

Abstract
We have previously reported that human cytomegalovirus (HCMV) infection induces large-scale changes to host cell glycolytic, nucleic acid, and phospholipid metabolism. Here we explore the viral mechanisms involved in fatty acid biosynthetic activation. Our results indicate that HCMV targets ACC1, the rate-limiting enzyme of fatty acid biosynthesis, through multiple mechanisms. HCMV infection was found to activate ACC1 expression, increasing the abundance of both ACC1 mRNA and protein. Viral gene expression but not viral DNA replication was found to be necessary for HCMV-mediated induction of ACC1 levels. HCMV infection was also found to increase the proteolytic processing of SREBP-2, a transcription factor whose proteolytic cleavage is known to activate a variety of phospholipid metabolic genes. Processing of SREBP-2 was found to be dependent on mTOR activity; pharmaceutical inhibition of mTOR blocked HCMV-induced SREBP-2 processing and prevented the induction of fatty acid biosynthesis and ACC1 expression. Independent of the increases in ACC1 expression, HCMV infection also induced ACC1's enzymatic activity. Inhibition of ACC1 through either RNA interference (RNAi) or inhibitor treatment was found to attenuate HCMV replication, and HCMV replication was sensitive to ACC1 inhibition even at the later stages of infection, suggesting a late role for fatty acid biosynthesis during HCMV replication. These findings indicate that HCMV infection actively modulates numerous functional aspects of a key metabolic regulatory enzyme that is important for high-titer viral replication.
INTRODUCTION
For decades, numerous reports have indicated that infection with a wide variety of evolutionarily divergent viruses results in a general activation of host cell metabolism (7, 15, 16, 26, 29, 39, 42). This metabolic activation can be therapeutically useful; for example, increased or divergent nucleotide metabolism is often clinically targeted to treat various viral infections, such as hepatitis B virus, HIV, human cytomegalovirus (HCMV), and herpes simplex virus (3, 13, 18, 28). Despite the successes of these antiviral strategies, relatively little is known about the specific metabolic activities induced by viral infection and the mechanisms responsible for their activation. Given the viral reliance on the host cell metabolic network for the production of viral progeny, elucidating the mechanisms of viral metabolic manipulation will likely highlight novel avenues for therapeutic development.
HCMV is a widespread opportunistic pathogen that can cause severe disease in various immunosuppressed populations, including the elderly, cancer patients receiving immunosuppressive chemotherapy, transplant recipients, and AIDS patients (17, 38). Additionally, congenital HCMV infection occurs in 1 to 2% of all live births (3) and can result in multiple organ system abnormalities, with central nervous system damage occurring in the majority of symptomatic newborns (11, 38).
HCMV is a large, double-stranded DNA virus that contains an ∼240-kb genome encoding over 200 open reading frames (ORFs). The HCMV genome is encased within a protein capsid which itself is surrounded by a protein layer named the tegument. The capsid and tegument are then enclosed within a phospholipid envelope containing glycoproteins.
We have previously found that infection with HCMV induces numerous changes to the host cell metabolic network (34, 35). Specifically, HCMV induces much of central carbon metabolism, including glycolysis and the tricarboxylic acid (TCA) cycle, but reduces the activity of the pentose-phosphate pathway (35). In addition, HCMV infection results in notable increases in phospholipid biosynthesis, which when inhibited results in attenuated HCMV replication (35).
Acetyl-coenzyme A (CoA) carboxylase (ACC) catalyzes the first committed step in fatty acid biosynthesis, the carboxylation of acetyl-CoA to form malonyl-CoA. There are two major isoforms of ACC, ACC1 (265 kDa) and ACC2 (280 kDa), which are encoded by two distinct genes (1, 2). ACC1 is ubiquitously expressed, whereas ACC2 is expressed primarily in strongly fatty acid-oxidative tissues, such as skeletal and heart muscle (reviewed in reference 33). ACC1 is heavily regulated by diverse upstream signals. For example, its activity is inhibited by signal transduction cascades that result in phosphorylation at ser79 (reviewed in reference 9).
Here, we have begun to analyze the mechanisms responsible for HCMV-induced activation of fatty acid biosynthesis. We find that HCMV infection increases the expression and specific activity of ACC1. Taken together, our data suggest that HCMV infection targets a cellular metabolic enzyme whose activity is important for viral replication. We propose that the interplay between viruses and the host cell metabolic machinery is a fundamental host-pathogen interaction whose continued elucidation may prove fertile ground for novel therapeutic development.
MATERIALS AND METHODS
Biological reagents and cell culture.
MRC-5 fibroblasts were maintained in Dulbecco's modified Eagle medium (DMEM) (Gibco) containing 10% fetal bovine serum and 4.5 g liter−1 glucose. Prior to infection, fibroblasts were grown to confluence, resulting in ∼3.2 × 104 cells per cm2. In all infections, viral inocula were added to cells for a 2-h adsorption period and then aspirated. In experiments utilizing UV-irradiated virus, the viral inocula were exposed to 254 nm light at various doses (indicated in Fig. 3) in a Stratalinker UV cross-linker 2400 prior to infection. In the event of serum starvation, serum-containing medium was removed and serum-free medium added. Cells were then maintained in serum-free DMEM for 24 h prior to infection. For experiments involving measurement of viral titers via plaque assay, unbound virus was inactivated through a sodium citrate wash (40 mM sodium citrate, 10 mM KCl, and 135 mM NaCl, pH 3.0) followed by a DMEM wash immediately following viral adsorption. Unless indicated otherwise, BADwt was the strain utilized in all studies. BADwt was derived from a bacterial artificial chromosome (BAC) clone of the AD169 laboratory strain of HCMV (52). A green fluorescent protein (GFP)-expressing virus (HCMV-GFP), BADsubUL21.5, expresses GFP under the control of the simian virus 40 (SV40) early promoter and replicates to similar titers and with the same growth kinetics as wild-type virus (49).
Fig. 3.

(A) Analysis of the delivery of pp65 upon infection with UV-irradiated virus. Serum-starved MRC-5 fibroblasts were infected with HCMV (MOI of 3.0) that had been UV irradiated (254 nm) with 0, 240, or 480 mJ/cm2 prior to infection. Cells were fixed and processed for immunofluorescence at 4 hpi using a pp65-specific monoclonal antibody (green). (B) Accumulation of ACC1 protein after infection with UV-irradiated virus. Mock-infected (M) or HCMV-infected (H) cells were treated as described above and harvested for protein analysis at 72 hpi. The abundance of ACC1, α-tubulin, IE2, and UL26 was measured by Western analyses. (C) Accumulation of phosphorylated ACC1 and ACC1 protein after treatment with PAA. Serum-starved MRC-5 fibroblasts were mock infected or infected with HCMV (MOI of 3.0). After adsorption, the cellular medium was removed and fresh medium containing 50 μg/ml PAA was added. Cells were harvested for protein analysis at 72 hpi. The abundance of phosphorylated ACC1, ACC1, and α-tubulin was measured by Western analysis.
Antibodies used were specific for ACC (Cell Signaling), phospho-ACC (ser79) (Cell Signaling), α-tubulin (Epitomics), polyclonal SREBP-2 (Abcam), monoclonal SREBP-2 (MBL), and ATP-dependent citrate lyase (ACL) (Cell Signaling). Monoclonal antibodies to the following viral proteins were also utilized: IE2 (T. Shenk laboratory, unpublished), UL26 (36), and pp65 (37).
Analysis of RNA and protein.
RNA accumulation was assayed by quantitative real-time PCR. RNA was prepared using Trizol (Invitrogen) as recommended by the manufacturer. cDNA synthesis was performed utilizing SuperScript First-Strand (Invitrogen) reagents with random hexamers as per the manufacturer's protocol. Quantitative PCR (qPCR) was performed using Fast SYBR green PCR master mix, 7500 Fast real-time PCR system sequence detection, and 7500 software version 2.0.1 (Applied Biosystems) following the manufacturer's instructions. Transcripts were quantified with specific primer pairs as follows: the ACC1 transcript with 5′ TTAACAGCTGTGGAGTCTGGCTGT 3′ and 5′ AACACTCGATGGAGTTTCTCGCCT 3′, the ACC2 transcript with 5′ GGTGCTTATTGCCAACAACGGGAT 3′ and 5′ TCTTGATGTACTCTGCGTTGGCCT 3′, the ACL transcript with 5′ TGACAGCACCATGGAGACCATGAA 3′ and 5′ TTGCCAATCTTAAAGCACCCAGGC 3′, the fatty acid synthase (FAS) transcript with 5′ AGAACTTGCAGGAGTTCTGGGACA 3′ and 5′ TCCGAAGAAGGAGGCATCAAACCT 3′, and the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) transcript, used for normalization, with 5′ CATGTTCGTCATGGGTGTGAACCA 3′ and ATGGCATGGACTGTGGTCATGAGT 3′. Quantitative PCR was performed using the ΔΔCT (threshold cycle) method, with GAPDH as a control.
Protein accumulation was assayed by Western blotting. Cells were washed with phosphate-buffered saline (PBS), scraped, and solubilized in disruption buffer containing 50 mM Tris (pH 7.0), 2% SDS, 5% 2-mercaptoethanol, and 2.75% sucrose. Resulting extracts were sonicated, boiled for 5 min, and centrifuged briefly at 14,000 × g to pellet insoluble material. Extracts were then subjected to electrophoresis in an SDS-8% or -10% polyacrylamide gel and transferred to a nitrocellulose sheet. Blots were then stained with Ponceau S to ensure equivalent protein loading and transfer, blocked by incubation in 5% milk, and reacted with primary and subsequently secondary antibodies. Protein bands were visualized through enhanced chemifluorescent detection, according to the instructions of the manufacturer (Amersham), and signals were obtained with a Typhoon 9410 phosphorimager. For SREBP-2 Western analysis, protein bands were visualized using an ECL system (Pierce) and scientific imaging film (Kodak). In certain instances, the brightness and contrast of the resulting images were adjusted in Microsoft PowerPoint to improve image clarity. All such processing was applied uniformly to the entire image.
For analysis of the phospho-ACC (ser79) antibody, fibroblasts were scraped in phosphatase buffer containing 50 mM Tris-HCl, pH 7.9, 100 mM NaCl, 10 mM MgCl2, 1 mM dithiothreitol, and 1% Triton X-100 with or without phosphatase inhibitors (10 mM NaF, 100 μM sodium orthovanadate). Cells were then sonicated and the resulting extracts incubated at 37°C with or without 200 units of calf intestinal phosphatase (CIP) (New England BioLabs) for 1 h. The sample volume was then brought up to 250 μl with disruption buffer, and samples were processed for Western analysis as described above.
For immunofluorescence, fibroblasts were grown on glass coverslips. At various times postinfection, cells were washed once with PBS, fixed with 2% paraformaldehyde in PBS for 15 min, and washed three times with PBS. Cells were subsequently blocked by overnight incubation in PBS containing 2% bovine serum albumin (BSA), 5% goat serum, 5% human serum, and 0.3% Triton X-100. We found that blocking with human serum is essential to prevent artifactual staining of the viral assembly zone. Initial experiments were performed without human serum but containing goat serum in the blocking solution, which led to assembly zone staining (see Fig. S1 in the supplemental material). This artifactual staining was likely the result of HCMV-encoded Fc receptor binding nonspecifically to antibody (4, 10). Cells were incubated with primary antibody diluted in PBS plus 0.3% Triton X-100 overnight, washed three times in PBS, incubated with fluorochrome-conjugated secondary antibody, and washed again three times with PBS. Coverslips were mounted in SlowFade Gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI) (Molecular Probes). Confocal images were captured with an FV1000 Olympus laser scanning confocal microscope. For Fig. 6, all images were captured under identical confocal settings.
Fig. 6.

Time course of ACC1 localization. Serum-starved MRC-5 fibroblasts were mock infected or infected with HCMV (MOI of 3.0). Cells were fixed and processed for confocal immunofluorescence analysis at 24, 48, and 72 hpi using an ACC1-specific antibody (green). DNA was stained with DAPI (blue). All fields were captured with identical confocal settings.
RNAi assay.
The following small interfering RNA (siRNA) duplexes were obtained from Dharmacon Research. For ACC1 targeting, 5′ CAAUGGCAUUGCAGCAGUG 3′ and its complementary sequence were utilized. For a non-protein-targeting control, 5′ CGUAAGCGACAUACUUACAUU 3′ and its complement were utilized. MRC-5 fibroblasts at 30% confluence were transfected with 75 nM RNA interference (RNAi) duplex using Oligofectamine (Invitrogen) as per the manufacturer's instructions.
Pharmaceuticals.
5-Tetradecyloxy-2-furoic acid (TOFA) (Cayman) was maintained as a 5 mg ml−1 stock in dimethyl sulfoxide (DMSO). Potential toxicity of TOFA was analyzed by a Live/Dead assay (MGT) according to the manufacturer's protocol. Phosphonoacetic acid (PAA) (MP Biomedicals) was maintained as a 20 mg ml−1 stock in water. Torin1 was maintained as a 250 μM stock in DMSO.
Measurement of acetyl-CoA carboxylase activity.
ACC activity in fibroblasts was evaluated by measuring the relative amount of malonyl-CoA produced during a given time via liquid chromatography-tandem mass spectrometry (LC–MS-MS). The ACC assays were conducted largely as reported previously (30), with the exception that malonyl-CoA production was detected by mass spectrometry instead of fixed radioactivity. For the ACC assay, fibroblasts were plated in 15-cm dishes at 5 × 106 cells dish−1. Cells were scraped in PBS, pelleted by low-speed centrifugation, and solubilized in 200 μl reaction buffer containing 20 mM HEPES, pH 7.4, 10 mM MgCl2, 10 mM sodium citrate, 5 mM sodium bicarbonate, 0.5% DMSO, and 0.038% fatty acid-free BSA. To lyse cells, samples were frozen at −80°C and thawed at 37°C a total of three times. Samples were then centrifuged at 13,000 × g for 10 min, and the supernatant was preincubated at 37°C for 10 min. To begin the reaction, 3.75 mM ATP and 9.3 μM acetyl-CoA (final concentrations) were simultaneously added to the sample, and the sample was incubated at 37°C. Twenty-microliter aliquots were then taken at 30, 60, and 120 s postincubation. Sample aliquots were quenched with 120 μl 80% methanol (MeOH), centrifuged at 13,000 × g for 5 min, and analyzed via LC–MS-MS. The LC–MS-MS conditions were as follows. LC separation was coupled by negative-mode electrospray ionization (ESI) to a triple-quadrupole mass spectrometer operating in multiple reaction monitoring mode. The LC method was reverse-phase chromatography using a Synergi 4-μm Hydro-RP 80-Å column coupled to a SecurityGuard guard cartridge (Phenomenex). LC solvents were as follows: solvent A, 100% methanol; solvent B, 10 mM tributylamine and 15 mM acetic acid in 97:3 water-methanol. The LC method parameters were as follows (t in minutes): t = 0, 15% A; t = 0.5, 15% A; t = 4, 97% A; t = 5, 97% A; t = 5.1, 15% A; t = 7, 15% A. For LC, we used an LC-20 AD high-pressure liquid chromatography (HPLC) system (Shimadzu) with an autosampler temperature of 4°C, oven temperature of 40°C, and injection volume of 10 μl. For MS, we used a TSQ Quantum Ultra triple-quadrupole mass spectrometer (Thermo Fisher Scientific). The mass spectrometer parameters for malonyl-CoA in negative mode were a 12C parent mass of 852 m/z, a collision cell energy of 24 eV, and a product mass of 808 m/z.
The resulting ACC activity data were fit to a single exponential curve of product formation, P(t) = Pmax(1−e−kt). Initial velocities of product production, k × Pmax, were plotted as ACC activity.
Measurement of fatty acid biosynthesis.
Fatty acid biosynthesis was evaluated largely as indicated previously (21). Briefly, fatty acid biosynthesis in fibroblasts was assayed by measuring the incorporation of [1-14C]acetic acid (NEN Radiochemicals) into cellular lipids. Fibroblasts were plated in 12-well plates at 1.2 × 105 cells well−1. To begin the assay, 500 μl of DMEM containing 1 μCi of 56.2 mCi mmol−1 [1-14C]acetic acid was added to each well, and cells were incubated at 37°C for 2 h. After incubation, cells were washed with PBS and dissolved in 0.5 ml of 0.2 M KOH. Samples were then saponified by the addition of 0.3 ml of 50% KOH and 2.0 ml of ethanol, followed by a 2-h incubation at 75°C and then an overnight incubation at room temperature. After saponification, the samples were extracted two times with 4.5 ml of hexane. The hexane-containing fractions were discarded, and the remaining fractions containing fatty acids were acidified to pH <2 by the addition of 0.5 ml 12 M HCl. After acidification, the samples were extracted one time with 4.5 ml hexane. The organic fractions were dried under N2, resuspended in 50 μl of 1:1 chloroform-hexane, dissolved in 1 ml of Ecoscint O liquid scintillation fluid, and assessed for radioactivity using a Beckman LS6500 liquid scintillation counter.
RESULTS
Impact of HCMV infection on fatty acid biosynthesis and acetyl-CoA carboxylase activity.
We have previously utilized LC–MS-MS to globally measure the impact of HCMV infection at 48 h postinfection (hpi), a time of robust viral DNA synthesis. We found that HCMV infection induced phospholipid biosynthesis, increasing the production of both phospholipid fatty acids and glycerol head groups (35). To more thoroughly explore how HCMV impacts fatty acid biosynthesis during the course of viral infection, we assayed fatty acid biosynthetic activity at numerous points throughout the HCMV life cycle. As shown in Fig. 1A, HCMV-infected fibroblasts and uninfected cells showed similar levels of fatty acid biosynthesis at 24 hpi. However, at 48 hpi, there was a substantial increase in fatty acid biosynthesis in HCMV-infected fibroblasts compared to that in mock-infected fibroblasts, ∼6-fold (Fig. 1A). This elevation in fatty acid biosynthetic activity continued to increase at 72 hpi. The fatty acid biosynthetic increase occurs after the bulk of viral immediate early gene expression has occurred and corresponds to a time of rapid viral DNA replication and virion assembly, i.e., 48 to 72 hpi.
Fig. 1.
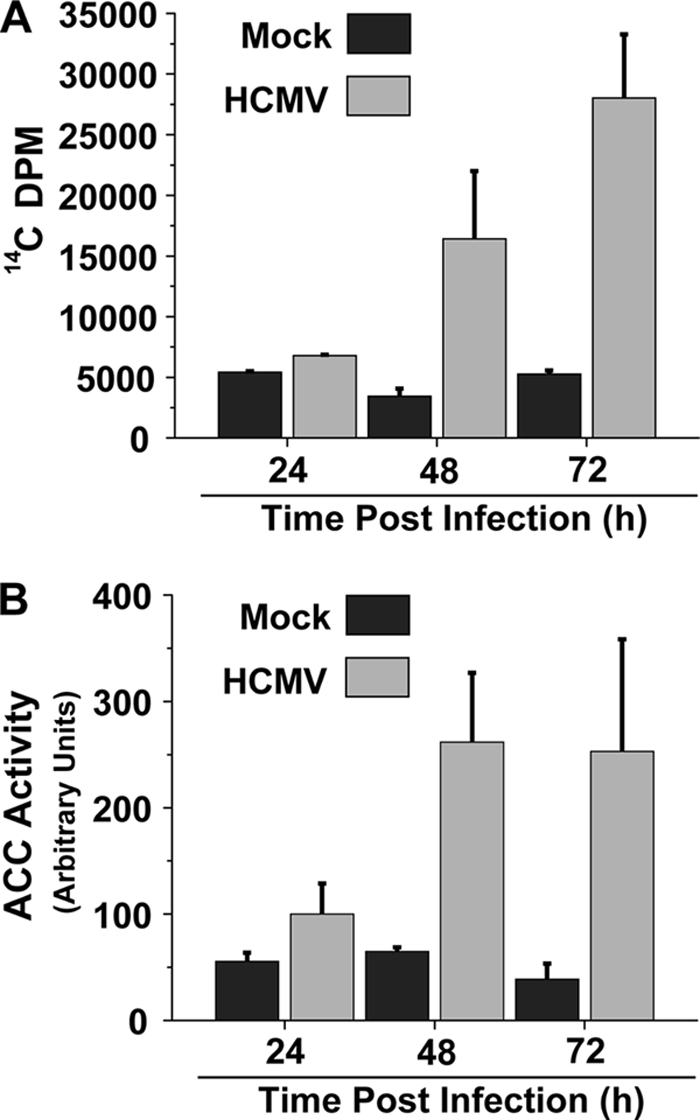
Impact of HCMV infection on fatty acid biosynthesis and ACC activity. (A) Time course of fatty acid biosynthesis during HCMV infection. Serum-starved MRC-5 fibroblasts were mock infected or infected with HCMV (multiplicity of infection [MOI] of 3.0). Cells were labeled with 14C-acetate at 24, 48, and 72 hpi for 2 h, at which time lipids were extracted and saponified and the radioactivity in the fatty acid fraction was scintillation counted. Values are means + standard errors (SE) (n = 2). (B) Time course of ACC activity during HCMV infection. Serum-starved MRC-5 fibroblasts were mock infected or infected with HCMV (MOI of 3.0). Cells were harvested at 24, 48, and 72 hpi, and the ACC activity of the resulting cellular lysates was measured via an LC–MS-MS–based ACC activity assay. ACC activity data are from a representative experiment of four (means + SE, n = 2).
ACC catalyzes the first committed step of fatty acid biosynthesis and is heavily regulated to control the rate of fatty acid biosynthesis (reviewed in reference 9). The observation that viral infection activates fatty acid biosynthesis raised the possibility that HCMV infection increases cellular ACC activity. To test the impact of HCMV infection on ACC activity, we utilized an in vitro LC–MS-MS–based ACC assay that measures the accumulation rate of malonyl-CoA, the product of the reaction catalyzed by ACC. At various times postinfection, we harvested mock- or HCMV-infected fibroblasts and analyzed the resulting lysates for ACC activity. At 24 hpi, there was only a slight increase in ACC activity, but at 48 and 72 hpi, there was a more substantial increase in ACC activity, a greater than 5-fold increase (Fig. 1B). These results indicate that HCMV infection increases cellular ACC activity upon infection at times that correspond to increased fatty acid biosynthetic activity, consistent with ACC being a major regulator of fatty acid biosynthesis (9).
HCMV infection induces an increase in ACC1 mRNA and protein expression.
Given the increase in fatty acid biosynthesis and ACC activity observed during HCMV infection, we hypothesized that the expression of lipogenic enzymes might be upregulated by HCMV infection. To investigate this possibility, we analyzed the mRNA transcript abundance of several fatty acid biosynthetic enzymes via quantitative PCR. The enzymes analyzed included ACL, ACC1, ACC2, and FAS. While the levels of ACL, ACC2, and FAS were elevated only marginally by HCMV infection compared to those for mock-infected controls, the levels of ACC1 were increased more substantially, ∼3-fold (Fig. 2A). The accumulation of ACC1 over ACC2 is consistent with the literature, as ACC1 is ubiquitously expressed in numerous tissues, while the ACC2 isoform is expressed primarily in highly metabolic/fatty acid oxidative tissues, such as liver and muscle (33).
Fig. 2.

Impact of HCMV infection on ACC expression. (A) Abundance of fatty acid biosynthetic enzyme mRNA. Serum-starved MRC-5 fibroblasts were mock infected or infected with HCMV (MOI of 3.0). Cells were harvested at 48 hpi and analyzed by quantitative PCR. The resulting data were normalized to GAPDH and plotted with respect to mock infection (means + SE, n = 3). (B) Accumulation of fatty acid biosynthetic enzymes during the course of HCMV infection. Serum-starved MRC-5 fibroblasts were mock infected (M) or infected with HCMV (H) (MOI of 3.0). Cells were harvested for protein analysis at 4, 24, 48, and 72 hpi and processed for Western analysis using the indicated antibodies. Data are from a representative experiment of four. (C) Quantification of ACC1 levels during HCMV infection. ACC1- and α-tubulin-specific signals were scanned by phosphorimager. The resulting HCMV ACC1 signal was normalized by its respective α-tubulin signal and plotted with respect to mock infection (means + SE, n ≥ 3).
The results indicating HCMV-induced increases in ACC1 mRNA abundance suggested that HCMV may induce accumulation of the ACC1 enzyme. To address this possibility, the accumulation of ACC1 was analyzed over the course of viral infection. ACC1 protein levels increased throughout HCMV infection, starting at 24 hpi, and reached a maximal amount compared to levels in mock-infected cells at 72 hpi (Fig. 2B). Quantification of ACC1 levels, after normalization to tubulin as a loading control, showed a greater than 2-fold and 3-fold increase in HCMV-infected cells at 48 and 72 hpi, respectively, compared to levels in mock-infected cells (Fig. 2C). In contrast to the increases in ACC1 protein, the levels of ACL protein, another enzyme involved in fatty acid biosynthesis, were not affected by HCMV infection (Fig. 2B). The observed increases in ACC1 mRNA and protein abundance indicate that HCMV infection increases the expression of ACC1 and are consistent with the increases in fatty acid biosynthesis and ACC1 activity observed during HCMV infection.
HCMV gene expression but not viral DNA replication is necessary for HCMV-induced increases in ACC1 abundance.
HCMV infection consists of a highly ordered program of viral gene expression. However, prior to the expression of viral genes, HCMV virion binding to cellular receptors results in the activation of numerous signal transduction pathways (44, 45). Furthermore, viral envelope fusion releases into the cytoplasm numerous viral tegument proteins that engage in diverse activities to initiate viral infection (5, 8, 25, 48). With these considerations in mind, we utilized UV-irradiated virus to determine whether viral binding/tegument protein delivery was sufficient for induction of ACC1 accumulation or whether viral gene expression was necessary. While UV irradiation is preferentially absorbed by nucleic acids over proteins, at higher doses proteins also become UV damaged. We therefore set out to determine a dose that would inhibit viral gene expression but not substantially impact viral tegument protein function. It is impossible to control for every function of every tegument protein, so we decided on an assay for pp65 transport to the nucleus as an estimate for functional tegument protein activity. The pp65 protein is an abundant tegument protein that upon infection is released to the cytosol and traffics to the nucleus (43). We empirically determined a UV dose that would attenuate HCMV viral gene expression but not impact pp65 nuclear trafficking. As shown in Fig. 3A, UV-irradiating HCMV virions with 480 or 240 mJ/cm2 did not impact the transport of pp65 to the nuclei of infected cells. At these same dosages, the accumulation of two viral genes, IE2 and UL26, was substantially decreased (Fig. 3B). Furthermore, UV irradiation completely blocked the HCMV-induced accumulation of ACC1 (Fig. 3B). These results indicate that viral binding and tegument protein delivery is not sufficient for induction of ACC1 levels, suggesting that viral gene expression is required.
As we found that the induction of ACC1 levels started at approximately 24 to 48 hpi and peaked at 72 hpi (Fig. 2B), we were interested to determine if late gene expression was necessary for ACC1 induction. Since abundant late gene expression depends on viral DNA synthesis, we sought to determine whether viral DNA synthesis was necessary for induction of ACC1 by treatment with PAA, which specifically inhibits viral DNA replication (23). As shown in Fig. 3C, addition of 50 μg/ml PAA at the time of adsorption did not block the HCMV-induced accumulation of total ACC1. Treatment with PAA also did not prevent the accumulation of phosphorylated ACC1 (pACC1) (Fig. 3C), a process which is discussed below. This result indicates that viral DNA synthesis is not required for induction of ACC1 levels during HCMV infection, suggesting that production of late proteins is not necessary for the induction of ACC1 protein accumulation.
HCMV infection increases the specific activity of ACC.
HCMV-infected cell lysates possessed increased ACC activity compared to mock-infected lysates (Fig. 1B). An increase in ACC activity in cell lysates can result from an increased concentration of an enzyme or from an increase in the specific activity of the enzyme. The observed increases in ACC1 protein levels during HCMV infection (Fig. 2B) likely contribute to the increase in ACC activity in HCMV-infected cellular lysates (Fig. 1B). To determine if HCMV infection increases the specific activity of ACC, we performed in vitro ACC assays that were normalized to the relative concentration of ACC1 protein present. At 72 hpi, the specific activity of ACC in HCMV-infected cell lysates was approximately 3-fold higher than that in mock-infected cell lysates (Fig. 4). These results suggest that HCMV infection induces the specific activity of ACC in addition to increasing its expression.
Fig. 4.
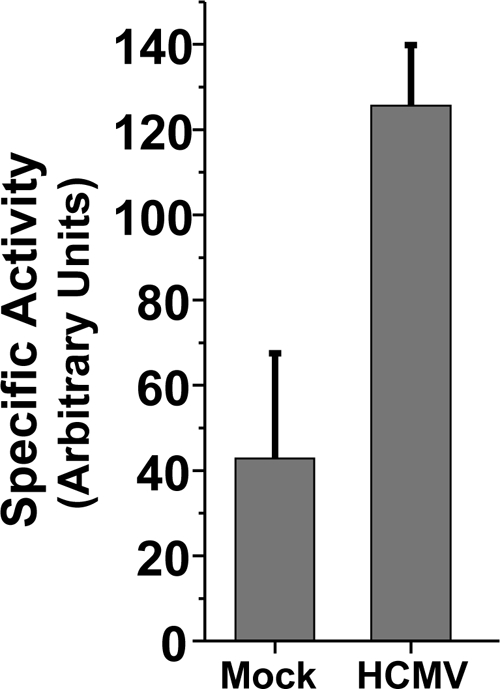
Impact of HCMV infection on ACC specific activity. Serum-starved MRC-5 fibroblasts were mock infected or infected with HCMV (MOI of 3.0). Cells were harvested at 72 hpi, and the ACC activity of the resulting cellular lysates was measured and normalized by the relative concentration of ACC1 protein (means + SE, n ≥ 3).
HCMV infection and the ser79 phosphorylation status of ACC1.
ACC1 specifically, and fatty acid biosynthesis consequently, is negatively regulated by phosphorylation of ACC1 at ser79 (20, 33). After finding that HCMV infection induces an increase in ACC specific activity (Fig. 4), we hypothesized that HCMV infection may result in decreased phosphorylation at ser79. To examine this possibility, we analyzed HCMV-infected cells at various times postinfection utilizing an ACC phospho-ser79-specific antibody. Surprisingly, HCMV infection induced an increase in the amount of ser79-phosphorylated ACC (Fig. 5A). Given the similarities between the increase in ACC1 abundance and the increase in phospho-ser79 abundance (Fig. 2 and 5), we wanted to confirm that the phospho-ACC-specific antibody was indeed specific for phosphorylated ACC. To address this issue, we utilized calf intestinal phosphatase (CIP) to determine if the pACC1-specific signal was phosphatase sensitive. As shown in Fig. 5D, phosphorylated ACC1 was not detectable in lysates treated with CIP, in contrast to results with non-CIP-treated lysates. Total ACC1 was still detectable regardless of CIP treatment. These results indicate that the phospho-ACC-specific antibody is specific for phosphorylated ACC. To examine the increases in pACC1 more closely, we utilized a phosphorimager to quantify the increases in total pACC1, i.e., after normalizing the pACC signal by the tubulin signal (Fig. 5B), as well as to quantify the relative ratio of pACC1/ACC1, i.e., after normalizing the pACC1 signal by the total ACC1 signal (Fig. 5C). HCMV infection increased the total amount of ser79 ACC1 phosphorylation starting at 4 hpi (Fig. 5B) and continuing throughout the time course, peaking with an approximate 3-fold increase at 72 hpi (Fig. 5B). Analysis of the ratio of pACC1/ACC1 suggests an increase in pACC1 relative to total ACC1 at 4 hpi in HCMV-infected cells (Fig. 5C). However, at later time points the HCMV-mediated increase in ser79 pACC1 is equivalent to the HCMV-mediated increase in total ACC1, with the ratio of pACC1 to total ACC1 not changing (Fig. 5C). Our results indicate that while the total amount of ser79 pACC is increased during HCMV infection, the relative proportion of pACC remains largely unchanged. These results indicate that HCMV infection does not mediate ACC1 activation through a reduction of ser79 ACC1 phosphorylation.
Fig. 5.

(A) Accumulation of phosphorylated ACC1 over the course of HCMV infection. Serum-starved MRC-5 fibroblasts were mock infected (M) or infected with HCMV (H) (MOI of 3.0). Cells were harvested for protein analysis at 4, 24, 48, and 72 hpi. The abundance of phosphorylated ACC1, ACC1, and α-tubulin was measured by Western analysis. Data are from a representative experiment of four. (B) Quantification of total phosphorylated ACC1 during HCMV infection. Phosphorylated-ACC1- and α-tubulin-specific Western signals were scanned by phosphorimager. The resulting HCMV phosphorylated ACC1 signal was normalized by its respective α-tubulin signal and plotted with respect to mock infection (means + SE, n ≥ 3). (C) Quantification of phosphorylated ACC1 relative to total ACC1 during HCMV infection. Phosphorylated-ACC1-, ACC1-, and α-tubulin-specific Western signals were scanned by phosphorimager. The resulting HCMV phosphorylated ACC1 and ACC1 signals were normalized by their respective α-tubulin signals. Phosphorylated ACC1 was subsequently normalized to ACC1 and plotted with respect to mock infection (means + SE, n ≥ 3). (D) Serum-starved MRC-5 fibroblasts were mock infected or infected with HCMV (MOI of 3.0). Cells were harvested at 72 hpi and immediately treated with calf intestinal phosphatase (CIP). After CIP treatment, the abundance of phosphorylated ACC1, ACC1, and α-tubulin was measured by Western analyses.
Analysis of ACC1 localization.
ACC1 is typically located in the cellular cytoplasm (19). As HCMV infection frequently targets the localization of numerous cellular proteins, we sought to determine how HCMV infection might impact the localization of ACC1. Of particular note in the analysis of ACC1 localization was the importance of using human serum in the confocal blocking buffer, the absence of which resulted in localization artifacts likely due to the presence of HCMV-encoded Fc receptors (see Materials and Methods; also see Fig. S1 in the supplemental material). Utilizing confocal microscopy, we found ACC1 to be localized to the cytoplasm during mock infection (Fig. 6). During HCMV infection, ACC1 was also primarily cytoplasmic at all time points tested (Fig. 6). While the localization of ACC did not change during the course of HCMV infection, the fluorescent intensity of ACC1-specific signal (utilizing identical confocal settings) was increased at 48 and 72 hpi compared to that for mock-infected cells (Fig. 6), consistent with our previous data indicating an increase in ACC1 expression.
Impact of ACC1 inhibition at various times postinfection.
We have previously reported that pharmaceutical inhibition of various fatty acid biosynthetic enzymes attenuates HCMV replication without affecting the accumulation of selected viral proteins representative of all three stages of viral gene expression, i.e., immediate early, early, and late (35). These results suggested that inhibition of fatty acid biosynthesis attenuates viral replication at a late stage of infection. If ACC1 activity is required at the later stages of infection, addition of an ACC1 inhibitor at later points of infection should still attenuate viral replication. To test this possibility, we employed TOFA, an ACC inhibitor (27). As shown in Fig. 7, addition of 5 μg/ml or 10 μg/ml of TOFA at the time of adsorption resulted in a greater than 10-fold or 100-fold attenuation of viral replication, respectively. TOFA treatment at 24 hpi still resulted in a greater than 10-fold reduction with addition of 5 μg/ml TOFA and 100-fold reduction with addition of 10 μg/ml TOFA (Fig. 7A). Addition of 5 μg/ml TOFA at 48 hpi, a time point where fatty acid biosynthesis is already substantially induced by infection (Fig. 1A), resulted in an attenuation of viral growth (∼10-fold) similar to that seen with TOFA treatment at earlier time points (Fig. 7A). Addition of 10 μg/ml TOFA at 48 hpi also attenuated viral growth more than 10-fold, still a substantial reduction but a smaller inhibitory effect than when TOFA was added at earlier times postinfection (Fig. 7A). Addition of 5 or 10 μg/ml TOFA at 72 hpi had no inhibitory effect on viral growth (Fig. 7A). The attenuation of viral production induced by addition of TOFA at later time points, i.e., 24 and 48 hpi, suggests that TOFA works at later times of infection, consistent with a role in late viral processes, such as envelopment. That treatment of cells with TOFA at 72 hpi does not impact production of HCMV virions suggests that TOFA does not attenuate infectious virions by binding or interfering with the activity of newly formed enveloped virions. It does, however, suggest that the window of sensitivity to fatty acid biosynthetic inhibition closes prior to 72 hpi.
Fig. 7.

(A) Growth characteristics of HCMV in cells treated with TOFA. Confluent MRC-5 fibroblasts were mock infected or infected with HCMV-GFP (MOI of 3.0). After adsorption, free virus was sodium citrate inactivated. At various times, as indicated, the cellular medium was removed and fresh medium with DMSO or TOFA was added. For all applicable cultures, at 48 hpi, the medium was changed, and fresh medium containing DMSO or TOFA was added. At 96 hpi, cells were harvested and viral titers were determined via standard plaque assay (means + SE, n = 4). (B) Analysis of the impact of TOFA treatment on fatty acid biosynthesis during HCMV infection. Confluent MRC-5 fibroblasts were infected with HCMV (MOI of 3.0). After a 2-h adsorption period, the inoculum was aspirated and fresh medium containing DMSO or the indicated TOFA dose was added. At 48 hpi, the medium was changed, and fresh DMSO or TOFA was added. Cells were labeled with 14C-acetate at 72 hpi for 2 h. Lipids were extracted and saponified, and the radioactivity in the fatty acid fraction was scintillation counted. Values are means + SE (n = 2). (C) Analysis of the potential cytotoxicity associated with TOFA treatment. Confluent MRC-5 fibroblasts were mock infected or infected with HCMV (MOI of 3.0). After a 2-h adsorption period, the inoculum was aspirated and fresh medium containing 10 μg/ml TOFA was added. At 48 hpi, the medium was changed, and fresh TOFA was added. At 72 hpi, cell viability was measured via a Live/Dead cell viability assay according to the manufacturer's instructions (MGT). Green indicates the presence of esterase activity, associated with viable cells, while red indicates a loss of cellular membrane integrity, associated with cell death. As a positive control for staining of a breakdown in membrane integrity, cells were treated with ethanol (+ Control). The images were obtained by inverted fluorescence microscopy.
To gauge the impact of TOFA treatment on virally induced fatty acid biosynthesis, we treated HCMV-infected cells with DMSO or 5 or 10 μg/ml TOFA and assayed for fatty acid biosynthesis. As expected, treatment of HCMV-infected cells with increasing concentrations of TOFA resulted in a dose-dependent reduction in fatty acid biosynthesis (Fig. 7B). To analyze how TOFA treatment impacts the cellular viability of mock- or HCMV-infected cells, we treated cells with 10 μg/ml TOFA and assayed for cell viability utilizing the Live/Dead assay, which stains live cells based on a their esterase activity (green) and stains the nucleic acids of dead cells based on the breakdown of membrane integrity (red). As shown in Fig. 7C, treatment of mock-infected or HCMV-infected cells with 10 μg/ml TOFA did not induce cellular toxicity, as made evident by the vast majority of cells staining green (>95%) versus red (<5%). These results suggest that TOFA treatment inhibits fatty acid biosynthesis and does not induce cellular toxicity.
Analysis of SREBP-2 processing and mTOR's contribution to HCMV-induced ACC1 expression and fatty acid biosynthesis.
Our results indicate that HCMV infection induces the levels of ACC1 mRNA and protein (Fig. 2). One potential mechanism behind this increase in ACC1 mRNA levels is transcriptional activation. Many cholesterol and fatty acid biosynthetic genes, including ACC1, contain sterol response elements (SRE) in their promoters which are activated upon binding by sterol regulatory element binding proteins (SREBPs) (reviewed in reference 6). Members of this family, e.g., SREBP-2, are endoplasmic reticulum (ER)-resident proteins that are transcriptionally inactive unless proteolytically cleaved, resulting in N-terminal translocation to the nucleus, whereupon they can activate SRE-containing genes (22, 41). To test whether HCMV infection activates the processing of SREBP-2, we employed an antibody that recognizes both the full-length SREBP-2 precursor (∼125 kDa) and the activated/cleaved N-terminal fragment (∼65 kDa) (50). Mock- and HCMV-infected cells were analyzed at various times postinfection for the accumulation of both the full-length SREBP-2 precursor and its N-terminal proteolytic fragment by using two independently derived antibodies directed against SREBP-2, a mouse monoclonal and a rabbit polyclonal antibody. As shown in Fig. 8A, Western blot analysis with the polyclonal antibody indicated a band of approximately 65 kDa, a size consistent with the N-terminal proteolytic fragment, that was present specifically during HCMV infection at 24, 48, and 72 hpi. Western analysis utilizing a monoclonal antibody directed against SREBP-2 also demonstrated an identically sized HCMV-specific band consistent with SREBP-2 processing; however, this band was absent at 24 hpi, present at 48 hpi, and greatly diminished but detectable at 72 hpi (Fig. 8B). HCMV infection did not appreciably impact the accumulation of full-length SREBP-2 (Fig. 8); however, Western analysis with the polyclonal antibody indicated the potential presence of a full-length SREBP-2 doublet specifically in HCMV-infected lanes (Fig. 8A). This could be indicative of a posttranslational modification of the full-length protein. This doublet, however, was not detected in the extracts assayed with the monoclonal antibody (Fig. 8B). Taken together, the combined results using both antibodies suggest that HCMV induces proteolytic processing of SREBP-2, which is consistent with transcriptional activation of SRE-containing genes, including ACC1.
Fig. 8.

Accumulation of SREBP-2 over time during HCMV infection. (A and B) Serum-starved MRC-5 fibroblasts were mock infected (M) or infected with HCMV (H) (MOI of 3.0). Cells were harvested for protein analysis at 24, 48, and 72 hpi. The abundance of SREBP-2 was measured by Western analyses using a rabbit polyclonal antibody (A) or a mouse monoclonal antibody (B). α-Tubulin was also blotted as a loading control. Western data for each antibody are from a representative experiment of two. (C) Analysis of the impact of Torin1 treatment on SREBP-2 and ACC1 accumulation over time during HCMV infection. Serum-starved MRC-5 fibroblasts were mock infected (M) or infected with HCMV (H) (MOI of 3.0). After a 2-h adsorption period, the inoculum was aspirated and fresh medium containing DMSO or 250 nM Torin1 was added. Cells were harvested for protein analysis at 48 and 72 hpi. The abundance of SREBP-2, ACC1, and α-tubulin was measured by Western analysis. (D) Analysis of the impact of Torin1 treatment on fatty acid biosynthesis during HCMV infection. Confluent MRC-5 fibroblasts were infected with HCMV (MOI of 3.0). After a 2-h adsorption period, the inoculum was aspirated and fresh medium containing DMSO or 250 nM Torin1 was added. At 48 hpi, the medium was changed, with fresh DMSO or 250 nM Torin1 added. Cells were labeled with 14C-acetate at 72 hpi for 2 h. Lipids were extracted and saponified, and the radioactivity in the fatty acid fraction was scintillation counted.
The mammalian target of rapamycin (mTOR) has been linked to SREBP expression and proteolytic processing (14, 40). In addition, mTOR activity is important for expression of fatty acid biosynthetic enzymes and fatty acid biosynthesis (14), as well as for HCMV infection (12, 32). We hypothesized that mTOR activity may be required for HCMV-induced SREBP-2 processing. Furthermore, if SREBP-2 processing is required for HCMV-mediated induction of ACC1 expression, inhibition of mTOR is predicted to block the HCMV-mediated induction of ACC1 expression. To test this possibility, we employed Torin1, an mTOR inhibitor (47). To gauge the impact of mTOR inhibition on virally induced SREBP-2 as well as ACC1 expression, we treated mock- and HCMV-infected cells with DMSO or 250 nM Torin1 and assayed for SREBP-2 processing and ACC1 expression via Western analysis. Treatment of HCMV-infected cells with Torin1 resulted in a reduction in full-length SREBP-2 expression and complete ablation of SREBP-2 processing (Fig. 8C). Furthermore, Torin1 treatment blocked HCMV-induced ACC1 expression at both 48 and 72 hpi (Fig. 8C). These results are consistent with a scenario in which mTOR-dependent SREBP-2 processing is important for HCMV-mediated induction of ACC1 expression. To determine the impact of mTOR inhibition on virally induced fatty acid biosynthesis, we again treated mock- and HCMV-infected cells with DMSO or 250 nM Torin1 and assayed for fatty acid biosynthesis. As shown in Fig. 8D, Torin1 treatment completely abrogated the induction of fatty acid biosynthesis observed upon HCMV infection. Taken together, these results suggest that mTOR activity is necessary for HCMV-mediated SREBP-2 processing, induction of ACC1 expression, and activation of fatty acid biosynthesis.
Analysis of the impact of ACC1 siRNA treatment during HCMV infection.
To further explore the role of ACC1 during HCMV infection, we utilized RNAi to target ACC1 expression levels. ACC1 siRNA treatment resulted in an approximately 32% reduction in ACC1 expression in both mock- and HCMV-infected cells compared to that seen with a control siRNA (Fig. 9A). Fatty acid biosynthesis was also measured following siRNA-mediated knockdown of ACC1. ACC1 knockdown resulted in a reduction in fatty acid biosynthesis in both mock- and HCMV-infected cells (Fig. 9B). While the impact of ACC1-specific RNAi on ACC1 protein levels was modest (Fig. 9A), we were interested to determine if RNAi-mediated targeting of ACC1 would impact HCMV replication. As shown in Fig. 9C, treatment with an ACC1-specific siRNA resulted in significant reduction in viral replication, ∼10-fold. These results further support the importance of ACC1 for production of wild-type levels of HCMV progeny.
Fig. 9.

Analysis of the impact of ACC1 siRNA treatment on ACC1 expression, fatty acid biosynthesis, and viral growth during HCMV infection. (A) MRC-5 fibroblasts at 30% confluence were transfected with 75 nM siRNA oligonucleotides specific for ACC1 and an unspecific control (Neg). At 24 h posttransfection, the cells were serum starved, and 24 h later, cells were mock infected (M) or infected with HCMV (H) (MOI of 3.0). Cells were harvested for protein analysis at 72 hpi, and the abundance of ACC1 and α-tubulin was measured by Western analysis. Data are from a representative experiment of two. (B) Cells were labeled with 14C-acetate at 72 hpi for 2 h, at which time lipids were extracted and saponified and the radioactivity in the fatty acid fraction was scintillation counted. Values are means + SE (n = 2). (C) After adsorption, free virus was sodium citrate inactivated. At 72 hpi, cells were harvested and viral titers were determined via standard plaque assay (means + SE, n = 2).
DISCUSSION
Viruses are entirely dependent on their host cells to provide the energy and biomass necessary for the production of viral particles. However, the mechanisms through which viruses ensure that their metabolic needs are met are mostly unclear. We find that HCMV targets multiple facets of a central regulator of fatty acid biosynthesis, ACC1. Our results indicate that HCMV infection increases the abundance of ACC1 mRNA and protein (Fig. 2). Our data show that viral binding and tegument protein delivery is not sufficient for the induction of ACC1 protein levels (Fig. 3). This result suggests that expression of a downstream viral gene(s) is required for increased ACC1 levels. Additionally, we find that viral DNA replication is not necessary for increased ACC1 levels, suggesting that true late gene expression is not necessary for induction of ACC1 levels. While our results argue that this induction of ACC1 expression is dependent on viral protein synthesis but not on viral DNA replication, the exact viral mechanisms responsible for these increases require further investigation. We also find that HCMV infection results in the proteolytic activation of SREBP-2, a known transcriptional activator of phospholipid metabolic genes containing SRE promoter elements (such as ACC1). SREBP-2 processing and ACC1 expression are both simultaneously blocked by pharmaceutical inhibition of mTOR (Fig. 8), a known SREBP activator (14). Taken together, these results are consistent with a scenario in which HCMV transcriptionally activates ACC1 through increased SREBP-2 processing, resulting in binding and activation of the ACC1 promoter. With respect to the dependence of ACC1 expression on mTOR activity, given mTOR's well-established role in translational regulation, we cannot rule out a potential impact of mTOR inhibition on ACC1 translation. Further experiments analyzing SREBP-2 occupancy at the ACC1 promoter and its impact on ACC1 promoter activity will provide insight into SREBP-2's role in the activation of ACC1 expression.
The inhibitory effect of Torin1 treatment on HCMV infection could in part reflect the inhibition of fatty acid biosynthesis observed upon Torin1 treatment. However, Torin1 treatment has an additional effect on DNA replication and viral gene synthesis that is not observed upon pharmaceutical inhibition of ACC (12, 32, 35). This suggests that there are other additional roles for mTOR during HCMV replication.
HCMV infection also resulted in an increase in the specific activity of ACC1 (Fig. 4). As ACC activity is negatively regulated by phosphorylation, we hypothesized that ACC would be dephosphorylated at ser79 during HCMV infection. On the contrary, we find that the total amount of ser79-phosphorylated ACC increased during infection, concurrent with increases in total ACC, resulting in similar ratios of ser79 phospho-ACC to total ACC in infected and uninfected cells (Fig. 5). These results suggest that dephosphorylation of ACC1 ser79 is not the mechanism through which HCMV activates ACC specific activity. The increase in ACC enzymatic activity observed during HCMV infection could be the result of an increase in the activity of ser79-phosphorylated ACC, nonphosphorylated ACC, or both. One possibility is that HCMV infection induces association with viral or cellular factors resulting in its activation. Various ACC-associated cellular regulators have been reported previously (reviewed in reference 9). Another possibility for activation of cellular ACC is through increasing the concentrations of an allosteric activator, such as citrate (20, 46). We have previously found that HCMV infection increases the concentrations of citrate substantially (34, 35), which is predicted to result in cellular ACC activation. However, increases in cellular citrate concentrations do not completely explain the in vitro activity differences we observe, as citrate is a necessary component of the activity assay. Other studies have suggested that ACC multimerization is correlated with ACC activity (31), which could be affected by HCMV infection. Further experimentation with respect to the impact of HCMV infection on the activity of phosphorylated and dephosphorylated ACC as well as ACC multimerization will likely provide insights into how HCMV induces the activity of this protein.
Most of our experiments were performed in serum-starved cells. We would argue that in many cases HCMV infects quiescent cells that are not actively growing. If so, infection of quiescent, serum-starved cells would reflect this possibility better than infection of rapidly growing cells stimulated with fetal growth factors. Regardless, we find that HCMV infection in the presence of serum still induces fatty acid biosynthesis, albeit to a lesser extent, and that pharmaceutical inhibition of ACC inhibits HCMV growth regardless of the presence of serum (data not shown), suggesting that these phenotypes are not dependent on serum starvation.
One unanswered question is how inhibition of fatty acid biosynthesis attenuates HCMV replication. Our data suggest that fatty acid biosynthesis is required at a relatively late stage during infection (Fig. 7). The simplest explanation is that fatty acid biosynthesis is necessary for the bulk production of fatty acids required for viral envelope phospholipids and that inhibition of this pathway limits the production of viral envelope. A related, but slightly different scenario is that the viral envelope consists of specific phospholipids whose production is activated and important for production of infectious virions. If this were the case, specific phospholipid-modifying enzymes working downstream of ACC1 would be potential targets for antiviral therapy. To our knowledge, no one has reported a detailed biochemical analysis of the HCMV phospholipid envelope, which would identify the relevant lipid species and thereby illuminate phospholipid metabolic enzymes of interest.
An alternative possibility is that HCMV replication depends on fatty acid biosynthesis for the modification of specific proteins. Fatty acid-based protein modifications, for example, myristoylation, are often essential for protein localization and function. This could be the case for the HCMV pp28 protein, an essential protein whose myristoylation is necessary for its proper localization and incorporation into virions (24, 51). Whether fatty acid biosynthesis is required for production of phospholipids or fatty acid protein modifications, its requirement at late times postinfection suggests that HCMV targeting of ACC1 is important for virion assembly and production. It is clear that HCMV infection targets this regulatory enzyme in multiple ways, and future work will elucidate the specific mechanisms involved and identify the specific fatty acid biosynthetic requirements for HCMV replication.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant from the National Institute of Allergy and Infectious Diseases (R01AI081773). J.M. is a Damon Runyon-Rachleff Innovator supported (in part) by the Damon Runyon Cancer Research Foundation (DRR-09-10).
We thank Thomas Shenk for the generous gift of antibodies. We also thank Nathanael Gray at the Dana-Farber Cancer Institute and David Sabatini at the Whitehead Institute for Biomedical Research for supplying us with Torin1.
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
Published ahead of print on 6 April 2011.
REFERENCES
- 1. Abu-Elheiga L., Almarza-Ortega D. B., Baldini A., Wakil S. J. 1997. Human acetyl-CoA carboxylase 2. Molecular cloning, characterization, chromosomal mapping, and evidence for two isoforms. J. Biol. Chem. 272:10669–10677 [DOI] [PubMed] [Google Scholar]
- 2. Abu-Elheiga L., Jayakumar A., Baldini A., Chirala S. S., Wakil S. J. 1995. Human acetyl-CoA carboxylase: characterization, molecular cloning, and evidence for two isoforms. Proc. Natl. Acad. Sci. U. S. A. 92:4011–4015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andrei G., De Clercq E., Snoeck R. 2008. Novel inhibitors of human CMV. Curr. Opin. Invest. Drugs 9:132–145 [PubMed] [Google Scholar]
- 4. Antonsson A., Johansson P. J. 2001. Binding of human and animal immunoglobulins to the IgG Fc receptor induced by human cytomegalovirus. J. Gen. Virol. 82:1137–1145 [DOI] [PubMed] [Google Scholar]
- 5. Bechtel J. T., Shenk T. 2002. Human cytomegalovirus UL47 tegument protein functions after entry and before immediate-early gene expression. J. Virol. 76:1043–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bengoechea-Alonso M. T., Ericsson J. 2007. SREBP in signal transduction: cholesterol metabolism and beyond. Curr. Opin. Cell Biol. 19:215–222 [DOI] [PubMed] [Google Scholar]
- 7. Bradley P. L. 1957. Metabolism of pyruvate and alpha-ketoglutarate in virus-infected mouse brain. Nature 180:1418–1419 [DOI] [PubMed] [Google Scholar]
- 8. Browne E. P., Shenk T. 2003. Human cytomegalovirus UL83-coded pp65 virion protein inhibits antiviral gene expression in infected cells. Proc. Natl. Acad. Sci. U. S. A. 100:11439–11444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brownsey R. W., Boone A. N., Elliott J. E., Kulpa J. E., Lee W. M. 2006. Regulation of acetyl-CoA carboxylase. Biochem. Soc. Trans. 34:223–227 [DOI] [PubMed] [Google Scholar]
- 10. Buchkovich N. J., Maguire T. G., Paton A. W., Paton J. C., Alwine J. C. 2009. The endoplasmic reticulum chaperone BiP/GRP78 is important in the structure and function of the human cytomegalovirus assembly compartment. J. Virol. 83:11421–11428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Burny W., Liesnard C., Donner C., Marchant A. 2004. Epidemiology, pathogenesis and prevention of congenital cytomegalovirus infection. Expert Rev. Anti Infect. Ther. 2:881–894 [DOI] [PubMed] [Google Scholar]
- 12. Clippinger A. J., Maguire T. G., Alwine J. C. 2011. The changing role of mTOR kinase in the maintenance of protein synthesis during human cytomegalovirus infection. J. Virol. 85:3930–3939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cochrane A. B. 2006. Antiviral dosing and efficacy for prophylaxis of cytomegalovirus disease in solid organ transplant recipients. Am. J. Health Syst. Pharm. 63:S17–S21 [DOI] [PubMed] [Google Scholar]
- 14. Duvel K., et al. 2010. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 39:171–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fioretti A., Furukawa T., Santoli D., Plotkin S. A. 1973. Nonproductive infection of guinea pig cells with human cytomegalovirus. J. Virol. 11:998–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Furukawa T., Tanaka S., Plotkin S. A. 1975. Restricted growth of human cytomegalovirus in UV-irradiated WI-38 human fibroblasts. Proc. Soc. Exp. Biol. Med. 148:1249–1251 [DOI] [PubMed] [Google Scholar]
- 17. Gerna G., Baldanti F., Revello M. G. 2004. Pathogenesis of human cytomegalovirus infection and cellular targets. Hum. Immunol. 65:381–386 [DOI] [PubMed] [Google Scholar]
- 18. Goodrich J. M., et al. 1993. Ganciclovir prophylaxis to prevent cytomegalovirus disease after allogeneic marrow transplant. Ann. Intern. Med. 118:173–178 [DOI] [PubMed] [Google Scholar]
- 19. Gratzner H. G., Ahmad P. M., Zegadlo J., Ahmad F. 1980. Immunofluorescent localization of acetyl CoA carboxylase, fatty acid synthetase and pyruvate carboxylase during the adipocyte conversion of 3T3 fibroblasts. Cell Biol. Int. Rep. 4:497–508 [DOI] [PubMed] [Google Scholar]
- 20. Halestrap A. P., Denton R. M. 1974. Hormonal regulation of adipose-tissue acetyl-coenzyme A carboxylase by changes in the polymeric state of the enzyme. The role of long-chain fatty acyl-coenzyme A thioesters and citrate. Biochem. J. 142:365–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Harwood H. J., Jr., et al. 2003. Isozyme-nonselective N-substituted bipiperidylcarboxamide acetyl-CoA carboxylase inhibitors reduce tissue malonyl-CoA concentrations, inhibit fatty acid synthesis, and increase fatty acid oxidation in cultured cells and in experimental animals. J. Biol. Chem. 278:37099–37111 [DOI] [PubMed] [Google Scholar]
- 22. Hua X., et al. 1993. SREBP-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc. Natl. Acad. Sci. U. S. A. 90:11603–11607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang E. S. 1975. Human cytomegalovirus. IV. Specific inhibition of virus-induced DNA polymerase activity and viral DNA replication by phosphonoacetic acid. J. Virol. 16:1560–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jones T. R., Lee S. W. 2004. An acidic cluster of human cytomegalovirus UL99 tegument protein is required for trafficking and function. J. Virol. 78:1488–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kalejta R. F., Bechtel J. T., Shenk T. 2003. Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol. Cell. Biol. 23:1885–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kaplan A. S., Ben-Porat T. 1961. The action of 5-fluorouracil on the nucleic acid metabolism of pseudorabies virus-infected and noninfected rabbit kidney cells. Virology 13:78–92 [DOI] [PubMed] [Google Scholar]
- 27. Kariya T., Wille L. J. 1978. Inhibition of fatty acid synthesis by RMI 14,514 (5-tetradecyloxy-2-furoic acid). Biochem. Biophys. Res. Commun. 80:1022–1024 [DOI] [PubMed] [Google Scholar]
- 28. Lee W. A., Martin J. C. 2006. Perspectives on the development of acyclic nucleotide analogs as antiviral drugs. Antiviral Res. 71:254–259 [DOI] [PubMed] [Google Scholar]
- 29. Litman R. M., Pardee A. B. 1956. Production of bacteriophage mutants by a disturbance of deoxyribonucleic acid metabolism. Nature 178:529–531 [DOI] [PubMed] [Google Scholar]
- 30. Locke G. A., et al. 2008. Differential activation of recombinant human acetyl-CoA carboxylases 1 and 2 by citrate. Arch. Biochem. Biophys. 475:72–79 [DOI] [PubMed] [Google Scholar]
- 31. Meredith M. J., Lane M. D. 1978. Acetyl-CoA carboxylase. Evidence for polymeric filament to protomer transition in the intact avian liver cell. J. Biol. Chem. 253:3381–3383 [PubMed] [Google Scholar]
- 32. Moorman N. J., Shenk T. 2010. Rapamycin-resistant mTORC1 kinase activity is required for herpesvirus replication. J. Virol. 84:5260–5269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Munday M. R. 2002. Regulation of mammalian acetyl-CoA carboxylase. Biochem. Soc. Trans. 30:1059–1064 [DOI] [PubMed] [Google Scholar]
- 34. Munger J., Bajad S. U., Coller H. A., Shenk T., Rabinowitz J. D. 2006. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog. 2:e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Munger J., et al. 2008. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 26:1179–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Munger J., Yu D., Shenk T. 2006. UL26-deficient human cytomegalovirus produces virions with hypophosphorylated pp28 tegument protein that is unstable within newly infected cells. J. Virol. 80:3541–3548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nowak B., et al. 1984. Characterization of monoclonal antibodies and polyclonal immune sera directed against human cytomegalovirus virion proteins. Virology 132:325–338 [DOI] [PubMed] [Google Scholar]
- 38. Pass R. F. 2001. Cytomegalovirus, p. 2675–2705 In Knipe D. M., et al. (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 39. Pearson H. E., Winzler R. J. 1949. Oxidative and glycolytic metabolism of minced day-old mouse brain in relation to propagation of Theiler's GD VII virus. J. Biol. Chem. 181:577–582 [PubMed] [Google Scholar]
- 40. Porstmann T., et al. 2008. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 8:224–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sakai J., et al. 1996. Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell 85:1037–1046 [DOI] [PubMed] [Google Scholar]
- 42. Salzman N. P., Lockart R. Z., Jr., Sebring E. D. 1959. Alterations in HeLa cell metabolism resulting from poliovirus infection. Virology 9:244–259 [DOI] [PubMed] [Google Scholar]
- 43. Schmolke S., Drescher P., Jahn G., Plachter B. 1995. Nuclear targeting of the tegument protein pp65 (UL83) of human cytomegalovirus: an unusual bipartite nuclear localization signal functions with other portions of the protein to mediate its efficient nuclear transport. J. Virol. 69:1071–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Simmen K. A., et al. 2001. Global modulation of cellular transcription by human cytomegalovirus is initiated by viral glycoprotein B. Proc. Natl. Acad. Sci. U. S. A. 98:7140–7145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Soroceanu L., Akhavan A., Cobbs C. S. 2008. Platelet-derived growth factor-alpha receptor activation is required for human cytomegalovirus infection. Nature 455:391–395 [DOI] [PubMed] [Google Scholar]
- 46. Thampy K. G., Wakil S. J. 1988. Regulation of acetyl-coenzyme A carboxylase. I. Purification and properties of two forms of acetyl-coenzyme A carboxylase from rat liver. J. Biol. Chem. 263:6447–6453 [PubMed] [Google Scholar]
- 47. Thoreen C. C., et al. 2009. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 284:8023–8032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Varnum S. M., et al. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J. Virol. 78:10960–10966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang D., Bresnahan W., Shenk T. 2004. Human cytomegalovirus encodes a highly specific RANTES decoy receptor. Proc. Natl. Acad. Sci. U. S. A. 101:16642–16647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Waris G., Felmlee D. J., Negro F., Siddiqui A. 2007. Hepatitis C virus induces proteolytic cleavage of sterol regulatory element binding proteins and stimulates their phosphorylation via oxidative stress. J. Virol. 81:8122–8130 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 51. Yu D., Silva M. C., Shenk T. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. U. S. A. 100:12396–12401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yu D., Smith G. A., Enquist L. W., Shenk T. 2002. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J. Virol. 76:2316–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.