNTG), which functions as the cognate-binding site for all bHLH factors and is present in most muscle-specific enhancers (Ludolph and Konieczny 1995; Molkentin and Olson 1996; Yun and Wold 1996). Skeletal muscle differentiation and the activation of myogenic-specific gene expression involves the concerted action of another bHLH factor, myogenin, and the MADS protein (MCM1-agamous deficiens-serum response factor), myocyte enhancer factor-2 (MEF-2; Gossett et al. 1989; Cserjesi and Olson 1991), which function in a cooperative manner (Molkentin et al. 1995; Black et al. 1998).
Members of the MEF-2 family of transcription factors play a critical role in the activation of contractile protein transcription. The MEF-2 family is encoded by four independent genes, mef2a, mef2b, mef2c, and mef2d (for review, see Black et al. 1998). MEF2 factors belong to the MADS box family and share a highly conserved 86-amino-acid region that encodes the MADS and MEF2 domains, which mediate DNA binding and dimerization, respectively (Molkentin et al. 1996). Gene targeting in Drosophila supports the critical role of MEF-2 in terminal muscle differentiation (Bour et al. 1995). Interestingly, MEF-2 proteins can be recruited by DNA-bound bHLH factors to synergistically regulate transcription by cooperative mechanisms that involve direct physical association of the MADS–bHLH regions and the transmission of an activating signal (Molkentin et al. 1995; Black et al. 1998). The bHLH protein Twist (Spicer et al. 1996) inhibits MEF-2-mediated transactivation, which has been demonstrated to inhibit the acetyltransferase activity of p300 and PCAF (Hamamori et al. 1999). MEF2A, MEF2B, and MEF2D are ubiquitously expressed, whereas MEF2C is restricted to skeletal muscle, brain, and spleen. However, MEF2C DNA-binding activity is highly enriched in muscle and neural tissue.
Investigation of myogenesis in culture suggests contractile-specific gene expression occurs in a coordinate manner. Within 24 hr of serum deprivation, proliferating myoblasts initiate myogenin expression, closely followed by the activation of the cyclin-dependent kinase inhibitor-p21 (Guo et al. 1995; Halevy et al. 1995; Parker et al. 1995) and the concomitant repression of cyclinD (Skapek et al. 1995, 1996; Guo and Walsh 1997), which results in withdrawal from the cell cycle. The post-mitotic cells then begin to express sarcomeric and enzymatic genes within 36–48 hr, followed by fusion into multinucleated myotubes (Walsh and Perlman 1997).
The retinoblastoma protein pRb has a central role in cell cycle exit and the establishment of the post-mitotic state (Schneider et al. 1994; Novitch et al. 1996). Recent studies support the notion that pRb is necessary for the latter steps in skeletal myogenesis and suggest that pRb and myoD cooperate to activate the functional transactivation properties of MEF2 (in the absence of direct protein–protein interaction; Novitch et al. 1999). Hence, it is apparent that control of the latter stages of differentiation is regulated by the pRB-dependent functional formation of transcriptionally competent bHLH and MEF-2 complexes.
CBP/p300 and PCAF are coactivators for MyoD and MEF-2C during myogenic commitment and differentiation (Eckner et al. 1996; Puri et al. 1997a,b; Sartorelli et al. 1997). The amino-terminal acid-rich activation domain of MyoD interacts directly with p300 and recruits PCAF to form a ternary multimeric complex. Moreover, p300 and PCAF coactivate myoD-mediated transactivation of the p21 gene and are necessary for MyoD-mediated cell cycle arrest. Studies with ligand-regulated transcription factors (e.g., nuclear hormone receptors) that have served as prototypic models of coactivator recruitment (Westin et al. 1998) suggest that recruitment of the coactivators p300 and PCAF is dependent on the presence of the steroid receptor coactivator (SRC) class of cofactors (Onate et al. 1995; for review, see McKenna et al. 1999). Furthermore, the SRCs coactivate several different classes of activators, for example, NFκB (Na et al. 1998; for review, see McKenna et al. 1999). The involvement of SRCs in a functional complex with p300 and PCAF that coactivates many different classes of transcription factors prompted us to investigate the expression and functional role of these cofactors during skeletal muscle differentiation. The structurally related, but genetically distinct, SRC family (for review, see McKenna et al. 1999) has three members variously denoted as SRC-1/N-CoA-1 (Onate et al. 1995; Kamei et al. 1996; Torchia et al. 1997), GRIP-1/TIF-2/N-CoA-2/SRC-2 (Hong et al. 1996; Voegel et al. 1996), and SRC-3/ACTR/pCIP/RAC-3/AIB1/TRAM1 (Anzick et al. 1997; Chen et al. 1997; Li et al. 1997; Takeshita et al. 1997; Torchia et al. 1997). Here, we report that the SRC GRIP-1 potentiates the latter stage of skeletal muscle differentiation by acting as a critical coactivator for MEF-2C-dependent gene expression.
Results
SRC-2/GRIP-1 mRNA is expressed during skeletal muscle differentiation
Recently, p300 and PCAF, have been demonstrated to function as critical coactivators for the muscle-specific basic HLH protein, MyoD, and the MADs protein, MEF-2C, during myogenesis. However, the latest studies firmly suggest that recruitment of p300 and PCAF is dependent on the presence of the SRC/p160 cofactors. Hence, we examined the functional role of the SRC family in muscle differentiation, an ideal paradigm for the analysis of the determinative events that govern the cell's decision to divide or differentiate.
To gain insight into which of the p160 cofactor(s), SRC-1, SRC-2/GRIP-1/TIF-2, and SRC-3/RAC-3/ACTR/pCIP/AIB-1 are expressed during skeletal muscle differentiation, we initially investigated the expression pattern of the mRNAs encoding the SRC family during the conversion of mouse myoblast C2C12 cells into multinucleated myotubes. Proliferating C2C12 myoblasts were induced to biochemically and morphologically differentiate into post-mitotic multinucleated myotubes by serum withdrawal in culture over a 4–72 hr period. The transition from a non-muscle phenotype to a contractile phenotype is associated with the activation/expression of (1) the myoD gene family (myoD, myogenin, myf-5, and MRF-4), (2) the MADs box protein, MEF-2C, and (3) a structurally diverse group of genes that encode a functional sarcomere responsible for contraction. Concomitant with these events is terminal cell cycle exit, characterized by the repression of cyclin D1 and the activation of the cell cycle inhibitor, p21.
Poly(A)+ RNA was isolated from proliferating myoblasts (PMB), confluent myoblasts (CMB), and post-mitotic myotubes after 4, 8, 24, and 72 hr of serum withdrawal and examined by Northern blot analysis. The SRC-2/GRIP-1 mRNA was constitutively expressed in proliferating myoblasts and as the cells exit the cell cycle and fuse to form post-mitotic differentiated multinucleated myotubes that have acquired a muscle-specific phenotype (Fig. 1A). The hybridization signal corresponded to the expected transcript size of ∼7.5 kb. It was necessary to use polyadenylated RNA, because the level of expression was quite low relative to GAPDH or myogenin; exposure times required for the detection of SRC-2/GRIP-1 were ∼10-fold longer than that of GAPDH. Western analysis demonstrated that the levels of the SRC-2/GRIP-1 protein increased during myogenesis (Fig. 1B). The identity of the signal was confirmed by the use of an SRC-2/GRIP-1-positive control (Transduction Laboratories) from Jurkat cells (Fig. 1B). The SRC-1 mRNA was very weakly expressed; however, we could not detect the mRNA transcript that encoded SRC-3 (Fig. 1A).
Figure 1.
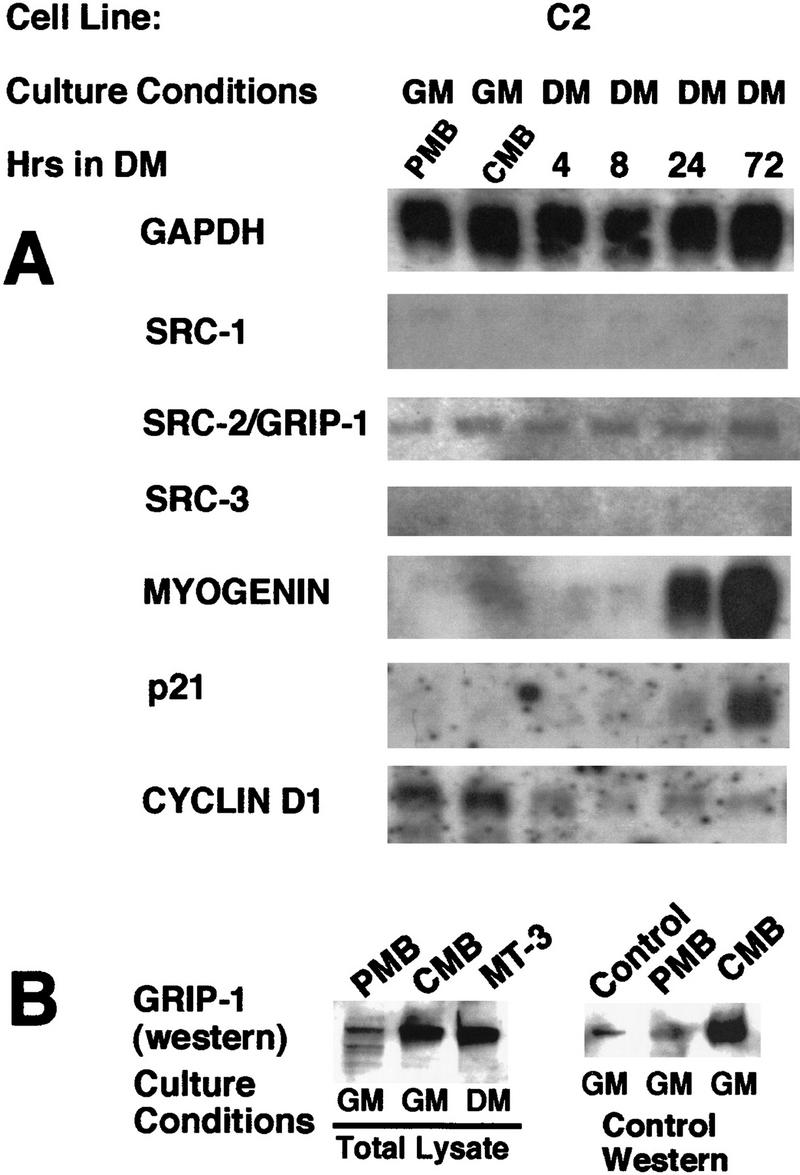
(A) GRIP-1 mRNA is constitutively expressed during skeletal muscle differentiation. Total RNA was isolated from PMBs (∼50% confluent), CMBs (100% confluent) cultured in growth medium, and developing myotubes 4, 8, 24, and 72 hr after serum withdrawal (i.e., propagated in differentiation medium). After blotting, RNA was probed with 32P-radiolabeled cDNA encoding GAPDH, myoD, myogenin, cyclinD1, p21, SRC-1, SRC-2/GRIP-1, and SRC-3/RAC-3/ACTR. (GM) Growth medium (DMEM containing 20% FCS); (DM) differentiation medium (DMEM containing 2% horse serum). The induction of myogenin and p21 mRNAs confirms that these cells have undergone terminal differentiation. (B) Western analysis of GRIP-1 levels during skeletal muscle differentiation and GRIP-1 Western control.
Concomitant with the constitutive expression of GRIP-1 mRNA was the induction of myogenin and p21 mRNAs, relative to the GAPDH mRNA, which confirmed that these cells had terminally differentiated (Fig. 1A). Moreover, the repression of cyclinD1 confirmed that these cells were exiting the cell cycles. The expression of GRIP-1 mRNA, which encodes an SRC, suggested that this cofactor may regulate the action of the MyoD/bHLH family and/or MEF-2/MADS box-mediated transcription during the activation of muscle-specific gene expression and skeletal muscle differentiation.
Exogenous expression of GRIP-1 sense and antisense in myogenic cells: the SRC is required for skeletal muscle differentiation
To delineate the functional role of the SRC GRIP-1 in myogenesis and to identify the putative target(s) of this coactivator in muscle, we proceeded to examine the effect of knocking out the coactivator function by exogenous expression of GRIP-1 antisense in the C2C12 cell line.
C2 muscle cells were transfected with the plasmid encoding full-length GRIP-1 cloned into the sense and antisense orientation of pcDNA3.1 (pcDNA3.1-GRIP-1 sense and pcDNA3.1-GRIP-1 antisense) and with the expression vehicle pcDNA3.1 alone. Then, after G418 selection stable transfectants were isolated as a number of individual clonal cell lines. Clonal cell lines were subsequently analyzed by Northern hybridization to identify clonal lines that efficiently expressed antisense and sense GRIP-1 (relative to endogenous GRIP-1 mRNA; data not shown). Wild-type C2 cells, vehicle-only transfected cells, and two clonal cell lines that abundantly expressed antisense GRIP-1 denoted as C2:GRIP-1 antisense #1 and #2 were subjected to preliminary Northern analysis with myogenin (an ideal marker for terminal differentiation in culture) and GAPDH probes. Northern analysis suggested antisense expression of GRIP-1 compromised the potential of the cells to differentiate in culture relative to wild-type and vehicle-only transfected cells (Fig. 2A). Then, we used Western analysis to investigate whether GRIP-1 protein is down-regulated in the C2:GRIP-1 antisense #2 line. Total cellular extract was isolated from wild-type C2, GRIP-1 antisense #2, and C2 cells transfected with the expression vehicle, pcDNA3.1. GRIP-1 protein levels were significantly reduced relative to the wild-type levels (Fig. 2B). The vehicle-only transfected cells expressed GRIP-1 protein, although the levels were slightly lower than wild type (Fig. 2B).
Figure 2.

(A) Northern analysis of wild-type C2 cells, and vehicle-only and GRIP-1 antisense-transfected cells. (B) Western analysis of GRIP-1 levels in wild-type C2 cells, GRIP-1 antisense, and vehicle-only transfected cells. (C) Northern analysis of wild-type C2 cells and GRIP-1 sense-transfected cells. (D) Northern analysis demonstates the ectopic expression of GRIP-1 in C2:GRIP-1 sense cells. Total RNA was isolated from both normal C2C12 and stably transfected C2:GRIP-1 sense and antisense cells at the PMB and CMB stage (∼50% and 100% confluent, respectively) cultured in growth medium, and at a stage 72 hr/3 days after the withdrawal of serum (i.e., propagated in differentiation medium, MT-3). RNA samples were blotted and probed with 32P-radiolabeled cDNA encoding GAPDH and myogenin. GRIP-1 proteins were detected by Western blot with 30 μg of nuclear proteins from each sample using T73620, a monoclonal antibody to human GRIP-1/TIF-2 that cross-reacts with the mouse species. (GM) Growth medium (DMEM containing 20% FCS); (DM) differentiation medium (DMEM containing 2% horse serum).
To investigate further the role played by the SRC GRIP-1 in myogenesis, we proceeded to examine the effect of exogenous sense expression of GRIP-1 on C2 skeletal muscle cell differentiation. Wild-type C2 cells, and two clonal cell lines that abundantly expressed sense GRIP-1 denoted as C2:GRIP-1 sense #1 and #2 were subjected to preliminary Northern analysis with myogenin and GAPDH probes (Fig. 2C). Northern analysis suggested sense expression of GRIP-1 resulted in precocious terminal differentiation of cells in culture, relative to wild-type cells (Fig. 2C). The C2:GRIP-1 sense #1 cells expressed exogenous GRIP-1 at levels similar to the endogenous transcript (Fig. 2D).
To examine rigorously the effect of exogenous GRIP-1 antisense expression on factors involved in determination (e.g., myoD), cell cycle regulation (e.g., cyclin D1 and p21), and differentiation (myogenin and MEF-2), we isolated total RNA from C2:GRIP-1 antisense #2 line and the wild-type C2 cells from PMBs, CMBs, and myotubes (MT-3) before and 3 days after serum withdrawal. After blotting, the RNA was probed with GAPDH, myoD, myogenin, cyclinD1, and p21 labeled cDNAs (Fig. 3). Comparisons of C2:GRIP-1 antisense to native C2C12 cells showed that the level of myoD mRNA after serum withdrawal was suppressed in the C2:GRIP-1 antisense cell line. Furthermore, the induction of the p21 mRNA after serum withdrawal was completely ablated, and the activation of the myogenin mRNA (and MEF-2 proteins) was dramatically reduced in the C2-GRIP-1 antisense cells (Fig. 3). Interestingly, exogenous expression of GRIP-1 antisense increased the steady-state levels of cyclinD1 mRNA in proliferating cells relative to the equivalent levels of GAPDH mRNA (Fig. 3).
Figure 3.
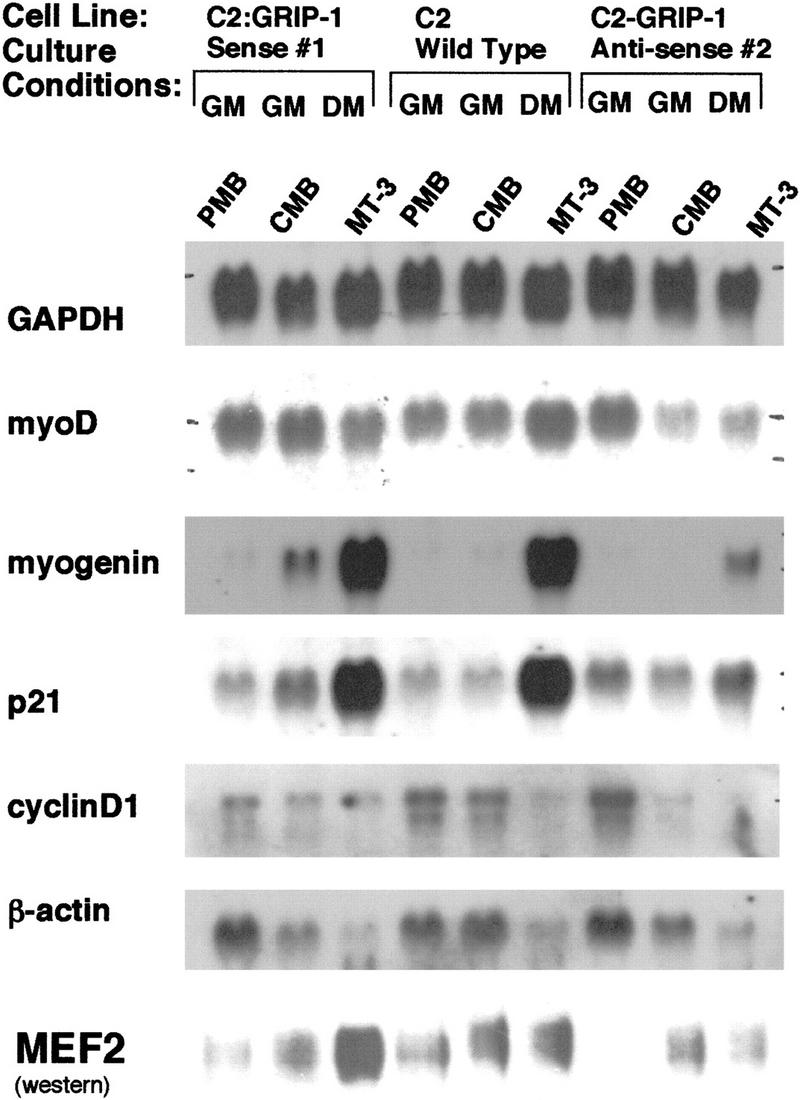
Exogenous stable expression of GRIP-1 sense and antisense in C2C12 myogenic cells demonstrates that GRIP-1 expression is necessary for terminal skeletal muscle cell differentiation. Total RNA was isolated from both normal C2C12 and stably transfected C2:GRIP-1 sense and antisense cells at the PMB and CMB stage (∼50% and 100% confluent, respectively) cultured in growth medium, and at a stage 72 hr/3 days after the withdrawal of serum (i.e., propagated in differentiation medium, MT-3). RNA samples were blotted and probed with 32P-radiolabeled cDNA encoding GAPDH, myoD, myogenin, cyclinD1, and p21. MEF-2 proteins were detected by Western blot with 30 μg of nuclear proteins from each sample using a rabbit antibody to MEF-2 (Santa Cruz C-21), which was generated against MEF-2A but which cross-reacts with mouse/human MEF-2A, MEF-2C, and MEF-2D. (GM) Growth medium (DMEM containing 20% FCS); (DM) differentiation medium (DMEM containing 2% horse serum).
Similarly, we rigorously examined the effect of exogenous GRIP-1 sense expression on factors involved in determination, cell cycle regulation (e.g., cyclin D1 and p21), and differentiation (myogenin). Then, we isolated total RNA from cell line C2:GRIP-1 sense #1 and wild-type C2 cells before and after serum withdrawal. After blotting, the RNA was probed with GAPDH, myoD, myogenin, p21, cyclinD1, and β-actin labeled cDNAs (Fig. 3). Comparisons of C2:GRIP-1 sense and native C2C12 cells clearly demonstrated the precocious induction of the p21 and myogenin mRNAs prior to serum withdrawal in confluent myoblasts. In fact, myogenin and p21 mRNAs are spontaneously induced by cell contact in the C2:GRIP-1 sense cell line. Moreover, we observed the hyperinduction of MEF-2 proteins after serum withdrawal. Consistent with the precocious differentiation of this cell line (i.e., C2:GRIP-1 sense), the steady-state levels of cyclinD1 mRNA in proliferating myoblasts growing in growth medium (DMEM supplemented with 20% FCS) was suppressed relative to the levels of cyclinD1 in native C2 cells and the C2:GRIP-1 antisense line. Moreover, the very different cyclinD1 mRNA levels in the proliferating myoblasts from sense and antisense cells is concordant with the ability of these cell lines to differentiate in culture. The impact of GRIP-1 expression on cyclinD1 mRNA levels is validated by the unaffected levels of non-muscle cytoskeletal β-actin.
Because the potential of the C2:GRIP-1 antisense cells to differentiate in culture was compromised, we examined a later step in myogenesis, the activation/induction of contractile/structural proteins that encode a functional sarcomere. We investigated this latter process in skeletal muscle cell differentiation by immunostaining with a monoclonal antibody directed against the fast isoform of the major thick filament protein, skeletal myosin heavy chain (MHC). MHC is associated with the contractile phenotype and is expressed during the process of muscle differentiation (Fig. 4). The immunohistochemistry dramatically demonstrates the difference between the C2:GRIP-1 sense (Fig. 4A,B) and antisense lines (Fig. 4C,D) and reveals that antisense GRIP-1 expression significantly compromises (1) the activation of contractile protein gene(s), (2) the formation of post-mitotic myotubes, and (3) morphological differentiation.
Figure 4.
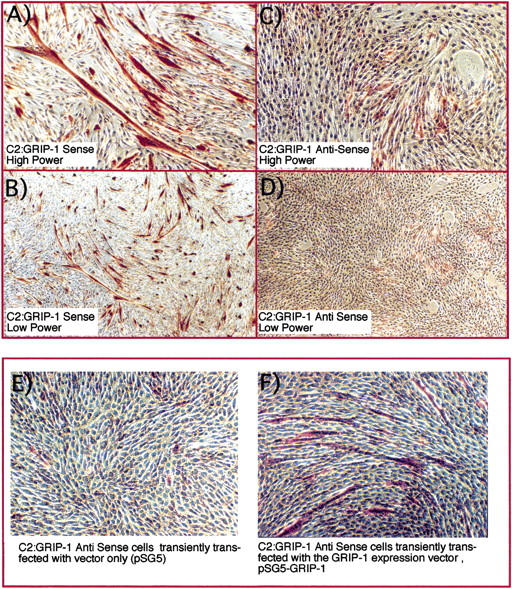
GRIP-1 expression is necessary for (1) the activation of contractile protein gene(s), (2) formation of post-mitotic myotubes, and (3) morphological differentiation. We examined the C2 GRIP-1 sense (A,B) and antisense lines (C,D) by immunostaining with a monoclonal antibody directed against the fast isoform of the major thick filament protein, skeletal MHC after 72 hr of culture in differentiation medium. Immunohistochemistry reveals that antisense GRIP-1 expression significantly compromises the activation of contractile protein gene(s) and formation of post-mitotic multinucleated myotubes. Sense and antisense cell lines were immunostained with a monoclonal antibody directed toward the fast isoform of the major thick filament protein MHC. MHC-positive cells are stained red and nuclei counterstained blue by Mayer's hematoxylin. (E) GRIP-1 antisense cells transiently transfected with vector/vehicle alone. (F) Rescue of the GRIP-1 antisense phenotype by transfection of pSG5-GRIP-1.
Then, we examined whether the GRIP-1 antisense cell differentiation-defective phenotype could be rescued by overexpression of GRIP-1. The antisense cells were transiently transfected with either the expression vector/vehicle alone (pSG5; Fig. 4E) or pSG5-GRIP-1 (Fig. 4F) with a cocktail of two liposomes to increase the efficiency of transfection. Following immunohistochemistry with the MHC antibody, we observed that GRIP-1 overexpression could rescue the antisense phenotype. This result further reinforced our hypothesis that down-regulation of GRIP-1 protein attentuates the activation of the contractile protein gene program and phenotypic acquisition.
These cell culture studies suggest that the function of GRIP-1 is necessary for the expression of myogenin and the cyclin-dependent kinase inhibitor, p21, which mediates cell cycle exit. Moreover, GRIP-1 is required for the formation of post-mitotic cells that express myofibrillar proteins. These studies suggest that GRIP-1 may function as a critical coactivator for the latter stages of skeletal muscle differentiation that are dependent on MEF-2C and myogenin-mediated events/cascades.
GRIP-1 coactivates MEF-2C-mediated transactivation of muscle-specific gene expression in the presence and absence of its cognate-binding site
The cell culture experiments presented above suggest that GRIP-1 is involved in the activation/regulation of myogenic differentiation. Therefore, we examined whether GRIP-1 was involved in the coactivation of myogenic-specific transcription by the muscle-specific bHLH factors, myoD, myf-5, myogenin, and MRF-4, which are, respectively, involved in myogenic commitment and differentiation. Moreover, we also investigated the ability of GRIP-1 to regulate the activity of MEF-2C. MEF-2C belongs to the MADS box family of transcription factors, which cooperate with the myogenic bHLH proteins in the activation of the contractile protein gene expression and function within a regulatory network that establishes the differentiated phenotype. We cotransfected pluripotent 10T1/2 cells with the native muscle creatine kinase (MCK) promoter linked to a LUC reporter (MCK–LUC) with a range of plasmids encoding bHLH factors MyoD, Myf-5, Myogenin, and MRF-4, and the MADS box factor, MEF-2C, in the presence and absence of GRIP-1. This promoter and cell line have been routinely used to assay myogenic bHLH factor and MEF-2-mediated transactivation of muscle-specific gene expression. Interestingly, GRIP-1 failed to coactivate MyoD, Myf-5, Myogenin, and MRF-4-mediated transactivation of the MCK–LUC reporter (Fig. 5A). Myogenic bHLH factors activate muscle transcription through several steps, which include heterodimerization with the ubiquitous E class of bHLH proteins, (products of the E2A gene, E12, E47, and E2–5). Furthermore, GRIP-1 also failed to coactivate bHLH factor-mediated activation of MCK–LUC in the presence of its ubiquitous heterodimeric partner, the E2A class of bHLH factors (data not shown). However, coexpression of GRIP-1 significantly enhanced MEF-2C-mediated transactivation of the MCK–LUC reporter (Fig. 5B). This specificity of coactivation was tested by cotransfection of MEF-2C and GRIP-1 with the pGL2-basic vector (i.e., LUC backbone only) and the pGL2-control plasmid (i.e., the SV40 early promoter linked to the LUC reporter). MEF-2C and GRIP-1 could not transactivate the empty LUC reporter vehicle or the SV40 promoter-driven LUC reporter (Fig. 5C). These results validated the nature and specificity of the MEF-2C/GRIP-1-mediated transactivation of the MCK–LUC reporter.
Figure 5.
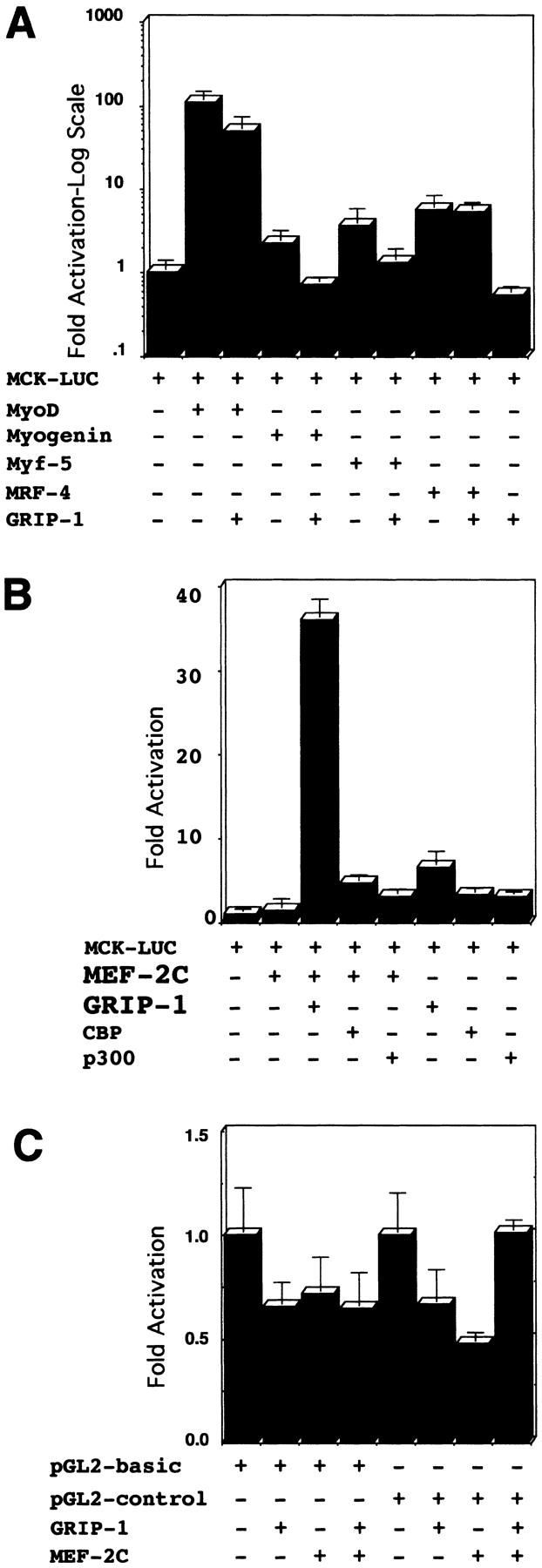
GRIP-1 coactivates MEF-2C and myogenin-mediated transactivation of muscle-specific gene expression. (A) GRIP-1 does not coactivate bHLH factor-mediated transactivation of the MCK promoter in pluripotent C3H10T1/2 cells. (B) GRIP-1 coactivates MEF-2C-mediated transactivation of the MCK promoter in pluripotent C3H10T1/2 cells. Fold activation is expressed relative to LUC activity obtained after cotransfection of the MCK–LUC reporter and vector alone. Activation mediated by vector alone is arbitrarily set at 1. Transfection results were derived from at least two sets of independently transfected triplicates.
It has been reported by Sartorelli et al. (1997) that the cofactors p300 and CBP coactivate MEF-2C-mediated transactivation of a MEF-2-dependent reporter. Therefore, we compared the relative ability of GRIP-1, p300, and CBP to coactivate MEF-2C-mediated transactivation of the MCK–LUC reporter. GRIP-1 and MEF-2C synergistically stimulated transcription; however, in comparison, p300/CBP weakly coactivated MEF-2C-mediated transcription in an additive manner (Fig. 5B).
To substantiate further these results, we tested the ability of GRIP-1 to coactivate MEF-2c-dependent activation of a LUC reporter with three tandem copies of the MEF-2-binding sites upstream of a basal E1b promoter (Fig. 6A). These experiments clearly demonstrated again that GRIP-1 synergistically coactivates MEF-2C-mediated transactivation of a MEF-2-dependent reporter (Fig. 6A) as was the case for the MCK promoter. The specificity of this coactivation was supported by the inability of GRIP-1 alone to activate the MEF-2-dependent reporter (mef2 × 3-E1b-LUC) and the failure of MEF-2C and GRIP-1 to regulate the expression of the vehicle, that is, the basal E1b promoter linked to LUC (Fig. 6B).
Figure 6.
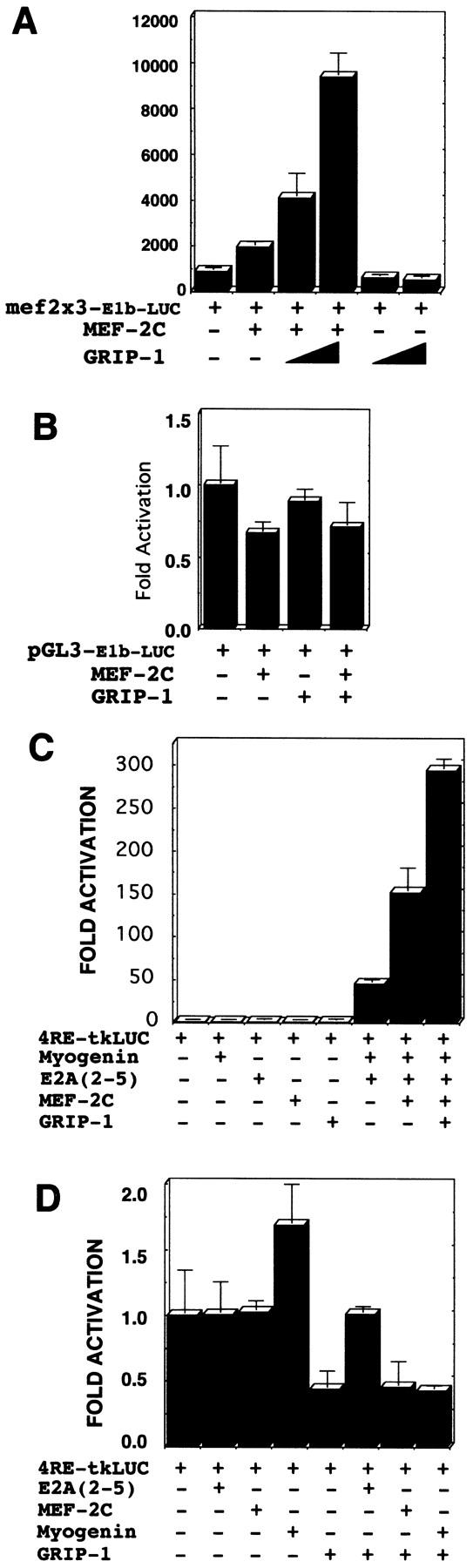
(A) GRIP-1 coactivates MEF-2-dependent activation of a reporter comprised of 3× mef-2 sites linked to a basal E1b–LUC reporter. (B) MEF-2C and GRIP-1 do not regulate the basal E1b promoter. (C) GRIP-1 coactivates myogenin-dependent/E box-driven transcription in the presence of tethered MEF-2C in pluripotent C3H10T1/2 cells. (D) E2A, MEF-2C, and GRIP-1 either independently, or in concert, do not transactivate the 4RE–LUC promoter in the absence of myogenin. Fold activation is expressed relative to LUC activity obtained after cotransfection of the 4RE–LUC reporter and vector alone. Activation mediated by vector alone is arbitrarily set at 1. Transfection results were derived from at least two sets of independently transfected triplicates.
The MADS box factor, MEF-2C, and the myogenic bHLH factors can activate muscle transcription by direct binding to their cognate response elements or by protein–protein interactions that mediate tethering. Moreover, MEF-2C and the bHLH factor, myogenin, cooperatively function in the post-commitment stage of myogenesis, and we observed that their expression was particularly altered in the ectopic GRIP-1 sense and antisense expression studies in cell culture. We tested the potential of GRIP-1 to coactivate myogenin/E2A-mediated transactivation of an E box/bHLH factor-dependent reporter, 4RE-tkLUC (which contains four tandem copies of the right E box from the MCK enhancer upstream of the basal thymidine kinase-promoter) in the presence of MEF-2C (Fig. 6C). As expected, we observed the activation of the E box-dependent reporter, 4RE-LUC, by the coexpression of myogenin and E2A (e2-5); addition of MEF-2C (in the absence of its cognate binding site) further activated the promoter in agreement with the published literature (Fig. 6C). Interestingly, addition of GRIP-1 increased transactivation of the E box-dependent promoter in the context of tethered MEF-2C (Fig. 6C). The assay was conducted with equimolar concentrations of the four factors; we observed additional coactivation with increasing amounts of GRIP-1 (data not shown). GRIP-1 expression did not have any effect on 4RE-tkLUC activity in the presence of E2A, MEF-2C, or myogenin alone (Fig. 6D).
In summary, we observed that GRIP-1 mediated the coactivation of (1) MEF-2-dependent transcription, and (2) E box/bHLH-dependent transcription in the presence of tethered MEF-2C (i.e., in the absence of a MEF-2C cognate-binding site).
GRIP-1 stimulates the activity of GAL4–MEF-2C: GRIP-1 interacts with MEF-2C
The experiments presented above suggest that GRIP-1 coactivates MEF-2C-mediated transcription. Because MEF-2C can activate transcription directly and/or by protein–protein interaction-mediated tethering to DNA-bound bHLH factors, we examined the effect of GRIP-1 expression on MEF-2C-mediated transactivation in the GAL4 hybrid system (Fig. 7A). In these assays the activity of MEF-2C is independent of its binding to its cognate binding motif, the A/T-rich MEF-2 site. If GRIP-1 regulates the transcriptional activity, then the potential of the GAL4–myogenic factor fusions to transactivate gene expression should be greatly increased in this assay (Fig. 7A).
Figure 7.
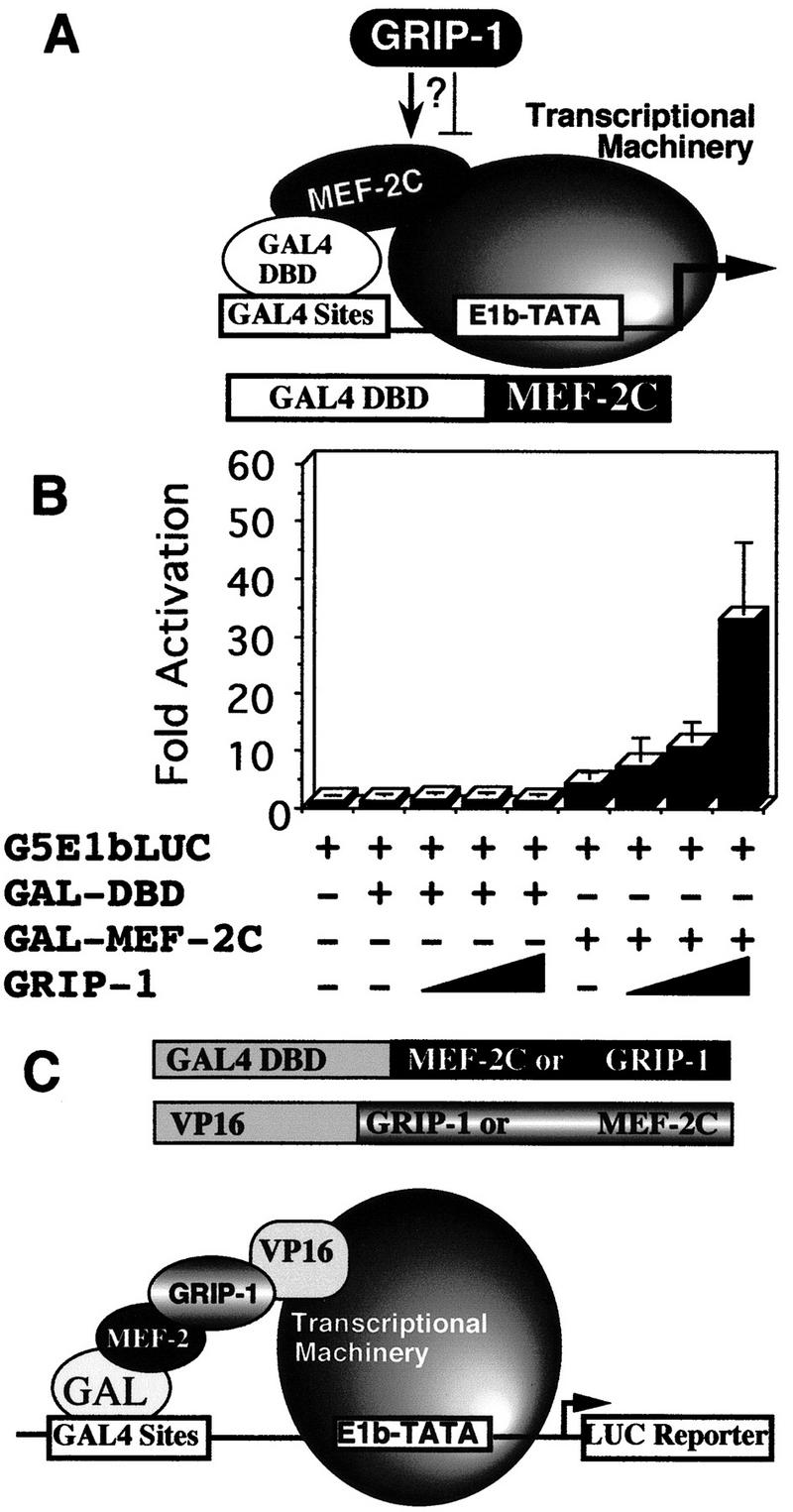


GRIP-1 potentiates the activity of MEF-2C, GRIP-1, and MEF-2C, which interact in a cellular context. (A) Diagrammatic representation of the GAL4 hybrid assay. This assay was used to determine the effect of GRIP-1 expression on MEF-2C transcriptional activity. Cells were transfected with a plasmid encoding GAL4–MEF-2C (i.e., the yeast GAL4 DBD fused in frame with full-length MEF-2C) and a reporter construct in which five copies of the GAL4-binding site are placed upstream of the minimal E1b promoter (i.e., pG5E1b–LUC). (B) GRIP-1 potentiates MEF-2C-mediated transactivation in C3H10T1/2 non-muscle cells in the absence of the MEF-2 cognate-binding site. Fold activation is expressed relative to LUC activity obtained after cotransfection of the GAL4 DBD and vector alone. Activation mediated by the GAL4 DBD and vector alone is arbitrarily set at 1. (C) Diagrammatic representation of the mammalian two-hybrid assay used to determine the interaction and cooperativity of GRIP-1 and MEF-2C in a cellular context. (D,E) GRIP-1 and MEF-2C significantly interact and cooperate in a cellular context. Fold activation is expressed relative to LUC activity obtained after cotransfection of the GAL4 DBD and the VP16 cassette alone. Activation mediated by the GAL4 DBD and the empty VP16 cassette is arbitrarily set at 1.
C3H10T1/2 pluripotent cells were cotransfected with GAL–MEF-2C and the G5E1b–LUC reporter, in the presence and absence of a GRIP-1 expression vector. G5E1b–LUC contains five copies of the GAL4-binding site placed upstream of a minimal E1b promoter. Transfection of GAL–MEF-2C alone modestly induced transcription relative to the GAL4 DNA-binding domain (DBD) alone; this level of activity was dramatically and very significantly stimulated by addition of GRIP-1, as was observed with the MCK and MEF-2-dependent LUC reporter genes (Fig. 7B).
To characterize further transcriptional regulation by MEF-2C, we investigated whether these transcription factors interact with GRIP-1 in a cellular context in vivo. Protein–protein-interaction assay systems were developed initially in yeast and further refined for the study of protein–protein interactions in transfected mammalian cells. In these experiments, the yeast GAL4 DNA-binding domain is fused to either MEF-2C (or GRIP-1) and expressed in transfected cells with either GRIP-1 or MEF-2C linked to the transactivation domain of herpes simplex virus VP16. Transactivation of a LUC reporter gene downstream of GAL4-binding sites fused to the E1b promoter is achieved only when the coexpressed proteins interact physically (Fig. 7C).
Chimeric GAL4–MEF-2C regulated transcription of an E1b promoter cloned downstream of five copies of the GAL4-binding site linked to the LUC reporter (as demonstrated above previously; Fig. 7). As shown in Figure 7D, GAL4–MEF-2C alone activated transcription (∼8-fold), relative to the GAL4 DBD. Then, we examined the ability of coexpressed VP16–GRIP-1 to transactivate gene expression in this mammalian two-hybrid system. We observed that VP16–GRIP-1 effectively and significantly induced transcription (∼7-fold) resulting in a 50-fold transactivation of gene expression. Similarly, we performed the experiment with GAL4–GRIP-1, which regulated transcription of the E1b promoter cloned downstream of five copies of the GAL4-binding site linked to the LUC reporter (Fig. 7E). As shown in Figure 7E, GAL4–GRIP-1 alone activated transcription ∼10-fold relative to the GAL4 DBD. Then, we examined the ability of coexpressed VP16–MEF-2C to transactivate gene expression in this mammalian two-hybrid system. We observed that VP16–MEF-2C effectively and significantly induced transcription a further ∼20-fold, resulting in a >200-fold activation of gene expression. In summary, these mammalian two-hybrid experiments demonstrate clearly that MEF-2C significantly interacts with GRIP-1 in a cellular context.
In summary, we observed that MEF-2C-mediated transcription in the absence of its cognate binding motif is increased by the SRC GRIP-1. We conclude that GRIP-1 can function as a coactivator of MEF-2C activity/function. Furthermore, the mammalian two-hybrid experiments demonstrate clearly that MEF-2C interacts with GRIP-1 in a cellular context and suggest that the mechanism of activation involves direct physical association.
GRIP-1 coactivates GAL4–myogenin in an E2Adependent manner
The experiments presented above (Fig. 6D) suggested that GRIP-1 potentiates myogenin/E2A-mediated transactivation in the presence of tethered MEF-2C. Because myogenin can activate transcription either by using its own activation domain or by acting as a tether to the DNA, we examined the effect of GRIP-1 expression on myogenin-mediated transactivation in the GAL4 hybrid system (Fig. 8A). In these assays, the activity of the bHLH factor myogenin is independent of its binding to its cognate-binding motif, the E box (CANNTG). If GRIP-1 regulates the transcriptional activity, then the potential of the GAL4–myogenin fusion to transactivate gene expression should be greatly increased in this assay. However, GAL4–myogenin-dependent transcription was not coactivated by GRIP-1 (data not shown).
Figure 8.

GRIP-1 potentiates the activity of myogenin in an E2A-dependent manner. GRIP-1 and myogenin interact in a cellular context. (A) Diagrammatic representation of the GAL4 hybrid assay. This assay was used to determine the effect of GRIP-1 expression on myogenin transcriptional activity in the presence and absence of exogenous E2A expression. Cells were transfected with a plasmid encoding GAL4–myogenin, an E2A expression vector, and a reporter construct in which five copies of the GAL4-binding site were placed upstream of the minimal E1b promoter (i.e., G5E1b–LUC). (B) GRIP-1 potentiates myogenin-mediated transactivation in C3H10T1/2 non-muscle cells in the presence of its heterodimeric bHLH partner. Fold activation is expressed relative to LUC activity obtained after cotransfection of the GAL4 DBD and vector alone. Activation mediated by the GAL4 DBD and vector alone is arbitrarily set at 1. (C) E2A and GRIP-1 do not interact in a cellular context in the absence of myogenin. (D) Myogenin and GRIP-1 do not interact in a cellular context in the absence of E2A. (E) Diagrammatic representation of the modified mammalian two-hybrid assay used to determine the interaction and cooperativity of GRIP-1 and the myogenin–E2A complex in a cellular context. (F) GRIP-1 and the myogenin–E2A complex significantly interact and cooperate in a cellular context. Fold activation is expressed relative to LUC activity obtained after cotransfection of the GAL4 DBD and the VP16 cassette alone. Activation mediated by the GAL4 DBD and the empty VP16 cassette is arbitrarily set at 1.
The bHLH factors function in heterodimeric complexes with the E proteins; hence, we examined the potential of GRIP-1 to coactivate myogenin in the presence of an expression vector encoding an E2A protein (E2–5). Myogenin-dependent activation was significantly stimulated by addition of E2A. Furthermore, GRIP-1 significantly coactivated myogenin-mediated transcription in the presence of exogenous E2A (Fig. 8B).
GAL4–E2A-dependent transcription (in the absence of myogenin) was not coactivated by GRIP-1 (Fig. 8C). The failure of GRIP-1 to coactivate either myogenin or E2A independently, correlated with the inability of E2A and myogenin to interact with GRIP-1 in the mammalian two-hybrid assay (Fig. 8, C and D, respectively) in contrast to MEF-2C.
In summary, we observed that myogenin-mediated transcription in the presence of exogenous E2A (and in the absence of its cognate binding motif) is coactivated by GRIP-1. In this GAL4 hybrid assay, GRIP-1 coactivated the myogenin/E2A heterodimeric complex in the absence of MEF-2C, raising the possibility that in certain contexts GRIP-1 could potentially regulate myogenin-mediated transcription in the absence of MEF-2C. However, as shown earlier, this effect was not observed in the context of the native muscle-specific MCK promoter.
GRIP-1 interacts with myogenin in an E2Adependent manner
Because coactivation of myogenin-dependent transcription by GRIP-1 required exogenous E2A expression, we used a modified mammalian two-hybrid system (Fig. 8E) to investigate further the molecular basis of myogenin/GRIP-1-mediated activation. We examined the effect of GRIP-1 expression on the interaction between GAL4–E2A (E12) and VP16–myogenin. GAL4–E2A (E12) recruited the cotransfected VP16–myogenin and dramatically stimulated transcription relative to GAL–E2A (E12) and the GAL4 DBD (Fig. 8F). We observed that addition of the GRIP-1 expression vector effectively and significantly induced transcription a further order of magnitude (∼10-fold) and resulted in a >300-fold transactivation of gene expression. In conclusion, the mammalian two-hybrid experiments demonstrate clearly that myogenin/E2 complex interacts with GRIP-1 in a cellular context.
In summary, the mammalian two-hybrid experiments demonstrate clearly that the functional myogenin/E2A complex has the potential to interact with GRIP-1 in the cellular context and suggest that the mechanism of activation involves direct physical association.
MEF-2C interacts directly with GRIP-1: the MADS-box domain of MEF-2C mediates the physical association with GRIP-1
The regulation of MEF-2C and myogenin function in the context of transcription by the coactivator GRIP-1 and the demonstration of interaction between these factors in the cellular context suggest strongly that these proteins interact by a direct mechanism. However, these observations do not eliminate completely the possibility of an indirect mechanism in which additional factor(s) mediate the interaction. We postulated that GRIP-1 could be functioning to stimulate MEF-2C and myogenin activity/function by direct interaction. We tested this hypothesis using a biochemical approach, in vitro GST-pulldown assays, to confirm a direct interaction between MEF-2C and GRIP-1 and to identify the domain in MEF-2C that mediates the interaction with the coactivator.
Glutathione–agarose-immobilized GST–GRIP-1 was tested for direct interaction with in vitro 35S-radiolabeled native MEF-2C, MEF-2C-amino acids 58–465 (M1), MEF-2C-amino acids 90–465 (M2), MEF-2C-amino acids 1–223, and MEF-2C-amino acids 1–177. Native MEF-2C showed a very strong interaction with GST–GRIP-1 relative to GST alone. In contrast, MEF-2C-M1 and MEF-2C-M2, which lack the MADS box and MADS-box/MEF-2 domains, respectively, failed to interact with GRIP-1 (Fig. 9A). These results suggest that the MADS box mediates the interaction with GRIP-1 (Fig. 9A). In support of this conclusion, carboxy-terminal deletions MEF-2C-amino acids 1–223 and MEF-2C-amino acids 1–177, which contain the MADS box, interacted efficiently with GRIP-1. Furthermore, glutathione–agarose-immobilized GST–GRIP-1 was tested for direct interaction with in vitro 35S-radiolabeled MEF-2C-amino acids 1–58, which encodes the MADS domain of MEF-2C. The MADS-box region alone interacted significantly with GST–GRIP-1 relative to GST alone (Fig. 9B).
Figure 9.



The MADS box of MEF-2C mediates the direct interaction with GRIP-1. (A) Diagrammatic representation showing the MEF-2C regulatory domains; the MADS domain of MEF-2C mediates the interaction with GRIP-1. Glutathione–agarose-immobilized GST and GST–GRIP-1 proteins were incubated with either 35S-radiolabeled full-length MEF-2C, or fragments of MEF-2C between amino acid positions 58 and 465, 90 and 465, 1 and 223, and 1 and 177. Deletion of the MADS box between amino acid positions 1 and 58 ablates binding to GRIP-1 in vitro. The input lanes represent ∼10% of the total protein. (B) Glutathione–agarose-immobilized GST and GST–GRIP-1 proteins were incubated with either 35S-radiolabeled MEF-2C–MADS box amino acids 1–58. The input lanes represent ∼10% of the total protein. (C,D) Mammalian two-hybrid assay used to determine the interaction between GRIP-1 and MEF-2C (carrying a MADS box/MEF-2 domain deletion) in a cellular context. Fold activation is expressed relative to LUC activity obtained after cotransfection of the GAL4 DBD and the VP16 cassette alone. Activation mediated by the GAL4 DBD and the empty VP16 cassette is arbitrarily set at 1.
The requirement of the MADS box to mediate the interaction between MEF-2C and GRIP-1 was investigated further by use of the mammalian two-hybrid system in a cellular context. In these experiments, the yeast GAL4 DBD fused to native GRIP-1 was expressed in transfected cells with either native MEF-2C, MEF-2C-M1, or MEF-2C-M2 linked to the transactivation domain of herpes simplex virus VP16. As shown in Figure 9C, GAL4–GRIP-1 independently activated transcription (∼30-fold) relative to the GAL4 DBD (as demonstrated previously; Fig. 7). As expected, coexpressed VP16–MEF-2C effectively and significantly induced transcription ∼10-fold, resulting in an ∼300-fold activation of gene expression. However, the deleted MEF-2C-M1–VP16 and MEF-2C-M2–VP16 chimeras lacked the ability to transactivate gene expression further in this system (Fig. 9C). This result clearly demonstrates that the MADS-box domain between amino acids 1 and 58 is necessary to mediate the physical interaction and subsequent transactivation of the LUC reporter. Similarly, we investigated the ability of VP16–GRIP-1 to regulate either GAL4–MEF-2C-M1 or MEF-2C-M2-mediated activation of an E1b promoter cloned downstream of five copies of the GAL4-binding site linked to the LUC reporter. GAL4–MEF-2C (wild type) independently activated transcription, ∼8-fold, relative to the GAL4 DBD (Fig. 9D, as demonstrated previously; Fig. 7). As expected, coexpressed VP16–GRIP-1 effectively and significantly induced transcription (∼6-fold) resulting in a 50-fold transactivation of gene expression. Similarly, we performed the experiment with GAL4–MEF-2C-M1 and MEF-2C-M2. Deletion of the MADS box (amino acids 1–58) and the MADS box/MEF2 domain (amino acids 1–89) significantly impaired the ability of MEF-2C to activate transcription independently, relative to the GAL4 DBD (Fig. 9D). Furthermore, coexpressed VP16–GRIP-1 failed to induce transcription significantly, resulting in <5-fold transactivation of gene expression (compared with 50-fold with wild-type MEF-2C).
To validate rigorously the direct physical association of GRIP-1 and MEF-2C in an additional assay, GRIP-1 and MEF-2C were translated in a rabbit reticulocyte lysate, which produces native proteins. We observed that immunoprecipitation of a rabbit reticulocyte lysate programmed with GRIP-1 in the presence and absence of MEF-2C with an antibody directed against MEF-2C resulted in the coimmunoprecipitation of GRIP-1 (Fig. 10A). This result clearly demonstrated further that these two proteins directly interact.
Figure 10.
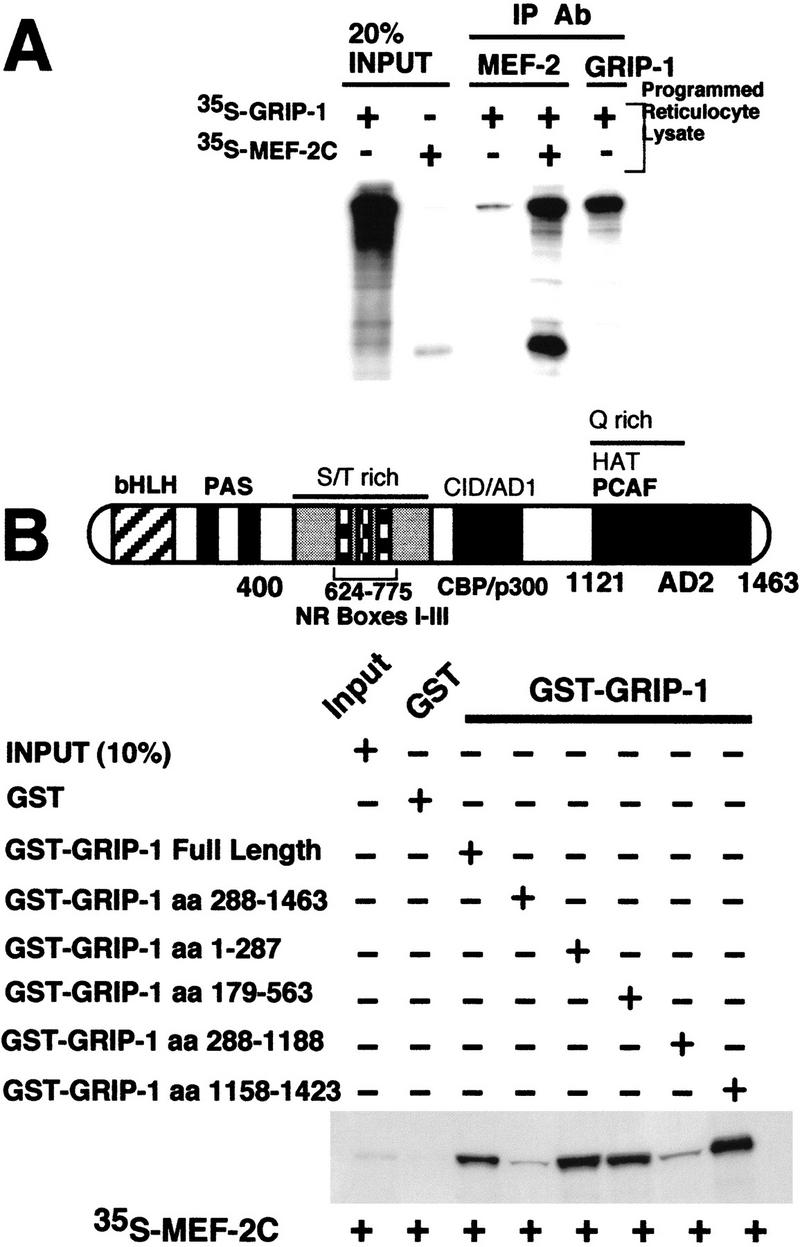
(A) Coimmunoprecipitation of GRIP-1 with MEF-2C. GRIP-1 and MEF-2C expression plasmids were transcribed/translated in rabbit reticulocyte lysate and mixed as indicated above the lanes. Translation reactions were immunoprecipitated with MEF-2 and GRIP-1 antibodies and the 35S-labeled proteins were analyzed by SDS-PAGE. GRIP-1 and MEF-2C were coimmunoprecipitated with MEF-2 antibodies. (B) GRIP-1 directly interacts with MEF-2. Diagrammatic representation showing the GRIP-1 regulatory domains; the GRIP-1 amino-terminal bHLH–PAS domain and carboxy-terminal activation domain (AD2) with HAT activity, which interacts with PCAF, are necessary for the interaction with MEF-2C. Glutathione–agarose-immobilized GST and GST–GRIP-1 proteins were incubated with 35S-radiolabeled full-length MEF-2C. The GRIP-1 amino-terminal region between amino acid positions 1 and 287, and the carboxy-terminal region between amino acid positons 1158 and 1423, support the MEF-2C interaction.
The above experiments demonstrate the direct interaction between MEF-2C and GRIP-1 and are in accordance with GRIP-1 coactivation of MEF-2C-mediated transcription and the interaction of GRIP-1 and MEF-2C in the mammalian two-hybrid assay. Furthermore, the in vitro and cellular protein–protein-interaction studies verified that the MADS domain of MEF-2C is necessary to mediate the interaction between GRIP-1 and MEF-2C.
The amino- and carboxy-terminal regions of GRIP-1 interact directly with MEF-2C
To delimit the region within GRIP-1 (see Fig. 10B) that interacts with MEF-2C, we examined the ability of a number of GST–GRIP-1 fusion chimeras containing various functional subdomains of GRIP-1 immobilized on glutathione–agarose beads [i.e., native GST–GRIP-1 (amino acids 1–1463), GST–GRIP-1 (amino acids 288–1463), GST–GRIP-1 (amino acids 1–287), GST–GRIP-1 (amino acids 179–563), GST–GRIP-1 (amino acids 287–1188), and GST–GRIP-1 (amino acids 1158–1423)] to interact with the 35S-radiolabeled MEF-2C (see Fig. 10B). As expected, native MEF-2C interacted with full-length GRIP-1. The amino-terminal deletion of GRIP-1 (288–1463), which lacked the bHLH–PAS domain, linked to GST failed to interact with in vitro-translated MEF-2C (Fig. 10B). However, the amino-terminal region of GRIP-1 (amino acids 1–287) linked to GST supported a significant interaction with MEF-2C. Both of these results suggest that the bHLH–PAS domain of GRIP-1 is required for MEF-2C binding. Interestingly, the segment of GRIP-1 between amino acid positions 179 and 563 also supported a significant interaction with MEF-2C. This data, in the above context, further delimits the region of GRIP-1 that supports MEF-2C interaction to amino acids 179–288, which encodes the PAS domain. As expected, and in accordance with the data above, the other amino- and carboxy-terminal deletion of GRIP-1 (288–1188) (which lacks the bHLH–PAS domain) linked to GST failed to interact with MEF-2C. Surprisingly, the carboxy-terminal region of GRIP-1 (amino acids 1158–1423) also supported a significant interaction with MEF-2C.
In summary, these data suggest that the amino- and carboxy-terminal regions of GRIP-1, which encode the bHLH–PAS domain and an activation domain (AD2), respectively, directly interact with MEF-2C. Furthermore, these data ascribe a function to the previously undefined bHLH–PAS domain of SRCs.
Myogenin/E2 interacts directly with GRIP-1: the HLH region mediates the physical interaction with GRIP-1
The regulation of myogenin/E2A-mediated transactivation by GRIP-1 and the demonstration of interaction between myogenin/E2 and GRIP-1 in the cellular context strongly suggest that these proteins physically associate (in the presence and absence of MEF-2C). However, the remote possibility of an indirect mechanism in which additional factor(s) mediate the interaction still existed. As demonstrated with MEF-2C, we tested this hypothesis using a biochemical approach in vitro GST-pulldown assays, to confirm the direct interaction between myogenin and GRIP-1 and to identify the domain in myogenin that mediates the interaction with the coactivator.
Glutathione–agarose-immobilized GST–GRIP-1 was tested for direct interaction with in vitro 35S-radiolabeled native myogenin, myogenin amino acids 1–169, and myogenin amino acids 1–99. Native myogenin showed a very strong interaction with GST–GRIP-1 relative to GST alone (Fig. 11A). Similarly, myogenin amino acids 1–169 (which lacked the carboxy-terminal transactivation domain TAD, but retained the bHLH region) supported the interaction with GRIP-1. This result suggests that the myogenin amino-terminal TAD or bHLH domain mediates GRIP-1 binding. We observed that myogenin amino acids 1–99, which included the N-TAD and the basic region (but lacked the HLH domain), failed to interact with GRIP-1 (Fig. 11A). This result demonstrates that the region between amino acids 100 and 169 and mediates the physical association with GRIP-1. Furthermore, the data strongly suggest that the HLH domain of myogenin mediates the association with GRIP-1.
Figure 11.



GRIP-1 directly interacts with myogenin. (A) Diagrammatic representation showing the regulatory domains in myogenin; the HLH domain of myogenin mediates the interaction with GRIP-1. Glutathione–agarose-immobilized GST and GST–GRIP-1 proteins were incubated with either 35S-radiolabeled full-length myogenin, or fragments of myogenin between amino acid positions 1 and 169 and 1 and 99, respectively. Deletion of the myogenin HLH region between amino acid positions 100 and 169 ablates binding to GRIP-1 in vitro. The input lanes represent ∼10% of the total protein. (*) Residual full-length protein produced after incomplete restriction digestion to produce the amino acid 1–99 truncation. (B) Coimmunoprecipitation of myogenin, e12–HA, and GRIP-1 with HA antibodies. GRIP-1 was translated in a rabbit reticulocyte lysate in the presence and absence of myogenin and E2A-e12 as depicted. Translation reactions were immunoprecipitated with HA and GRIP-1 antibodies and the 35S-labeled proteins were analyzed by SDS-PAGE. GRIP-1 and myogenin–E2A were coimmunoprecipitated with HA antibodies. (C) Diagrammatic representation showing the GRIP-1 regulatory domains, the GRIP-1 amino-terminal bHLH–PAS domain and the carboxy-terminal activation domain (AD2) with HAT activity, which interacts with PCAF, are necessary for the interaction with myogenin. Glutathione–agarose immobilized GST and GST–myogenin proteins were incubated with 35S-radiolabeled full-length GRIP-1, or fragments of GRIP-1 between amino acid positions 1 and 1124, 1 and 765, 1 and 577, and 1125 and 1423, respectively. The GRIP-1 amino-terminal region between amino acid positions 1 and 577, and the carboxy-terminal region between amino acid positions 1125 and 1463, support the interaction with myogenin.
We verified the direct physical association of GRIP-1 with the myogenin/E2A complex in an additional assay. Myogenin and E2A were translated in a rabbit reticulocyte with either MEF-2C or GRIP-1. We observed that immunoprecipitation with an HA antibody of rabbit reticulocyte lysate transcribed/translated with MEF-2C, myogenin, and E2A-e12-HA resulted in the coimmunoprecipitation of MEF-2C, myogenin, and E2A-e12 as described previously by Olson and colleagues (Molkentin et al. 1995; Black et al. 1998; Fig. 11B). Similarly, we observed that immunoprecipitation of a rabbit reticulocyte lysate programmed with GRIP-1, myogenin, and E2A-e12-HA resulted in the coimmunoprecipitation of GRIP-1, myogenin, and E2A-e12-HA (Fig. 11B). These data further reinforced the coactivation of GAL–myogenin by GRIP-1 in the presence of E2A proteins and stimulation of the interaction between myogenin and E2A-e12 in the mammalian two-hybrid assay by GRIP-1.
The amino- and carboxy-terminal regions of GRIP-1 interact independently with myogenin
To delimit the region within GRIP-1 (see Fig. 11C) that interacts with myogenin, we examined the ability of a variety of native, amino- and carboxy-terminal-deleted 35S-labeled GRIP-1 proteins to interact with GST–myogenin immobilized on glutathione–agarose beads. As expected, native GRIP-1 interacted with myogenin. All of the carboxy-terminal deletions of GRIP-1 (i.e., amino acids 1–1124, 1–765, and 1–577) supported a significant interaction with myogenin. However, the carboxy-terminal region of GRIP-1 between amino acid positions 1125 and 1463 also interacted strongly with myogenin. These results suggest that the amino-terminal (amino acids 1–577) and carboxy-terminal (amino acids 1125–1423) regions of GRIP-1, which encode the bHLH–PAS domain and an activation domain (AD2) with HAT activity that interacts with PCAF, respectively, mediate the interaction with myogenin (and MEF-2C).
Detection and direct physical association of a complex comprised of myogenin, E2A-e12, MEF-2C, and GRIP-1
We had observed that GRIP-1-mediated coactivation of E box-dependent gene expression was dependent on the cooperative function between myogenin, E2A, MEF-2C, and GRIP-1 in transfection experiments. We investigated whether these proteins are physically associated in a large complex. We verified the direct physical association of a complex comprised of E2A-e12, myogenin, MEF-2C, and GRIP-1 in an immunoprecipitation assay. MEF-2C, myogenin, and E2A-e12 were translated in a rabbit reticulocyte lysate. We observed that immunoprecipitation with an antibody directed against MEF-2C of rabbit reticulocyte lysate-transcribed/translated myogenin, E2A-e12, and MEF-2C, resulted in the coimmunoprecipitation of the myogenin–E2A heterodimeric complex (Fig. 12), as demonstrated previously by Olson and colleagues (Molkentin et al. 1995; Black et al. 1998). Subsequently, we observed that immunoprecipitation with an antibody directed against MEF-2C of rabbit reticulocyte lysate-transcribed/translated myogenin, E2A-e12, MEF-2C, and GRIP-1, resulted in the coimmunoprecipitation of MEF-2C, myogenin, E2A-e12, and GRIP-1 (Fig. 12A). The appropriate controls for these experiments are found in Figure 12 (B,C). The nonspecific cross-reactivity of the MEF-2 antibody with GRIP-1 and E2A under the immunoprecipitation conditions is shown in Figure 12B. There is minor nonspecific interaction with GRIP-1 and E2A (Fig. 12B). We addressed this issue by directly comparing immunoprecipitation (with the MEF-2 Ab) of rabbit reticulocyte lysate-transcribed/translated myogenin, E2A-e12, and GRIP-1 in the presence and absence of MEF-2C (Fig. 12C). This experiment showed that the the coimmunoprecipitation of MEF-2C, myogenin, E2A-e12, and GRIP-1 in Figure 12A was not due to nonspecific cross-reactivity.
Figure 12.

Coimmunoprecipitation of GRIP-1, MEF-2C, and myogenin and E12 with MEF-2C. MEF-2C, myogenin, and E12 were translated in a rabbit reticulocyte lysate in the presence and absence of GRIP-1 as depicted. (A) Translation reactions were immunoprecipitated with a MEF-2 antibody, and the 35S-labeled proteins were analyzed by SDS-PAGE. MEF-2C antibodies coimmunoprecipitated myogenin, E2A-e12, and GRIP-1 in the presence of MEF-2C. (B,C) Translation reactions were immunoprecipitated with MEF-2, GRIP-1, and HA antibodies to assess nonspecific cross-reactivity against other proteins. (D) Translation reactions were immunoprecipitated with an HA antibody and the 35S-labeled proteins were analyzed by SDS-PAGE. The HA antibody coimmunoprecipitated myogenin, MEF-2C, and GRIP-1 in the presence of E2A-e12.3Corresponding author.
Further validation was gained from translation of myogenin, E2A-e12, and MEF-2C in rabbit reticulocyte lysate in the presence and absence of GRIP-1 (Fig. 12D). We observed that immunoprecipitation of rabbit reticulocyte lysate programmed with MEF-2C, myogenin, and E2A-e12-HA, with an HA antibody directed against E2A-e12-HA, resulted in the coimmunoprecipitation of MEF-2C, myogenin, and E2A-e12-HA as described previously by Olson and colleagues (Molkentin et al. 1995; Fig. 12D). Moreover, as above (with the MEF-2C antibody), we observed that immunoprecipitation with an HA antibody of rabbit reticulocyte lysate-transcribed/translated GRIP-1, MEF-2C, and myogenin in the presence of E2A-e12-HA, resulted in the coimmunoprecipitation of GRIP-1, MEF-2C, myogenin, and E2A-e12-HA (Fig. 12D). These data support a cooperative interaction between myogenin/E2, MEF-2C, and GRIP-1 in the regulation of muscle-specific gene expression.
Discussion
The control of skeletal muscle transcription and differentiation is regulated by the muscle specific bHLH factors and the MADS-box protein, MEF-2C. The cofactors, p300 and PCAF, have been demonstrated to function as critical coactivators of MyoD during transcription, the activation of the myogenic program, and the induction of cell cycle arrest (Eckner et al. 1996; Puri et al. 1997a,b; Sartorelli et al. 1997). Here, we demonstrate that muscle cells express the SRC GRIP-1, which functions as a cofactor for the nuclear receptors ER, GR, TR, and PPAR and several other factors (Moras and Gronemyer 1998; Xu et al. 1999). Furthermore, our study strongly suggested that terminal skeletal muscle differentiation, contractile protein gene expression, and withdrawal from the cell cycle are contingent on GRIP-1-mediated coactivation of myogenin and MEF-2-dependent gene expression. Direct physical association with GRIP-1, which is dependent on the HLH and MADS domains of myogenin and MEF-2C, respectively, mediates the effects on myogenic factor-mediated transcription. Moreover, our study suggests that cooperative MEF-2/bHLH interactions with respect to transactivation involve coactivation by GRIP-1 and direct physical association. Nuclear receptor-mediated transactivation involves coactivation by SRCs and subsequent recruitment of p300 and PCAF (Glass and Rosenfeld 2000). Hence, the function of the SRC GRIP-1 in myogenesis is also consistent with the involvement of p300 and PCAF in muscle differentiation.
Recent evidence suggests that myogenic bHLH factors activate transcription and regulate myogenesis by a functional and cooperative interaction with the MEF-2 family of MADS-domain transcription factors. Olson and colleagues (Molkentin et al. 1995; Black et al. 1998) hypothesized that MEF-2 proteins function as cofactors required for bHLH factor-mediated transactivation. bHLH and MEF-2 family members associate by direct physical association mediated by their DNA-binding and dimerization motifs (Molkentin 1996). The synergistic activation of transcription requires direct DNA binding by either/both factor(s) and subsequent protein–protein interaction between the factors. Moreover, MEF-2 proteins synergistically activate muscle-specific transcription with the myogenic/neurogenic-specific bHLH-E protein heterodimeric complexes. This transactivation event does not occur with E-protein homodimers. Our study demonstrates that GRIP-1 increases myogenin and MEF-2C activity in transcriptional assays and potentiates MEF-2C-mediated transactivation of a native muscle promoter and a MEF-2-dependent reporter. Furthermore, consistent with the bHLH/MEF-2 cooperativity hypothesis, our study demonstrates that GRIP-1 functions as a coactivator for the synergistic activation of E box-dependent transcription by MEF-2c and the bHLH factor, myogenin. This coactivation of bHLH/MEF-2C-mediated gene expression involves independent direct interactions between GRIP-1 and each of the myogenic factors. Moreover, immunoprecipitation with either MEF-2C or E-protein antibodies coprecipitated a complex including myogenin, MEF-2C, E2A-e12, and GRIP-1/SRC-2. In summary, our investigation suggests that GRIP-1 functions as a cofactor for the bHLH/MEF-2 complex, and as a coactivator for the independent functions of myogenin and MEF-2C during the commitment process.
GRIP-1 is necessary for myogenic differentiation
Specifically, our investigation demonstrates that the expression of the mRNA encoding GRIP-1 is expressed in proliferating myoblasts and post-mitotic myotubes; however, protein levels increase during myogenesis. GRIP-1 gain- and loss-of-function analysis by exogenous/ectopic expression of GRIP-1 sense and antisense vectors suggested that this SRC is critical to the activation/induction of mRNAs encoding myogenin, the cyclin-dependent kinase inhibitor, p21, and the MEF-2 proteins. Moreover, GRIP-1 function is required for the expression of the myofibrillar proteins in post-mitotic multinucleated cells. These studies suggest that GRIP-1 may function as a critical coactivator for the latter stages of skeletal muscle differentiation, which are dependent on MEF-2C and myogenin-mediated events/cascades.
Consistent with these observations, our study showed that GRIP-1 potentiated myogenin and mef-2-mediated activation of the native MCK promoter, MEF-2C, and E box-dependent reporters. These observations suggest that GRIP-1 functions as a coactivator of myogenin and MEF-2C-mediated events during the latter/commitment stages of skeletal muscle cell differentiation and in congruence with the cell culture analysis.
Puri, Sartorelli and colleagues (Puri et al. 1997b) have demonstrated that exogenous expression of the coactivators p300 and PCAF potentiates myoD and MEF-2-mediated transcription, stimulates differentiation, and promotes myoD-mediated p21 activation and cell cycle arrest. Our observations are consistent with the above studies and firmly imply that GRIP-1, p300, and PCAF have a central role in myogenic commitment and differentiation.
Recently, MEF-2A and MEF-2C have been shown to interact with HDAc-4 (Miska et al. 1999; Wang et al. 1999). Furthermore, Xenopus MEF2A and MEF2D have been demonstrated to interact with a corepressor, MITR (i.e., related to HDAc-4, HDAc-5, and HDAc-6), which subsequently recruits HDAc-1 during repression of MEF-2-dependent gene expression (Sparrow et al. 1999).
The opposing effects of p300/GRIP-1 and MITR/HDAc-1 and MITR/HDAc-4 on MEF2 are analogous to the differential effects of coactivators (p300/PCAF) and corepressors (Nuclear Receptor Corepressor, N-CoR) on MyoD-mediated events. Furthermore, they are a corollary to the opposing actions of the SRC/p300/PCAF coactivator complex and the N-CoR/Sin3/HDAc corepressor complex on nuclear receptor-mediated transcriptional regulation (Xu et al. 1999, and references therein) and the similar affects mediated by the p300/CBP coactivator complex and the RB/histone deacetylase-corepressor complex on E2F1-mediated transcription.
GRIP-1 coactivates the activity of myogenin and MEF-2C directly
We demonstrated that myogenin and MEF-2C-mediated transactivation of the native MCK promoter, the mef-2, and E box-dependent reporters were potentiated by GRIP-1 coexpression. Moreover, the activity of GAL–MEF-2C and GAL–myogenin were activated by GRIP-1 expression. Interestingly, these results suggest that GRIP-1 can function as a coactivator in the presence or absence of the cognate-binding site for myogenin and MEF-2C, which correlates with the ability of these factors to function by direct binding or tethering to the DNA and in a synergistic/cooperative manner. Our experiments suggest that one of the mechanisms involved in this process of activation was the direct interaction between either the bHLH region of myogenin or the MADS domain of MEF-2C with GRIP-1.
Interestingly, the interaction with the myogenic factors (i.e., myogenin and MEF-2) is mediated by two regions of GRIP-1, an amino-terminal bHLH–PAS domain region and a carboxy-terminal region between amino acids 1158 and 1423. The amino-terminal region contains a highly conserved domain; however, the function of this domain in the SRC family has not been well characterized to date (Voegel et al. 1996; Hong et al. 1997; Li et al. 1997; Torchia et al. 1997; Suen et al. 1998).
The carboxy-terminal region encodes an activation domain and has HAT activity. This study ascribes a function to the bHLH–PAS domain that is strongly conserved between the members of the p160 coactivator family. This domain mediates interaction with the myogenic bHLH and MADS group of proteins, which are key regulators of cell/lineage differentiation. Many other classes of transcription factors that contain bHLH–PAS domains have critical roles in homeostasis and function as important links between environment, physiology, and development (Crews and Fan 1999). Structural analysis of the bHLH–PAS domain has implicated this region to be an environmental sensor and signal transducer (Crews and Fan 1999, and references therein). Our study demonstrates that this region mediates, and is necessary for, the physical association of GRIP-1 with either MEF-2 or myogenin, and we hypothesize that the bHLH–PAS domain in the SRC superfamily of cofactors mediates the tissue-specific functions of these ubiquitously expressed coactivators.
Activation domain 1 (AD1), located between amino acids 1040 and 1120 in GRIP-1, recruits p300/CBP, activates transcription and is responsible for downstream signaling. However, recent studies suggest a second activation domain in GRIP-1, AD-2, located downstream of amino acid 1122 in the carboxyl terminus, encodes HAT activity. Whether this activity is involved in SRC function is unclear. Stallcup and colleagues (Chen et al. 1999) have identified and characterized a novel protein, CARM-1 (coactivator-associated methyltransferase 1), which binds to the carboxyl terminus of the SRC family, stimulates activity, and is involved in AD-2 function. Because our study has demonstrated that the carboxyl terminus/AD2 of GRIP-1 also mediates the interaction with MEF-2C and myogenin, we speculate that coactivator-associated methyltransferases will play key roles in the transmission of myogenic–specific signals during differentiation.
HLH and MADS domains are mediators of key protein–protein interactions and critical targets during myogenesis
This study reinforced further the critical role of the HLH motif and the MADS domain in the regulation of myogenesis, transactivation, and physical association with regulatory cofactors. Previous studies demonstrated that the MADS domain of MEF-2 mediated interaction with the coactivator p300, consistent with the MADS domain-mediated interaction with GRIP-1. The corepressor MITR and the histone deacetylase HDAc-4 similarly interact with the MADS domain of MEF-2C. This domain is similar to the conserved carboxy-terminal ligand-binding domain of the nuclear receptors that mediates the interaction with several coactivators (e.g., SRCs and p300) and corepressors (e.g., N-CoR, SMRT, and RIPs). In contrast, the amino-terminal activation domain, and not the bHLH region of MyoD, was involved in the recruitment of p300; however, the bHLH region of MyoD mediated the interaction with the corepressor N-CoR. Whether these observations reflect isoform-specific differences in MyoD versus myogenin or p300 versus SRC mediated interactions will require further investigation and clarification.
The targeting of the HLH region in myogenin is quite interesting in light of several observations. Firstly, the pocket proteins, pRb, p107, and p130, which interact with many classes of transcription factors and viral oncoproteins, participate in the induction and maintenance of the post-mitotic state (Schneider et al. 1994; Kiess et al. 1995; Zackenhaus et al. 1996). A single controversial paper has reported that these pocket proteins in their hypophosphorylated forms, specifically and directly interact with the bHLH region of the products of the myoD gene family and mediate cell cycle withdrawal and the activation of the myogenic program (Gu et al.1993). Moreover, MyoD-mediated myogenic conversion, growth arrest, and activation of MEF-2C is dependent on the function of pRB.
We suggest that during cell cycle exit and the activation of the myogenic program, multiprotein complexes that includes GRIP-1, myogenin, E-proteins, and MEF-2C function to complete the later stages of myogenic commitment in a background of hypophosphorylated pRB. pRb functions in a concomitant manner to promote cell cycle arrest and activate the function of MEF-2C (Novitch et al. 1999).
Coactivators and corepressors, key regulators of skeletal muscle differentiation
This work contributes to an emerging and complex story about the crucial role of cofactors in myogenesis. The evidence to date supports the notion that corepressors and coactivators function in both the proliferative phases and post-mitotic states of myogenesis. It has been observed that the coactivator complex p300 mediates E2F-dependent S-phase gene expression, DNA synthesis, and cell cycle progression (Trouche et al. 1996). This process is dependent on and mediated by the phosphorylation of RB or the other pocket proteins by the cyclin/cdk pathway. During withdrawal from the cell cycle and myogenic differentiation, a number of key events take place, (1) pRb becomes hypophosphorylated and recruits histone deacetylase (in a pocket domain-dependent fashion) to directly repress E2F-dependent gene expression (Brehm et al. 1998; Ferreira et al. 1998; Magnaghi-Jaulin et al. 1998), (2) hypophosphorylated pRb and MyoD function in a cooperative manner consistent with the effects of MyoD on the myogenic pathway and cell cycle withdrawal, and (3) concomitant with the above events, p300 and PCAF directly interact with MyoD and potentiate myogenic differentiation and permanent withdrawal from the cell cycle (Puri et al. 1997a,b). Our previous work demonstrated that the N-CoR corepressor complex can prevent activation of p21 and myogenesis. Moreover, it can directly mediate the repression of MyoD function (Bailey et al. 1999) and this N-CoR-mediated repression is sensitive to inhibitors of histone deacetylation. Our current observations on GRIP-1 are consistent with a role for the cofactors p300 and PCAF in myogenesis. We speculate that GRIP-1 functions with p300 and PCAF to potentiate the transactivation activity and myogenic effects of the muscle-specific transcription factors during commitment. Finally, in a broader context, our studies paint a picture that suggests that the HLH and MADS domains are both key targets of corepressors and coactivators during the control of differentiation.
Materials and methods
Construction of stable cell lines
C2C12 cells were stably transfected at ∼40% confluence using a liposome-mediated (Boehringer-Mannheim) procedure as described previously (Bailey et al. 1999). Briefly, a DNA/DOTAP/DOSPER mixture containing 5–10 μg of pcDNA3.1–NEO driving the expression of sense or antisense GRIP-1 in 20 mm HEPES, 150 mm NaCl (pH 7.4) was added to the cells in a 100-mm dish. Cells were incubated for 24 hr in 15 ml of fresh DMEM, 20% FCSat 6% CO2. The cells were then grown for a further 24 hr to allow cell recovery and for high-level NEO expression before selection. Stable transfectants/monoclonal cell lines were isolated after 14 days of selection in DMEM, 20% FCS, 800 μg/ml G418. Monoclonal cell lines expressing the transfected constructs were further confirmed and selected by Northern blot analysis.
RNA extraction, Northern hybridization, and probe preparation
Total RNA was extracted by the acid guanidinium thiocyanate-phenol-chloroform method. Northern blots, random priming, and hybridizations were performed as described previously (Burke et al. 1995; Downes et al. 1995). The cDNA probes used have been described previously (Burke et al.1996; Downes et al. 1995; Bailey et al. 1999).
Mouse myogenin cDNAs were excised from the pEMSVscribe (Moloney Sarcoma Virus) expression vectors. Mouse cyclinD1 was excised from pGEX-3X-CYL1 (60) and mouse p21 was excised from pCMW35, an unpublished clone encoding mouse p21 from the Vogelstein laboratory. The cDNA probe used for SRC-1 encompassed the region downstream of amino acid 1138 and included the 3′ UTR, the GRIP-1/SRC-2 probe spanned amino acids 544–1157, and the SRC-3 probe corresponded to amino acids 507–937.
cDNA probes were radioactively labeled by random priming. DNA fragments (50–100 ng) were boiled with 20 ng of random primers (pdN6; Pharmacia). The DNA was then incubated at room temperature overnight with RPB (50 mm Tris at pH 7.5, 10 mm MgCl2, 200 μm dATP, dGTP, dTTP), 10 μl of [α-32P]dCTP (Bresatec), and 5–10 units of Klenow polymerase (New England Biolabs). Probes were purified using a NICK column (Pharmacia) according to the manufacturer's instructions.
Cell culture and transient transfections
10T1/2, or JEG-3 (human choriocarcinoma) cells were cultured for 24 hr in DMEM supplemented with 10% FCS in 6% CO2 before transfection. Mouse myogenic C2C12 cells were grown in DMEM supplemented with 20% FCS in 6% CO2. Cells grown in 12-well dishes to 60%–70% confluence were transiently transfected with 0.3–1 μg of the MCK–LUC, 4RE–tkLUC, or MEF-2C-e1b–LUC reporter plasmid and 0.5–1.5 μg of a mixture of the bHLH factor or MEF-2C in the presence or absence of the coactivator with 10 μl of a DOTAP/DOSPER liposome mixture (Boehringer-Mannheim) as described previously (total DNA 2–2.5 μg/well).
The DNA/DOTAP/DOSPER mixture was added to the cells in 1 ml of fresh medium, and incubated for 24 hr. Post transfection, the medium was replaced and the cells grown a further 24–48 hr. Cells were harvested and assayed for luciferase activity as described previously. Each experiment represented at least two sets of independent triplicates to overcome the variability inherent in transfections.
GAL4 hybrid assay in C3H10T1/2 pluripotent cells
The GAL4 DBD alone, GAL–MEF-2C, and GAL–myogenin were cotransfetced with the pG5E1b–LUC reporter in the presence and absence of GRIP-1 in C3H10T1/2 by the DOTAP/DOSPER (Boehringer Mannheim)-mediated procedure, using a total DNA concentration of 2–2.5 μg per transfection/well. Cells were transfected in DMEM supplemented with 10% FCS 24 hr after transfection, during which time the cells reached confluency, the medium was changed to DMEM supplemented with 2% horse serum, and cells were harvested and assayed for luciferase activity as described previously after a further 24–48 hr of culture. Each experiment represented at least two sets of independent triplicates to overcome the variability inherent in transfection experiments.
Immunostaining of myocytes
Cells were fixed and stained as described previously. Then, immunohistochemical analysis was performed with a monoclonal antibody directed toward the fast isoform of the major thick filament protein, skeletal MHC (Sigma; clone MY32). This procedure is described in detail elsewhere (Sartorelli et al. 1997; Bailey et al. 1999).
Mammalian two-hybrid assays
Each well of a 12-well plate of JEG-3 cells (60%–70% confluence) was cotransfected with 1 μg of pG5E1b–LUC reporter, 0.3 μg of GAL chimeras, and 0.3 μg of VP16 chimeras in 1 ml of DMEM containing 5% charcoal-stripped FCS by the DOTAP-mediated procedure (Boehringer Mannheim). After 24 hr, the medium was replaced, and cells were harvested for the assay of luciferase activity 36–48 hr after transfection. Each experiment represented at least two sets of independent triplicates to overcome the variability inherent in transfections.
Luciferase assays
Luciferase activity was assayed using a Luclite kit (Packard) according to the manufacturer's instructions. Briefly, cells were washed once in PBS and resuspended in 150 μl of phenol red-free DMEM and 150 μl of Luclite substrate buffer. Cell lysates were transferred to a 96-well plate and relative luciferase units were measured for 5 sec in a Wallac Trilux 1450 microbeta luminometer.
GST pulldowns
GST and GST-fusion proteins were expressed in Escherichia coli (BL21) and purified using glutathione–agarose affinity chromatography as described previously (Downes et al. 1995; Muscat et al. 1995). The GST-fusion proteins were analyzed on 10% SDS-polyacrylamide gels for integrity and to normalize the amount of each protein. The TNT-coupled transcription–translation system (Promega) was used to produce [35S]methionine-labeled proteins that were used in the pulldown assays. In vitro binding assays were performed with glutathione–agarose beads (Sigma) coated with ∼500 ng of GST-fusion protein and 2–30 μl [35S]methionine-labeled protein in 200 ml of HEMG buffer containing 100 mm KCl, 40 mm HEPES (pH 7.4), 0.2 mm EDTA, 0.1% Nonidet P-40, 1.5 mm DTT, 10% glycerol, freshly supplemented 5% glycine, and 1% BSA. The reaction was allowed to proceed for 1–2 hr at 4°C with rocking. Then, affinity beads were collected by centrifugation and washed three times with 1 ml of binding buffer with BSA and glycine and two times with HEMG buffer without BSA and glycine. Beads were resuspended in 30 μl of SDS-PAGE sample buffer and boiled for 5 min. The denatured proteins were run on a 7%, 10%, or 12% SDS–polyacrylamide gel, according to the molecular weight of the probes, that were subsequently treated with Amersham Amplify fluor (Amersham), dried, and autoradiographed.
Immunoprecipitation
35S-labeled in vitro-translated proteins or nuclear proteins were incubated overnight at 4°C with 1 μg of antibody in 300 μl of TIP buffer (25mm HEPES at pH 7.2, 300 mm KCl, 0.5 mm EDTA, 5 mm DTT, 0.1% NP-40, 5 mm MgCl2, and 10% glycerol) supplemented with freshly made 5% glycine, 1% BSA, and protease inhibitor. Then, protein A/G–agarose beads were added and incubated for another 90 min. After washing four times in TIP buffer, bound proteins were eluted with 2× SDS sample buffer.
Plasmids
MCK–LUC, 4REtk–LUC, and pG5E1b–LUC are described elsewhere (Bailey et al. 1999; Novitch et al. 1999). pCDNA3.1–MyoD, pCDNA3.1–myogenin, pCDNA3.1–MYF-5, and pCDNA3.1–MRF-4 were constructed by excising cDNAs from pEMSVscribe (Moloney Sarcoma Virus) expression vectors and recloning into the EcoRI site of the pCDNA3.1vector.
GRIP-1 cDNA was released from SG5–GRIP-1 by EcoRI digestion and cloned into the pCDNA3.1 and PGEX1N vectors in both EcoRI sites. GRIP-1 constructs pmGRIP-1 and pVP16–GRIP-1 used in the mammalian two-hybrid assay were generous gifts from Dr. Michael Stallcup. The CBP expression plasmid was a generous gift from Dr. Marc Montminy, and the p300 expression plasmid was a generous gift from Dr. David Livingston.
The plasmids pGALO, pNLVP16, pCMX-VP16-N, pCMX-GAL4-N, and EMSV–myogenin have been described elsewhere (Edmondson et al. 1989; Umesono et al. 1991; Casanova et al. 1994; Willy et al. 1995; Sartorelli et al. 1997; Bailey et al. 1999). All PCR was done with Pfu DNA polymerase (Stratagene), using the manufacturer's buffer. The pGALO–myogenin was constructed by PCR with two primers, 5′-GCGGAATTCACCATGGAGCTGTATGAGACATC-3′ and 5′-GCGGAATTCCTACAGAAGTGATGGCTTTTG-3′ that were synthesized to allow PCR amplification of the entire 246-amino-acid coding sequence of mouse myogenin from the parent plasmid, EMSV–myogenin. Then, the myogenin PCR product was digested with EcoRI and cloned into the EcoRI site of pGALO. To make pNLVP16–myogenin, the myogenin sequence was cut out of pGALO–myogenin with EcoRI, blunt ended, and cloned into the NdeI site of pNLVP16. The pGALO–MEF2C and pNLVP16–MEF2C plasmids were constructed by PCR with two primers, 5′-CCGAGCTCACCATGGGGAGAAAAAAGATTCAG-3′ and 5′-CCGAGCTCTCATGTTGCCCATCCTTCAG-3′, that were synthesized to allow PCR amplification of the entire 466-amino-acid coding sequence of mouse MEF2C from the parent plasmid, pGEX2T–MEF2C. Then, the MEF2C PCR product was digested with SacI and cloned into the SacI site of pGALO and pNLVP16. MEF2C DNA was isolated from pGALO–MEF2C by SacI digestion and cloned into pGL2-Basic (Promega) via the SacI site. After restriction digestion of the pGL2-Basic-MEF2C plasmids to determine correct orientation, the MEF2C sequence was isolated from a correct clone by Asp718/NheI digestion and cloned into pCMX-GAL4-N and pCMX-VP16-N via the Asp718/NheI sites. The pCMX-VP16-N plasmid was digested with HindIII, isolated to remove the VP16 DNA sequence, and religated. Then, the resulting plasmid (called pCMX-D) was cut with HindIII and Asp718 and isolated. The oligonucleotides 5′-AGCTTGCCACCATGGACTACAAGGACGACGATGACAAGG-3′ and 5′-GTACCCTTGTCATCGTCGTCCTTGTAGTCCATGGTGGCA-3′ were annealed, phosphorylated with T4 polynucleotide kinase, and ligated into the HindIII- and Asp718-cut pCMX-D vector to create the plasmid pCMX-KF, which contains a FLAG epitope preceded by a Kozak sequence and ATG start codon. The Asp718/NheI MEF2C DNA fragment isolated from pGL2-Basic-MEF2C was then cloned into the Asp718/NheI sites of pCMX-KF to create pCMX-KF-MEF2C. The oligonucleotides M1, 5′-GCGGGTACCGCCAGCACTGACATGGATAAGG-3′, and M3, 5′-GTCTATGTGTTACACCAGGAG-3′, were used to PCR amplify the MEF2C DNA ORF from base 178 to 568. The PCR product was digested with Asp718/PstI and ligated into Asp718/PstI-digested and isolated pCMX-KF-MEF2C, to create pCMX-KF-MEF2C-ΔM1, which has amino acids 1–57 of the original MEF2C protein deleted.
Isolation of the DNA fragment containing the E1b TATA box from pG5E1b–Luciferase was done using SalI and SphI restriction enzymes. The SalI–SphI E1b TATA-box DNA fragment was then cloned into pGL3-Basic (Promega) via XhoI/SphI restriction sites to create pGL3-E1b–Luciferase. Oligonucleotides 5′-ctagCTCGCTCTAAAAATAACCCTGTCG-3′ and 5′-ctagCGACAGGGTTATTTTTAGAGCGAG-3′ were annealed, phosphorylated with T4 polynucleotide kinase, and self-ligated with T4 DNA ligase. These products were then cloned into the NheI site in the pGL3-E1b–Luciferase plasmid and initially screened by Asp718/HindIII digestion. Clones containing putative inserts were sequenced by double-stranded sequencing to determine the orientation and number of oligonucleotide copies cloned. pGL3-MEF2C-1x-E1b–Luciferase and pGL3-MEF2C-3x-E1b–Luciferase contained one and three copies, respectively, of the MEF2C response element in a 3′ to 5′ orientation with respect to the luciferase transcriptional start site.
Acknowledgments
We thank Drs. Michael Stallcup, J. Don Chen, and Andrew Lassar for the genrous gift of plasmids. Special thanks to Dr. David Moore for supplying plasmids prior to publication and helpful discussions. This investigation was supported by the National Health and Medical Research Council (NHMRC) of Australia. The Centre for Molecular and Cellular Biology is part of the Special Research Centre for Functional and Applied Genomics that is supported by Australia Research Council (ARC). G.E.O.M. is an NHMRC senior research fellow.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL G.Muscat@cmcb.uq.edu.au; FAX 61 7 3365 4388.
References
- Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan XY, Sauter G, Kailioniemi OP, Trent JM, Meltzer PS. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Nature. 1997;277:965–968. doi: 10.1126/science.277.5328.965. [DOI] [PubMed] [Google Scholar]
- Bailey P, Downes M, Lau P, Harris J, Chen SL, Hamamori Y, Sartorelli V, Muscat GE. The nuclear receptor corepressor N-CoR regulates differentiation: N-CoR directly interacts with MyoD. Mol Endocrinol. 1999;13:1155–1168. doi: 10.1210/mend.13.7.0305. [DOI] [PubMed] [Google Scholar]
- Black BL, Molkentin J, Olson EN. Multiple roles for the MyoD basic region in transmission of transcriptional activation signals and interaction with MEF2. Mol Cell Biol. 1998;18:69–77. doi: 10.1128/mcb.18.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bour BA, O'Brien MA, Lockwood WL, Goldstein ES, Bodner R, Taghert PH, Abmayr SM, Nguyen HT. Drosophila MEF-2C, a transcription factor required for myogenesis. Genes & Dev. 1995;9:730–741. doi: 10.1101/gad.9.6.730. [DOI] [PubMed] [Google Scholar]
- Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- Burke L, Downes M, Carozzi A, Giguere V, Muscat GE. Nucleic Acids Res. 1996;24:3481–3489. doi: 10.1093/nar/24.18.3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova J, Helmer E, Selmi-Ruby S, Qi J-S, Au-Fliegner M, Desai-Yajnik V, Koudinova N, Yarm F, Raaka BM, Samuels HH. Functional evidence for ligand-dependent dissociation of thyroid hormone and retinoic acid receptors from an inhibitory cellular factor. Mol Cell Biol. 1994;14:5756–5765. doi: 10.1128/mcb.14.9.5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, Aswad DW, Stallcut MR. Regulation of transcription by a protein methyltransferase. Science. 1999;284:2174–2177. doi: 10.1126/science.284.5423.2174. [DOI] [PubMed] [Google Scholar]
- Chen H, Lin RJ, Schiltz RL, Chakravarti D, Nash A, Nagy L, Privalsky ML, Nakatani Y, Evans RM. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell. 1997;90:569–580. doi: 10.1016/s0092-8674(00)80516-4. [DOI] [PubMed] [Google Scholar]
- Crews ST, Fan C-M. Remberance of things PAS: Regulation of development by bHLH-PAS proteins. Curr Opin Genet Dev. 1999;9:580–587. doi: 10.1016/s0959-437x(99)00003-9. [DOI] [PubMed] [Google Scholar]
- Cserjesi P, Olson EN. Myogenin induces muscle specific enhancer binding factor MEF-2 independently of other muscle specific gene products. Mol Cell Biol. 1991;11:4854–4862. doi: 10.1128/mcb.11.10.4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downes M, Carozzi AJ, Muscat GE. Constitutive expression of the orphan receptor, Rev-erbA alpha, inhibits muscle differentiation and abrogates the expression of the myoD gene family.Mol. Endocrinol. 1995;9:1666–1678. doi: 10.1210/mend.9.12.8614403. [DOI] [PubMed] [Google Scholar]
- Eckner R, Yao TP, Oldread E, Livingston DM. Interaction and functional collaboration of p300/CBP and bHLH proteins in muscle and B-cell differentiation. Genes & Dev. 1996;10:2478–2490. doi: 10.1101/gad.10.19.2478. [DOI] [PubMed] [Google Scholar]
- Edmondson DG, Olson EN. A gene with homology to the myc similarity region of MyoD1 is expressed during myogenesis and is sufficient to activate the muscle differentiation program. Genes & Dev. 1989;3:628–640. doi: 10.1101/gad.3.5.628. [DOI] [PubMed] [Google Scholar]
- Ferreira R, Maghaghi-Jaulin L, Robin P, Harel-Bellan A, Trouche D. The three members of the pocket proteins family share the ability to repress E2F activity through recruitment of a histone deactylase. Proc Natl Acad Sci. 1998;95:10493–10498. doi: 10.1073/pnas.95.18.10493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes & Dev. 2000;9:140–147. [PubMed] [Google Scholar]
- Gossett LA, Kelvin DJ, Sternberg EA, Olson EN. A new myocyte specific enhancer binding factor that recognizes a conserved element associated with muscle specific genes. Mol Cell Biol. 1989;9:5022–5033. doi: 10.1128/mcb.9.11.5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Schneider JW, Condorelli G, Kaushal S, Mahdavi V, Nadal-Ginard B. Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation. Cell. 1993;72:309–324. doi: 10.1016/0092-8674(93)90110-c. [DOI] [PubMed] [Google Scholar]
- Guo K, Walsh K. Inhibition of myogenesis by multiple cyclin-cdk complexes. J Biol Chem. 1997;272:791–797. doi: 10.1074/jbc.272.2.791. [DOI] [PubMed] [Google Scholar]
- Guo K, Wang J, Andres V, Smith RC, Walsh K. MyoD-induced expression of p21 inhibits cyclin-dependent kinase activity upon myocyte terminal differentiation. Mol Cell Biol. 1995;15:3823–3829. doi: 10.1128/mcb.15.7.3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halevy O, Novitch BG, Spicer DB, Skapek SX, Rhee J, Hannon GJ, Beach D, Lassar AB. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science. 1995;267:1018–1021. doi: 10.1126/science.7863327. [DOI] [PubMed] [Google Scholar]
- Hamamori Y, Sartorelli V, Ogryzko V, Puri PL, Wu H-Y, Wang J, Nakatani Y, Kedes L. Regulation of histone acetyltransferases, p300 and PCAF, by the bHLH protein twist and adenoviral oncoprotein E1A. Cell. 1999;96:405–413. doi: 10.1016/s0092-8674(00)80553-x. [DOI] [PubMed] [Google Scholar]
- Hasty P, Bradley A, Morris J, Edmondson D, Venutti J, Olson E, Klein W. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature. 1993;364:501–506. doi: 10.1038/364501a0. [DOI] [PubMed] [Google Scholar]
- Henthorn P, Kiledjian M, Kadesch T. Two distinct transcription factors that bind the immunoglobulin enhancer μE5/κE2. Science. 1990;247:467–470. doi: 10.1126/science.2105528. [DOI] [PubMed] [Google Scholar]
- Hong H, Kohli K, Trivedi A, Johnson DL, Stallcup MR. GRIP-1 a novel mouse protein that serves as transcriptional coactivator in yeast for the hormone binding domains of steroid receprtors. Proc Natl Acad Sci. 1996;93:4946–4952. doi: 10.1073/pnas.93.10.4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadesch T. Consequences of heteromeric interactions among helix loop helix proteins. Cell Growth Diff. 1993;4:49–55. [PubMed] [Google Scholar]
- Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin SC, Heyman R, Rose DW, Glass CK, Rosenfeld MG. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- Kiess M, Gill M, Hamel P. Expression and activation of the retinoblastoma protein (pRb)-family proteins, p107 and p130 during L6 myoblast differentiation. Cell Growth Differ. 1995;6:1287–1298. [PubMed] [Google Scholar]
- Lassar AB, Davis RL, Wright WE, Kadesch T, Murre C, Voronova A, Baltimore D, Weintraub H. Functional activity of myogenic HLH proteins requires hetero-oligomerization with E12/E47-like proteins in vivo. Cell. 1991;66:305–315. doi: 10.1016/0092-8674(91)90620-e. [DOI] [PubMed] [Google Scholar]
- Li H, Gomes PJ, Chen JD. RAC-3, a steroid receptor associated coactivator that is related to SRC-1 and RAC-3. Proc Natl Acad Sci. 1997;94:8479–8484. doi: 10.1073/pnas.94.16.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludolph D, Koniesczny S. Transcription factor families: Muscling in on the myogenic program. FASEB J. 1995;9:1595–1604. doi: 10.1096/fasebj.9.15.8529839. [DOI] [PubMed] [Google Scholar]
- Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P, Lorain S, Le Villain JP, Troalen F, Trouche D, Harel-Bellan A. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature. 1998;391:601–605. doi: 10.1038/35410. [DOI] [PubMed] [Google Scholar]
- McKenna NJ, Lanz RB, O'Malley BW. Nuclear receptor coregulators: Cellular and molecular biology. Endoc Rev. 1999;20:321–344. doi: 10.1210/edrv.20.3.0366. [DOI] [PubMed] [Google Scholar]
- Miska E, Karlsson C, Langley E, Nielson SJ, Pines J, Kouzarides T. HDAc4 deacetylase associates with and represses the MEF-2 transcription factor. EMBO J. 1999;18:5099–5107. doi: 10.1093/emboj/18.18.5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin JD, Olson EN. Defining the regulatory network for muscle development. Curr Opin Genet Dev. 1996;6:445–453. doi: 10.1016/s0959-437x(96)80066-9. [DOI] [PubMed] [Google Scholar]
- Molkentin J, Black BL, Martin JF, Olson EN. Cooperative activation of muscle gene expression by MEF-2 and myogenic bHLH proteins. Cell. 1995;83:1125–1136. doi: 10.1016/0092-8674(95)90139-6. [DOI] [PubMed] [Google Scholar]
- ————— Mutational analysis of the DNA binding, dimerization and transcriptional activation domains of MEF-2C. Mol Cell Biol. 1996;16:2627–2636. doi: 10.1128/mcb.16.6.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moras D, Gronemeyer H. The nuclear receptor ligand binding domain: Structure and function. Curr Opin Cell Biol. 1998;10:384–391. doi: 10.1016/s0955-0674(98)80015-x. [DOI] [PubMed] [Google Scholar]
- Murre C, Voronova A, Baltimore D. B-cell and myocyte specific E2-box binding factors binding factors contain E12/E47-like subunits. Mol Cell Biol. 1991;11:1156–1160. doi: 10.1128/mcb.11.2.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscat GE, Rea S, Downes M. Identification of a regulatory function for an orphan receptor in muscle: COUP-TF II affects the expression of the myoD gene family during myogenesis. Nucleic Acids Res. 1995;23:1311–1318. doi: 10.1093/nar/23.8.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na SY, Lee SSK, Han SJ, Choi HS, Im SSY, Lee JW. Steroid receptor coactivator-1 interacts with the p50 subunit and coactivates nuclear factor kappaB-mediated transactivation. J Biol Chem. 1998;2273:10831–10834. doi: 10.1074/jbc.273.18.10831. [DOI] [PubMed] [Google Scholar]
- Novitch BG, Mulligan GJ, Jacks T, Lassar AB. Skeletal muscle cells lacking the retinoblastoma protein display defects in muscle gene expression and accumulate in S and G2 phases of the cell cyle. J Cell Biol. 1996;135:441–456. doi: 10.1083/jcb.135.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novitch BG, Spicer DB, Kim PS, Cheung WL, Lassar AB. pRb is required for MEF-2 dependent gene expression as well as cell cycle arrest during skeletal muscle differentiation. Curr Biol. 1999;9:449–459. doi: 10.1016/s0960-9822(99)80210-3. [DOI] [PubMed] [Google Scholar]
- Olson EN. Interplay between proliferation and differentiation within the myogenic lineage. Dev Biol. 1992;154:261–272. doi: 10.1016/0012-1606(92)90066-p. [DOI] [PubMed] [Google Scholar]
- Olson EN, Arnold HH, Rigby PW, Wold BJ. Know your neighbors: Three phenotypes in null mutants of the myogenic bHLH gene MRF4. Cell. 1996;85:1–4. doi: 10.1016/s0092-8674(00)81073-9. [DOI] [PubMed] [Google Scholar]
- Olson EN, Klein WH. bHLH factors in muscle development: Dead lines and commitments, what to leave in and what to leave out. Genes & Dev. 1994;8:1–8. doi: 10.1101/gad.8.1.1. [DOI] [PubMed] [Google Scholar]
- Onate SA, Tsai SY, Tsai MJ, O'Malley BW. Sequence and characterization of a coactivator for the steroid receptor superfamily. Science. 1995;270:1354–1357. doi: 10.1126/science.270.5240.1354. [DOI] [PubMed] [Google Scholar]
- Parker SB, Eichele G, Zhang P, Rawls A, Sands AT, Bradley A, Olson EN, Harper JW, Elledge SJ. p53-independent expression of p21Cip1 in muscle and other terminally differentiating cells. Science. 1995;267:1024–1027. doi: 10.1126/science.7863329. [DOI] [PubMed] [Google Scholar]
- Puri PL, Avantaggiati ML, Balsano C, Sang N, Graessmann A, Giordano A, Levrero M. p300 is required for MyoD-dependent cell cycle arrest and muscle-specific gene transcription. EMBO J. 1997a;16:369–383. doi: 10.1093/emboj/16.2.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri PL, Sartorelli V, Yang X, Hamamori Y, Ogryzko VV, Howard BH, Kedes L, Wang JYL, Graessmann A, Nakatani Y, Levrero M. PCAF histone acetyl transferase promotes muscle differentiation. Mol Cell. 1997b;1:35–45. doi: 10.1016/s1097-2765(00)80005-2. [DOI] [PubMed] [Google Scholar]
- Rudnicki M, Schnegelsberg P, Stead R, Braun T, Arnold H-H, Jaenisch R. MyoD or Myf-5 is required for the formation of skeletal muscle. Cell. 1993;75:1351–1359. doi: 10.1016/0092-8674(93)90621-v. [DOI] [PubMed] [Google Scholar]
- Sartorelli V, Huang J, Hamamori Y, Kedes L. Molecular mechanisms of myogenic coactivation by p300: Direct interaction with the activation domain of MyoD and with the MADS box of MEF2C. Mol Cell Biol. 1997;17:1010–1026. doi: 10.1128/mcb.17.2.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JW, Gu W, Zhu L, Mahdavi V, Nadal-Ginard B. Reversal of terminal differentiation mediated by p107 in Rb−/− muscle cells. Science. 1994;264:1467–1471. doi: 10.1126/science.8197461. [DOI] [PubMed] [Google Scholar]
- Skapek SX, Rhee J, Spicer DB, Lasser AB. Inhibition of myogenic differentiation in proliferating myoblasts by cyclinD1-dependant kinase. Science. 1995;267:1022–1024. doi: 10.1126/science.7863328. [DOI] [PubMed] [Google Scholar]
- Skapek SX, Rhee J, Kim PS, Novitch BG, Lasser AB. Cyclin-mediated inhibition of muscle gene expression via a mechanism that is independant of pRB hyperphosphorylation. Mol Cell Biol. 1996;16:7043–7053. doi: 10.1128/mcb.16.12.7043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow DB, Miska EA, Langley E, Deonauth SR, Kotecha AS, Towers N, Spohr G, Kouzaridies T, Mohun TJ. MEF-2 function is modified by a novel co-repressor, MITR. EMBO J. 1999;18:5085–5098. doi: 10.1093/emboj/18.18.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spicer DB, Rhee J, Cheung WL, Lassar AB. Inhibition of myogenic bHLH and MEF-2 transcription factors by the bHLH protein, twist. Science. 1996;272:1476–1480. doi: 10.1126/science.272.5267.1476. [DOI] [PubMed] [Google Scholar]
- Suen CS, Berrodin TJ, Mastroeni R, Cheskis BJ, Lytlle CR, Frail DE. A transcriptional coactivator, SRC-3, selectively augments steroid receptor activity. J Biol Chem. 1998;273:27645–27653. doi: 10.1074/jbc.273.42.27645. [DOI] [PubMed] [Google Scholar]
- Sun X-H, Baltimore D. An inhibitory domain of E12 transcription factor prevents DNA binding in E12 homodimers, but not in E12 heterodimers. Cell. 1991;64:459–470. doi: 10.1016/0092-8674(91)90653-g. [DOI] [PubMed] [Google Scholar]
- Takeshita A, Cardona GR, Koibuchi N, Suen CS, Chin WW. TRAM-1, a novel 160-kDa thyroid hormone receptor activator molecule, exhibits distinct properties from SRC-1. J Biol Chem. 1997;272:27629–27634. doi: 10.1074/jbc.272.44.27629. [DOI] [PubMed] [Google Scholar]
- Tapscott SJ, Weintraub H. MyoD and the regulation of myogenesis by helix-loop-helix proteins. J Clin Invest. 1991;87:1133–1138. doi: 10.1172/JCI115109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torchia J, Rose DW, Inostroza J, Kamei Y, Westin S, Glass CK, Rosenfeld MG. The transcritional coactivator pCIP binds CBP and mediates nuclear receptor function. Nature. 1997;387:677–684. doi: 10.1038/42652. [DOI] [PubMed] [Google Scholar]
- Trouche D, Cook A, Kouzarides T. The CBP co-activator stimulates E2F1/DP1 activity. Nucleic Acids Res. 1996;24:4139–4145. doi: 10.1093/nar/24.21.4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umesono K, Murakami KK, Thompson CC, Evans RM. Direct repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin D3 receptors. Cell. 1991;65:1255–1266. doi: 10.1016/0092-8674(91)90020-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voegel J, Heine M, Zechel C, Chambon P, Gronemeyer H. TIF2, a 160 kDa transcriptional mediator for the ligand dependent activation function AF-2 of nuclear receptors. EMBO J. 1996;15:3667–3675. [PMC free article] [PubMed] [Google Scholar]
- Walsh K, Perlman H. Cell cycle exit upon myogenic differentiation. Curr Opin Genet Dev. 1997;7:597–602. doi: 10.1016/s0959-437x(97)80005-6. [DOI] [PubMed] [Google Scholar]
- Wang AH, Bertos NR, Vezmar M, Pelletier N, Crosato M, Heng HH, Ng JT-H, Han J, Yang X-J. HDAC4, a human histone deacetylase related to HDA1, is a transcriptional corepressor. Mol Cell Biol. 1999;19:7816–7827. doi: 10.1128/mcb.19.11.7816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub H, Dwarki VJ, Verma I, Davis R, Hollenberg S, Snider L, Lassar A, Tapscott SJ. Muscle-specific transcriptional activation by MyoD. Genes & Dev. 1991;5:1377–1386. doi: 10.1101/gad.5.8.1377. [DOI] [PubMed] [Google Scholar]
- Westin S, Kurokawa R, Nolte RT, Wisely GB, McInerney EM, Rose DW, Milburn MV, Rosenfeld MG, Glass CK. Interactions controlling the assembly of nuclear receptor heterodimers and coactivators. Nature. 1998;395:199–202. doi: 10.1038/26040. [DOI] [PubMed] [Google Scholar]
- Willy PJ, Umesono K, Ong ES, Evans RM, Heyman RA, Mangelsdorf DJ. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes & Dev. 1995;9:1033–1045. doi: 10.1101/gad.9.9.1033. [DOI] [PubMed] [Google Scholar]
- Xu L, Glass CK, Rosenfeld MG. Coactivator and corepressor complexes in nuclear receptor function. Curr Opin Genet Dev. 1999;9:140–147. doi: 10.1016/S0959-437X(99)80021-5. [DOI] [PubMed] [Google Scholar]
- Yun K, Wold B. Skeletal muscle determination and differentiation: Story of a core regulatory network and its context. Curr Opin Cell Biol. 1996;8:877–889. doi: 10.1016/s0955-0674(96)80091-3. [DOI] [PubMed] [Google Scholar]
- Zacksenhaus E, Jiang Z, Chung D, Marth JD, Phillips RA, Gallie BL. pRb controls proliferation, differentiation, and death of skeletal muscle cells and other lineages during embryogenesis. Genes & Dev. 1996;10:3051–3064. doi: 10.1101/gad.10.23.3051. [DOI] [PubMed] [Google Scholar]