

Abstract
A nonredundant database of 2312 full-length human 5′-untranslated regions (UTRs) was carefully prepared using state-of-the-art experimental and computational technologies. A comprehensive computational analysis of this data was conducted for characterizing the 5′ UTR features. Classification and regression tree (CART) analysis was used to classify the data into three distinct classes. Class I consists of mRNAs that are believed to be poorly translated with long 5′ UTRs filled with potential inhibitory features. Class II consists of terminal oligopyrimidine tract (TOP) mRNAs that are regulated in a growth-dependent manner, and class III consists of mRNAs with favorable 5′ UTR features that may help efficient translation. The most accurate tree we found has 92.5% classification accuracy as estimated by cross validation. The classification model included the presence of TOP, a secondary structure, 5′ UTR length, and the presence of upstream AUGs (uAUGs) as the most relevant variables. The present classification and characterization of the 5′ UTRs provide precious information for better understanding the translational regulation of human mRNAs. Furthermore, this database and classification can help people build better computational models for predicting the 5′-terminal exon and separating the 5′ UTR from the coding region.
Gene expression is regulated at each step from DNA to RNA to protein. Regulation of translational initiation is a central control point in mammalian cells, and the rate of initiation limits the translation of most mRNAs. Mechanistically, cap-dependent ribosomal scanning occurs on the majority of cellular 5′ UTRs. This process is severely hampered on long 5′ UTRs, containing upstream AUGs (uAUGs), upstream open reading frames (uORFs), and secondary structure. These features are often found in mRNAs encoding regulatory proteins like proto-oncogenes, growth factors, their receptors, and homeodomain proteins. Some of these mRNAs use an alternative mechanism of translation initiation, involving an internal ribosomal entry site (IRES). Cellular mRNAs containing a complex 5′ UTR or an IRES share an intriguing characteristic: Their translational efficiency can be very specifically regulated by their 5′ UTR, providing post-transcriptional regulation. Despite the fact that the modulation of translation by these multiple control elements has been studied by researchers in many individual mRNAs on a case by case basis (for review, see Kaufman 1994; Kozak 1996, 1999; Gray and Wickens 1998; Preiss and Hentze 1999), the detailed mechanisms involved in 5′ UTR-mediated control are not well understood. The binding of trans-acting factors could mediate translation stimulation or repression. The precise localization of uAUGs and the activity of the cap-binding initiation factor 4E are suggested to be important for translation regulation of these mRNAs.
As completing the human genome sequencing is imminent, systematic study of regulatory noncoding regions has become a pressing need. We need to know not only where the genes are and what they do but also when, where, and how they are expressed. Functional analysis of gene expression at the translational level requires a knowledge of 5′ UTR. During embryonic development, the 5′ UTRs of Antp, Ubx, RARβ2, c-mos, and c-myc regulate protein expression in a spatiotemporal manner. Translation initiation on a number of growth factor mRNAs (IGFII, PDGF2, TGFβ, FGF-2, and VEGF) is specifically regulated during differentiation, growth, and stress. Furthermore, 5′ UTR activity, mutations in the 5′ UTR, or the occurrence of alternative 5′ UTRs have been implicated in the progression of various forms of cancer (for review, see Clemens and Bommer 1999; van der Velden and Thomas 1999). Here, we attempt a comprehensive characterization of the 5′ UTR features by computational analysis of a large collection of full-length (i.e., from transcription start site to translation start site) 5′ UTR sequences. As far as we know, this work is the first classification of 5′ UTRs in a rigorous way. Kochetov et al. (1998, 1999) did a computational prediction of eukaryotic mRNA translational properties using partial 5′ UTRs for only two classes (high and low overall expression). However, our analysis includes a third class [terminal oligopyrimidine tract (TOP) mRNAs] and is more comprehensive in terms of the size of the database and the number and nature of the feature variables. Furthermore, all the 5′ UTRs in our database are of full length.
A high-quality database of 2312 full-length 5′ UTRs was prepared for the analysis. Three classes of genes were considered for comparing and contrasting the 5′ UTR features. Class I consists of mRNAs encoding transcription factors, growth factors, their receptors, proto-oncogenes, and other regulatory proteins that are poorly translated under normal conditions. Class II consists of TOP mRNAs whose translation is regulated in a growth-dependent manner. Class III consists of mRNAs of highly expressed genes, whose expression is controlled mainly at the transcriptional level and may be candidates for efficient translation. We compared the three classes with respect to their 5′ UTR features and identified those features that discriminate most. Classification and regression tree (CART) analysis (Breiman et al. 1984) was used to develop a classification model for segregating these three classes with significantly different 5′ UTR features. CDS length and codon bias were also added as the additional feature variables to improve the model.
The CART model indicated that secondary structure (free energy estimate by Zuker's mfold program; Mathews et al. 1999) was the most predictive variable. This was followed by the presence of TOP, UTR length, the number of stable free energies, the presence of stable secondary structure within the first 100 bp from the cap site, CDS length, A/T ratio, G/C ratio, the presence of uAUGs, the G+C percentage (GC%), the presence of uORFs, and codon bias, in the order of relative importance for predictive classification. Most of the 5′ UTR features, which are inhibitory for translation, were commonly observed in the 5′ UTRs of class I transcripts, whereas the 5′ UTRs of classes II and III are comparatively short and free from these inhibitory features. The presence of TOP, secondary structure, UTR length, and uAUGs remained as the most relevant variables for the final classification model that facilitated a clear-cut separation into the three classes.
RESULTS
We constructed 5′-end enriched cDNA libraries based on the oligo-capping method. By clustering these cDNAs, a set of 954 5′ UTR sequences was prepared see Methods). Eighty-two percent of these sequences were, on an average, 45 bp longer than any other sequences previously reported. The overall sequence quality of these 5′ UTRs was 99.2% with 0.8% of ambiguity base N (for further details, see Suzuki et al. 2000). This set was expanded with another set of 5′ UTRs retrieved from UTRdb (Pesole et al. 2000) database. Finally, a nonredundant high-quality database of 2312 human 5′ UTRs was prepared for the analysis.
The data collected on all the 12 variables were analyzed by CART analysis (for details, see Methods). Multivariate analysis by CART indicated that free energy estimate was the most discriminative variable for the three classes. This was followed by the presence of TOP, 5′ UTR length, the number of stable free energies, the presence of stable secondary structure within the first 100 bp from the cap site, CDS length, A/T ratio, G/C ratio, the number of uAUGs, GC% ,the number of uORFs, and codon bias, in the order of relative importance for predictive classification. The summary statistics on the important variables are presented in Table 1.
Table 1.
Summary Statistics for Four Categorical Variables
| Number of UTRs with | Class I (226)a | Class II (70)a | Class III (76)a | I + II + III (372)a | Overall (2312)a |
|---|---|---|---|---|---|
| Stable folds within first 100 bp from the cap site | 136 (60.2) | 0 (0) | 1 (1.3) | 137 (36.8) | 635 (27.5) |
| TOP | 10 (4.4) | 70 (100) | 2 (2.6) | 83 (22.3) | 152 (6.6) |
| uAUGs | 96 (42.5) | 0 (0) | 4 (5.3) | 100 (26.9) | 465 (20.1) |
| uORFs | 73 (32.3) | 0 (0) | 1 (1.3) | 74 (19.9) | 347 (15.0) |
| Start site good context | 111 (49.1) | 46 (65.7) | 44 (57.9) | 201 (54.0) | 1153 (49.9) |
Size of the class.
Values in the parentheses are percentages.
As UTR length and free energy estimate were identified as the two most discriminating features, we presented their distributions in Figure 1. Ninety-five percent of the 5′ UTRs of class I transcripts have a length of >100, whereas the transcripts of classes II and III have much shorter 5′ UTRs, with mean lengths of 45 and 73, respectively. Similarly, >90% of class I 5′ UTRs are embedded with stable secondary structures with average free energies less than −50 kcal/mole. It is reported that a structure with a free energy of −50 kcal/mole is sufficient to impose a strong block on ribosomal scanning (Pelletier and Sonenberg 1985; Kozak 1989). 5′ UTRs of classes II and III are almost free from this translational inhibitory feature. An exception to this is HBQ1, a hemoglobin, θ1 (from class III) gene whose 5′ UTR contained a highly stable secondary structure with an estimated free energy of −87.3 kcal/mole. Also, 60% of the class I 5′ UTRs have stable secondary structures within the proximity of the cap site, and only one (HBQ1) from the other two classes has this inhibitory feature.
Figure 1.

(Top) 5′ UTR length distributions. (Bottom) Free energy distributions (ΔG in kcal/mole).
The presence of uAUGs and uORFs was observed as a common feature in class I 5′ UTRs. We counted only those uAUGs and uORFs that are in good initiation context (see Methods), and ∼42% of the class I 5′ UTRs have uAUGs, and 32% have uORFs. Class II and III are quite free from these features, and the few outliers that have these features are presented in Table 3, below. On an average, we observed three uAUGs in class I 5′ UTRs and one uAUG in class III 5′ UTRs for every 1000 bp; class II 5′ UTRs did not contain any uAUGs. The ratios A/T and G/C are close to 1 in the case of class 1 5′ UTRs than classes II and III. This is consistent with the fact that the 5′ UTRs have more secondary structures than the other two classes. In the case of start site context, 65% of class II transcripts are in good context followed by class III with 57% and class I with 49%.
Table 3.
Misclassified Genes
| Cases in class I classified in class III | |
| MITF (AF034755) | quiescin (Q6): bone-derived growth factor (L42379) |
| BTF3 (X74070) | c-fms proto-oncogene (X03663) |
| ZNF143 (U09850) | CDKN3: CD11/KAP (L25876) |
| STAT4 (L78440) | AREB6 (D15050) |
| TFIIIC α-chain (AC002551) | PDGF platelet-derived growth-factor receptor β-like tumor suppressor (D37965) |
| GOS24 (M92844) | TGF-β superfamily protein (AB000584) |
| HIC-1 (zinc finger TF) (L41919) | hepatocyte growth factor-like protein (U37055) |
| Stat2 (U18671) | |
| Cases in class III classified in class I | |
| Immunoglobulin κ light chain, subclass II, A3 gene (X12690) ΔG AUG | TBG: thyroxin-binding globulin (Z83850) ΔG AUG |
| Myosin VIIA (Usher syndrome 1B (autosomal recessive, severe) (U39226) ΔG | immunoglobulin from VH4 family (from a patient with X-linked agammaglobulinemia) (X56158) TOP AUG |
| Myosin I beta (X98507) ΔG | histone H3.3 (Z48950) ΔG |
| A2M: α2-macroglobulin (Z11711) ΔG AUG | fructose–bisphosphatase, aldolase A (D28356) ΔG |
| HBQ1: hemoglobin, θ1 (M91453) ΔG | myosin regulatory light chain (U26162) ΔG |
| Cases in classes I & III classified in class II | |
| Mammaglobin (AF015224) | Phosphogluconate dehydrogenase (U30255) |
| QM-tumor suppressor gene (U37218) | |
(ΔG) Presence of stable secondary structure; (AUG) presence of uAUGs; (TOP) presence of 5′ TOP.
We applied a standard two-sample Z-test (Snedecor and Cochran 1980) to test the significant difference in mean GC% and mean codon bias between the three classes. The Z values for comparing the GC% of classes I and II, classes I and III, and classes II and III were 1.12, 1.61, and 0.47, respectively. These values suggest that there was no significant difference in the case of GC%, though class I 5′ UTRs have slightly higher GC content than the other two classes. Similarly the Z values for comparing the mean codon bias between classes I and II, classes I and III, and classes II and III were 1.02, 2.6, and 1.48, respectively. These values too were not significant at the 1% level of significance and suggest that there wasn't any significant difference in mean codon bias between the three classes. This indicates that the codon usage and expression level in human genes are not correlated. Duret and Mouchiroud (1999) also reported the same. In contrast, codon bias plays an important role in translational efficiency in some lower eukaryotes, such as yeast (Sharp and Li 1987).
Multivariate analysis of CART gave the classification model that is presented in the form of a decision tree (Fig. 2). The presence of TOP, secondary structure, UTR length, and the presence of uAUGs remained as the most relevant variables in the final classification model that facilitated a clear-cut separation into the three classes. The misclassification errors of the CART model by class were presented in Table 2. The most accurate tree we found has 92.5% classification accuracy as estimated by cross validation. Furthermore, the model correctly classified all the class II transcripts and misclassified 7% of class I and 16% of class III transcripts. The second part of Table 2 gives cross validation classification by class. For example, the first row explains that 210 (93%), 1 (0.4%), and 15 (6.6%) of 226 class I transcripts were classified as class-I, II, and III, respectively. The transcripts that were misclassified are presented in Table 3. The full CART classification of all 2312 human 5′ UTRs is available at the ftp site provided in Methods.
Figure 2.

CART model.
Table 2.
Misclassification Estimates by Class
| Class | Prior probability | Cross validation | Learning sample | ||||
|---|---|---|---|---|---|---|---|
| N (class size) | N misclassified | percent of N misclassified | N (class size) | N misclassified | percent of N misclassified | ||
| I | 0.333 | 226 | 16 | 7.1 | 226 | 8 | 3.5 |
| II | 0.333 | 70 | 0 | 0 | 70 | 0 | 0 |
| III | 0.333 | 76 | 12 | 15.8 | 76 | 11 | 14.5 |
| Total | 1.0 | 372 | 28 | 7.5 | 372 | 19 | 5.1 |
| Cross Validation Classification |
| Actual class | Predicted class | Actual total | ||
|---|---|---|---|---|
| I | II | III | ||
| I | 210 (93.0) | 1 (0.4) | 15 (6.6) | 226 |
| II | 0 (0) | 70 (100) | 0 (0) | 70 |
| III | 10 (13.2) | 2 (2.6) | 64 (84.2) | 76 |
| Predicted total | 73 | 220 | 79 | 372 |
Values in the parentheses are percentages.
DISCUSSION
The rate-limiting step in protein synthesis is thought to be at translation initiation (Merrick 1992), and various classes of mRNAs differ considerably in their translational efficiency. The mechanisms related to 5′ UTR features play an important role in translation regulation, and there are many articles in recent years that reported individual cases of translational regulation. However, most of these experimental reports are about mRNAs that are translationally repressed and the mechanisms involved in it, and little experimental evidence is available for efficiently translated transcripts. Garcia-Sanz et al. (1998) estimated that the number of translationally controlled mRNAs, following T-cell activation, is close to 13% (7.9% are activated and 4.7% are repressed), whereas the transcriptionally activated is 36%. They showed that a subset of individual mRNA species were translationally controlled and indicated that translational control might contribute significantly to the changes in gene expression that result in T-cell activation. Recently, Zong et al. (1999) used human cancer cDNA expression arrays to identify those mRNAs undergoing active translation. They identified populations of cellular mRNAs that are either efficiently or poorly translated in human foreskin fibroblast cell lines. Other than these two, we haven't come across any other experimental reports about translationally efficient mRNAs, especially for wild-type cells under normal conditions.
In this article we made a rigorous computational analysis of full-length 5′ UTRs, by taking advantage of the 5′-end-enriched cDNA library and UTRdb database. We compared three different classes of transcripts that perform completely different functions. Class I consists of genes involved in cell growth regulation and differentiation, regulation of metabolic pathways, and protection of cells from external damage. The transcripts encoding these proteins are poorly translated under normal conditions (e.g., in cells in the resting state). Class II consists of TOP mRNAs that participate in protein synthesis. These are known to be translationally regulated in a growth-dependent manner (Meyuhas et al. 1996) and contain a cis-regulatory element called 5′ TOP at the cap site. Class III might be considered as a control set, predominantly consisting of highly expressed gene transcripts. Most of these genes are either efficiently translated or not regulated at the (default) translational level. Our results show that these three classes of transcripts are significantly different in many of their 5′ UTR features.
Class I Transcripts Have Long 5′ UTRs Filled with Stable Secondary Structures, uAUGs, and uORFs
Kozak (1991) presented a comprehensive review on 5′ UTR features involved in translation control and predicted that many of the growth-related proteins would be poorly translated. Substantial experimental evidence has been accumulated in recent years that supports this prediction. Some of the well-studied transcripts that are poorly translated because of the presence of stable secondary structures or the presence of uAUGs in the 5′ UTR are ornithine decarboxylase (ODC), TGF-β3, β1,4-galactosyltransferase (β4GalT-I), cyclin D1, p53, AdoMetDC, RARβ2, and potassium channel ROM-K3. Our classification model classified all these transcripts (not included in the training set) in class I along with many other that have highly stable secondary structures and uAUGs. In most of these cases, translation occurs by the cap-dependent scanning model (Merrick and Hershey 1996). The cytoplasmic cap-binding protein, eIF-4E, participates in unwinding the secondary structures, and hence, its availability is crucial for the translation of these highly structured transcripts. When the availability of active eIF-4E is limiting, these transcripts are poorly translated. One way to overcome this problem is the overexpression of eIF-4E. Elevated levels of eIF-4E have been found in many tumor cell lines and almost all breast carcinomas. As a consequence, some of these poorly translated transcripts in class I might be efficiently translated in cells with eIF-4E overexpression. ODC is a good example for this as its levels were found to be drastically increased in eIF4-E transformed cells (Shantz et al. 1996).
One of the other ways these poorly translated transcripts can get rid of these inhibitory features is by a shift in the transcription start site and alternative splicing. TGF-β3 and β4GalT-I are good examples for this mechanism. Enhanced translational efficiency of TGF-β3 was observed in human breast cancer cells, and its 5′ UTR lacks the 5′ end of ∼870 nucleotides (Arrick et al. 1994) that contained inhibitory secondary structure. The β4GalT-I gene results in two transcripts with different 5′ UTRs. Charron et al. (1998) showed that mammary gland-specific β4GalT-I transcript, with truncated 5′ UTR that lacks extensive secondary structure, was efficiently translated both in vitro and in vivo. Both these transcripts that were not included in the training set have been successfully classified as class III transcripts by our classification model.
Class II and III Transcripts Have Small 5′ UTRs Free from Stable Secondary Structures, uAUGs, and uORFs
Class II mRNAs contain a 5′ TOP that regulates the translation of these transcripts in a growth-dependent manner. 5′ UTRs of this class were relatively short and almost completely free from the inhibitory features that were commonly observed in class I 5′ UTRs. However, we found a few transcripts from the other two classes that contain this regulatory element. Avni et al. (1997) showed that elongation factor 2 (EF2) and β1-tubulin, which contain 5′ TOP, are not regulated in a growth-dependent manner but regulated in a cell type-specific manner. They showed that the downstream sequences suppressed the regulatory features of the 5′ TOP and suggested that the mRNAs with longer 5′ UTRs might not be regulated in the same way as ribosomal proteins. Our classification model correctly classified all those transcripts of class I and III even though some of them contained 5′ TOP.
In the classic review, Merrick (1992) suggested the optimal characteristics for efficient mRNA translation, and most of the transcripts in class III have the favored characteristics. Hence, we suggest that most of these mRNAs are likely to be efficiently translated or, at least not repressed at the translational level. For a definite proof, we would have to wait for the experimental results. In a personal communication, Dr. David Morris provided the list of highly translated genes in human foreskin fibroblast cell lines by using a method called sucrose gradient analysis (Zong et al. 1999). Some of the genes in the list that were not in our training set are vimentin, desmin, CD59, caveolin-1, decorin, Ku80, and cytokeratin 8. Our classification model was able to correctly classify all these into class III.
Why CART Is Good for the Present Analysis
We analyzed large multivariate data that included both continuous and categorical variables. The CART technique is particularly applicable for studies like this, in which many of the variables considered do not seem to follow any particular distribution. In other words, we didn't make any parametric assumptions regarding the distributions of the variables under study. Moreover, our analysis was pattern driven rather than model driven; rather than building a coherent global model that includes all variables of interest, our classification algorithm produced a set of statements about local dependencies among predictor variables (in rule form with yes or no answers).
Also, CART uses predictor variables independently. That is, initially the entire data is partitioned into two subgroups according to the variable that produces the best split, for example, presence of TOP. Then, in each of the resulting strata, the process is repeated recursively until none of the selected variables shows significant influence on the split or the size of the subgroup is too small. In the final process, subgroups of cases that do not differ in any of the characteristics under study are joined together to form homogeneous classes.
CART also picks the best discriminating variables and ranks all the variables according to the relative discriminating power. We tried other classical methods such as linear discriminant analysis (LDA) and quadratic discriminant analysis (QDA) by considering the top three relevant variables picked by CART. The models of LDA and QDA didn't seem to give any better prediction than the CART model (data not shown). This might be due to the non-normality of the data and forcing parametric assumptions that didn't seem to exist.
Our experience shows CART as a useful data-mining tool for analyzing large data with many variables, where conventional statistical methods like LDA or QDA are not effective.
Limitations of CART
CART exhibits its greatest strengths in classification trees with a highly nonlinear structure (e.g., the 5′ UTR data in the present study). The closer the model is to linear, the less useful CART will be. When data exhibit a genuinely linear structure, CART is not a particularly useful analytical technique. Another important problem with CART is heteroscedasticity (within class variance). If the cases within a node genuinely belong together but have a high variance because of heteroscedasticity, CART may select a spurious split to partition the data. Although cross validation is designed to protect against the retention of such splits, some do survive the pruning process. In the present study class II is highly homogeneous, followed by class II and class III. The classification model could clearly segregate class II from the other two. However, 6.6% of class I were misclassified in class III, and 13.2% of class III were misclassified in class I (Tables 2 and 3). This might be due to the heteroscedasticity present in classes I and III. On the other hand, experimentalists are encouraged to look into these misclassified transcripts for proper reasons for their misclassification.
Conclusion
We made a comprehensive analysis of a large collection of 5′ UTRs and broadly classified the data into three functional groups. The class I transcripts seem to be very poorly translated under normal conditions, and those from class III might be candidates for efficient translation. Our classification model and the data we have generated may provide valuable information for experimentalists engaged in translational control and regulation studies. For example, the next natural step is to look for IRES or internal entry points within class I 5′ UTRs experimentally as well as computationally.
One of the main goals of our study is to develop a complete gene prediction system. As a first step toward this goal, we recently added a 3′-terminal exon-recognition module (J. Tabaska, R.V. Davuluri, and M.S. Zhang, in prep.) to the internal exon finder, MZEF (Zhang 1997). Our next step in achieving this task is the development of a 5′-terminal exon-detection module. We are presently working on the extension of the results of this work to the 5′-terminal exon-prediction program. The feature variables studied here would be valuable to identify the correct start site and separate 5′ UTR from the coding region. The drastic differences in 5′ UTR features observed between the three classes indicate that distinct models could be used to predict 5′-terminal exons. Hence, the classification of the 5′ UTRs into homogenous classes would facilitate the building of separate models for each class so that the overall quality of the 5′-terminal-exon prediction is expected to be higher than the prediction based on a single mixture model.
METHODS
5′ UTR Database
A set of 954 human 5′ UTR sequences was obtained from the 5′-end-enriched cDNA library (Suzuki et al. 1997, 2000) with their mRNA start sites. The 5′-end-enriched cDNA library was constructed to isolate the mRNA start site of long mRNA, by using a method called oligo-capping (Maruyama and Sugano 1994) with some modifications. We collected 5′ UTRs from this library as follows: First, cDNA sequences were clustered with DYNACLUST (Dynacom) after removing the oligo-capped 5′-oligonucleotide sequence from each 5′ end.DYNACLUST is a database management software, which clusters the sequences using BLAST with the score of e − 40 for 400 bp. The position of the translation start site (ATG) was marked for each sequence according to the annotation in GenBank. Then, the sequence between the oligo-capped 5′-oligonucleotide sequence and the translation start site (ATG) was extracted from each cluster. If alternative mRNA start sites or translation start sites were observed, then the cDNAs containing the longest 5′ UTRs at both the 5′ and 3′ boundaries were selected as the representative. (for details, see Suzuki 2000).
The experimentally derived set of 954 5′ UTRs was augmented with a second set of 1613 full-length 5′ UTR sequences retrieved from the UTRdb (Pesole et al. 2000) database. Only those sequences with UT feature tag as complete 5′ UTR are considered. These sequences were extensively verified by going through their corresponding GenBank records, and only those records with evidence = experiment were considered. All the redundant and ambiguous sequences were eliminated, and finally, a nonredundant set of 2312 5′ UTR sequences was prepared for the analysis. A sequence was considered redundant if it has 90% similarity and 90% overlapping with a larger sequence in the database. However, there may be more than one 5′ UTR for some genes because of alternative splicing and usage of different transcription start site. From this database the following three classes of 5′ UTRs were considered for analysis: Class I, the first class, consists of 5′ UTRs of growth factors, their receptors, transcription factors, proto-oncogenes, cytokine receptors, and tumor suppressor genes. Most of these are understood to be translationally repressed mRNAs. Class II, the second class, consists of TOP mRNAs. TOP mRNAs are vertebrate transcripts with a C residue at the cap site, followed by an uninterrupted stretch of 4–13 pyrimidines, called 5′ TOP, encode for ribosomal proteins and elongation factors 1α and 2α. The translation of this class of mRNAs is regulated in a growth-dependent manner. Class III, the third class, consists of 5′ UTRs of highly expressed genes, tubulins, globins, globulins, myosins, caseins, glycolytic enzymes, β-actin, γ-actin, and histones. The expression of these genes is controlled mainly at the transcriptional level, and their transcripts are believed to be efficiently translated. In other words, these genes are either translationally efficient or (at least) not repressed at the translational level. In contrast, the first two classes of genes are tightly regulated at the translational level in stringent ways. There are 226 5′ UTRs in the first class, 70 in the second, and 76 in the third class. The complete data set is available at ftp://cshl.org/pub/science/mzhanglab/ramana.
Data Analysis
CART
CART is a nontraditional algorithm developed by Berkeley and Stanford statisticians (Breiman et al. 1984). Tree-based models are becoming increasingly important in many fields. They are quite useful for uncovering structural relationships between a response/classification variable and a set of predictor variables in large multivariate data sets and in devising prediction rules that are both easy to interpret and easy to evaluate. Tree-based methods have numerous advantages. They produce decision rules that can readily accommodate both continuous and categorical predictor variables, and they readily capture nonlinear and nonadditive behavior and very general sorts of interactions among the predictors. CART is a decision tree structured statistical analysis and data mining tool that partitions a data set into discrete classes based on the value of a user-defined classification variable. The predictor variables in the database are selected as to whether or not they provide a predictive segregation of the data between different values of the classification variable. This restriction presupposes that there is a causal relationship between the predictor variables and the classification variable. The CART software segregates the different values of a classification variable through the growth of a binary decision tree, composed of a progression of binary splits on the values of the predictor variables. Each split is chosen such that the segregation of different values of the classification variable is improved. The resulting tree has multiple branches, of various complexities, each of which represents a path to a particular value of the classification variable.
Procedure for Constructing a CART Tree
The key components of tree-structured data analyses are tree growing, tree pruning, and optimal tree selection. Tree growing depends on splitting rules and stopping criteria. CART begins with all the data points in the learning sample, L. The CART classification tree initially consists of one node—the parent node of the tree, which contains all the points in L. The CART program searches through all possible values of all the variables, looking for the split that best separates the classes. The first split creates two child nodes. CART takes each of the child nodes and recursively partitions each child node in the same way that it partitioned the parent node. CART evaluates the goodness of any candidate split using an impurity function. A node that contains members of only one class is perfectly pure, and the node that contains an equal proportion of every class is least pure. Given a node t with estimated class probabilities p(j/t), j = 1,…,J, and a measure of node impurity, CART searches for the split that most reduces node, or equivalently, tree impurity. CART provides different impurity functions, for example, Gini Measure, Twoing criterion, etc. (for more details, see Breiman et al. 1984). We tried all the impurity functions and finally selected Gini Measure, which best suited our data. For a node t with estimated class probabilities p(j/ t), j = 1, … , J, Gini Measure is defined as
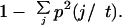 |
The next important step in tree growing is stopping. However, stopping is not an essential component of CART. CART grows the tree until no further growth is possible, that is, terminal nodes have only one case, or terminal nodes with more than one case are identical on the classification variable. The resulting maximal tree is called Tmax. Because Tmax usually overfits the data (i.e., being overly sensitive to irregularities in data) especially when it is noisy, CART applies a pruning and evaluation procedure to find the optimum-sized classification tree,Topt. The pruning procedure generates a nested sequence of smaller and smaller subtrees, {Tmax,…,T2,T1}, from which CART selects the Topt that has the lowest or near lowest cost of misclassification as determined by a cross validation procedure. In other words, CART performs postpruning, whereby as many child nodes as possible are eliminated as long as the overall estimated accuracy of the tree is not significantly reduced.
Cross Validation
A learning sample of 372 cases (226, 70, and 76 from classes I, II, and III, respectively) was considered for the CART analysis. A 10-fold cross validation was used for estimating the misclassification rates. That is, CART divides the learning sample into 10 roughly equal parts, each containing similar distribution for the classification variable. CART takes the first nine parts of the data, constructs the largest possible treeTmax, and uses the remaining one-tenth of the data to obtain initial estimates of the error rate of selected subtrees. The same process is then repeated on another nine-tenths of the data and uses a different one-tenth part as the test sample. The process continues until each part of the data has been held in reserve one time as a test sample. The results of the 10 minitest samples are then combined to form the best estimates of true error rates for trees of each possible size; these estimated error rates are applied to the tree based on the entire learning sample. This cross validation estimate is used in CART for two important functions: (1) to determine the degree to which the final tree should be pruned and (2) to estimate the true misclassification rate of the final tree.
Feature Variables
The following variables were used as predictor variables in CART analysis:
1. |UTR length − 80|: Kozak (1991) has shown that the rate of mRNA translation increases proportionally as the length of the 5′ UTR is increased from 17 to 80. Hence, taking 80 as the optimal 5′ UTR length for efficient translation, we considered |UTR length − 80| as one of the variables.
2. Free energy estimate of secondary structure (ΔG): The latest version of the mfold (Mathews et al. 1999) program was obtained from Michael Zuker (mfold v.3.1). The mfold program minimizes a free energy function, which sums contributions from stacking, loop lengths, etc. It actually estimates the difference between the free energy of the unfolded state and folded state. For any given RNA sequence length, the lower the energy estimate the more stable the predicted secondary structure. Local free energy estimates were worked out for each 5′ UTR. This was done by cutting the longer 5′ UTRs into sequences 200 bp in length with an overlapping window of 100-bp size between successive sequence fragments. The most stable secondary structure predicted in each sequence fragment was considered, and its free energy estimate is recorded in the database.
3. Number of stable folds: The number of predicted folds with minimum folding energy of less than −50 kcal/mole were counted.
4. GC percentage: G + C percentage was calculated for each 5′ UTR.
5. G/C ratio: The absolute value of G/C − 1 was calculated. If the value of G/C is in the neighborhood of 1, then the chance of forming stable secondary structures is high, and it is less otherwise.
6. A/T ratio: Similar argument holds good for the absolute value of A/T − 1.
7. Number of uAUGs: The most important positions for efficient translation are a purine at the −3 position and a G at position +4, where A of the AUG codon is position +1 (Kozak 1997). If either of these main features is unfavorable, positions +5 and +6 can influence the start site efficiency, with a A or C being preferred at position +5 and a U at position +6 (Boeck and Kolakofsky 1994). An AUG is said to be in good context if it satisfies these criteria, and all those uAUGs that were in good context are counted for each 5′ UTR.
8. Number of uORFs: Some mRNAs with a long 5′ UTR contain one or more uORFs, and these uORFs are often inhibitory for the translation of the downstream coding region. All those uORFs with good initiation context, as defined above, were counted for each 5′ UTR.
9. TOP: This is a categorical variable with value 1 if 5′ TOP exits and 0 otherwise.
10. Start site context: This is another categorical variable taking value 1 if the start codon (AUG) is in good context and 0 otherwise. Along with these 5′ UTR features, we have also included the following two more variables that may also influence the mRNA translation:
11. CDS length: The length of the coding sequence in terms of the number of amino acids was calculated.
12. Codon bias: Codon bias was calculated according to the Karlin formula (Karlin and Mrazek 1996) with slight modifications.
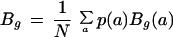 |
where p(a) are the amino acid frequencies, N is the total amino acid count and Bg(a) are calculated as follows:
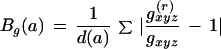 |
with g(r)xyz being the relative codon frequencies for the gene gr and gxyz the relative codon frequencies of a reference set. Here the reference set constitutes a set of 30 genes with poor translational efficiency.
Acknowledgments
Work in the Zhang lab was partly supported by NIH grants HG01696 and CA81152. Work in the Sugano lab was partly supported by a Grant-in-Aid for Scientific Research on Priority Areas from the Ministry of Education, Science, Sports and Culture of Japan and by Special Coordination Funds for Promoting Science and Technology (SCF) from the Science and Technology Agency (STA) of Japan. We thank Prof. David R. Morris (Department of Biochemistry, University of Washington, Seattle, WA) and Dr. Jose A. Garcia-Sanz (Department of Immunology and Oncology, Centro Nacional de Biotechnologia CNG—CSIC, Madrid, Spain) for their advice and for providing the lists of highly translated genes from their cDNA expression array experiments. We also thank Prof. Michael Zuker (Washington University, St. Louis, MO) for providing the mfold program.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL mzhang@cshl.org; FAX (516) 367-8461.
Article published online before print: Genome Res., 10.1101/gr.146000.
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.146000.
REFERENCES
- Arrick BA, Grendell RL, Griffin LA. Enhanced translational efficiency of a novel transforming growth factor beta 3 mRNA in human breast cancer cells. Mol Cell Biol. 1994;14:619–628. doi: 10.1128/mcb.14.1.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avni D, Biberman Y, Meyuhas O. The 5′ terminal oligopyrimidine tract confers translational control on TOP mRNAs in a cell type- and sequence context-dependent manner. Nucleic Acids Res. 1997;25:995–1001. doi: 10.1093/nar/25.5.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeck R, Kolakofsky D. Positions +5 and +6 can be major determinants of the efficiency of non-AUG initiation codons for protein synthesis. EMBO J. 1994;13:3608–3617. doi: 10.1002/j.1460-2075.1994.tb06668.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L, Friedman JH, Olshen RA, Stone CJ. Classification and regression trees. New York, NY: Chapman & Hall; 1984. [Google Scholar]
- Charron M, Shaper JH, Shaper NL. The increased level of beta1,4-galactosyltransferase required for lactose biosynthesis is achieved in part by translational control. Proc Natl Acad Sci. 1998;95:14805–14810. doi: 10.1073/pnas.95.25.14805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens MJ, Bommer UA. Translational control: The cancer connection. Int J Biochem Cell Biol. 1999;31:1–23. doi: 10.1016/s1357-2725(98)00127-7. [DOI] [PubMed] [Google Scholar]
- Duret L, Mouchiroud D. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis. Proc Natl Acad Sci. 1999;96:4482–4487. doi: 10.1073/pnas.96.8.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Sanz JA, Mikulits W, Livingstone A, Lefkovits I, Mullner EW. Translational control: A general mechanism for gene regulation during T cell activation. FASEB J. 1998;12:299–306. doi: 10.1096/fasebj.12.3.299. [DOI] [PubMed] [Google Scholar]
- Gray NK, Wickens M. Control of translation initiation in animals. Annu Rev Cell Dev Biol. 1998;14:399–458. doi: 10.1146/annurev.cellbio.14.1.399. [DOI] [PubMed] [Google Scholar]
- Karlin S, Mrazek J. What drives codon choices in human genes? J Mol Biol. 1996;262:459–472. doi: 10.1006/jmbi.1996.0528. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ. Control of gene expression at the level of translation initiation. Curr Opin Biotechnol. 1994;5:550–557. doi: 10.1016/0958-1669(94)90073-6. [DOI] [PubMed] [Google Scholar]
- Kochetov AV, Ischenko IV, Vorobiev DG, Kel AE, Babenko VN, Kisselev LL, Kolchanov NA. Eukaryotic mRNAs encoding abundant and scarce proteins are statistically dissimilar in many structural features. FEBS Lett. 1998;440:351–355. doi: 10.1016/s0014-5793(98)01482-3. [DOI] [PubMed] [Google Scholar]
- Kochetov AV, Ponomarenko MP, Frolov AS, Kisselev LL, Kolchanov NA. Prediction of eukaryotic mRNA translational properties. Bioinformatics. 1999;15:704–712. doi: 10.1093/bioinformatics/15.7.704. [DOI] [PubMed] [Google Scholar]
- Kozak M. Circumstances and mechanisms of inhibition of translation by secondary structure in eucaryotic mRNAs. Mol Cell Biol. 1989;9:5134–5142. doi: 10.1128/mcb.9.11.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Structural features in eukaryotic mRNAs that modulate the initiation of translation. J Biol Chem. 1991;266:19867–19870. [PubMed] [Google Scholar]
- ————— Interpreting cDNA sequences: Some insights from studies on translation. Mamm Genome. 1996;7:563–574. doi: 10.1007/s003359900171. [DOI] [PubMed] [Google Scholar]
- ————— Recognition of AUG and alternative initiator codons is augmented by G in position +4 but is not generally affected by the nucleotides in positions +5 and +6. EMBO J. 1997;16:2482–2489. doi: 10.1093/emboj/16.9.2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Initiation of translation in prokaryotes and eukaryotes. Gene. 1999;234:187–208. doi: 10.1016/s0378-1119(99)00210-3. [DOI] [PubMed] [Google Scholar]
- Maruyama K, Sugano S. Oligo-capping: A simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides. Gene. 1994;138:171–174. doi: 10.1016/0378-1119(94)90802-8. [DOI] [PubMed] [Google Scholar]
- Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- Merrick WC. Mechanism and regulation of eukaryotic protein synthesis. Microbiol Rev. 1992;56:291–315. doi: 10.1128/mr.56.2.291-315.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick WC, Hershey JB. The pathway and mechanism of eukaryotic protein synthesis. In: Hershey JB, Mathews MB, Sonenberg N, editors. Translational control. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1996. pp. 31–69. [Google Scholar]
- Meyuhas O, Avni D, Shama S. Translational control of ribosomal protein mRNAs in eukaryotes. In: Hershey JB, Mathews MB, Sonenberg N, editors. Translational control. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1996. pp. 363–388. [Google Scholar]
- Pelletier J, Sonenberg N. Insertion mutagenesis to increase secondary structure within the 5′ noncoding region of a eukaryotic mRNA reduces translational efficiency. Cell. 1985;40:515–526. doi: 10.1016/0092-8674(85)90200-4. [DOI] [PubMed] [Google Scholar]
- Pesole G, Liuni S, Grillo G, Licciulli F, Larizza A, Makalowski W, Saccone C. UTRdb and UTRsite: Specialized databases of sequences and functional elements of 5′ and 3′ untranslated regions of eukaryotic mRNAs. Nucleic Acids Res. 2000;28:193–196. doi: 10.1093/nar/28.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preiss T, Hentze MW. From factors to mechanisms: translation and translational control in eukaryotes. Curr Opin Genet Dev. 1999;9:515–521. doi: 10.1016/s0959-437x(99)00005-2. [DOI] [PubMed] [Google Scholar]
- Shantz LM, Hu RH, Pegg AE. Regulation of ornithine decarboxylase in a transformed cell line that overexpresses translation initiation factor eIF-4E. Cancer Res. 1996;56:3265–3269. [PubMed] [Google Scholar]
- Sharp PM, Li WH. The codon adaptation index—A measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snedecor GW, Cochran WG. Statistical methods. 7th ed. Ames, IA: Iowa State University Press; 1980. [Google Scholar]
- Suzuki Y, Yoshitomo-Nakagawa K, Maruyama K, Suyama A, Sugano S. Construction and characterization of a full length-enriched and a 5′-end-enriched cDNA library. Gene. 1997;200:149–156. doi: 10.1016/s0378-1119(97)00411-3. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Ishihara D, Sasaki M, Nakagawa H, Hata H, Tsunoda T, Watanabe M, Komatsu T, Ota T, Isogai T, et al. Statistical analysis of the 5′ untranslated region of human mRNA using “Oligo-Capped” cDNA libraries. Genomics. 2000;64:286–297. doi: 10.1006/geno.2000.6076. [DOI] [PubMed] [Google Scholar]
- van der Velden AW, Thomas AA. The role of the 5′ untranslated region of an mRNA in translation regulation during development. Int J Biochem Cell Biol. 1999;31:87–106. doi: 10.1016/s1357-2725(98)00134-4. [DOI] [PubMed] [Google Scholar]
- Zhang MQ. Identification of protein coding regions in the human genome by quadratic discriminant analysis. Proc Natl Acad Sci. 1997;94:565–568. doi: 10.1073/pnas.94.2.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong Q, Schummer M, Hood L, Morris DR. Messenger RNA translation state: The second dimension of high-throughput expression screening. Proc Natl Acad Sci. 1999;96:10632–10636. doi: 10.1073/pnas.96.19.10632. [DOI] [PMC free article] [PubMed] [Google Scholar]