

Abstract
The Herpesviridae are a large group of well-characterized double-stranded DNA viruses for which many complete genome sequences have been determined. We have extracted protein sequences from all predicted open reading frames of 19 herpesvirus genomes. Sequence comparison and protein sequence clustering methods have been used to construct herpesvirus protein homologous families. This resulted in 1692 proteins being clustered into 243 multiprotein families and 196 singleton proteins. Predicted functions were assigned to each homologous family based on genome annotation and published data and each family classified into seven broad functional groups. Phylogenetic profiles were constructed for each herpesvirus from the homologous protein families and used to determine conserved functions and genomewide phylogenetic trees. These trees agreed with molecular-sequence-derived trees and allowed greater insight into the phylogeny of ungulate and murine gammaherpesviruses.
Viruses contain relatively small genomes and the gene products encoded by the genomes are typically involved in a restricted number of functions, including recognition and entry into cells, specific replication of the viral genome, and formation of new virus particles. Some viruses with very small genomes contain <10 open reading frames (e.g., retroviruses and papillomaviruses), whereas others are relatively large and encode for a few hundred gene products (e.g., poxviruses). Among viruses with large genomes, some of the best characterized are members of the Herpesviridae. Herpesviruses are double-stranded DNA viruses known to infect mammals, fish, and birds. On the basis of differences in the cellular tropism, genome organization, and gene content, herpesviruses have been classified into three subfamilies: the Alphaherpesvirinae, Betaherpesvirinae, and Gammaherpesvirinae. A large number of completely sequenced genomes are available covering all three herpesvirus subfamilies (Table 1). A typical herpesvirus genome consists of ∼70 to 120 ORFs, although human cytomegalovirus (HCMV, HHV-5) may encode over 220 gene products (Cha et al. 1996). The three subfamilies are estimated to have arisen 180 to 220 million years ago (McGeoch et al. 1995), before the major mammal radiation, and as such are a diverse group of viruses. Apart from a number of essential, or core, genes, contained on seven conserved gene blocks, each genome has a subset of genes characteristic of the subfamily and a variable number of ORFs, which are specific to one or a few closely related viruses.
Table 1.
Herpesvirus Genomes Used to Construct Homologous Protein Families
| Subfamily and sublineage | Virus name (strain) | Acronyms | GenBank Accession no. | ORFs* | Length (Kb) |
|---|---|---|---|---|---|
| Alphaherpesviruses | |||||
| α1 | Human herpesvirus 1 (17) | HSV-11/HHV-1 | X14112 | 77 | 152 |
| α1 | Human herpesvirus 2 (HG52) | HSV-22/HHV-2 | Z86099 | 77 | 154 |
| α2 | Human herpesvirus 3 | VZV3/HHV-3 | X04370 | 71 | 124 |
| α2 | Equine herpesvirus 1 (Ab4p) | EHV-1 | M86664 | 80 | 150 |
| α2 | Equine herpesvirus 4 (NS80567) | EHV-4 | AF030027 | 79 | 145 |
| α2 | Bovine herpesvirus 1 (K22) | BHV-1 | AJ004801 | 73 | 135 |
| α3 | Gallid herpesvirus 2 (HPR524) | GHV-2 | AB024414 | 65 | 110 |
| Betaherpesviruses | |||||
| β1 | Human herpesvirus 5 (AD169) | HHV-5/HCMV4 | X17403 | 203 | 229 |
| β2 | Human herpesvirus 6 A (U1102) | HHV-6 (A) | X83413 | 121 | 159 |
| β2 | Human herpesvirus 6 B (HST) | HHV-6 (B/HST) | AB021506 | 115 | 161 |
| β2 | Human herpesvirus 7 (JI) | HHV-7 (JI) | U43400 | 107 | 144 |
| Gammaherpesviruses | |||||
| γ1 | Human herpesvirus 4 (B95-8) | HHV-4/EBV5 | V01555 | 86 | 172 |
| γ2 | Alcelaphine herpesvirus 1 (C500) | AHV-1 | AF005370 | 70 | 130 |
| γ2 | Ateline herpesvirus 3 (73) | HVA-36 | AF083424 | 71 | 108 |
| γ2 | Macaca mulatta rhadinovirus (17577) | RRV7 | AF083501 | 80 | 133 |
| γ2 | Saimiriine herpesvirus 2 | HVS8 | X64346 | 76 | 112 |
| γ2 | Equine herpesvirus 2 (86/87) | EHV-2 | U20824 | 79 | 184 |
| γ2 | Human herpesvirus 8 | HHV-8/KSHV9 | U75698 | 82 | 137 |
| γ2 | Murine herpesvirus 68 (WUMS) | MHV-68 | U97553 | 80 | 119 |
Number of open reading frames as extracted from GenBank entry (Benson et al. 1999).
HSV-1; herpes simplex virus-1
HSV-2; herpes simplex virus-2
VZV; Varicella Zoster virus
HCMV; human cytomegalovirus
EBV; Epstein-Barr Virus
HVA-3; herpesvirus ateles-3
RRV; rhesus rhadinovirus
HVS; herpesvirus saimiri
KSHV; Kaposi's sarcoma associated herpesvirus
The determination of sequence homology in genes from different organisms is key in identifying conserved functions or pathways (Tatusov et al. 1997; Andrade et al. 1999; Pellegrini et al. 1999). Functionally related proteins often share sequence similarity as conserved sequence motifs. Such information has been used to construct phylogenetic trees based on the number of shared genes between different completely sequenced cellular genomes (Fitz-Gibbon and House 1999; Snel et al. 1999; Tekaia et al. 1999) and recently to build a gene-content herpesvirus phylogeny using 13 herpesvirus genomes (Montague and Hutchison 2000). We have also used such a whole-genome approach to gain insight into herpesvirus function conservation and evolution. A larger number of herpesvirus genomes (19) have been included in our study and both gene content and sequence-alignment-derived phylogenies have been constructed and compared.
Sequence similarities between the ORFs in the currently available complete genomes have been mapped and used to obtain herpesvirus homologous protein families (HPFs). We have used the phylogenetic distribution of these homologous families (phylogenetic profiles) to determine the level of gene conservation between the viruses at different levels of the Herpesviridae taxonomy. This has enabled the assignment of homologous families to known functions and the study of how these functions are distributed within the herpesviruses. The phylogenetic profiles have been used to construct phylogenetic trees based on conserved gene function, reflecting the gain and loss of functions that underlay herpesvirus taxonomy.
RESULTS
Identification of Homologous Protein Families and Function Assignment
Sequence homology among all proteins derived from complete herpesvirus genomes (Table 1) was determined and used as a basis to construct HPFs (Fig. 1). We identified 243 homologous families that contained two or more proteins, comprising 1496 proteins out of a total of 1692 predicted ORFs in the 19 genomes studied. We observed that ∼80% of the total herpesvirus proteins had homologs in a different herpesvirus, whereas 20% appeared to be unique to particular genomes, sometimes existing as multiple copies (paralogs). Three-dimensional structural information for a subset of herpesvirus proteins validated the homologous family groups. It was not possible to collapse the homologous families into smaller groups based on such structural information. We used GenBank header files to manually assign functions to the different HPFs, including those with only one protein member. Functions consisted of both a short definition, such as DNA polymerase, and a broad functional class, for example, replication. Uncharacterized proteins were assigned to the unknown class.
Figure 1.

Schematic representation of a homologous protein family (HPF). Identically shaded boxes represent identified regions of each protein that have sequence homology. HPFs are constructed computationally by identifying one (or more) region(s) of sequence homology (i.e., unfilled oval) that builds the largest group of sequences. The HPF-conserved sequence region can be found in all proteins in the HPF.
All HPFs that belong to different functional classes can be retrieved from http://www.biochem.ucl.ac.uk/bsm/virus_database. In addition, the HPFs can be searched using virus name, functional annotation, keywords, or GenBank protein entry numbers. Each HPF has been assigned a distinct family number (HPF 1, HPF 2, etc.).
Phylogenetic Distribution of Protein Homologous Families
We used the homologous families to build protein phylogenetic profiles (Pellegrini et al. 1999), in which for each homologous family the presence or absence in every genome was recorded in the form of a binary matrix, where 1 means presence of at least one protein from the genome and 0 means no protein. In this type of analysis, paralogous proteins, resulting from multiple copies of a gene in the same genome, will only be counted once. The profiles were used to determine the number of gene functions conserved in pairs of genomes and to construct phylogenetic trees. In addition, the profiles were used to determine the minimum number of functions conserved at the subfamily/lineage level and to study the degree of conservation with respect to the functional class of the gene.
The distribution of the number of shared functions, based on sequence homology, between any two genomes across the different Herpesviridae was in accordance with the main evolutionary herpesvirus lineages (subfamilies) and sublineages (individual viruses). A relatively high number of homologous families were conserved within subfamilies and a much lower number conserved between members of different subfamilies (Table 2, lower triangle). Using our sequence-comparison algorithm, the minimum number of shared homologous families was 26 conserved between the Alpha- and Betaherpesvirinae, and the maximum, 96, was found between the closely related HHV6-A and HHV6-B. The number of shared homologous families was more variable within subfamilies than between members of different subfamilies. For example, the number of shared homologous families within any two Alphaherpesvirinae viruses ranged from 52 to 77, but between members of this subfamily and the other two subfamilies, the range of conserved homologous families was much narrower, between 26 and 30. We also calculated the percentage of homologous families conserved between any two genomes, taken relative to the genome with a smaller number of different families (Table 2, upper triangle). At least one-third of the homologous families were conserved between any two genomes with respect to the smallest genome of the pair. Within subfamilies the percentage varied between 54% (HHV-5 vs. HHV-6) and 100% (HSV-1 vs. HSV-2).
Table 2.
Number of Homologous Families Shared between Any Two Genomes
| HSV-1 | HSV-2 | VZV | EHV-1 | EHV-4 | BHV-1 | GHV-2 | HHV-5 | HHV-6A | HHV-6B | HHV-7 | HHV-4 | AHV-1 | HVA-3 | RRV | HVS | EHV-2 | HHV-8 | MHV-68 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HSV-1 | 74 | 100 | 86 | 82 | 82 | 82 | 80 | 35 | 36 | 36 | 36 | 38 | 41 | 40 | 38 | 38 | 39 | 38 | 38 |
| HSV-2 | 74 | 75 | 86 | 82 | 82 | 83 | 80 | 35 | 36 | 36 | 36 | 38 | 41 | 40 | 38 | 37 | 39 | 37 | 37 |
| VZV | 58 | 58 | 67 | 94 | 94 | 89 | 81 | 39 | 40 | 40 | 40 | 42 | 43 | 43 | 43 | 43 | 45 | 43 | 42 |
| EHV-1 | 61 | 62 | 63 | 77 | 100 | 92 | 83 | 34 | 35 | 35 | 35 | 39 | 41 | 40 | 38 | 37 | 39 | 36 | 36 |
| EHV-4 | 61 | 62 | 63 | 77 | 77 | 92 | 83 | 34 | 35 | 35 | 35 | 39 | 41 | 40 | 38 | 37 | 39 | 36 | 36 |
| BHV-1 | 59 | 60 | 60 | 66 | 66 | 72 | 81 | 36 | 37 | 37 | 37 | 39 | 41 | 40 | 39 | 39 | 40 | 39 | 39 |
| GHV-2 | 52 | 52 | 53 | 54 | 54 | 53 | 65 | 40 | 42 | 42 | 42 | 43 | 45 | 43 | 43 | 43 | 45 | 43 | 43 |
| HHV-5 | 26 | 26 | 26 | 26 | 26 | 26 | 26 | 167 | 54 | 54 | 58 | 45 | 51 | 51 | 49 | 47 | 48 | 44 | 46 |
| HHV-6A | 27 | 27 | 27 | 27 | 27 | 27 | 27 | 56 | 104 | 92 | 85 | 45 | 51 | 51 | 49 | 47 | 48 | 44 | 46 |
| HHV-6B | 27 | 27 | 27 | 27 | 27 | 27 | 27 | 56 | 96 | 104 | 83 | 43 | 50 | 50 | 47 | 46 | 47 | 43 | 45 |
| HHV-7 | 27 | 27 | 27 | 27 | 27 | 27 | 27 | 56 | 82 | 81 | 96 | 45 | 51 | 51 | 49 | 47 | 48 | 44 | 46 |
| HHV-4 | 28 | 28 | 28 | 29 | 29 | 28 | 28 | 33 | 33 | 32 | 33 | 74 | 71 | 69 | 68 | 67 | 68 | 68 | 63 |
| AHV-1 | 29 | 29 | 29 | 29 | 29 | 29 | 29 | 36 | 36 | 36 | 36 | 50 | 70 | 80 | 80 | 81 | 80 | 80 | 76 |
| HVA-3 | 28 | 28 | 29 | 28 | 28 | 28 | 28 | 36 | 36 | 36 | 36 | 48 | 56 | 70 | 90 | 97 | 80 | 90 | 83 |
| RRV | 28 | 28 | 29 | 28 | 28 | 28 | 28 | 36 | 36 | 36 | 36 | 50 | 56 | 63 | 74 | 89 | 77 | 96 | 81 |
| HVS | 28 | 28 | 29 | 28 | 28 | 28 | 28 | 36 | 36 | 36 | 36 | 50 | 57 | 68 | 66 | 76 | 76 | 87 | 79 |
| EHV-2 | 29 | 29 | 30 | 29 | 29 | 29 | 29 | 36 | 36 | 36 | 36 | 50 | 56 | 56 | 57 | 57 | 75 | 76 | 71 |
| HHV-8 | 28 | 28 | 29 | 28 | 28 | 28 | 28 | 36 | 36 | 36 | 36 | 50 | 56 | 63 | 71 | 66 | 57 | 81 | 78 |
| MHV-68 | 28 | 28 | 28 | 28 | 28 | 28 | 28 | 36 | 36 | 36 | 36 | 47 | 53 | 58 | 60 | 60 | 53 | 61 | 78 |
Absolute number (lower triangle and diagonal, shaded) or percentage with respect to the genome with the lowest number of homologous functions (upper triangle)
Conservation of Function Within and Across Subfamilies
We next focused our attention at the number of homologous families, which formed the core set of proteins in the different herpesvirus subfamilies. We detected 26 different ORFs that were conserved across the Herpesviridae (Table 3), which is close to previous estimations of the minimal herpesvirus genome on the basis of clear sequence homology (Hannenhalli et al. 1995; McGeoch and Davison 1999a). Each of these ORFs formed a separate homologous family, except for the major and the minor capsid proteins that share a region of ∼66 amino acids and, therefore, are part of the same homologous family. Apart from this common set of genes, other ORFs were conserved in two subfamilies but were absent in the third. In particular, we found three homologous families that were specific for Alpha- and Gammaherpesviruses and 10 specific for Beta- and Gammaherpesviruses. We did not identify any homologous families present in all members of the Alpha- and Betaherpesviruses but not present in the Gammaherpesviruses. According to this, the Gamma and Beta lineages clearly share more genes with detectable sequence homology than either of the two with the Alphaherpesviruses. By computing the ORFs that were only conserved in all members of one subfamily but in no other herpesvirus, we determined the subfamily-specific homologous families. There were 22 such homologs for the Alphaherpesviruses, 23 for the Betaherpesviruses, and only 8 for the Gammaherpesviruses. By adding the homologous families conserved at the level of two or three subfamilies, we obtained 51 families totally conserved for Alphaherpesviruses, 59 for Betaherpesviruses, and 46 for Gammaherpesviruses.
Table 3.
List of Herpesviridae Open Reading Frames with Clear Sequence Conservation in the 19 Genomes
| Gene block1 | Length of conserved sequence2 | GenBank number (HSV-1) | Gene name (HSV-1) | Function3 | Functional class4 |
|---|---|---|---|---|---|
| A | 750 | gi:59530 | UL30 | DNA polymerase | Rep |
| A | 86 | gi:59531 | UL31 | unknown | Unk |
| A | 436 | gi:59533 | UL32 | virion protein | Str |
| A | 42 | gi:59536 | UL36 | tegument protein | Str |
| A | 285 | gi:59539 | UL39 | ribonucleotide reductase large subunit | Nuc |
| B | 667 | gi:59527 | UL27 | glycoprotein B | Gly |
| B | 599 | gi:59528 | UL28 | transport protein | Str |
| B | 960 | gi:59529 | UL29 | ssDNA binding protein | Rep |
| C | 714 | gi:59552 | UL52 | helicase-primase complex | Rep |
| C | 39 | gi:59554 | UL54 | immediate-early transactivator | Trf |
| D | 63 | gi:59522 | UL22 | glycoprotein H | Gly |
| D | 111 | gi:59523 | UL24 | fusion protein | Str |
| D | 131 | gi:59525 | UL25 | tegument protein | Str |
| D | 185 | gi:59526 | UL26 | capsid protease | Str |
| E | 102 | gi:59518 | UL18 | capsid protein | Str |
| E | 66 | gi:59519 | UL19 | major capsid protein | Str |
| F | 767 | gi:59507 | UL5 | helicase-primase complex | Rep |
| F | 66 | gi:59506 | UL6 | minor capsid protein | Str |
| F | 67 | gi:59508 | UL7 | unknown | Unk |
| F | 283 | gi:59510 | UL10 | glycoprotein M | Gly |
| F | 276 | gi:59513 | UL12 | deoxyribonuclease | Nuc |
| F | 140 | gi:59514 | UL13 | protein kinases | Oth |
| F | 315 | gi:59501 | UL152 | DNA packaging | Str |
| F | 143 | gi:59501 | UL151 | DNA packaging | Str |
| F | 90 | gi:59516 | UL16 | virion protein | Str |
| G | 218 | gi:59503 | UL2 | uracil-DNA glycosylase | Nuc |
Gene blocks are regions where the order of genes is conserved of which seven are present in all Herpesviridae genomes (A-G).
Conserved sequence regions where sequence homology was clearly detected. These corresponded to a single contigous sequence motif in all cases except for DNA polymerase, in which three different motifs were conserved.
Function as derived from GenBank annotations.
Functional classes: Rep (replication), Nuc (nucleotide metabolism and DNA repair), Str (structural), Trf (transcription), Gly (glycoprotein), Oth (other), Unk (unknown).
Analysis of Different Functional Classes
The number of homologous families with known function identified in viruses from different lineages was variable. The Alphaherpesviruses were the best characterized, with between 60% and 80% of the proteins of any virus having an assigned function. This percentage was between 55% and 70% for the Gammaherpesviruses. The Betaherpesviruses contained the largest number of uncharacterized proteins among the different lineages. Only about half of the predicted HHV-6 and HHV-7 ORFs and about one-fourth of the HHV-5 (human cytomegalovirus, HCMV) ORFs have a documented function.
Next we compared the degree of conservation of the different homologous families across the whole Herpesviridae. To do this, we analyzed separately the phylogenetic profiles of homologous families that belonged to different functional classes. The analysis is shown for the structural class (Fig. 2). The size distribution of the homologous family, taken as the number of different viruses represented, was markedly different for the seven functional classes (Fig. 3). Genes involved in nucleotide metabolism and DNA repair were the most conserved, with most of them being in large groups containing viruses from two or three subfamilies. Structural proteins, including capsid and tegument proteins, were also well conserved, as were proteins from the replication functional class. However, glycoproteins showed a much lower conservation and most of them belonged to families with a size of 1–3 viruses, clearly below the size of a herpesvirus subfamily. Proteins identified as being involved in transcription, as well as proteins in the others group, which included genes involved in virus-host interactions, were also poorly conserved. Finally, the majority of homologous families with an as-yet-unknown function (unknown class) fell into the 1–3 viruses size range.
Figure 2.

Phylogenetic profile of homologous proteins families (HPF) known to be involved in structural functions (capsid, tegument, virus assembly). The presence of the family in any genome is indicated by 1 and the absence by 0. Alpha, Alphaherpesviruses; Beta, Betaherpesviruses; and Gamma, Gammaherpesviruses. The HPF numbers are indicated and relate directly to accompanying data available at http://www.biochem.ucl.ac.uk/bsm/virus_database. HPF 1* is indicated twice because it represents a shared domain present in both the major and minor capsid proteins of all herpesviruses.
Figure 3.
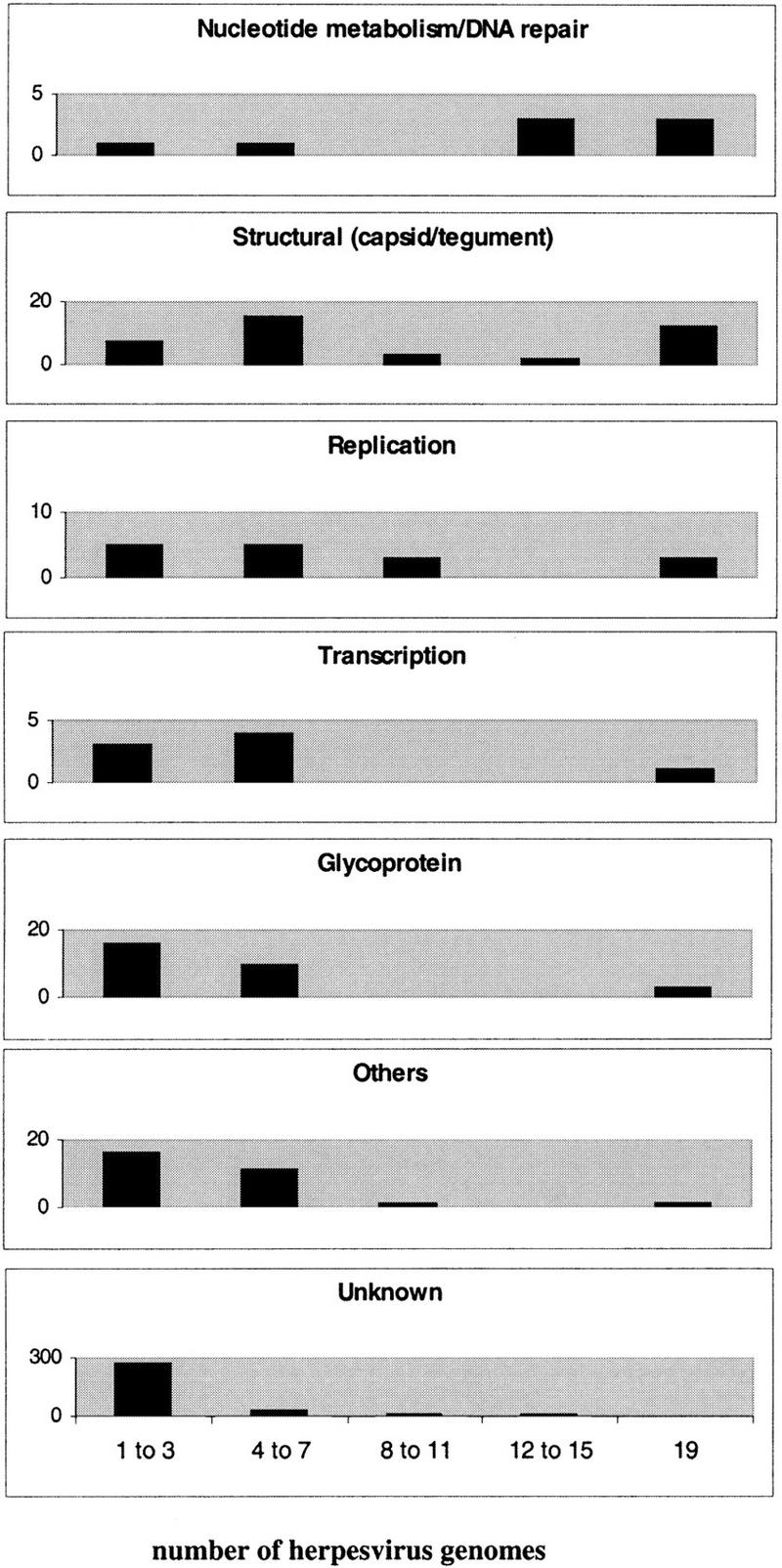
Distribution of functional classes of homologous families across the 19 herpesviruses considered. The number of herpesvirus genomes that contain the functional class are shown on the X-axis (lowest graph) and the number of homologous families in the functional class are shown on each Y-axis.
Interestingly, the three proteins that have been conserved in all Alpha- and Gammaherpesviruses but not in Betaherpesviruses belonged to the same functional group, nucleotide metabolism/DNA repair, namely ribonucleotide reductase small subunit, dUTPase, and thymidine kinase (HPF 28, 29, and 31, respectively). In contrast, homologous families that are exclusively conserved between the Beta- and Gammaherpesviruses were structural or of unknown function. One HPF, 9, contained the DNA origin-binding protein from the Alphaherpesviruses and the Betaherpesviruses HHV-6/HHV-7. However, this protein showed no homology to any Gammaherpesvirus protein or to proteins from the Betaherpesvirus HHV-5 (human cytomegalovirus). Homologous families that appeared to be exclusive to particular herpesvirus subfamilies occurred across different functional classes, although 12 homologous families corresponded to structural proteins in Alphaherpesviruses and 12 to genes of unknown function in Betaherpesviruses.
Phylogenetic Reconstruction Based on Function Conservation
Phylogenetic profiles were used to construct phylogenetic trees based on whole-genome homologous family content. In this type of tree, the distances between the different viruses are based on the degree of conservation of gene functions. Therefore, the topology of the tree will be affected by gene loss, gene capture (typically from the host genome in herpesviruses), and extensive sequence divergence beyond the recognition by the sequence comparison methods used here. The phylogenetic profiles were bootstrapped 100 times before constructing the trees. To build neighbor-joining trees, we explored the use of two types of intergenomic distance, the fraction of nonshared functions, and the fraction of dissimilar functions (Fig. 4A,B, respectively). The branching order of the two trees was the same for the two approaches and the main differences were in the branch lengths. As expected, the distance method that used the total of dissimilar functions, which was not standardized to the size of the smaller genome, reflected the difference in the number of genes per genome much better (Fig. 4B). For example, the branch length for HHV-5, which has approximately twice as many genes than any other herpesvirus genome, was longer than branches in other parts of the tree. Bootstrap supports, in general, were very high, with the exception of the split of the two ungulate herpesviruses, alcelaphine herpesvirus 1 (AHV-1) and equine herpesvirus 2 (EHV-2), with bootstrap values of 44% and 37%, respectively. In addition to neighbor-joining trees, we built up a maximum parsimony tree from the same set of data (Fig. 4C). Again the branching pattern was the same, except for the independent split of AHV-1 and EHV-2 as sisters, although, again, the bootstrap value was relatively low (64%).
Figure 4.
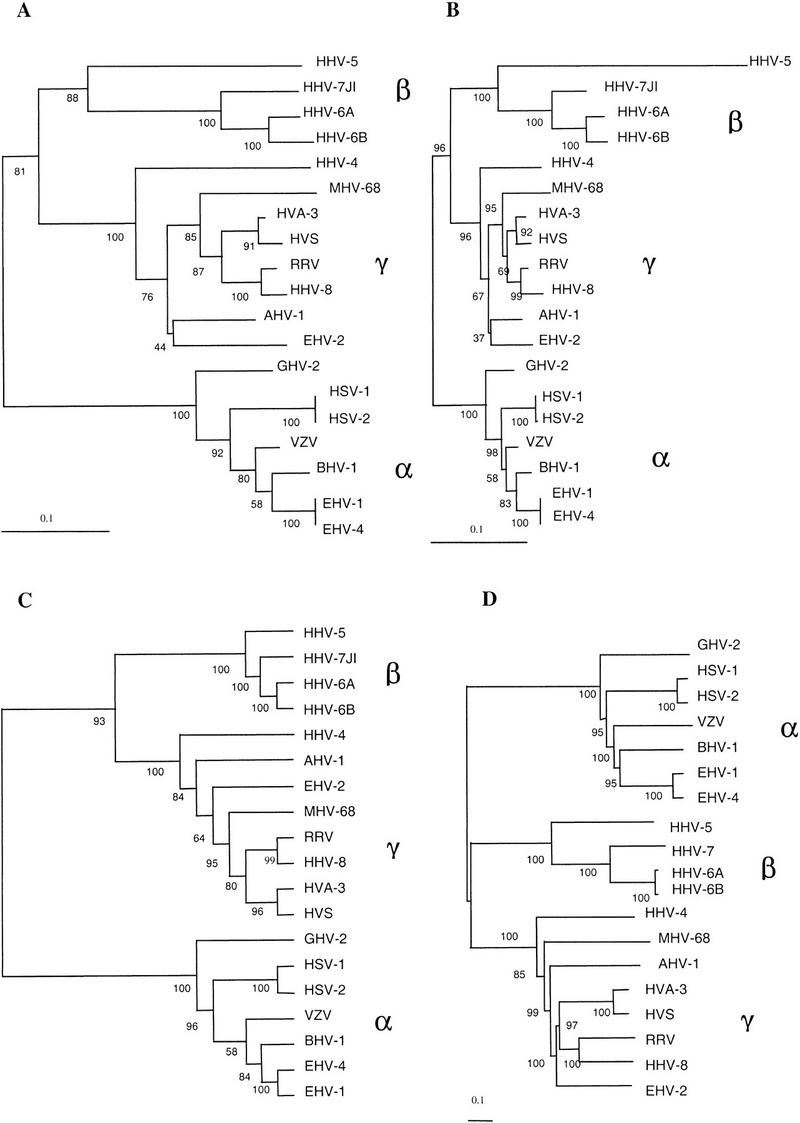
Phylogenetic trees based on protein family phylogenetic profiles (A,B,C) or on sequence comparison of herpesvirus-conserved domains (D). The initial data or alignments were bootstrapped 100 times. Neighbor-joining trees were constructed using the fraction of nonshared homologous families (A) or the fraction of dissimilar homologous families (B), as described in the text. Maximum parsimony cladogram built from the phylogenetic profiles (C). Neighbor-joining tree constructed using conserved regions in 26 herpesvirus open reading frames (D). α, Alphaherpesviruses; β, Betaherpesviruses; and γ, Gammaherpesviruses. The rhadinovirus subgroup of the Gammaherpesviruses consists of the viruses MHV-68, AHV-1, HVA-3, HVS, RRV, HHV-8, and EHV-2.
The trees based on the phylogenetic profile clearly resolved the splits between herpesvirus subfamilies and sublineages (Table 1). In addition, our data regarding the number of shared functions between different subfamilies supported previous observations of an early split of the Beta- and Gammaherpesviruses from the Alphaherpesviruses. This branching pattern was observed when we simulated a root by using an artificial outgroup genome that had none of the homologous proteins, that is, a row of 0s in the phylogenetic profile. To compare these trees with a sequence-comparison-based tree, we constructed an alignment of all conserved domains in the 26 ORFs identified as clear homologs in all herpesviruses. These genes have been preserved throughout herpesvirus evolution and are present in one copy per genome. The alignment contained 8900 positions and, using 100 bootstrapped data sets, neighbor-joining, UPGMA, and maximum parsimony trees were constructed. All trees showed the same topology and the neighbor-joining tree is shown in Figure 4D. The trees were representative and agreed with previous phylogenetic trees produced using a smaller set of highly conserved herpesvirus proteins (McGeoch and Davison 1999a).
There was complete consistency between the trees based on either function conservation or on sequence alignment, for the Alpha- and Betaherpesviruses, with all trees producing the same branching pattern. Among the Gammaherpesviruses, branch differences occurred in the positions of the ungulate herpesviruses (AHV-1 and EHV-2) and the murine herpesvirus MHV-68 when comparing the various trees. The position of the MHV-8, previously unresolved (McGeoch and Davison 1999b), appeared basal to the rhadinoviruses (all Gammaherpesviruses except HHV-4 in this study) in the alignment-based tree with a bootstrap value of 99%. Instead MHV-68 clustered together with the human and primate viruses in the other trees (bootstrap values of 87% and 69%). AHV-1 and EHV-2 formed a cluster in the neighbor-joining trees based on homologous family conservation. This association is in accordance with the hypothesis that herpesviruses have coevolved with their hosts (McGeoch and Cook 1994; McGeoch and Davison 1999b). However, the bootstrap values were low and the cluster was not observed in the other two trees. Therefore, the result is suggestive but requires further investigation.
DISCUSSION
The evolution of herpesviruses has been studied by sequence-comparison methods using a subset of conserved proteins (McGeoch and Cook 1994; McGeoch et al. 1995; McGeoch and Davison 1999a), by genome compositional properties such as dinucleotide frequency and CG content (Karlin et al. 1994), and by rearrangements of conserved gene blocks within the different genomes (Hannenhalli et al. 1995). This study of the molecular functions shared in 19 complete genomes in the form of phylogenetic profiles from herpesvirus HPFs has provided additional information on the degree of gene conservation at different levels of the Herpesviridae taxonomy. The complete genome approach has been successfully used to construct a phylogenetic tree that, although being in agreement with alignment-derived trees with respect to the best-supported branching events, provides additional insights into Gammaherpesvirus evolution.
The rate of gene turnover in herpesviruses appears to be quite high outside the core of conserved genes. This is reflected in a high number of genes that are unique to a particular herpesvirus and do not have counterparts in other herpesviruses. This group represents ∼20% of the total herpesvirus ORFs. The majority of these genes are of unknown function, although it seems likely that many of them were captured from the host genome during a relatively recent time. Virus-specific genes, including some multigene families, are not distributed evenly across the Herpesviridae but are particularly abundant in some subfamilies or viruses. For example, within the Betaherpesviruses, ∼70% of the HHV-5 genes appear to be virus specific. A similar feature is seen for the Gammaherpesvirus MHV-68, for which ∼20% of the genes have no sequence homologs in any other herpesvirus.
According to the sequence comparison algorithm used, the Herpesviridae share a set of 26 different ORFs and, therefore, about one-third of their functions are common (except for the large HHV-5 genome). These common functions include replication and nucleotide metabolism proteins, some structural proteins and glycoproteins, and a virus gene expression regulatory factor, designated UL54 in HSV-1. The less-well-conserved functional groups belong to the transcription, glycoproteins, and proteins classified as others. These observations, applied to the whole of the herpesvirus family, confirm similar conclusions as those derived from a protein functional analysis of the well-characterized herpes simplex virus 1 and its relatives in other host species (McGeoch and Davison 1999a). Within subfamilies, the conservation of function is always >50%, establishing a clear demarcation between subfamilies. Functions that are selectively conserved or eliminated in certain subfamilies are clearly visible, for example, the conservation of certain enzymes involved in nucleotide metabolism in the Alpha- and Gammaherpesviruses but not in the Betaherpesviruses. This has been previously interpreted as the Betaherpesvirus subfamily having abandoned the strategy of supplying enzymes of nucleotide synthesis for the replication of their genomes (McGeoch and Davison 1999a). From this study, we found that the Beta- and Gammaherpesviruses share more functions than either of these subfamilies do with the Alphaherpesviruses. Although many of these proteins are as yet uncharacterized, it seems likely that some will have a virus-structure functional role. This is supported by the fact that Alpha-specific genes are mostly from the structural class and, therefore, may be distant relatives of the Beta- and Gamma-specific genes. This level of relationship may be undetectable at the amino acid sequence level but may become apparent by secondary and three-dimensional structure prediction methods.
Taking into account the estimates for herpesvirus divergence (McGeoch et al. 1995) and the differences in the number of shared functions in the different herpesvirus genomes, we have calculated that, on average, a decrease of ∼7% in shared functions corresponds to 20 Myrs. From this we could extrapolate a rate of decrease of shared gene fraction between two herpesvirus genomes of about 3.5 × 10−3/Myr. In reality, this is an estimate of the minimum gene turnover, as recent gene duplications, represented as several proteins in the same homologous family from the same genome, would not enter into this equation. The rate of decrease of shared gene fraction between prokaryotic genomes can be estimated to be about 1 × 10−4 to 3 × 10−4/Myr from prokaryotic genome comparison data (Snel et al. 1999). Therefore, the gene turnover in herpesvirus genomes is an order of magnitude higher than in prokaryotic genomes. Similarly, amino acid mutation rates in herpesvirus proteins have been estimated to be higher (∼10–100 times) than in corresponding proteins in the host genomes (McGeoch and Cook 1994).
The construction of phylogenetic trees from gene content is a relatively new method of phylogenetic inference (Fitz-Gibbon and House 1999; Snel et al. 1999; Teichmann and Mitchison 1999; Tekaia et al. 1999) that we have applied to the study of viral genomes. Classical molecular methods, based on the alignment of individual gene sequences, are subject to the fact that different genes may have different evolutionary histories and undergo different types of selective pressure. As a consequence, the trees derived from such genes or proteins often differ. Instead, phylogenetic trees derived from gene content or molecular function conservation capture a broader picture and may accommodate some of the gene-specific biases. However, phylogeny based on gene content are affected by horizontal gene transfer and by differences in the number of genes in the genomes. Despite these potential problems, we have successfully applied homologous-family conservation-based methods to reconstruct a phylogeny of the Herpesviridae. The tree-branching pattern is in excellent agreement with phylogenies derived from alignments of conserved amino acid regions.
Differences exist at the level of the murine and ungulate rhadinoviruses. The position of MHV-68 could not previously be resolved by sequence-comparison-based methods (McGeoch and Davison 1999b). MHV-68 appears basal to the rhadinovirus clade in our alignment-based tree, representing the general trend of sequence divergence in the conserved domains for this virus. However, MHV-68 clusters with a relatively high confidence with primate Gammaherpesviruses in the three different trees based on homologous family conservation. In addition, a common split for the two ungulate Gammaherpesviruses (AHV-1 and EHV-2) is suggested by using the distance-based methods with phylogenetic profile data. This latter split would be expected by the hypothesis of coevolution of herpesviruses with their hosts (McGeoch and Davison 1999b) but is not detectable from sequence-comparison-based methods. Analysis of the homologous families within rhadinoviruses provides further insight into the evolution of this clade. The cluster of the murine and primate viruses is supported by two different genes present in these viruses but absent from the rest of herpesviruses, namely the viral-cyclin D homolog and the latent nuclear antigen (HPF 110 and HPF 111, respectively). These genes are involved in latency or interactions with the host and have corresponding locations within the different genomes. In addition, there are no genes exclusive to the ungulate and murine herpesviruses or to the ungulate and primate rhadinoviruses. However, two homologous families (HPF 81 and HPF 89, structural and glycoprotein groups, respectively) are present in all Gammaherpesviruses (including HHV-4/EBV) but absent from MHV-68, possibly reflecting specific gene losses in MHV-68.
The evidence for a common branch for AHV-1 and EHV-2 is not strongly supported by high bootstrap values for the number of shared genes, but specific genes do give support for the tree topology. A homologous family of a putative transmembrane protein (HPF 232) is only present in AHV-1 and EHV-2 and, therefore, could have been present in a common ancestor of these two viruses. Also in support of an early branching of the ungulate viruses is the existence of one gene of unknown function present in EBV (ORF BZLF2), AHV-1, and EHV-2 but absent from the rest of the rhadinoviruses (HPF 153). Furthermore, a homologous family including ORF BRRF1 from EBV (HPF 97) is present in all rhadinoviruses except the two ungulate viruses. The first two genes, therefore, could have been lost in a branch common to murine and primate herpesviruses, whereas the latter could have been lost in the ungulate branch.
Trees based simply on sequence alignment may not be able to successfully reconstruct distant branching events, especially if the proteins have diverged quickly. Rates of mutation are not uniform between different organisms and, in the case of pathogens, infection of new hosts may lead to accelerated sequence change in some or all proteins. The basal position of MHV-68 in the alignment-based tree could be due to an early ancestry of this virus within the rhadinoviruses or alternatively to a high rate of amino acid sequence divergence. If MHV-68 is truly basal to the rhadinoviruses, the proximity to the primate Gammaherpesviruses in the trees based on shared genes would imply that MHV-68 and primate viruses have been under similar selection pressures for the conservation and loss of gene sets, distinct from those conserved or lost in the ungulate Gammaherpesvirus. An alternative way to explain the differences between the two types of trees is that the murine and primate Gammaherpesviruses are evolutionarily closer, as supported by gene content trees, but that a high rate of amino acid change in MHV-68 results in an underestimation of their relationship in the alignment-based tree. For large genome viruses, trees based on homologous family conservation may capture other phylogenetic signatures, such as gene loss and acquisition that although prone to the errors associated with horizontal gene transfer and secondary losses, may provide higher resolution in cases such as the ones discussed.
Two additional cytomegalovirus genome sequences, murine cytomegalovirus 1 and rat cytomegalovirus, were not included in this study. The genome of murine cytomegalovirus was sequenced in 1996 (Rawlinson et al. 1996), but, unfortunately, the translated protein sequences are not available. The sequence of rat cytomegalovirus genome (Vink et al. 2000) appeared at a late stage of the revision of this paper. These two viruses belong to the Betaherpesvirus subfamily and have been reported to be evolutionarily closer to human cytomegalovirus than to Betaherpesviruses 6 and 7 (Rawlinson et al. 1996; Vink et al. 2000). The main conclusions of this study, therefore, do not change significantly. For example, the number of functions shared within the Betaherpesvirus lineage is unlikely to be significantly different, as these are the genes that the cytomegalovirus and the HHV-6/HHV-7 branches share among each other. Another herpesvirus complete genome that was not included is that of the channel catfish herpesvirus, as this virus is a very distant relative to the Alpha-, Beta-, and Gammaherpesviruses (McGeoch and Davison 1999a).
During the preparation of this paper, a cross-genome comparison of gene content applied to a more restricted subset of herpesvirus genomes (13) was published (Montague and Hutchison 2000). As in the present analysis, sequence similarity was initially detected by BLASTP (Altschul et al. 1990), but families were constructed by a different procedure and different stringency levels were tested. At the lowest stringency level, the authors detected 104 multiprotein families, a result that cannot be directly compared to our 243 families because our study includes more genomes (19). However, the sensitivity of the two methods appears to be very similar as the number of genes identified as conserved in all herpesvirus is essentially the same. Although the results appear consistent, the data presented here provide a greater depth and insight into herpesvirus phylogeny.
One of the objectives of this study was to establish a formal framework through the construction of homologous families and phylogenetic profiles for the study of gene function in large families of viruses. The production of a database of virus genomes and HPFs (VIDA, Virus Database) will greatly facilitate such future studies. This approach has proven useful in the interpretation of herpesvirus homologous family content and evolution and should also yield interesting results when applied to other virus families. The future characterization of new virus gene functions, together with protein structure and gene expression data, will further strengthen the importance of genomewide integrative approaches in the understanding of virus biology.
METHODS
Identification of Homologous Families
A total of 19 complete genomes representative of viruses in the Herpesviridae were retrieved from GenBank (see Table 1). Protein sequences from all identified ORFs were extracted and used to build up a protein-sequence dataset containing a total of 1692 proteins. XDOM (Gouzy et al. 1997) was used to identify homology between the proteins and to identify regions of sequence similarity that were common to related proteins. XDOM is based on BLASTP (Altschul et al. 1990) and had previously been used to identify regions of protein-sequence similarity in different complete genomes from bacteria, archaea, and eukarya (Gouzy et al. 1999). Initially, we empirically tested several parameters of the program so as to maximize sensitivity without compromising accuracy. After the initial observations, XDOM was used with the parameters SCORE = 75 and SCORE2 = 40 instead of the default values (90 and 50, respectively). We found that these parameters increased sensitivity although they still prevented the appearance of spurious matches between functionally unrelated proteins. A C++ program, PSC BUILDER, was written to cluster protein sequence domains together into HPFs. We clustered all proteins that shared at least one sequence domain, so that in each HPF there is at least one conserved region that is present in all proteins (Fig. 1). The method used identifies all proteins that share sequence similarity. Therefore, orthologous and paralogous sequences, derived from recent gene duplications, may be found in the same HPF. Proteins that did not share sequence homology to any other protein were treated as single-protein families. In these cases, the equivalent of the HPF-conserved sequence region will be the complete protein sequence.
Function Identification
Protein function, if known, was extracted for each herpesvirus protein from the original sequence-entry annotations. As no major disagreements were found in the annotated function of different proteins in the same homologous family, we considered that a function could be used to define most herpesvirus HPFs. Functions were simple definitions such as DNA polymerase or capsid protein. All protein functions were classified into seven major pathways or functional classes: replication, nucleotide metabolism and DNA repair, transcription, structural (including capsid, tegument, and virus assembly proteins), glycoproteins, others (including proteins involved in host-virus interactions such as immune modulation proteins), and unknowns.
Phylogenetic Profiles of the Homologous Families
Phylogenetic profiles can be defined from the presence or absence of a HPF in each virus genome (Pellegrini et al. 1999). A matrix was constructed, which for each homologous family, the presence of proteins from each given genome was expressed as 1 (presence) or 0 (absence). The matrix consisted of 439 columns for the total of homologous families, including those with only one protein, and 19 rows for the number of herpesvirus genomes. The presence of more than one protein from the same genome in the same homologous family (presumably due to paralogous genes) was not taken into account for the purpose of matrix construction. For the separate analysis of functional class conservation, the complete matrix was split into class submatrices. The number of shared gene functions across all genomes was determined as a whole number, representing all homologous families in which both genomes were present and also as a percentage of the number of shared functions.
Phylogenetic Analysis of Herpesvirus Genomes on a Functional Basis
The phylogenetic profiles were used to conduct phylogenetic analysis of the different viruses. The different protein families can be considered as molecular function characters for which the different viruses are positive (1) or negative (0). The data was bootstrapped 100 times using our own scripts and maximum parsimony, and distance methods (neighbor-joining) were applied.
For the distance methods, two distance measures were used: (1) Fraction of nonshared functions dx,y = 1−[(positive in X and in Y)/(minimum between total positives in X and total positives in Y)] and (2) fraction of dissimilar functions dx,y = [(positive in X but not in Y) + (positive in Y but not in X)]/total of homologous families.
In both cases, a positive refers to a 1 in the matrix (presence of a gene from the homologous family in that genome).
The first measure was previously used to build trees from gene content in unicellular organisms (Snel et al. 1999); the second was chosen because it may better satisfy the property of additivity of distance (Rzhetsky and Nei 1993). We used the programs NEIGHBOR and DNAPARS from the PHYLI8P package (Felsenstein 1993) for neighbor-joining and maximum parsimony methods, respectively. Consensus trees were derived using CONSENSE from the same package. The final trees were drawn with TREEVIEW (Page 1996).
Phylogenetic Analysis Based on Protein Sequence Alignments
We used the 26 ORFs identified as homologous in all Herpesviridae to construct a phylogeny based on sequence similarity. Alignments from a total of 28 conserved domains from the 26 ORFs and derived with MKDOM (Gouzy et al. 1997) were concatenated to form a single alignment of 8900 amino acids, including gaps. The alignment was bootstrapped 100 times and distances were computed with CLUSTALX default metric based on the Gonnet matrices (Benner et al. 1994) and corrected for multiple substitutions. Neighbor-joining trees were constructed using CLUSTALX (Thompson et al. 1997); UPGMA and maximum parsimony trees were constructed using NEIGHBOR and PROTPARS, respectively, from the PHYLIP package (Felsenstein 1993). Consensus trees were obtained with CONSENSE from PHYLIP and trees visualized with TREEVIEW (Page 1996).
Acknowledgments
We thank Robin A. Weiss and Sylvia Nagl for their advice on this project. This work is funded by the Biotechnology and Biological Sciences Research Council (BBSRC; M.A.) and the Medical Research Council (MRC; C.O. and P.K.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL p.kellam@ucl.ac.uk; FAX. 02-07-6799555.
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.149801.
REFERENCES
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Andrade MA, Ouzounis C, Sander C, Tamames J, Valencia A. Functional classes in the three domains of life. J Mol Evol. 1999;49:551–557. doi: 10.1007/pl00006576. [DOI] [PubMed] [Google Scholar]
- Benner SA, Cohen MA, Gonnet GH. Amino acid substitution during functionally constrained divergent evolution of protein sequences. Prot Eng. 1994;7:1323–1332. doi: 10.1093/protein/7.11.1323. [DOI] [PubMed] [Google Scholar]
- Benson DA, Boguski MS, Lipman DJ, Ostell J, Ouellette BF, Rapp BA, Wheeler DL. GenBank. Nucleic Acids Res. 1999;27:12–17. doi: 10.1093/nar/27.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha TA, Tom E, Kemble GW, Duke GM, Mocarski ES, Spaete RR. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J Virol. 1996;70:78–83. doi: 10.1128/jvi.70.1.78-83.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP (Phylogeny Inference Package), version 3.5c. Distributed by the author. Seattle: Department of Genetics, University of Washington; 1993. [Google Scholar]
- Fitz-Gibbon ST, House CH. Whole genome-based phylogenetic analysis of free-living microorganisms. Nucleic Acids Res. 1999;27:4218–4222. doi: 10.1093/nar/27.21.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouzy J, Eugene P, Greene EA, Kahn D, Corpet F. XDOM, a graphical tool to analyse domain arrangements in any set of protein sequences. Comput Appl Biosci. 1997;13:601–608. doi: 10.1093/bioinformatics/13.6.601. [DOI] [PubMed] [Google Scholar]
- Gouzy J, Corpet F, Kahn D. Whole genome protein domain analysis using a new method for domain clustering. Comput Chem. 1999;23:333–340. doi: 10.1016/s0097-8485(99)00011-x. [DOI] [PubMed] [Google Scholar]
- Hannenhalli S, Chappey C, Koonin EV, Pevzner PA. Genome sequence comparison and scenarios for gene rearrangements: A test case. Genomics. 1995;30:299–311. doi: 10.1006/geno.1995.9873. [DOI] [PubMed] [Google Scholar]
- Karlin S, Mocarski ES, Schachtel GA. Molecular evolution of herpesviruses: Genomic and protein sequence comparison. J Virol. 1994;68:1886–1902. doi: 10.1128/jvi.68.3.1886-1902.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeoch DJ, Cook S. Molecular phylogeny of the Alphaherpesvirinae subfamily and a proposed evolutionary timescale. J Mol Biol. 1994;238:9–22. doi: 10.1006/jmbi.1994.1264. [DOI] [PubMed] [Google Scholar]
- McGeoch DJ, Davison AJ. Origin and Evolution of Viruses. UK: London Academic Press; 1999a. The molecular evolutionary history of herpesviuses. [Google Scholar]
- ————— The descent of human herpesvirus 8. Seminars in Cancer Biology. 1999b;9:201–209. doi: 10.1006/scbi.1999.0093. [DOI] [PubMed] [Google Scholar]
- McGeoch DJ, Cook S, Dolan A, Jamieson FE, Telford EAR. Molecular phylogeny and evolutionary timescale for the family of mammalian herpesviruses. J Mol Biol. 1995;247:443–458. doi: 10.1006/jmbi.1995.0152. [DOI] [PubMed] [Google Scholar]
- Montague MG, Hutchison CA., III Gene content phylogeny of herpesviruses. Proc Natl Acad Sci. 2000;97:5334–5339. doi: 10.1073/pnas.97.10.5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page RDM. TREEVIEW: An application to display phylogenetic trees on personal computers. Comput Appl Biosci. 1996;12:357–358. doi: 10.1093/bioinformatics/12.4.357. [DOI] [PubMed] [Google Scholar]
- Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO. Assigning protein functions by comparative genome analysis: Protein phylogenetic profiles. Proc Natl Acad Sci. 1999;96:4285–4288. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlinson DW, Farrell HE, Barrell BG. Analysis of the complete DNA sequence of murine cytomegalovirus. J Virol. 1996;70:8833–8849. doi: 10.1128/jvi.70.12.8833-8849.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzhetsky A, Nei M. Theoretical foundation of the minimum-evolution method of phylogenetic inference. Mol Biol Evol. 1993;10:1073–1095. doi: 10.1093/oxfordjournals.molbev.a040056. [DOI] [PubMed] [Google Scholar]
- Snel B, Bork P, Huynen MA. Genome phylogeny based on gene content. Nat Gen. 1999;21:108–110. doi: 10.1038/5052. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- Teichmann SA, Mitchison G. Making family trees from gene families. Nat Gen. 1999;21:66–67. doi: 10.1038/5001. [DOI] [PubMed] [Google Scholar]
- Tekaia F, Lazcano A, Dujon B. The genomic tree as revealed from whole proteome comparisons. Genome Res. 1999;9:550–557. [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The ClustalX windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;24:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vink C, Beuken E, Bruggeman A. Complete DNA sequence of the rat cytomegalovirus genome. J Virol. 2000;74:7656–7665. doi: 10.1128/jvi.74.16.7656-7665.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]