

Abstract
Malaria infection is initiated by sporozoite invasion of hepatocytes and asexual reproduction of liver stages, processes that are regarded to be “clinically and diagnostically silent.” Merozoites, which egress from hepatocytes, infect erythrocytes in periodic cycles and induce disease. How the host innate immune system contributes to disease outcomes and to the induction of effector cells during malaria remains unclear. Likewise, how the initial liver stages may shape responses to blood-stage parasites is unknown. Here, using both sporozoite- and blood-stage-induced infections with the rodent malaria parasite Plasmodium berghei ANKA, we show that the MyD88 and Toll-like receptor 2/4 (TLR2/4) pathways play critical roles in the development of experimental cerebral malaria (ECM). Strikingly, an absolute dependence on MyD88 and TLR2/4 was observed when infections were initiated with sporozoites. In addition, we show that caspase-1 activation of interleukin-1β (IL-1β) and IL-18, which is associated with the inflammasome pathway, does not contribute to P. berghei ANKA-induced immunopathology. Consistent with these data, prophylactic cover with the IL-1β antagonist anakinra did not reduce the incidence of ECM. Therefore, we propose that protection against ECM due to loss of TLR signaling functions is caused by effector mechanisms other than IL-1β activation.
INTRODUCTION
Malaria, which is caused by infection with Plasmodium parasites, is a significant global health problem that is responsible for approximately 250 million infections and 0.9 million deaths annually (51). Malaria infection is initiated following inoculation of sporozoites that invade and differentiate into preerythrocytic forms in the liver. Merozoites egress from hepatocytes and initiate a blood-stage infection. While the sporozoites and liver stages of the parasite are known to induce a “clinically silent” infection, blood stages elicit pathological responses associated with malaria.
Cerebral malaria (CM) is the most severe complication due to infection with the human parasite Plasmodium falciparum. Both the adhesion of infected red blood cells (RBCs) in the endothelial microvasculature of the brain and the dysregulation of the host's immune response to the parasite have been implicated in the pathogenesis of CM (34, 50). Infection of susceptible C57BL/6 mice with Plasmodium berghei ANKA reproduces the neurological signs associated with human CM; within 6 to 10 days of infection, animals develop ataxia, paraplegia, seizures, and coma and become moribund (12). This experimental model (referred to as experimental cerebral malaria [ECM]) has revealed important roles for proinflammatory cytokines such as gamma interferon (IFN-γ) (3), tumor necrosis factor alpha (TNF-α) (19), and lymphotoxin alpha (LT-α) (15) and for the intravascular arrest of “pathogenic” CD8+ T cells to the brain (23, 39) as vital effectors responsible for ECM onset. However, despite the progress made with rodent models, the nature of the innate immune pathways that direct the induction of inflammation and pathological sequelae remains poorly understood.
Innate immune signaling through Toll-like receptors (TLRs), which comprise several molecules using a common adaptor molecule, MyD88 (myeloid differentiation factor 88), invokes inflammation in response to microbial challenge (26). Numerous studies have provided conflicting information regarding the role of TLRs in ECM. Two studies have reported either partial or almost full protection from ECM in P. berghei ANKA-infected mice deficient in MyD88 (11, 20); partial protection from ECM was also reported in mice deficient in TLR2 and TLR9 (11, 20). While the ligands for TLR2 and TLR9 in P. berghei ANKA remain unknown, these results support in vitro studies in P. falciparum suggesting a role for glycosylphosphatidylinositol (GPI) and hemozoin as parasite-derived ligands for TLR2 and TLR9, respectively (10, 28). However, the identification of hemozoin as a TLR9 ligand remains controversial (36). In sharp contrast, two studies have reported that ECM development is independent of TLR signaling. Mice deficient in MyD88 or TLR1, -2, -3, -4, -6, -7, or -9 (48) and triple TLR2/4/9-deficient mice (29) have been described to be as susceptible to immune pathology as wild-type (WT) animals. The reasons for these discrepancies remain to be resolved.
The reported partial dependence or nondependence of ECM pathogenesis on MyD88 suggests that P. berghei ANKA infection stimulates inflammation utilizing some other innate immune pathway. The MyD88 adaptor molecule utilized by TLRs is shared with the interleukin-1 (IL-1) and IL-18 signaling pathways (1, 8). IL-1β and IL-18 are structurally related proinflammatory cytokines (5) that are central to the initiation of the inflammatory cascade during a host's innate response to microbial infection (9, 40). Both cytokines are synthesized as propeptides that are cleaved by active caspase-1 for full cytokine maturation (18, 22). TLR agonists are necessary for pro-IL-1β and IL-18 synthesis, and ATP stimulates caspase-1-dependent cleavage and secretion (33). Cytoplasmic “danger” signals, which are independent of TLR ligation, from microbes, microbe-derived products, and other endogenous stimuli have been shown to activate caspase-1 (from its inactive form pro-caspase-1) within a multiprotein complex termed the inflammasome (25).
In addition to reports of hemozoin being a TLR9 ligand, recent studies have identified its function as a “danger” signal for the Nalp3 inflammasome (13, 21, 42), which is composed of the Nod-like protein Nalp3, the PYRIN-CARD protein ASC, and caspase-1 (37). Additional known activators of the Nalp3 inflammasome include other crystalline structures such as asbestos and silica (14). In vitro studies utilizing bone-marrow derived macrophages (BMDM) have shown that pure hemozoin induces the cleavage of caspase-1 into its enzymatically active form, which leads to the production of IL-1β and IL-18 (13, 21, 42). Moreover, in vivo studies have highlighted a role for Nalp3, caspase-1, and IL-1 receptor (IL-1R) in a mouse model of hemozoin-induced peritonitis (13). Consequently, the Nalp3 inflammasome has recently been implicated in the development of ECM using Nalp3-deficient mice (13). However, a separate study refuted a role for the Nalp3 inflammasome by showing that mice deficient in caspase-1, ASC, or the IL-1 receptor succumbed to P. berghei ANKA infection as WT mice did. Mice deficient in Nalp3 remained susceptible to ECM despite displaying a delayed onset of neurological signs (38). A clarification of the role of caspase-1 dependent activation of IL-1β as well as IL-18 in ECM is therefore warranted.
A potentially critical issue which also needs to be addressed in malaria research is that almost all the studies mentioned above utilized an artificial method of parasite infection by either intraperitoneal or intravenous injection of asexual blood-stage parasites. This type of inoculation bypasses the obligate sporozoite and liver-stage phases of the life cycle. How and to what extent malaria preerythrocytic stages shape the immune responses to the subsequent blood-stage infection remain largely unknown. Thus, this lack of knowledge warrants a reevaluation of immune responses to blood stages initiated by sporozoite infection. Indeed, very few studies have investigated immune responses to blood-stage parasites that are initiated with sporozoites (4, 16).
In the present study, we reexamined the roles of MyD88 and caspase-1 activation of the IL-1β and IL-18 pathways during ECM and contrasted infections initiated with sporozoites versus parasitized erythrocytes. Finally, we ascertained whether the IL-1β pathway is a potential target for therapeutic and/or prophylactic intervention against severe disease caused by Plasmodium infection.
MATERIALS AND METHODS
Animals.
Procedures with experimental animals were performed according to the German Animal Protection Law, United Kingdom Home Office, and European regulations. C57BL/6AnNCr mice (6 to 10 weeks old) were obtained from Charles River Laboratories, Sulzfeld, Germany. Mice deficient in MyD88 (obtained from S. Akira [1]), TLR2/4 (originally obtained as singly deficient mice from S. Akira [47]), caspase-1 (obtained from BASF [30]), IL-1β (obtained from D. Chaplin [43]), or IL-18 (obtained from S. Akira [46]) in the C57BL/6 background were bred in a pathogen-free animal facility at the Max-Planck-Institute for Infection Biology and have been backcrossed to the C57BL/6 background for at least 10 generations.
P. berghei ANKA infection.
The complete life cycle of P. berghei ANKA was maintained by passage through naïve mice and Anopheles stephensi mosquitoes. The P. berghei ANKA parasites utilized in this study were clonal parasites which are passaged at regular intervals through the entire life cycle and originate from clone 15cy1, which had been engineered to express the green fluorescent protein (17). Infections were initiated by intravenous (i.v.) inoculation of 104 sporozoites, purified from mosquito salivary glands, or similar numbers of infected red blood cells (iRBCs), obtained from infected C57BL/6 mice. Infected mice were observed daily for the manifestation of ECM, which was defined as the sudden onset of ataxia, paralysis, convulsion, or coma (12). The presence of ECM signs served as the ethical endpoint criterion for culling of animals. Spleens and brains were removed for analysis by flow cytometry. Infected mice that did not develop ECM until day 18 after infection were considered resistant and were sacrificed in order to avoid suffering from high parasitemia and severe anemia. The presence of blood-stage parasites was determined by daily examination of Giemsa-stained blood smears by oil immersion microscopy. For all infection experiments, parasitemia was assessed every other day by counting up to 50 microscopy fields.
Quantification of parasite development in the liver.
Total RNA was extracted from mouse livers at 44 h after infection using the RNeasy kit (Qiagen) and treated with DNase I. cDNA was synthesized using the RETROscript kit (Ambion). Hepatic parasite development was assessed by quantitative reverse transcription-PCR (RT-PCR) using SYBR green I master mix (Applied Biosystems). Reverse transcripts of P. berghei ANKA 18S rRNA were amplified in parallel with murine glyceraldehyde-3-phosphate dehydrogenase (mGAPDH) in a final volume of 25 μl in 96-well plates (Applied Biosystems). Oligonucleotide primers (Eurofins MWG Operon) for mGAPDH were 5′-CGT CCC GTA GAC AAA ATG GT-3′ (forward) and 5′-TTG ATG GCA ACA ATC TCC AC-3′) (reverse), and those for P. berghei ANKA 18S rRNA were 5′-AAG CAT TAA ATA AAG CGA ATA CAT CCT TAC-3′ (forward) and 5′-GGA GAT TGG TTT TGA CGT TTA TGT G-3′ (reverse).
Cytokine measurement.
Cytokine measurement was performed using the mouse inflammation cytometric bead array kit from BD Biosciences according to the manufacturer's instructions on a LSR II cell analyzer (BD Biosciences). Raw data were analyzed with FlowJo software (Tree Star). Plasma samples for in vivo measurement of cytokines were prepared from heparinized blood collected from naïve and infected mice.
Preparation of brain-sequestered lymphocytes.
Brain-sequestered lymphocytes were isolated as described previously (6). Perfused brains were digested in collagenase D buffer and lymphocytes isolated on a 30% Percoll (GE Healthcare) gradient. The isolated brain-sequestered lymphocytes were surface stained according to standard protocols. Antibodies for cell surface staining were obtained from eBiosciences (anti-mouse CD4 [GK1.5] and CD8 [53.6-7]). Antibodies for intracellular staining of IFN-γ (XMG1.2) were obtained from eBioscience. Cells were analyzed using an LSR II cell analyzer (BD Biosciences) and FlowJo software (TreeStar).
Statistical analysis.
Statistical significance was assessed using the Mann-Whitney test, with a P value of <0.05 taken as indicating a significant difference. Survival curves were compared by using the log rank (Mantel-Cox) test. All statistical tests were computed with GraphPad Prism 5 (GraphPad Software).
RESULTS
ECM pathogenesis is dependent on MyD88 and TLR2/4 signaling.
We first evaluated whether the MyD88 and TLR2/4 signaling pathways are involved in the development of ECM. For this purpose, we compared the courses of Plasmodium berghei ANKA infection in WT, MyD88-deficient, and TLR2/4-deficient C57BL/6 mice and contrasted infections caused by inoculation of 104 sporozoites or the same number of infected red blood cells (iRBCs) (Fig. 1).
Fig. 1.

MyD88−/− and TLR 2/4−/− mice show an increased resistance to experimental cerebral malaria. WT (black, •) MyD88−/− (red, ▪), and TLR 2/4−/− (green, ▴) mice were infected with 104 P. berghei sporozoites (A to C) or iRBCs (D to F) by i.v. injection. (A and D) Average parasitemia during the course of infection. Error bars indicate the standard errors of the means (SEM). Results of a single representative experiment are displayed for one sporozoite-induced and one blood-stage-induced infection (n = 5). No significant differences were observed between any groups of mice. (B and E) Kaplan-Meier analysis of the time from infection to development of signs of ECM. Data were cumulative from multiple experiments. For sporozoite-induced infections: WT, n = 25; MyD88−/−, n = 18; TLR2/4−/−, n = 13. For iRBC-induced infections: WT, n = 15; MyD88−/−, n = 16; TLR 2/4−/−, n = 12. Survival rates were analyzed by using the log rank test. ***, P < 0.001. (C and F) Absence of ECM shown as the percentage of infected animals based on cumulative data. (G and H) MyD88−/− (G) and TLR2/4−/− (H) mice were infected with 104 P. berghei sporozoites. The parasite load in the liver was measured by real-time PCR at 42 h postinfection. No significant differences were observed between groups of mice.
All infections initiated with sporozoites in WT, MyD88−/−, and TLR2/4−/− mice became patent at 3 to 4 days after infection (Fig. 1A). The majority of the sporozoite-infected WT mice (n = 25) developed ECM on day 7 or 8 after sporozoite inoculation (Fig. 1B). These results indicate that a blood-stage infection of as short as 4 to 5 days is sufficient to induce ECM. In sharp contrast, none of the sporozoite-infected MyD88−/− (n = 18) and TLR2/4−/− (n = 13) mice developed ECM. All animals that did not develop ECM were carefully culled on day 18 postinfection to avoid severe anemia and high parasitemia. There was no difference in parasitemia levels during the first 7 days of infection among sporozoite-inoculated WT, MyD88−/−, and TLR2/4−/− mice (Fig. 1A). Thus, we conclude that the MyD88-dependent TLR2/4 pathway is critically involved in induction of ECM upon a sporozoite-induced malaria infection.
We next compared disease progression in infections initiated with iRBCs that bypasses the initial liver stage of infection (Fig. 1D to F). All of the WT mice developed ECM on day 7 or 8 after infection. In contrast, 38% (6/16) of the infected MyD88−/− mice had a significant delay in the onset of neurological signs (day 9 to 11 after infection; P < 0.0001 by log rank [Mantel-Cox] test), and 44% (7/16) did not develop ECM. Similarly, 33% (4/12) of the infected TLR2/4−/− mice had a significant delay in the onset of neurological signs (day 9 to 11 after infection; P = 0.0002), and 42% (5/12) did not develop ECM. The survival curves for infected MyD88−/− and TLR2/4−/− mice did not differ significantly (P = 0.8). We did not observe any difference in parasitemia levels during the first 7 days of infection between WT, MyD88−/−, and TLR2/4−/− mice (Fig. 1D). A follow-up of the remaining mutant mice, which stayed free of ECM signs, revealed an increasing variability of parasitemia levels after day 7. All animals developed severe anemia and parasitemia above 20% by day 18 postinfection (data not shown).
One possible explanation of why we obtained disparate ECM levels when infection was initiated by sporozoite versus blood stages is that liver-stage development is different in WT versus MyD88−/− or TLR2/4−/− mice. To examine this possibility, we infected WT, MyD88−/−, or TLR2/4−/− mice with 104 sporozoites. The livers of the animals were excised at 42 h postinfection and processed for quantitative real-time PCR to measure parasite development in the liver. As shown in Fig. 1G and H, we did not observe any difference in the development of the parasite in the liver.
Taken together, our results concur with reports indicating a partial dependency of ECM on MyD88 and TLR2/4 signaling following an iRBC-induced infection (11, 20). Strikingly, an absolute dependency is observed when infection is initiated with sporozoites, leading to a complete protection against ECM in the MyD88−/− and TLR2/4−/− mice. This difference cannot be attributed to differential development of parasites in the liver when infection is initiated with sporozoites.
Correlation of disease outcome with plasma cytokine levels.
ECM is characterized by excessive production of proinflammatory cytokines (41). We measured the systemic levels of various cytokines in the plasma of sporozoite-infected animals (Fig. 2). Infected WT mice had significantly elevated levels of plasma IFN-γ and monocyte chemotactic/chemoattractant protein 1 (MCP-1), which peaked on day 5 after sporozoite infection. Expression of TNF-α was significantly elevated on day 5 and even further increased on day 7. The anti-inflammatory cytokine IL-10 only showed a significant increase on day 7.
Fig. 2.

Plasma concentrations of cytokines correlate with the course of P. berghei infection. Plasma samples were collected before and on day 5 and day 7 after i.v. infection with 104 sporozoites, and the concentrations of IFN-γ, TNF-α, IL-10, and MCP-1 were measured. Data points represent individual animals; median and ranges are displayed for each group at the individual time points. Changes over the course of infection within a group (day 0 versus day 5 and day 5 versus day 7) as well as differences between WT and deficient mice on day 5 and day 7 after infection were assessed using the Mann-Whitney test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
In infected MyD88−/− mice, however, significantly lower IFN-γ and MCP-1 levels (P = 0.036 and P < 0.0001, respectively, by the Mann-Whitney test), which corresponded to significantly higher IL-10 levels (P = 0.0072), were observed on day 5 postinfection. In contrast, infected TLR2/4−/− mice presented significantly elevated plasma concentrations of IFN-γ, TNF-α, and IL-10 (P = 0.0046, P = 0.042, and P = 0.0097, respectively) that exceeded those of infected WT mice on day 5 after infection. While beyond the scope of the current study, it will be interesting to analyze the underlying molecular mechanisms that can explain these differential effects.
Inoculation of iRBCs induced plasma cytokine kinetics (see Fig. S1 in the supplemental material) that were comparable to those after infection initiated with sporozoites. However, while day 5 MCP-1 levels were significantly lower in MyD88−/− mice than in WT mice, no difference in the IFN-γ and IL-10 levels in the two groups was observed. The disparate IFN-γ and IL-10 levels in infections initiated by different stages of the Plasmodium parasite in WT and MyD88−/− mice may possibly explain the partial susceptibility of mutant mice to ECM when infected with blood stages. Overall, our data demonstrate that the innate host defense system contributes critically to ECM.
ECM pathogenesis is independent of caspase-1, IL-1β, and IL-18.
Other receptors that are known to signal through MyD88 are the IL-1 and IL-18 receptors (1). Synthesized as propeptides, mature IL-1β and IL-18 are products of activated caspase-1, which in turn is tightly regulated by the Nalp3 inflammasome, another innate immune pathway involved in inflammatory responses (32).
To determine a role for caspase-1 activation of IL-1β and IL-18 during ECM, we compared the courses of P. berghei ANKA infection in WT, caspase-1−/−, IL-1β−/−, and IL-18−/− mice inoculated with sporozoites (Fig. 3 A to C). Infected enzyme/cytokine-deficient mice developed ECM as WT mice did (Fig. 3B). Furthermore, we did not observe any difference in parasitemia levels during the first 7 days of infection in the different groups (Fig. 3A). Similar results were obtained when infections were initiated with iRBCs (Fig. 3D to F). Collectively, these results indicate that caspase-1 activation of IL-1β and IL-18 is dispensable for the development of effector responses associated with ECM.
Fig. 3.

P. berghei ANKA infections are unaltered in caspase-1−/−, IL-1β−/−, and IL-18−/− mice. WT (black, •), caspase-1−/− (red, ▪), IL-1β−/− (green, ▴), and IL-18−/− (yellow, ▾) mice were i.v. injected with 104 sporozoites (A to C) or iRBCs (D to F). (A and D) Average parasitemia during the course of infection. Error bars indicate the SEM. Results of 1 representative experiment from 2 experiments are displayed (n = 4 or 5 each). No significant differences were observed between any groups of mice. (C and E) Kaplan-Meier analysis of the time from infection to development of signs of ECM. Data were cumulative from two experiments. For sporozoite-induced infections: WT, n = 10; caspase 1−/−, n = 9; IL-1β−/−, n = 10; IL-18−/−, n = 10. For iRBC-induced infections: WT, n = 20; caspase 1−/−, n = 10; IL-1β−/−, n = 10; IL-18−/−, n = 10. Survival rates were analyzed by using the log rank test. No significant differences were observed between any groups of mice (P > 0.05). (C and F) Absence of ECM shown as the percentage of infected animals based on cumulative data.
Consistent with the above results, the plasma cytokine profiles of IFN-γ, MCP-1, TNF-α, and IL-10 in sporozoite-inoculated caspase-1−/−, IL-1β−/−, and IL-18−/− mice were indistinguishable from those in infected WT mice (Fig. 4). Similarly, using iRBCs for infection, we did not observe any significant differences in the cytokine profiles among the different groups (see Fig. S2 in the supplemental material).
Fig. 4.

Effect of deficiency in caspase-1, IL-1β, or IL-18 on the kinetics of cytokines over the course of P. berghei infections. Plasma samples were collected before and 5 and 7 days after i.v. infection with 104 P. berghei sporozoites, and levels of IFN-γ, TNF-α, IL-10, and MCP-1 were measured. Data points represent individual animals; medians and ranges are displayed for each group at the individual time points. Changes over the course of infection within a group (day 0 versus day 5 and day 5 versus day 7) as well as differences between WT and deficient mice on day 5 and day 7 after infection were assessed using the Mann-Whitney test. *, P < 0.05; **, P < 0.01.
To examine the induction of IFN-γ responses, we additionally performed short-term stimulation of day 7 splenocytes from WT and enzyme/cytokine-deficient mice in the presence of phorbol myristate acetate (PMA)/ionomycin. In good support of our findings from the plasma cytokine profiling, intracellular staining analysis of CD4+ and CD8+ cells from infected WT and enzyme/cytokine-deficient mice showed comparable frequencies of cells producing IFN-γ (see Fig. S3 in the supplemental material).
The IFN-γ-controlled trafficking of pathogenic CD8 T cells in the brain is a canonical feature of ECM (6, 7, 23). Therefore, we isolated brain-infiltrating lymphocytes from WT and enzyme/cytokine-deficient mice at the time the animals were committed to neurological signs (Fig. 5). As predicted from the clinical outcome, WT and enzyme/cytokine-deficient mice had similar proportions of brain-infiltrating CD8+ T cells. This infiltration was independent of the induction of infection by either sporozoites (Fig. 5A and B [left panel]) or iRBCs (Fig. 5B [right panel]). Therefore, we conclude that caspase-1 activation of IL-1β and IL-18 plays no pathological role in ECM in vivo.
Fig. 5.

CD8+ T cells sequester to the brains of caspase-1−/−, IL-1β−/−, and IL-18−/− mice suffering from ECM. (A) Representative dot plots of leukocytes from brains of naïve mice and of mice suffering from ECM at 7 days after P. berghei sporozoite injection. (B) Percentages of CD8+ cells among total brain-derived leukocytes from mice that developed ECM (+) and naïve age-matched control mice (−).
Interference with IL-1β signaling does not offer an experimental therapeutic approach.
Our findings of a dispensable role for IL-1β signaling in ECM further substantiate previous work showing that IL-1R-deficient mice succumb to ECM indistinguishably from WT mice (38). To obtain further independent evidence and to examine a potential drug target to prevent severe forms of malaria, we tested whether treatment with the IL-1β antagonist anakinra (24) modulates the clinical outcome of a malaria infection (Fig. 6). High doses of anakinra (250 mg/kg) were administered to mice for 7 consecutive days following infection with P. berghei ANKA sporozoites. In agreement with our data from the knockout mice, treated and untreated mice showed similar courses of infection (Fig. 6A). Importantly, animals in both groups displayed signs of ECM at 7 days after infection to similar degrees (Fig. 6B). As a consequence, no protection against ECM was detected in anakinra-treated animals (Fig. 6C). These findings independently reject the hypothesis that the IL-1β signaling pathway plays an important role in sporozoite-induced ECM.
Fig. 6.
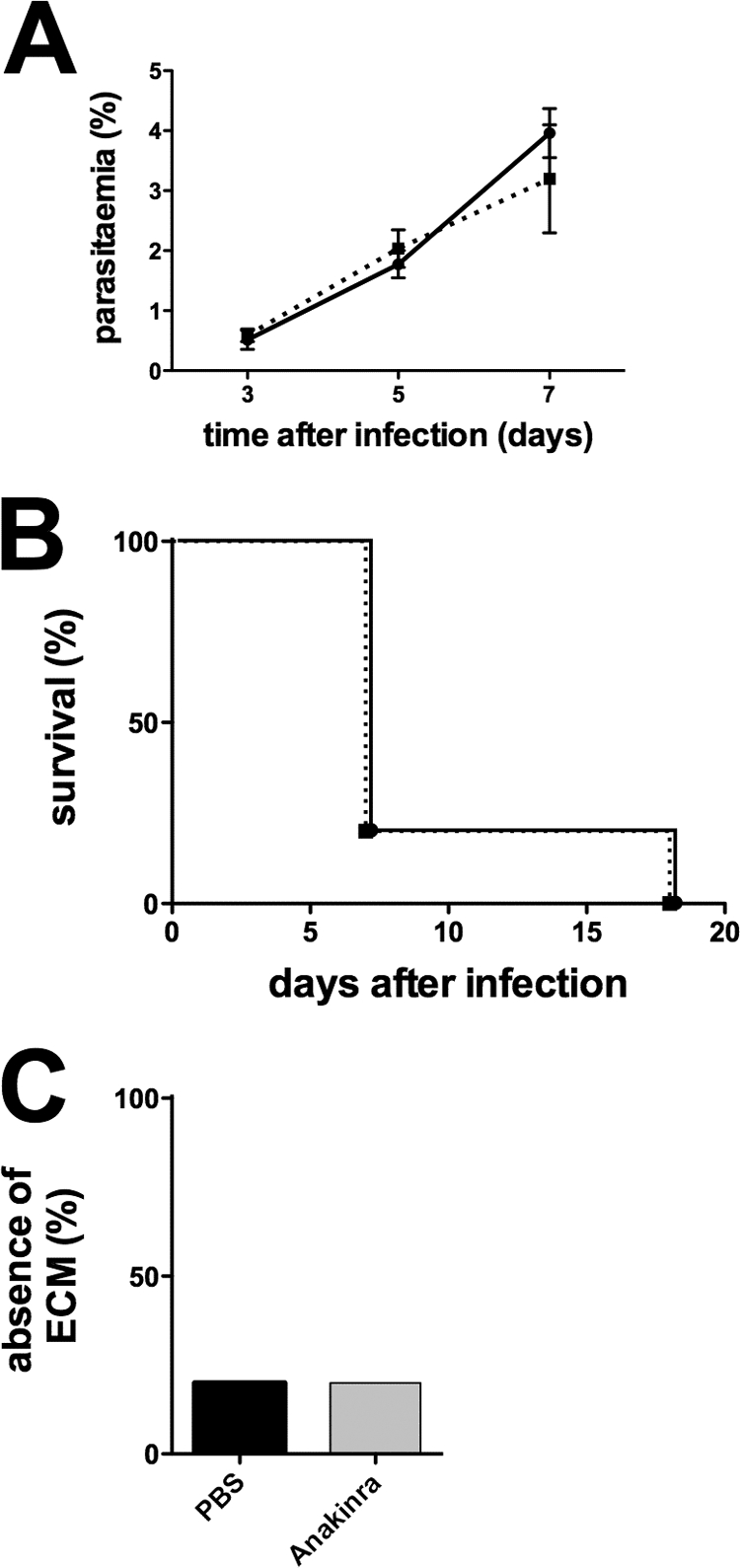
Treatment with anakinra does not improve the clinical course of P. berghei ANKA infection. WT C57BL/6 mice (n = 5) were infected with 104 P. berghei ANKA sporozoites i.v. and treated for 7 days with 250 mg/kg anakinra i.p. once daily (▪). Controls were infected with the same number of parasites and received injections of PBS (•). (A) Average parasitemia during the course of infection. Error bars indicate the SEM. (B) Kaplan-Meier analysis of the time from infection to development of signs of ECM. (C) Absence of ECM shown as the percentage of infected animals.
DISCUSSION
Innate immune pathways triggered by pathogen-associated molecular patterns or “danger” signals are pivotal for the first-line defense against various microbial infections, but their roles during blood-stage malaria are either poorly understood or controversial. Moreover, the potential of preerythrocytic stages, which precede a Plasmodium blood-stage infection, to modulate innate immune responses and disease outcomes has typically been neglected. Our present study addressed the pathological roles of central players of innate defense mechanisms in malaria infections initiated with the preerythrocytic parasite stages, i.e., sporozoites, in side-by-side comparisons to infections induced by transfer of infected blood. Inoculation of WT mice with iRBCs bypasses the 2 to 3 days of liver-stage development (Fig. 1 and 3) but does not translate into an altered onset of ECM. Additionally, all mice displayed the effector response characterized by induction of proinflammatory cytokines at 5 days after infection (Fig. 2 and 4; see Fig. S2 in the supplemental material) regardless of the route of infection.
In line with this result, it has been demonstrated previously that MyD88-deficient C57BL/6 mice have a higher hepatic parasite load than WT animals (49). In contrast to the published data, we did not observe any difference in the development of liver-stage parasites between WT and MyD88−/− (or TLR2/4−/−) mice. Consequently, we did not observe any difference in prepatency between WT animals and the different mice with deficiencies in innate defense mechanisms that we utilized in this study (Fig. 1A and 3A). It is conceivable that immune responses that direct the development of ECM may already be primed during the “clinically silent” liver stage. Therefore, it is possible that the pathological role of MyD88-dependent signaling in natural infections may have been underestimated.
Our infection experiments with MyD88−/− and TLR2/4−/− mice substantiate a role for TLR signaling in severe rodent model malaria. Protection against ECM was partial after inoculation of iRBCs, as reported previously (11, 20). Notably, protection in knockout mice was complete when infections were initiated by sporozoite inoculations. An unexpected finding of our study is that despite comparable clinical protection in MyD88−/− and TLR2/4−/− mice, the proinflammatory cytokine profiles were very distinct and hence may not qualify as signatures of pathogenesis. While inactivation of the downstream adaptor molecule MyD88 correlated with reduced IFN-γ and MCP-1 plasma levels, there was a corresponding significant increase in IL-10 levels. Similarly, a significant increase in IL-10 was associated with increased IFN-γ levels in TLR2/4−/− mice. These observations are consistent with the reported protective role for IL-10 in ECM (27). Slightly different results were obtained with infections initiated with blood stages in MyD88−/− mice, as significantly reduced levels of only MCP-1 were observed at day 5 postinfection. We conclude that MyD88 and TLR2/4 may trigger inflammatory responses that ultimately lead to ECM when infections are associated with blood stages. However, the associated cytokine response is largely dependent on MyD88 alone. In the absence of TLR2/4, high levels of proinflammatory cytokines can be compensated for by a yet-unknown mechanism that is beyond the scope of this study. Irrespective of the underlying mechanisms, the observed dichotomy of protection against ECM in cytokinehigh as well as cytokinelow infection models underscores the complexity of this acute neuroinflammatory complication, as documented for human cerebral malaria (2, 50).
The recent description of a role for the Nalp3 inflammasome in response to hemozoin as a malarial “danger signal” (13, 42) prompted us to test whether caspase-1-dependent activation of IL-1β and IL-18 is a central pathway which triggers ECM pathology. In this study, we found caspase-1, IL-1β, and IL-18 to be dispensable for the induction of ECM. Knockout mice succumbed to cerebral signs indistinguishably from WT mice. In support of earlier findings that IL-18 is dispensable for ECM caused by P. berghei ANKA blood stages (44), we now extend this conclusion to sporozoite-induced Plasmodium infections. We also demonstrated that IL-18 is not a key trigger for high IFN-γ production. Our results also support very recent findings that IL-1β receptor-deficient mice infected with 106 iRBCs are susceptible to ECM (38). Based on our findings, the validity of this study can likely be extended to sporozoite-induced infections. A potential translational aspect of the disruption of IL-1β signaling was ultimately assessed by protective treatment of P. berghei ANKA-infected mice with anakinra. We could rule out any beneficial effect on animal survival or reduced parasite growth, although the doses that we applied were several times higher than what proved to be efficient in a mouse model of acute gout (45). The combined results from the infection of knockout mice and pharmacological intervention in infected WT animals suggest that interference with IL-1β signaling is not a promising approach for adjunct therapy against neurological complications of malaria.
It is important to note that our study was not designed to test the role of hemozoin in activation of the Nalp3 inflammasome. However, our in vivo studies demonstrate that this pathway is not crucially involved in the immunopathology of Plasmodium infections. A reason for this apparent discrepancy may lie in the smaller amounts and accessibility of hemozoin following natural Plasmodium infections than after experimental inoculation of pure hemozoin. Another possible explanation is that hemozoin triggers a pleiotropic response that, in addition to activating of IL-1β and IL-18, induces other, yet-unidentified ECM-inducing innate immune pathways.
Most importantly, our findings also reflect inconclusive data from field studies in Africa and Asia, which have so far failed to convincingly correlate serum/plasma levels of IL-1β (31) or IL-18 (35) with the severity of disease.
In conclusion, using comparative experimental Plasmodium infections, we found differential immunopathological patterns associated with MyD88 and TLR2/4 and no evidence for an in vivo role of proinflammatory cytokines that are under the control of the Nalp3 inflammasome in experimental cerebral malaria.
Supplementary Material
ACKNOWLEDGMENTS
J.C.R.H. is supported by a Royal Society (United Kingdom) University Research Fellowship and a European Federation of Immunological Societies—Immunological Letters Short-Term Fellowship. This work was supported by the Max Planck Society and in part by grants from the European Commission (EviMalaR, no. 34) and the Chica and Heinz Schaller Foundation.
We thank Arturo Zychlinsky, Felix Meissner, Elyzana Dewi Putrianti, Katja Mueller, and Kawi Molawi for discussions.
Footnotes
Supplemental material for this article may be found at http://iai.asm.org/.
Published ahead of print on 27 June 2011.
REFERENCES
- 1. Adachi O., et al. 1998. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 9:143–150 [DOI] [PubMed] [Google Scholar]
- 2. Adams S., Brown H., Turner G. 2002. Breaking down the blood-brain barrier: signaling a path to cerebral malaria? Trends Parasitol. 18:360–366 [DOI] [PubMed] [Google Scholar]
- 3. Amani V., et al. 2000. Involvement of IFN-gamma receptor-medicated signaling in pathology and anti-malarial immunity induced by Plasmodium berghei infection. Eur. J. Immunol. 30:1646–1655 [DOI] [PubMed] [Google Scholar]
- 4. Bagot S., et al. 2004. Comparative study of brain CD8+ T cells induced by sporozoites and those induced by blood-stage Plasmodium berghei ANKA involved in the development of cerebral malaria. Infect. Immun. 72:2817–2826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bazan J. F., Timans J. C., Kastelein R. A. 1996. A newly defined interleukin-1? Nature 379:591. [DOI] [PubMed] [Google Scholar]
- 6. Belnoue E., et al. 2002. On the pathogenic role of brain-sequestered alphabeta CD8+ T cells in experimental cerebral malaria. J. Immunol. 169:6369–6375 [DOI] [PubMed] [Google Scholar]
- 7. Belnoue E., et al. 2008. Control of pathogenic CD8+ T cell migration to the brain by IFN-gamma during experimental cerebral malaria. Parasite Immunol. 30:544–553 [DOI] [PubMed] [Google Scholar]
- 8. Burns K., et al. 1998. MyD88, an adapter protein involved in interleukin-1 signaling. J. Biol. Chem. 273:12203–12209 [DOI] [PubMed] [Google Scholar]
- 9. Cao Z., Xiong J., Takeuchi M., Kurama T., Goeddel D. V. 1996. TRAF6 is a signal transducer for interleukin-1. Nature 383:443–446 [DOI] [PubMed] [Google Scholar]
- 10. Coban C., et al. 2005. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J. Exp. Med. 201:19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coban C., et al. 2007. Pathological role of Toll-like receptor signaling in cerebral malaria. Int. Immunol. 19:67–79 [DOI] [PubMed] [Google Scholar]
- 12. de Souza J. B., Hafalla J. C., Riley E. M., Couper K. N. 2010. Cerebral malaria: why experimental murine models are required to understand the pathogenesis of disease. Parasitology 137:755–772 [DOI] [PubMed] [Google Scholar]
- 13. Dostert C., et al. 2009. Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS One 4:e6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dostert C., et al. 2008. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320:674–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Engwerda C. R., et al. 2002. Locally up-regulated lymphotoxin alpha, not systemic tumor necrosis factor alpha, is the principle mediator of murine cerebral malaria. J. Exp. Med. 195:1371–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fonseca L., Seixas E., Butcher G., Langhorne J. 2007. Cytokine responses of CD4+ T cells during a Plasmodium chabaudi chabaudi (ER) blood-stage infection in mice initiated by the natural route of infection. Malaria J. 6:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Franke-Fayard B., et al. 2004. A Plasmodium berghei reference line that constitutively expresses GFP at a high level throughout the complete life cycle. Mol. Biochem. Parasitol. 137:23–33 [DOI] [PubMed] [Google Scholar]
- 18. Ghayur T., et al. 1997. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature 386:619–623 [DOI] [PubMed] [Google Scholar]
- 19. Grau G. E., et al. 1987. Tumor necrosis factor (cachectin) as an essential mediator in murine cerebral malaria. Science 237:1210–1212 [DOI] [PubMed] [Google Scholar]
- 20. Griffith J. W., et al. 2007. Toll-like receptor modulation of murine cerebral malaria is dependent on the genetic background of the host. J. Infect. Dis. 196:1553–1564 [DOI] [PubMed] [Google Scholar]
- 21. Griffith J. W., Sun T., McIntosh M. T., Bucala R. 2009. Pure hemozoin is inflammatory in vivo and activates the NALP3 inflammasome via release of uric acid. J. Immunol. 183:5208–5220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gu Y., et al. 1997. Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science 275:206–209 [DOI] [PubMed] [Google Scholar]
- 23. Hafalla J. C., Cockburn I. A., Zavala F. 2006. Protective and pathogenic roles of CD8+ T cells during malaria infection. Parasite Immunol. 28:15–24 [DOI] [PubMed] [Google Scholar]
- 24. Hannum C. H., et al. 1990. Interleukin-1 receptor antagonist activity of a human interleukin-1 inhibitor. Nature 343:336–340 [DOI] [PubMed] [Google Scholar]
- 25. Kanneganti T. D., et al. 2006. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440:233–236 [DOI] [PubMed] [Google Scholar]
- 26. Kopp E., Medzhitov R. 2003. Recognition of microbial infection by Toll-like receptors. Curr. Opin. Immunol. 15:396–401 [DOI] [PubMed] [Google Scholar]
- 27. Kossodo S., et al. 1997. Interleukin-10 modulates susceptibility in experimental cerebral malaria. Immunology 91:536–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Krishnegowda G., et al. 2005. Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of Plasmodium falciparum: cell signaling receptors, glycosylphosphatidylinositol (GPI) structural requirement, and regulation of GPI activity. J. Biol. Chem. 280:8606–8616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lepenies B., et al. 2008. Induction of experimental cerebral malaria is independent of TLR2/4/9. Med. Microbiol. Immunol. 197:39–44 [DOI] [PubMed] [Google Scholar]
- 30. Li P., et al. 1995. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell 80:401–411 [DOI] [PubMed] [Google Scholar]
- 31. Lyke K. E., et al. 2004. Serum levels of the proinflammatory cytokines interleukin-1 beta (IL-1beta), IL-6, IL-8, IL-10, tumor necrosis factor alpha, and IL-12(p70) in Malian children with severe Plasmodium falciparum malaria and matched uncomplicated malaria or healthy controls. Infect. Immun. 72:5630–5637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mariathasan S., et al. 2004. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430:213–218 [DOI] [PubMed] [Google Scholar]
- 33. Mariathasan S., et al. 2006. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440:228–232 [DOI] [PubMed] [Google Scholar]
- 34. Medana I. M., Turner G. D. 2006. Human cerebral malaria and the blood-brain barrier. Int. J. Parasitol. 36:555–568 [DOI] [PubMed] [Google Scholar]
- 35. Nagamine Y., et al. 2003. Involvement of interleukin-18 in severe Plasmodium falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 97:236–241 [DOI] [PubMed] [Google Scholar]
- 36. Parroche P., et al. 2007. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc. Natl. Acad. Sci. U. S. A. 104:1919–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Petrilli V., Dostert C., Muruve D. A., Tschopp J. 2007. The inflammasome: a danger sensing complex triggering innate immunity. Curr. Opin. Immunol. 19:615–622 [DOI] [PubMed] [Google Scholar]
- 38. Reimer T., et al. 2010. Experimental cerebral malaria progresses independently of the Nlrp3 inflammasome. Eur. J. Immunol. 40:764–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Renia L., et al. 2006. Pathogenic T cells in cerebral malaria. Int. J. Parasitol. 36:547–554 [DOI] [PubMed] [Google Scholar]
- 40. Robinson D., et al. 1997. IGIF does not drive Th1 development but synergizes with IL-12 for interferon-gamma production and activates IRAK and NFkappaB. Immunity 7:571–581 [DOI] [PubMed] [Google Scholar]
- 41. Schofield L., Grau G. E. 2005. Immunological processes in malaria pathogenesis. Nat. Rev. Immunol. 5:722–735 [DOI] [PubMed] [Google Scholar]
- 42. Shio M. T., et al. 2009. Malarial hemozoin activates the NLRP3 inflammasome through Lyn and Syk kinases. PLoS Pathog. 5:e1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shornick L. P., et al. 1996. Mice deficient in IL-1beta manifest impaired contact hypersensitivity to trinitrochlorobenzone. J. Exp. Med. 183:1427–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Singh R. P., et al. 2002. The role of IL-18 in blood-stage immunity against murine malaria Plasmodium yoelii 265 and Plasmodium berghei ANKA. J. Immunol. 168:4674–4681 [DOI] [PubMed] [Google Scholar]
- 45. So A., De Smedt T., Revaz S., Tschopp J. 2007. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res. Ther. 9:R28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Takeda K., et al. 1998. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity 8:383–390 [DOI] [PubMed] [Google Scholar]
- 47. Takeuchi O., et al. 1999. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11:443–451 [DOI] [PubMed] [Google Scholar]
- 48. Togbe D., et al. 2007. Murine cerebral malaria development is independent of Toll-like receptor signaling. Am. J. Pathol. 170:1640–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Torgler R., et al. 2008. Sporozoite-mediated hepatocyte wounding limits Plasmodium parasite development via MyD88-mediated NF-kappa B activation and inducible NO synthase expression. J. Immunol. 180:3990–3999 [DOI] [PubMed] [Google Scholar]
- 50. van der Heyde H. C., Nolan J., Combes V., Gramaglia I., Grau G. E. 2006. A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends Parasitol. 22:503–508 [DOI] [PubMed] [Google Scholar]
- 51. World Health Organization 2009. World malaria report. World Health Organization, Geneva, Switzerland [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.