

Abstract
Respiratory syncytial virus (RSV) is the most common cause of epidemic respiratory diseases in infants and young children. RSV infection of airway epithelial cells induces the expression of immune/inflammatory genes through the activation of a subset of transcription factors, including Nuclear Factor-κB (NF-κB) and AP-1. In this study, we have investigated the signaling pathway leading to activation of these two transcription factors in response to RSV infection. Our results show that IKKβ plays a key role in viral-induced NF-κB activation, while JNK regulates AP-1-dependent gene transcription, as demonstrated by using kinase inactive proteins and chemical inhibitors of the two kinases. Inhibition of TAK1 activation, by overexpression of kinase inactive TAK1 or using cells lacking TAK1 expression, significantly reduced RSV-induced NF-κB and AP-1 nuclear translocation and DNA-binding activity, as well as NF-κB-dependent gene expression, identifying TAK1 as an important upstream signaling molecule regulating RSV-induced NF-κB and AP-1 activation.
Keywords: RSV, airway epithelial cells, NF-κB, inflammation
INTRODUCTION
Respiratory syncytial virus (RSV) is an enveloped negative sense single-strand RNA virus belonging to the Paramyxoviridae family. It is the leading cause of epidemic respiratory tract illness in children worldwide (Hall, 2001). RSV has also been implicated in respiratory illnesses among elderly and immunocompromised patients (Falsey, 2007;Falsey, Hennessey et al., 2005). RSV induces a broad spectrum of clinical diseases ranging from otitis media to mild upper respiratory tract infection, acute laryngeo-tracheo-bronchitis and more severe lower respiratory tract infections, such as broncholitis and pneumonia. Several studies, both in vitro and in vivo, have suggested that the severity of RSV disease is the result of enhanced local inflammatory response. The pulmonary inflammation occurs in part through the ability of RSV-infected airway epithelial cells, the major target of infection, to synthesize chemokines, leading to the migration and activation of leucocytes at the site of infection (Garofalo & Haeberle, 2000). IL-8 is known to be a potent chemoattractant and activator for neutrophils, T cells, basophils, and primed eosinophils (Baggiolini, Dewald et al., 1994). High concentrations of IL-8 have been detected in nasal and bronchioalveolar lavage fluids of children with RSV respiratory infections (Sheeran, Jafri et al., 1999;Noah & Becker, 1993) and in middle ear effusions of children with viral otitis media (Chonmaitree, Patel et al., 1996). It is therefore likely that this cytokine plays a major role in the recruitment of inflammatory cells to the lung following infection by RSV.
Using the model of lung type II alveolar epithelial cells (A549), we have previously shown that RSV replication results in increased IL-8 gene expression and protein release (Garofalo, Sabry et al., 1996). Nuclear run-on assays have shown that the enhanced IL-8 synthesis was primarily due to increased gene transcription. Mutational analysis of the IL-8 promoter demonstrated that Nuclear Factor (NF)-κB binding was absolutely required for RSV-induced IL-8 gene transcription (Garofalo, Sabry et al., 1996). In addition, RSV-induced IL-8 activation also required the participation of a functional AP-1 binding site (Casola, Garofalo et al., 2000). The signaling intermediates leading to RSV-induced NF-κB and AP-1 activation have not been investigated. We have previously shown that in response to RSV infection, NF-κB translocation is mediated by two distinct pathways termed the canonical and crosstalk pathways (Jamaluddin, Casola et al., 1998;Liu, Li et al., 2008). The canonical pathway requires the “signalosome,” a multiprotein signaling complex composed of the catalytic kinases IKKα and β, and a third adapter subunit, termed IKKγ. Upon IKKγ association with RIG-I–MAVS, IKKβ is phosphorylated in its activation loop, an event that induces IκBα NH2-terminal phosphorylation, ubiquitylation, and degradation, releasing RelA to translocate into the nucleus. RSV infection induces IκBα proteolysis, which leads to NF-κB nuclear translocation and DNA-binding (Jamaluddin, Casola et al., 1998). Additionally, we found that RSV induces NF-κB by releasing p52 subunit from the p100 precursor through the cross-talk pathway, which involves IKKα–NF-κB-inducing kinase (NIK) activation (Liu, Li et al., 2008). Through coupling both the canonical and crosstalk pathways, RSV infection results in p65 release from several distinct cytoplasmic stores.
AP-1 activation occurs via phosphorylation by the c-jun N-terminal kinase (JNK), which belongs to the family of mitogen activated protein kinases (MAPKs), resulting in increased transactivation potential (Karin, 1995;Whitmarsh & Davis, 1996). The (MAP) kinase/extracellular signal regulated kinase kinase 1 (MEKK1) and TGF-β activating kinase 1 (TAK1) are serine/threonine kinases in the MAP kinase kinase kinase (MAPKKK) family that have been show to activate intracellular kinases such as p38 MAPK, JNK and IKKs, leading to NF-κB and AP-1 induction (Ninomiya-Tsuji, Kishimoto et al., 1999;Zhou, Tan et al., 2003).
In this study, we investigated the role of TAK1, IKK and JNK in NF-κB and AP-1 activation and in modulating IL-8 production upon RSV infection of airway epithelial cells. Our results show that RSV induced a time-dependent IKKβ activation and that IKKβ plays a fundamental role in RSV-induced NF-κB activation, as shown by the inhibition of NF-κB-dependent gene transcription and NF-κB nuclear translocation and DNA-binding in cells expressing a dominant negative mutant IKKβ or treated with the NEMO binding domain (NBD) peptide. Similarly, RSV infection induced JNK activity in airway epithelial cells and pharmacological inhibition of JNK kinase significantly reduced AP-1-driven gene transcription, leading to lower IL-8 mRNA levels and protein secretion in infected cells. Furthermore, we found that TAK1 is a major upstream kinase activating both NF-κB and AP-1 in response to RSV infection, as shown by overexpression of kinase inactive TAK1 or using cells lacking TAK1 expression, which demonstrated a significant reduction in RSV-induced NF-κB and AP-1 nuclear translocation and DNA-binding activity, as well as NF-κB-dependent gene expression.
RESULTS
IKKβ is necessary for RSV-induced NF-κB activation
In unstimulated cells, cytoplasmic NF-κB subunits, mainly p65/p50 dimers, interact with the inhibitory IκB molecules. The classical NF-κB activation cascade is initiated by the stimulus-induced phosphorylation of the IκBα inhibitor by IKKβ, that leads to IκBα degradation, liberation of the NF-κB dimers, nuclear accumulation, and transcription of the NF-κB target genes (Hayden & Ghosh, 2008). Although we have previously shown that RSV induces IκBα degradation, we did not directly investigate the role of IKKβ in RSV-induced NF-κB activation To determine if RSV infection of airway epithelial cells induced IKKβ activation, IKKβ was immunoprecipitated from control or RSV infected cells and tested for its ability to phosphorylate the amino terminus of GST-IκBα substrate (amino acids 1 to 54). RSV infection markedly increased IKKβ kinase activity for the GST-IκBα substrate, starting at 3 h post-infection (p.i.) and peaking between 6 and 12 h p.i. (Fig. 1A), with gradual decrease at later time points of infection (data not shown). The immunoprecipitated IKKβ failed to phosphorylate mutated GST-IκBα at all time points, indicating specificity of the assay.
Fig. 1. IKKβ regulates RSV-induced NF-κB activation.



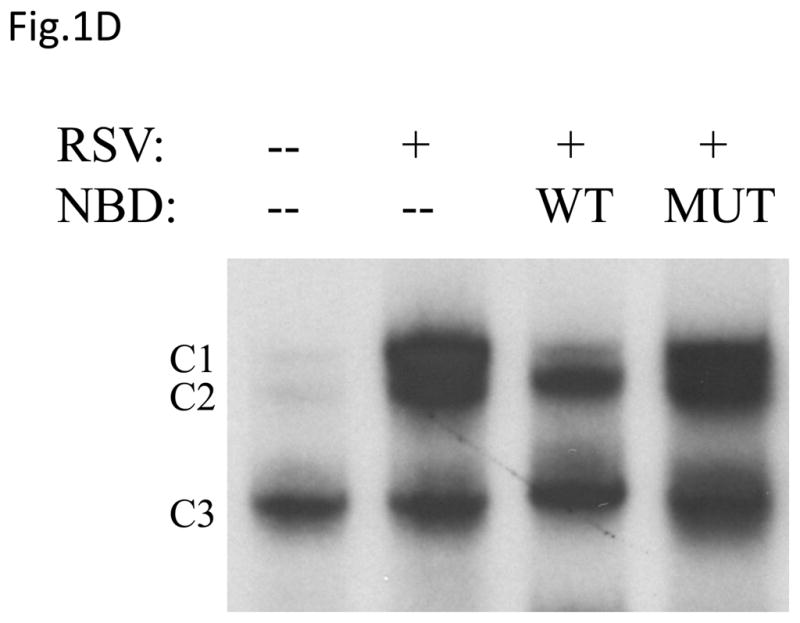
(A) Total cell lysates prepared from control and infected A549 cells were immunoprecipitated with an antibody against IKKβ for in vitro kinase assay using GST-IκBα wild type (WT) or a mutated form (MUT) as substrates. Samples were subjected to SDS-PAGE and transferred to PVDF membranes. Membranes were dried and exposed to an XAR film. A non-specific phosphorylated band is shown to demonstrate similar loading of the samples. N.S.: non-specific. (B) A549 cells were transiently transfected with a plasmid containing the human IL-8 promoter linked to the luciferase reporter gene and either a plasmid encoding dominant negative mutant IKKβ (SS≫AA), IKKβ (K44>M) or the empty vector pCMV2. The next day cells were infected with RSV and harvested at 24 h p.i. to measure luciferase activity. Uninfected plates served as controls. For each plate luciferase was normalized to the β-galactosidase reporter activity. Data are expressed as mean ± standard deviation of normalized luciferase activity. Data are representative of two independent experiments performed in triplicates, *P<0.05 relative to empty vector. (C and D) A549 cells were infected with RSV in the presence or absence of 200 μM NBD wild type (WT) or mutated (MUT) peptide and harvested at 24 h p.i. to prepare nuclear extracts. Nuclear proteins were used for Western Blot assays (C) and for binding to the IL-8 NF-κB site in EMSA (D). C stands for DNA-protein complex in the EMSA assay.
In order to determine whether IKKβ regulated RSV-induced NF-κB-dependent gene transcription, we used two IKKβ Dominant Negative (DN) mutants in gene reporter assays. A549 cells were co-transfected with a reporter plasmid containing the luciferase gene linked to the IL-8 promoter and plasmids expressing one of the two IKKβ DN, either the IKKβ [S177S181≫AA], in which the serine residues at position 177 and 181 have been replaced with alanine, or the IKKβ (K44>M), in which the lysine at residue 44 was replaced with methionine. Both mutants have been shown to inhibit TNF-α induced NF-κB mediated gene expression (Mercurio, Zhu et al., 1997). There was significant induction of IL-8 promoter activation in response to RSV infection, which was almost completely inhibited by the expression of either IKKβ DN mutants (Fig. 1B), indicating a fundamental role of this kinase in RSV-induced IL-8 gene transcription.
To confirm the role of IKKβ in viral-induced NF-κB activation, we treated airway epithelial cells with NBD peptide, a known inhibitor of IKKβ catalytic activity, which we have previously shown to block RSV-induced NF-κB activation in vivo (Haeberle, Casola et al., 2000). A549 cells were infected with RSV in the presence or absence of NBD wild type or mutated and harvested at 24 h p.i. to collect supernatants and prepare nuclear extracts. Our results show that NBD almost completely blocked p65 nuclear translocation (Fig.1C), whereas a mutant NBD peptide failed to do so, and it markedly inhibited p65 DNA-binding activity (Fig. 1D). Indeed, as shown in Fig. 1D, using an oligonucleotide corresponding to the IL-8 NF-κB site, a single nucleoprotein complex (C3) was primarily formed from nuclear extracts of uninfected cells, while two other complexes, C1 and C2, were faintly detected. RSV infection markedly increased the binding of C1 and C2, which represent p65 homodimer and p65-p50 heterodimers, respectively (Garofalo, Sabry et al., 1996). In cells treated with the NBD peptide there was a significant reduction in the formation of both RSV-inducible C1 and C2 (more pronounced for the C1 complex). In addition, cells treated with NBD showed a significantly reduced IL-8 secretion in response to RSV infection (2,500 pg in untreated versus 1,100 pg in treated cells). Altogether, these data indicate an essential role of IKKβ in RSV-induced NF-κB activation.
JNK activation regulates RSV-induced AP-1-dependent gene transcription
As stated before, RSV-induced IL-8 activation requires the participation of a functional AP-1 binding site (Casola, Garofalo et al., 2000). The AP-1 family of transcription factors consists of homodimers and heterodimers of Jun (c-Jun, JunB, JunD), Fos (c-Fos, FosB, Fra-1, Fra-2), or the activating transcription factor (ATF-2, ATF-3) proteins (Karin, Liu et al., 1997). We have previously shown that RSV infection induces the activation of several members of the AP-1 family, including c-Jun and ATF-2 (Casola, Garofalo et al., 2001). Overexpression of c-Jun dominant negative significantly inhibits IL-8 gene transcription (Casola A, personal communication), as well as RANTES gene transcription (Casola, Garofalo et al., 2001), indicating an important role of this family of transcription factors in RSV-induced gene expression. Regulation of AP-1 activation occurs at the level of its expression, as well as through post-translational modifications, specifically phosphorylation by the c-jun N-terminal kinase (JNK), which belongs to the mitogen-activated protein kinase family (MAPKs), resulting in increased transactivation potential of AP-1 (Karin, 1995;Whitmarsh & Davis, 1996). To determine whether RSV infection of A549 cells induced JNK kinase activity, we performed kinase assays. A549 cells were either mock- or RSV-infected and total cell lysates were immunoprecipitated using an anti-JNK antibody. The immunoprecipitates were then used to phosphorylate ATF-2 substrate and levels of phospho-ATF-2 were detected by Western Blot. RSV infection markedly increased JNK kinase activity in a time-dependent manner, as early as 3 h p.i. (Fig. 2A).
Fig 2. JNK regulates RSV induced AP-1 dependent gene transcription.





(A) Total cell lysates prepared from control and infected A549 cells were immunoprecipitated with an antibody against JNK for in vitro kinase assay using ATF2 as substrate. JNK activation was determined by levels of phosphorylated ATF2 by Western blot. (B) A549 cells were infected with RSV in the presence or absence of 20 μM of the JNK inhibitor SP600125 (SP) and cell supernatants were harvested at 24 h p.i. to measure IL-8 production by ELISA. Data are expressed as the mean + standard deviation of two different experiments performed in triplicate; *P < 0.05 relative to RSV alone. (C) Northern blot of IL-8 mRNA in A549 cells infected with RSV in the presence or absence of 20 μM SP600125. A549 cells were infected with RSV for 6, 12 and 24 h in the presence or absence of the JNK inhibitor. Total RNA was extracted from control and infected cells and 20 μg of RNA was fractionated on a 1.2% agarose-formaldehyde gel, transferred to nylon membrane, and hybridized to a radiolabeled IL-8 cDNA probe (top). The membrane was stripped and hybridized again with a probe against β-actin (bottom), to control for equal loading of the samples. (D and E) A549 cells were transiently transfected with a plasmid containing the human IL-8 promoter, linked to the luciferase reporter gene, and infected with RSV in the absence or presence of 20 μM SP600125 (left panel), or cotransfected with the IL-8 promoter and either a plasmid encoding JIP-1 or the empty vector pcDNA3 (right panel) and then infected with RSV (D). A549 cells were transiently transfected with a plasmid containing multimers of IL-8 NF-κB (NF-κB3) and AP-1 binding sites (AP-3) linked to the luciferase reporter gene and infected with RSV in the absence or presence of 20 μM SP600125 (E). At 24 h p.i., cells were harvested to measure luciferase activity. Uninfected plates served as controls. For each plate luciferase was normalized to the β-galactosidase activity. Data are expressed as mean ± standard deviation of normalized luciferase activity. Data are representative of two independent experiments performed in triplicate, *P<0.05 relative to RSV alone.
To determine the effect of inhibiting JNK activation on RSV-induced IL-8 secretion, A549 cells were infected with RSV in the presence or absence of the inhibitor SP600125 and cell supernatants were collected to measure IL-8 concentration by ELISA. There was a significant decrease in RSV-induced IL-8 secretion in cells treated with the inhibitor (Fig. 2B). Similarly, viral-induced IL-8 mRNA levels tested by Northern Blot assay (Fig. 2C), as well as IL-8 gene transcription measured by reporter gene assay (Fig. 2D, left panel), were markedly reduced in the presence of the JNK inhibitor. When A549 cells were transfected with plasmids containing multimers of either IL-8 NF-κB or AP-1 sites linked to the luciferase reporter gene, RSV-induced NF-κB-driven reporter activity was not significantly reduced by the presence of SP600125, which on the other hand greatly reduced the AP-1-driven promoter activation (Fig. 2E). To confirm the results obtained with the JNK inhibitor, A549 cells were co-transfected with a reporter plasmid containing the luciferase gene linked to the IL-8 promoter or multimers of the IL-8 AP-1 site and a plasmid expressing the JNK interacting protein (JIP)-1, a scaffold protein of JNK, whose overexpression leads to inhibition of JNK signal transduction pathway (Dickens, Rogers et al., 1997). Overexpression of JIP-1 significantly inhibited RSV-induced IL-8 promoter activation (Fig.2D, right panel), as well as AP-1 dependent gene transcription (data not shown), indicating that indeed JNK plays a major role in regulating RSV-induced AP-1 activation.
TAK1 Regulates NF-κB and AP-1 Activation Following RSV Infection
MEKK1 and TAK1 are serine/threonine kinases that have been show to activate JNK and IKKs, leading to NF-κB and AP-1 induction (Ninomiya-Tsuji, Kishimoto et al., 1999;Zhou, Tan et al., 2003). To determine whether TAK1 or MEKK1 would play a role in RSV-induced gene expression, A549 cells were transfected with the IL-8 promoter, linked to the luciferase reporter gene, together with various doses of plasmids expressing kinase inactive, also known as dominant negative mutant (DN) TAK1 or MEKK1, or the empty vector, and then infected with RSV for 24 h. Cells were harvested to measure luciferase activity. There was a significant reduction in RSV-induced IL-8 promoter activation in cells expressing TAK1 DN but not MEKK1 DN, compared to cells transfected with the empty vector (Fig.3A), suggesting that TAK1 plays a critical role in regulating RSV-induced IL-8 gene transcription.
Fig 3. TAK1 Regulates NF-κB and AP-1 Activation Following RSV Infection.





(A) A549 cells were transiently transfected with a plasmid containing the human IL-8 promoter and increasing concentrations of either TAK1 dominant negative (DN) or MEKK1 DN expression plasmids or their control vector (pcDNA3). Cells were infected with RSV and harvested at 24 h p.i. to measure luciferase activity. (B) A549 cells were transiently transfected with a plasmid containing multimers of either the IL-8 NF-κB (NF-κB3) or AP-1 binding sites (AP-3) linked to the luciferase reporter gene, and TAK1 DN plasmid or the empty vector (pCDNA3). Cells were infected and harvested at 24 h p.i. to measure luciferase activity. Uninfected plates served as controls. For each plate luciferase was normalized to the β-galactosidase activity. Data are expressed as mean ± standard deviation of normalized luciferase activity. Data are representative of two independent experiments performed in triplicate, *P<0.05 relative to empty vector. (C) Wild type (WT) and TAK1−/− MEFs were infected with RSV for 24 h or treated with TNFα (10 ng/ml) for 6 h. Cell culture supernatants were assayed for KC production by ELISA. Data is expressed as mean ± standard deviation of triplicate samples, *P<0.05 relative to WT. (D) Nuclear extracts were prepared from control and RSV-infected WT and TAK1−/− MEFs and probed for p65, phospho c-Jun and total c-Jun levels by Western Blot. Time p.i. (in hours) is shown on top. Membranes were stripped and reprobed with lamin B to control for equal loading of the samples. (E) Nuclear extracts were prepared from control and RSV-infected WT and TAK1−/− MEFs and used for binding to radiolabeled IL-8 NF-κB and AP-1 oligonucleotides in EMSA. Time p.i. (in hours) is shown on top. C stands for DNA-protein complex.
To investigate whether TAK1 controls both AP-1 and NF-κB activation, A549 cells were transfected with plasmids containing multimers of either IL-8 NF-κB or AP-1 sites, linked to the luciferase reporter gene, and a plasmid expressing TAK1 DN, and infected with RSV for 24 h. Expression of TAK1 DN significantly inhibited both AP-1- and NF-κB-dependent promoter activation (Fig.3B), suggesting that TAK1 modulates induction of both transcription factors.
To confirm the role of TAK1 in RSV-induced NF-κB and AP-1 activation, mouse embryonic fibroblasts (MEFs) from wild type (WT) and TAK1 deficient mice (TAK1−/−) were infected with RSV or treated with TNFα, as control. There was a robust induction of KC, a mouse homologue of human IL-8, whose expression is NF-κB dependent (Ohmori, Fukumoto et al., 1995), in response to RSV or TNFα stimulation in WT MEFs, which was significantly reduced in TAK1 −/− MEFs (Fig.3C). To determine whether the absence of TAK1 had an effect on p65 and c-Jun activation, nuclear extracts of RSV-infected WT MEFs and TAK1−/− MEFs were analyzed for p65 and phospho c-Jun levels by Western blot assay. Our results indicate that TAK1 deletion was associated with decreased p65, phospho c-Jun and total c-Jun nuclear translocation (Fig.3D). Finally, to investigate NF-κB and AP-1 DNA-binding activity in the absence of TAK1, nuclear extracts prepared from control and RSV-infected WT MEFs and TAK1−/− MEF cells were used for EMSA using oligonucleotides corresponding to the NF-κB and AP-1 binding sites of the IL-8 promoter. A single nucleoprotein complex (C3) was primarily formed from nuclear extracts of uninfected WT MEFs, while two other complexes, C1 and C2, were faintly detected. RSV infection markedly increased the binding of C1 and C2, starting at 6 h p.i., peaking at 12 h and decreasing in intensity at 24 h p.i. Both C1 and C2 formation was significantly reduced in nuclear extracts of TAK1−/− MEFs (Fig. 3E). Similarly, there was a single nucleoprotein complex (C1) formed from nuclear extracts of uninfected WT MEFs using the AP-1 binding site, which was enhanced by RSV infection and greatly reduced in nuclear extracts of RSV-infected TAK1−/− MEFs (Fig. 3E). We have previously shown that C1 represents the specific RSV-inducible complex formed on the IL-8 AP-1 binding site using nuclear extracts of infected airway epithelial cells (Casola, Garofalo et al., 2000). Taken together, these data indicate that TAK1 functions as a common upstream activator of both NF-κB and AP-1 pathway in the context of RSV infection of airway epithelial cells.
DISCUSSION
Pulmonary inflammation is a highly concerted series of events that includes the inducible expression of chemokines and adhesion molecules, which direct leukocyte transendothelial migration and movement through the extracellular matrix. Upon exposure to infectious agents or other agents like cigarette smoke, chemical pollutants and particulate matter, airway epithelial cells are able to secrete a variety of proinflammatory molecules, including chemokines, which are responsible for the selective recruitment of circulating leukocytes into the lung (Bacon & Schall, 1996;Zlotnik & Yoshie, 2000;Hashimoto, Gon et al., 2000;Jaspers, 1997). RSV is a potent stimulus for chemokine production in cultured human nasal, bronchial and alveolar epithelial cells (Garofalo, Sabry et al., 1996;Saito, Deskin et al., 1997)(Zhang Y, Luxon et al., 2001) as well as in an animal model of viral infection (Haeberle, Casola et al., 2004;Haeberle, Kuziel et al., 2001). Furthermore, in vivo studies have also demonstrated elevated chemokine concentrations in nasal washes of children infected with RSV (Garofalo, Olszewska-Pazdrak et al., 2001;Garofalo, Patti J et al., 2001), which correlates with the severity of the disease.
Differential levels of IL-8 gene expression are likely to play an important role in the pathogenesis of RSV-induced lung disease, as recently shown by the association of single gene polymorphisms of the IL-8 promoter with increased disease severity in children with RSV infection (Hull, Thomson et al., 2000;Puthothu, Krueger et al., 2006). Promoter deletion and mutagenesis experiments have shown the NF-κB and AP-1-binding sites play an important role in RSV-induced IL-8 gene transcription (Casola, Garofalo et al., 2000), however the pathway(s) leading to activation of these two important transcription factors in infected airway epithelial cells was not investigated. In this study we show that RSV-induced NF-κB and AP-1 activation proceeds through the canonical IKK complex and JNK, respectively, and that TAK1 functions as a common upstream of both pathways. Our finding that IKK is essential for RSV-induced NF-κB activation in airway epithelial cells is in agreement with previous work by our group showing that proteolysis of IκB subunits, mainly IκBα, is linked to NF-κB activation in infected cells (Jamaluddin, Casola et al., 1998). We have also shown that IKKβ plays a pivoral role in RSV-induced NF-κB activation in vivo, as functional blocking of IKKβ by IL-10 or the specific NBD peptide resulted in a marked attenuation of RSV-induced lung inflammation, due in part to the inhibition of inflammatory chemokine gene expression (Haeberle, Casola et al., 2004).
The observation that IKK is essential for RSV-induced NF-κB activation is not surprising, as most stimuli activate NF-κB through this kinase (Israel, 2000). Among viral infections, we have shown that rotavirus induces IKK activation in intestinal epithelial cells and overexpression of a dominant negative mutant of IKKβ greatly reduced rotavirus-induced IL-8 promoter activation and NF-κB driven transcription, indicating a major role of this kinase in rotavirus-induced NF-κB activation (Casola, Garofalo et al., 2002). Similarly, IKK is required to activate NF-κB in herpes virus (HSV)-infected cells (Mogensen, Melchjorsen et al., 2003).
Cells respond to a variety of stress conditions by activation of JNK, which regulates the activity of transcription factors of the AP-1 family (Karin, 1995;Wisdom, 1999). JNK activation has been shown to be critical in the innate response to acute viral infections, as AP-1 factors transactivate many antiviral and proinflammatory cytokine genes. For example, activation of JNK and p38 by influenza virus infection of lung epithelial cells has been linked to the induction of the proinflammatory cytokines TNF-α and RANTES (Kujime, Hashimoto et al., 2000;Lee, Cheung et al., 2005). JNK was also found to be activated in intestinal epithelial cells in response to rotavirus infection and regulate IL-8 and AP-1-driven gene transcription (Holloway & Coulson, 2006). Similarly, IL-8 secretion in response to varicella zoster virus infection was shown to be dependent in part on JNK activation (Desloges, Schubert et al., 2008). In line with these studies, our results also show that RSV strongly induced JNK activity in A549 cells and that viral-induced IL-8 expression and AP-1-driven gene transcription were greatly reduced in the presence of a specific JNK inhibitor. Interestingly, double-stranded RNA-induced JNK activation has been reported to negatively regulate TNFα induction in epithelial cells, possibly acting as a countermeasure to the proinflammatory signals generated during viral infections (Stewart, Kulkarni et al., 2006).
TAK1 is a serine/threonine kinase in the MAPKKK family (Yamaguchi, Shirakabe et al., 1995) whose activity is induced in response to a variety of stimuli, from cytokines to toll-like receptors and viral infections. Once activated, TAK1 regulates p38 MAPK, JNK and IKK (Landstrom, 2010). Among viruses, activation of NF-κB by HSV in macrophages has been shown to be dependent on IKK and the upstream kinases TAK1, MEKK1 and NIK (Mogensen, Melchjorsen et al., 2003). In fibroblasts and dendritic cells infected with Sendai virus, RIG-I activated NF-κB and p38 MAPK through a common upstream pathway, which involved TRAF2 and TAK1, and diverged downstream of TAK1 (Mikkelsen, Jensen et al., 2009). Similarly, TAK1 was found to be essential for retroviral Tax oncoprotein-stimulated persistent NF-κB activation, which is essential for HTLV-I-induced T-cell transformation (Wu & Sun, 2007). However, another study demonstrated that TAK1 was essential for Epstein Barr viral protein Latent Membrane Protein 1 (LMP1)-induced JNK activation but was dispensable for NF-κB activation (Uemura, Kajino et al., 2006). Our data indicate that TAK1 has a major role in regulating NF-κB and AP-1 activation in response to RSV, as in cells lacking TAK1 expression there was significantly reduced NF-κB and AP-1 nuclear translocation and DNA-binding activity, as well as NF-κB-dependent gene expression. The presence of residual NF-κB nuclear translocation in TAK1 −/− MEFs suggests that additional kinases are involved in RSV-induced IKK activation, possibly NIK, as we have previously shown that RSV infection rapidly activates the noncanonical NF-kappaB activation pathway in airway epithelial cells, prior to the more potent canonical pathway activation (Choudhary, Boldogh et al., 2005).
In conclusion, this study highlights the role of TAK1 in modulating viral-induced NF-κB and AP-1 activation and subsequent gene expression. Identification of pathways controlling viral-induced transcription factor activation is important for the design of novel approaches to treat pulmonary inflammation in the context of RSV infection.
MATERIAL AND METHODS
RSV preparation
The RSV A2 strain was grown in Hep-2 cells and purified by centrifugation on discontinuous sucrose gradients as described elsewhere (Ueba, 1978). The virus titer of the purified RSV pools was 8–9 log10 plaque forming units (PFU)/ml using a methylcellulose plaque assay. No contaminating cytokines were found in these sucrose-purified viral preparations (Patel, Kunimoto et al., 1995). LPS was not detected when assayed using the limulus hemocyanin agglutination assay. Virus pools were aliquoted, quick-frozen on dry ice/alcohol and stored at −70°C until used.
Cell culture and infection of epithelial cells with RSV
A549, human alveolar type II-like epithelial cells (ATCC, Manassas, VA), were maintained in F12K medium, containing 10% (v/v) FBS, 10 mM glutamine, 100 IU/ml penicillin and 100 μg/ml streptomycin. Cell monolayers were infected with RSV at multiplicity of infection (MOI) of 3, as described (Garofalo, Sabry et al., 1996). An equivalent amount of a 30% sucrose solution was added to uninfected A549 cells, as a control.
Kinase assays
To investigate IKKβ activation, A549 cells uninfected and infected with RSV for various time points were mechanically harvested in PBS and cells were collected in microcentrifuge tubes by centrifugation (1 min at 500 X g). Total cells extracts were prepared as previously described (Casola, Garofalo et al., 2002) and the protein content was measured. Five hundred micrograms of protein were gently rotated for 3 h at 4°C in the presence of 1 μg of anti-IKK-β antibody (Santa Cruz Biotechnology, Santa Cruz, CA). After adding 100 μl of sepharose protein-A beads (Sigma) the mixture was further incubated at 4°C for 3 h with gentle rotation. The immunoprecipitate was washed 3 times with RIPA buffer (50 mM TrisHCl, 150 mM NaCl, 1 mM EDTA, NP-40 1%, Deoxycholate 0.25%, 1mM Na3VO4, 1mM NaF, 1 μg/ml Leupeptin, 1 μg aprotinin, 1 mM PMSF) followed by 3 washes with 5X assay buffer [100 mM MOPS, 125 mM β-glycerophosphate (pH 7.2) plus 5 mM Na3VO4, 5 mM EGTA, 5 mM DTT, 1 μg/ml aprotinin, 100 μM ATP, 75 mM MgCl2]. The kinase reaction was performed in 40 μl kinase buffer in the presence of 10 μCi [γ32P] ATP (Du Pont NEN Research Products, Boston, MA) and 0.6 μg of the fusion protein GST-IκB-α (amino acid 1-54) or a mutated form of IκB-α in which the serine phosphorylation sites were mutated to alanine at position 32 and 36 (S32A, S36A). The reaction was incubated at 30°C for 30 min and was terminated by the addition of 20 μl 5x SDS-PAGE sample buffer. To visualize phosphorylated IκB-α, the reaction was subjected to 10% SDS-PAGE, transferred to a PVDF membrane and exposed to a XAR film (Eastman Kodak, Rochester, NY).
JNK kinase assay was performed according to the instruction of the kinase assay kit (Cell Signaling Technology Inc., Beverly, MA). Briefly, 200 μl of total cell lysates were incubated with an agarose-conjugate anti-JNK antibody and immunoprecipitated overnight. Immunocomplexes were washed and resuspended in kinase buffer containing 2 μg of recombinant ATF-2 and 200 μM ATP, as substrate. After 30 minutes, reactions were terminated by adding SDS sample buffer and samples were loaded on a 12% SDS-PAGE. Proteins were transferred to PVDF membrane and phosphorylated ATF2 was detected by Western blot.
Plasmid construction
The IL-8 Luc, containing the first -162 nucleotides of the IL-8 promoter, as well as the plasmid containing multiple copies of the IL-8 NF-κB or AP-1 site linked to the luciferase reporter gene, have been previously described (Casola, Garofalo et al., 2000;Casola, Garofalo et al., 2001;Casola, Henderson et al., 2002). The two plasmids expressing kinase inactive IKKβ Dominant Negative (DN) were a generous gift of Dr. Mercurio (Signal Pharmaceutics, San Diego, CA, (Mercurio, Zhu et al., 1997). The plasmids expressing MEKK1 DN and TAK1 DN were a generous gift of Søren R. Paludan, Aarhus University, Aarhus, Denmark. The control vector for MEKK1 DN and TAK1 DN was pCDNA3. The JNK interacting protein (JIP)-1 expression plasmid and its control vector (pcDNA3) were a generous gift of Dr. Roger Davis, University of Massachusetts, Worcester, MA.
Inhibitors
The NEMO binding domain (NBD) fusion peptide is an 11-amino-acid peptide spanning the region from T735 to E745 of IKKβ fused with a 17-amino-acid sequence of the Antennapedia homeodomain that has been previously described as a specific inhibitor of IKKβ catalytic activity (May, D'Acquisto et al., 2000). Based on the described sequence, the NBD peptide and a mutated control peptide were chemically synthesized in the Protein Chemistry Core at UTMB by 9-fluorenylmethoxy carbonyl chemistry and were purified by high-performance liquid chromatography with a C18 reverse phase column to 95% purity. The peptide sequence was confirmed by matrix assisted laser desorption ionization–time of flight mass spectrometry. Before use, the peptides were resuspended in 1 mM HCl to a stock solution of 10 mg/ml. Final concentrations were prepared in Dulbecco’s PBS, pH 7.2 (Haeberle, Casola et al., 2004).
SP600125 (Calbiochem) is a cell-permeable, selective and reversible inhibitor of JNK. The inhibition is competitive with respect to ATP and it exhibits over 300-fold greater selectivity for JNK as compared to other members of the MAP kinase family (Heo, Kim et al., 2004).
When inhibitors were used, cells were pretreated for one hour and then infected in the presence of the compound. Since SP600125 was diluted in DMSO, equal amount of diluent was added to untreated cells, as a control. Total number of cells, cell viability and viral replication, following inhibitor treatment, were measured by trypan blue exclusion and by plaque assay, respectively. No cell toxicity or differences in viral replication were observed for any compound at concentrations used for the experiments.
Luciferase Assay
Logarithmically growing A549 or HEK- 293 cells were transfected in triplicate in 60 mm dishes using Fugene 6 (Roche Diagnostic Corp., Indianapolis, Ind.). One μg of the reporter gene plasmid, 0.5 μg of β-galactosidase, varying quantities of control, IKKβ DN, MEKK1 or TAK1 DN expression plasmids were premixed with FuGene 6 and added to the cells in 3 ml of regular medium. The next morning, cells were infected with RSV and at 24 h p.i., cells were lysed to independently measure luciferase and β-galactosidase reporter activity, as previously described (Casola, Garofalo et al., 2000). Luciferase activity was normalized to the internal control β-galactosidase activity. Results are expressed in arbitrary units. All experiments were performed at least two to three times.
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts of uninfected and infected cells were prepared using hypotonic/nonionic detergent lysis as previously described (Bao, Indukuri et al., 2010). After the extraction, nuclear proteins were normalized by protein assay (Protein Reagent, Bio-Rad, Hercules, Calif.) and used to bind to duplex oligonucleotides corresponding to the IL-8 NF-κB or AP-1 binding site, as described previously (Garofalo, Sabry et al., 1996),(Casola, Garofalo et al., 2001). The resulting hybridized DNA–protein complexes were resolved using a 6% nondenaturing polyacrylamide gel in TBE buffer (22 mM Tris-HCl, 22 mM boric acid, 0.25 mM EDTA, pH 8.0). Gels were dried and exposed for autoradiography with Kodak XAR films at −70°C.
Western blotting
Total cell lysates were prepared by adding ice-cold lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1mM EGTA, 0.25% sodium deoxycholate, 1 mM Na3VO4, 1 mM NaF, 1% Triton X-100 and 1 μg/ml of aprotinin, leupeptin and pepstatin). After incubation on ice for 10 min, the lysates were collected and detergent insoluble materials were removed by centrifugation at 4°C at 14,000g. Cytoplasmic and nuclear extracts were prepared using hypotonic/nonionic detergent lysis, as previously described (Casola, Garofalo et al., 2000;Casola, Garofalo et al., 2001;Casola, Henderson et al., 2002). Proteins (20 to 50 μg per sample) were then boiled in 2X Laemmli buffer for 2 min and resolved on SDS-PAGE. Proteins were transferred for 6 h onto Hybond-ECL nitrocellulose membrane (Amersham Pharmacia Biotech, Piscataway NJ) and nonspecific binding sites were blocked by immersing the membrane in TBST blocking solution [10mM Tris-HCl, pH 7.6, 150 mM NaCl, 0.05% Tween-20 (v/v)] containing 5% skim milk powder or 5% bovine serum albumin for 30 minutes. After a short wash in TBST, the membranes were incubated with the primary antibody overnight at 4°C, followed by a host specific peroxidase-conjugated secondary antibody, diluted 1:10,000 in TBST for 30 min at room temperature. After washing, the proteins were detected using ECL (Amersham Pharmacia Biotech, Piscataway, NJ) according to manufacturer’s protocol. All primary and secondary antibodies used for Western blot were purchased from Santa Cruz Biotechnology, Santa Cruz, CA.
IL-8 and KC ELISA
Immunoreactive IL-8 and KC were quantified by ELISA (DuoSet, R&D Systems, Minneapolis, MN) following the manufacturer's protocol.
Northern Blot
Total RNA was extracted from control and infected A549 cells by the acid guanidium thiocyanate-phenol chloroform method (Chomczynski & Sacchi, 1987). Twenty micrograms of RNA were fractionated on a 1.2 % agarose-formaldehyde gel, transferred to nylon membranes and hybridized to a radiolabeled IL-8 cDNA, as previously described (Brasier, Jamaluddin et al., 1998;Garofalo, Sabry et al., 1996). After washing, membranes were exposed for autoradiography using Kodak XAR film at −70°C, using intensifying screens. After exposure, membranes were stripped and rehybridized with a β-actin probe for equal loading of the samples.
Statistical Analysis
Data from experiments involving multiple samples subject to each treatment, were analyzed by the Student Newman Keuls t-test for multiple pairwise comparisons. Results were considered significantly different at a p value <0.05.
Highlights.
IKKβ major kinase involved in RSV-induced NF-κB activation
JNK regulates AP-1-dependent gene transcription in RSV infection
TAK1 is a critical upstream signaling molecule for both pathways in RSV-infected cells
Acknowledgments
This work was supported by grants NIEHS 06676 and NIAID P01 062885. The authors would like to thank Sankar Gosh, Department of Microbiology and Immunology, Columbia University, NY, for the generous gift of WT and TAK1 −/− MEFs and Deborah Flaniken for her assistance in the manuscript submission.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bacon KB, Schall TJ. Chemokines as mediators of allergic inflammation. International Archives of Allergy & Immunology. 1996;109:97–109. doi: 10.1159/000237207. [DOI] [PubMed] [Google Scholar]
- Baggiolini M, Dewald B, Moser B. Interleukin-8 and related chemotactic cytokines -CXC and CC chemokines. Advances in Immunology. 1994;55:97–179. [PubMed] [Google Scholar]
- Bao X, Indukuri H, Liu T, Liao SL, Tian B, Brasier AR, Garofalo RP, Casola A. IKKepsilon modulates RSV-induced NF-kappaB-dependent gene transcription. Virology (New York NY) 2010;408:224–231. doi: 10.1016/j.virol.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasier AR, Jamaluddin M, Casola A, Duan W, Shen Q, Garofalo RP. A promoter recruitment mechanism for tumor necrosis factor-alpha-induced interleukin-8 transcription in type II pulmonary epithelial cells. Dependence on nuclear abundance of Rel A, NF-kappaB1, and c-Rel transcription factors. Journal of Biological Chemistry. 1998;273:3551–3561. doi: 10.1074/jbc.273.6.3551. [DOI] [PubMed] [Google Scholar]
- Casola A, Garofalo RP, Crawford SE, Estes MK, Mercurio F, Crowe SE, Brasier AR. Interleukin-8 gene regulation in intestinal epithelial cells infected with Rotavirus: role of viral-induced IkB kinase activation. Journal of Virology. 2002;298:8–19. doi: 10.1006/viro.2002.1475. [DOI] [PubMed] [Google Scholar]
- Casola A, Garofalo RP, Haeberle H, Elliott TF, Lin A, Jamaluddin M, Brasier AR. Multiple cis regulatory elements control RANTES promoter activity in alveolar epithelial cells infected with respiratory syncytial virus. Journal of Virology. 2001;75:6428–6439. doi: 10.1128/JVI.75.14.6428-6439.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casola A, Garofalo RP, Jamaluddin M, Vlahopoulos S, Brasier AR. Requirement of a novel upstream response element in RSV induction of interleukin-8 gene expression: stimulus-specific differences with cytokine activation. Journal of Immunology. 2000;164:5944–5951. doi: 10.4049/jimmunol.164.11.5944. [DOI] [PubMed] [Google Scholar]
- Casola A, Henderson A, Liu T, Garofalo RP, Brasier AR. Regulation of RANTES promoter activation in alveolar epithelial cells after cytokine stimulation. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1280–L1290. doi: 10.1152/ajplung.00162.2002. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Analytical Biochemistry. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Chonmaitree T, Patel JA, Sim T, Garofalo R, Uchida T, Sim T, Howie VM, Owen MJ. Role of leukotriene B4 and interleukin-8 in acute bacterial and viral otitis media. Ann Otol Rhinol Laryngol. 1996;105:968–974. doi: 10.1177/000348949610501207. [DOI] [PubMed] [Google Scholar]
- Choudhary S, Boldogh S, Garofalo R, Jamaluddin M, Brasier AR. Respiratory syncytial virus influences NF-kappaB-dependent gene expression through a novel pathway involving MAP3K14/NIK expression and nuclear complex formation with NF-kappaB2. The Journal of Virology. 2005;79:8948–8959. doi: 10.1128/JVI.79.14.8948-8959.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desloges N, Schubert C, Wolff MH, Rahaus M. Varicella-zoster virus infection induces the secretion of interleukin-8. Med Microbiol Immunol. 2008;197:277–284. doi: 10.1007/s00430-007-0060-3. [DOI] [PubMed] [Google Scholar]
- Dickens M, Rogers JS, Cavanagh J, Raitano A, Xia Z, Halpern JR, Greenberg ME, Sawyers CL, Davis RJ. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science (Washington DC) 1997;277:693–696. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- Falsey AR. Respiratory syncytial virus infection in adults. Semin Respir Crit Care Med. 2007;28:171–181. doi: 10.1055/s-2007-976489. [DOI] [PubMed] [Google Scholar]
- Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE. Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med. 2005;352:1749–1759. doi: 10.1056/NEJMoa043951. [DOI] [PubMed] [Google Scholar]
- Garofalo RP, Haeberle H. Epithelial regulation of innate immunity to respiratory syncytial virus. American Journal of Respiratory Cell and Molecular Biology. 2000;23:581–585. doi: 10.1165/ajrcmb.23.5.f204. [DOI] [PubMed] [Google Scholar]
- Garofalo RP, Olszewska-Pazdrak B, Ogra PL, Welliver RC. Beta-chemokines in nasal secretion of infants with respiratory syncytial virus-induced upper respiratory illness and bronchiolitis. Pediatric Asthma Allergy and Immunology. 2001;15:89–96. [Google Scholar]
- Garofalo RP, Patti J, Hintz KA, Hill V, Ogra PL, Welliver RC. Macrophage inflammatory protein 1-alpha, and not T-helper type 2 cytokines, is associated with severe forms of bronchiolitis. Journal of Infectious Diseases. 2001;184:393–399. doi: 10.1086/322788. [DOI] [PubMed] [Google Scholar]
- Garofalo RP, Sabry M, Jamaluddin M, Yu RK, Casola A, Ogra PL, Brasier AR. Transcriptional activation of the interleukin-8 gene by respiratory syncytial virus infection in alveolar epithelial cells: Nuclear translocation of the RelA transcription factor as a mechanism producing airway mucosal inflammation. Journal of Virology. 1996;70:8773–8781. doi: 10.1128/jvi.70.12.8773-8781.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeberle HA, Casola A, Dieterich H-J, Brasier AR, Garofalo RP. Respiratory Syncytial Virus-inducible activation of the transcription factor NF-kB mediatres pulmonary inflammation. Pediatr Res. 2000;47:17A. Ref Type: Abstract. [Google Scholar]
- Haeberle HA, Casola A, Gatalica Z, Petronella S, Dieterich HJ, Ernst PB, Brasier AR, Garofalo RP. IkappaB Kinase Is a Critical Regulator of Chemokine Expression and Lung Inflammation in Respiratory Syncytial Virus Infection. The Journal of Virology. 2004;78:2232–2241. doi: 10.1128/JVI.78.5.2232-2241.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeberle HA, Kuziel WA, Dieterich HJ, Casola A, Gatalica Z, Garofalo RP. Inducible expression of inflammatory chemokines in respiratory syncytial virus-infected mice: role of MIP-1alpha in lung pathology. Journal of Virology. 2001;75:878–890. doi: 10.1128/JVI.75.2.878-890.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CB. Respiratory syncytial virus and parainfluenza virus. N Engl J Med. 2001;344:1917–1928. doi: 10.1056/NEJM200106213442507. [DOI] [PubMed] [Google Scholar]
- Hashimoto S, Gon Y, Takeshita I, Matsumoto K, Jibiki I, Takizawa H, Kudoh S, Horie T. Diesel exhaust particles activate p38 MAP kinase to produce interleukin 8 and RANTES by human bronchial epithelial cells and N-acetylcysteine p38 MAP kinase activation. Am J Respir Crit Care Med. 2000;161:280–285. doi: 10.1164/ajrccm.161.1.9904110. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell (Cambridge MA) 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Heo YS, Kim SK, Seo CI, Kim YK, Sung BJ, Lee HS, Lee JI, Park SY, Kim JH, Hwang KY, Hyun YL, Jeon YH, Ro S, Cho JM, Lee TG, Yang CH. Structural basis for the selective inhibition of JNK1 by the scaffolding protein JIP1 and SP600125. EMBO J. 2004;23:2185–2195. doi: 10.1038/sj.emboj.7600212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway G, Coulson BS. Rotavirus activates JNK and p38 signaling pathways in intestinal cells, leading to AP-1-driven transcriptional responses and enhanced virus replication. The Journal of Virology. 2006;80:10624–10633. doi: 10.1128/JVI.00390-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull J, Thomson A, Kwiatkowski D. Association of respiratory syncytial virus bronchiolitis with the interleukin 8 gene region in UK families. Thorax. 2000;55:1023–1027. doi: 10.1136/thorax.55.12.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel A. The IKK complex: an integrator of all signals that activate NF-kappaB? Trends Cell Biol. 2000;10:129–133. doi: 10.1016/s0962-8924(00)01729-3. [DOI] [PubMed] [Google Scholar]
- Jamaluddin M, Casola A, Garofalo RP, Han Y, Elliott T, Ogra PL, Brasier AR. The major component of IkBa proteolysis occurs independently of the proteasome pathway in Respiratory Syncytial Virus-infected pulmonary epithelial cells. Journal of Virology. 1998;72:4849–4857. doi: 10.1128/jvi.72.6.4849-4857.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspers Ozone induced IL-8 expression and transcription factor binding in respiratory epithelial cells. American Journal of Physiology. 1997;272:L504–L511. doi: 10.1152/ajplung.1997.272.3.L504. [DOI] [PubMed] [Google Scholar]
- Karin M. The regulation of AP-1 activity by mitogen-activated protein kinase. Journal of Biological Chemistry. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9:240–246. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- Kujime K, Hashimoto S, Gon Y, Shimizu K, Horie T. p38 mitogen-activated protein kinase and c-jun-NH2-terminal kinase regulate RANTES production by influenza virus-infected human bronchial epithelial cells. Journal of Immunology (Baltimore MD) 2000;164:3222–3228. doi: 10.4049/jimmunol.164.6.3222. [DOI] [PubMed] [Google Scholar]
- Landstrom M. The TAK1-TRAF6 signalling pathway. Int J Biochem Cell Biol. 2010;42:585–589. doi: 10.1016/j.biocel.2009.12.023. [DOI] [PubMed] [Google Scholar]
- Lee DC, Cheung CY, Law AH, Mok CK, Peiris M, Lau AS. p38 mitogen-activated protein kinase-dependent hyperinduction of tumor necrosis factor alpha expression in response to avian influenza virus H5N1. The Journal of Virology. 2005;79:10147–10154. doi: 10.1128/JVI.79.16.10147-10154.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Li K, Garofalo RP, Brasier AR. Respiratory syncytial virus induces RelA release from cytoplasmic 100-kDa NF-kappa B2 complexes via a novel retinoic acid-inducible gene-I{middle dot}NF-kappa B-inducing kinase signaling pathway. Journal of Biological Chemistry. 2008;283:23169–23178. doi: 10.1074/jbc.M802729200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May MJ, D'Acquisto F, Madge LA, Glockner J, Pober JS, Ghosh S. Selective inhibition of NF-kappaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science (Washington DC) 2000;289:1550–1554. doi: 10.1126/science.289.5484.1550. [DOI] [PubMed] [Google Scholar]
- Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, Rao A. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science (Washington DC) 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- Mikkelsen SS, Jensen SB, Chiliveru S, Melchjorsen J, Julkunen I, Gaestel M, Arthur JS, Flavell RA, Ghosh S, Paludan SR. RIG-I-mediated activation of p38 MAPK is essential for viral induction of interferon and activation of dendritic cells: dependence on TRAF2 and TAK1. Journal of Biological Chemistry. 2009;284:10774–10782. doi: 10.1074/jbc.M807272200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogensen TH, Melchjorsen J, Hollsberg P, Paludan SR. Activation of NF-kappa B in virus-infected macrophages is dependent on mitochondrial oxidative stress and intracellular calcium: downstream involvement of the kinases TGF-beta-activated kinase 1, mitogen-activated kinase/extracellular signal-regulated kinase kinase 1, and I kappa B kinase. Journal of Immunology. 2003;170:6224–6233. doi: 10.4049/jimmunol.170.12.6224. [DOI] [PubMed] [Google Scholar]
- Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K. The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature (London) 1999;398:252–256. doi: 10.1038/18465. [DOI] [PubMed] [Google Scholar]
- Noah TL, Becker S. Respiratory syncytial virus-induced cytokine production by a human bronchial epithelial cell line. American Journal of Physiology. 1993;265:L472–L478. doi: 10.1152/ajplung.1993.265.5.L472. [DOI] [PubMed] [Google Scholar]
- Ohmori Y, Fukumoto S, Hamilton TA. Two structurally distinct kappa B sequence motifs cooperatively control LPS-induced KC gene transcription in mouse macrophages. Journal of Immunology. 1995;155:3593–3600. [PubMed] [Google Scholar]
- Patel JA, Kunimoto M, Sim TC, Garofalo R, Eliott T, Baron S, Ruuskanen O, Chonmaitree T, Ogra PL, Schmalstieg F. Interleukin-1 alpha mediates the enhanced expression of intercellular adhesion molecule-1 in pulmonary epithelial cells infected with respiratory syncytial virus. American Journal of Respiratory Cell & Molecular Biology. 1995;13:602–609. doi: 10.1165/ajrcmb.13.5.7576697. [DOI] [PubMed] [Google Scholar]
- Puthothu B, Krueger M, Heinze J, Forster J, Heinzmann A. Impact of IL8 and IL8-receptor alpha polymorphisms on the genetics of bronchial asthma and severe RSV infections. Clin Mol Allergy. 2006;4:2. doi: 10.1186/1476-7961-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Deskin RW, Casola A, Haeberle H, Olszewska B, Ernst PB, Alam R, Ogra PL, Garofalo R. Respiratory syncytial virus induces selective production of the chemokine RANTES by upper airway epithelial cells. Journal of Infectious Diseases. 1997;175:497–504. doi: 10.1093/infdis/175.3.497. [DOI] [PubMed] [Google Scholar]
- Sheeran P, Jafri H, Carubelli C, Saavedra J, Johnson C, Krisher K, Sanchez PJ, Ramilio mO. Elevated cytokine concentrations in the nasopharyngeal and tracheal secretions of children with respiratory syncytial virus disease. Pediatrics Infectious Diseases Journal. 1999;18:115–122. doi: 10.1097/00006454-199902000-00007. [DOI] [PubMed] [Google Scholar]
- Stewart MJ, Kulkarni SB, Meusel TR, Imani F. c-Jun N-terminal kinase negatively regulates dsRNA and RSV induction of tumor necrosis factor-alpha transcription in human epithelial cells. J Interferon Cytokine Res. 2006;26:521–533. doi: 10.1089/jir.2006.26.521. [DOI] [PubMed] [Google Scholar]
- Ueba O. Respiratory syncytial virus: I. concentration and purification of the infectious virus. Acta Medica Okayama. 1978;32:265–272. [PubMed] [Google Scholar]
- Uemura N, Kajino T, Sanjo H, Sato S, Akira S, Matsumoto K, Ninomiya-Tsuji J. TAK1 is a component of the Epstein-Barr virus LMP1 complex and is essential for activation of JNK but not of NF-kappaB. Journal of Biological Chemistry. 2006;281:7863–7872. doi: 10.1074/jbc.M509834200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmarsh AJ, Davis RJ. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J Mol Med. 1996;74:589–607. doi: 10.1007/s001090050063. [DOI] [PubMed] [Google Scholar]
- Wisdom R. AP-1: one switch for many signals. Exp Cell Res. 1999;253:180–185. doi: 10.1006/excr.1999.4685. [DOI] [PubMed] [Google Scholar]
- Wu X, Sun SC. Retroviral oncoprotein Tax deregulates NF-kappaB by activating Tak1 and mediating the physical association of Tak1-IKK. EMBO Rep. 2007;8:510–515. doi: 10.1038/sj.embor.7400931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, Taniguchi T, Nishida E, Matsumoto K. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science (Washington DC) 1995;270:2008–2011. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Luxon B, Casola A, Garofalo RP, Jamaluddin M, Brasier AR. Expression of RSV-induced chemokine gene networks in lower airway epithelial cells revealed by cDNA microarrays. Journal of Virology. 2001;75:9044–9058. doi: 10.1128/JVI.75.19.9044-9058.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Tan A, Iasvovskaia S, Li J, Lin A, Hershenson MB. Ras and mitogen-activated protein kinase kinase kinase-1 coregulate activator protein-1- and nuclear factor-kappaB-mediated gene expression in airway epithelial cells. Am J Respir Cell Mol Biol. 2003;28:762–769. doi: 10.1165/rcmb.2002-0261OC. [DOI] [PubMed] [Google Scholar]
- Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–127. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]