

Abstract
We have developed an accurate, yet inexpensive and high-throughput, method for determining the allele frequency of biallelic polymorphisms in pools of DNA samples. The assay combines kinetic (real-time quantitative) PCR with allele-specific amplification and requires no post-PCR processing. The relative amounts of each allele in a sample are quantified. This is performed by dividing equal aliquots of the pooled DNA between two separate PCR reactions, each of which contains a primer pair specific to one or the other allelic SNP variant. For pools with equal amounts of the two alleles, the two amplifications should reach a detectable level of fluorescence at the same cycle number. For pools that contain unequal ratios of the two alleles, the difference in cycle number between the two amplification reactions can be used to calculate the relative allele amounts. We demonstrate the accuracy and reliability of the assay on samples with known predetermined SNP allele frequencies from 5% to 95%, including pools of both human and mouse DNAs using eight different SNPs altogether. The accuracy of measuring known allele frequencies is very high, with the strength of correlation between measured and known frequencies having an r2 = 0.997. The loss of sensitivity as a result of measurement error is typically minimal, compared with that due to sampling error alone, for population samples up to 1000. We believe that by providing a means for SNP genotyping up to thousands of samples simultaneously, inexpensively, and reproducibly, this method is a powerful strategy for detecting meaningful polymorphic differences in candidate gene association studies and genome-wide linkage disequilibrium scans.
It has been proposed that association studies of polymorphic markers in genome-wide scans may be the most efficient way of identifying genetic regions or genes implicated in common, complex diseases and traits (Risch and Merikangas 1996). Association studies may further be useful when family-based samples are not available and to fine-map or confirm larger candidate regions identified by family-based, linkage analyses. Recently, single-nucleotide polymorphisms (SNPs) have been recognized as the marker of choice for gene mapping by linkage disequilibrium and association, in part because they appear throughout the genome with much greater frequency than other types of polymorphisms (Collins et al. 1997; Landegren et al. 1998; Brookes 1999). Efforts are currently underway to generate a collection of SNPs sufficiently large to saturate the human genome with an average spacing of ∼30 kb (Lai et al. 1998; Marshall 1997, 1999; Picoult-Newberg et al. 1999).
In genome-wide scans using case and control populations to investigate disease association, many thousands, and perhaps hundreds of thousands, of polymorphisms will need to be typed in a large number of individuals (Risch and Merikangas 1996; Kruglyak 1999). An approach that types one SNP for one sample at a time will most likely not have the necessary throughput (for a review of such methods, see Landegren et al. 1998). One approach to high-throughput genotyping of SNPs is to type multiple polymorphisms one individual at a time with high-density oligonucleotide hybridization arrays (Wang et al. 1998). The capacity of the first commercially available array is limited to 1500 SNPs. It will be a problem to increase this number of SNPs as the simultaneous (multiplex) PCR amplification required will become increasingly laborious and difficult to control.
An alternative way of typing large numbers of samples and markers is to pool equal amounts of DNA from all the individual samples and then type one marker at a time. Test statistics have recently been described for study designs using biallelic markers with pooled samples (Barcellos et al. 1997; Risch and Teng 1998). Pooling of DNA samples has been successfully used with both microsatellite markers and SNPs (Arnheim et al. 1985; Pacek et al. 1993; Syvänen et al. 1993; Kwok et al. 1994; Barcellos et al. 1997; Shaw et al. 1998). All of these methods, however, require substantial post-PCR processing. We describe here a novel method for the determination of SNP allele frequencies in pooled samples that has a number of advantages: It is not based on expensive fluorescently labeled primers or probes; it is a homogenous assay that requires no post-PCR processing; it operates under uniform conditions without the need for marker specific assay optimization; and it is accurate. It promises to be inexpensive, time-saving, and precise enough to allow detection of the relatively weak but important genetic associations expected for complex traits in outbred populations.
Principle of the Method
To measure a SNP allele frequency in a mixture of DNAs pooled from individual samples, equal aliquots of the pool are divided between two PCR reactions, each of which contains a primer pair specific to one or the other SNP allelic variant. The specificity of the PCR amplification is conferred by placing the 3′ end of one of the primers directly over and matching one or the other of the variant nucleotides (Newton et al. 1989; Sommer et al. 1989; Wu et al. 1989). This specificity can be enhanced particularly by using the Stoffel fragment of Taq DNA polymerase (Lawyer et al. 1993; Tada et al. 1993; Germer and Higuchi 1999). Ideally, only completely matched primers are extended, and only the matching allele is amplified. In practice, however, there will be amplification of the mismatched allele, but this will occur much less efficiently such that many more amplification cycles are needed to generate detectable levels of product. Mismatch amplification is frequently delayed by >10 cycles when amplification is monitored on a cycle-by-cycle basis (Higuchi et al. 1993), using fluorescent dsDNA binding dyes such as SYBR Green I. A delay of around six cycles is adequate for the determination of allele frequencies of SNPs for which the frequency of the minor allele is greater than a few percent.
When the allele frequency is 50%, one expects that each of the two PCR amplifications will require the same number of cycles to produce the same fluorescent signal, assuming that both allele-specific primers amplify with equal efficiency. The number of cycles before a reaction crosses a predetermined threshold, the Ct, can be fractional. When one allele is more frequent, amplification of that allele will reach the threshold at an earlier cycle, that is, have a smaller Ct. The difference in Ct's between the two PCR reactions, the ΔCt, is a measure of the bias and thus of the allele frequency. A one-cycle delay means that the ratio of the amount of one allele to the other is 1:2; a two-cycle delay, 1:4; or in general, 1:2ΔCt. Converting a ratio to a frequency by adding the numerator to the denominator results in
 |
1 |
 |
Note that ΔCt can be either positive or negative, depending on which specific PCR exhibits the lowest Ct. The “2” in the denominator is properly “1 + the initial replication efficiency”. However, the initial replication efficiency is usually close to 100% so that “2” is an adequate approximation (Higuchi and Watson 1999). The amplification efficiencies for the two allele-specific PCRs may differ slightly. As shown below, this can be measured and compensated for by performing the assay on a DNA known to be heterozygous for the SNP of interest. The ΔCt for this DNA should equal zero if the PCRs are equally efficient. Any deviation from zero indicates that they are not. This deviation can then be subtracted from all ΔCt measurements to compensate for differential amplification efficiencies.
Figure 1 shows kinetic growth curves for two separate PCR reactions (four replicates of each) performed on a sample of DNA, prepared by adding 1 part of a DNA homozygous for one allele to 19 parts of a DNA homozygous for the other allele, for a total mixture of 5% allele1 and 95% allele2. Reactions amplified with the primer specific to allele2 crossed the threshold at approximately cycle 26 (average 25.77), whereas reactions with the primer specific to allele1 crossed the threshold at approximately cycle 30 (average 30.45). The ΔCt is the difference between the two sets of Ct's, in this case an average of 4.68 cycles (see Table 1, below). The ΔCt was then used to calculate the allele frequency according to equation 1.
Figure 1.

The basis of allele frequency measurement using kinetic PCR. Shown are amplification growth curves of PCR reactions performed for the ApoB71 polymorphism. A sample was constructed from two DNAs each homozygous for the different alleles of the ApoB71 SNP and contains 5% of allele 1. Equal aliquots of the pool (20 ng of DNA each) were put into PCRs containing either of the two allele-specific primer sets. Four replicate reactions were performed with each primer set (eight PCRs total). The relative allele frequency is determined on the basis of the ΔCt using equation 1 (see text and Fig. 2).
Table 1.
Allele Frequency Measurements Across a Range of Values
| Allele 1 frequency (by OD260) | Expected ΔCt | PON | B71 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| average observed ΔCt | heterozygote corrected ΔCt | measured allele 1 frequency | standard deviation (±) | average observed ΔCt | heterozygote corrected ΔCt | measured allele 1 frequency | standard deviation (±) | ||
| 0.95 | −4.25 | −4.01 | −4.05 | 0.94 | 0.016 | −3.21 | −3.77 | 0.93 | 0.012 |
| 0.90 | −3.17 | −2.99 | −3.03 | 0.89 | 0.010 | −2.23 | −2.79 | 0.87 | 0.023 |
| 0.80 | −2.00 | −1.88 | −1.92 | 0.79 | 0.014 | −1.22 | −1.78 | 0.77 | 0.040 |
| 0.67 | −1.00 | −1.02 | −1.06 | 0.68 | 0.051 | −0.21 | −0.77 | 0.63 | 0.032 |
| 0.59 | −0.50 | −0.44 | −0.48 | 0.58 | 0.030 | 0.24 | −0.32 | 0.56 | 0.031 |
| 0.50 | 0.00 | 0.14 | 0.10 | 0.48 | 0.016 | 0.70 | 0.14 | 0.48 | 0.024 |
| Het. | 0.00 | 0.04 | 0.00 | 0.50 | 0.051 | 0.56 | 0.00 | 0.50 | 0.013 |
| 0.42 | 0.50 | 0.78 | 0.74 | 0.37 | 0.045 | 1.21 | 0.66 | 0.39 | 0.016 |
| 0.33 | 1.00 | 1.18 | 1.14 | 0.31 | 0.007 | 1.65 | 1.09 | 0.32 | 0.021 |
| 0.20 | 2.00 | 2.11 | 2.07 | 0.19 | 0.007 | 2.54 | 1.99 | 0.20 | 0.011 |
| 0.10 | 3.17 | 3.21 | 3.17 | 0.10 | 0.014 | 3.63 | 3.07 | 0.11 | 0.007 |
| 0.05 | 4.25 | 4.52 | 4.48 | 0.04 | 0.003 | 4.68 | 4.12 | 0.05 | 0.002 |
Ct measurements are average of four replicates.
In Figure 2, a representation of equation 1, allele1 frequency was plotted as a function of the ΔCt between the two PCR reactions (solid central line). The flanking solid lines represent the uncertainty in estimating the population allele frequency due to sampling error when the sample size = 1000. The dashed lines depict the predicted additional uncertainty contributed, on average, by the measurement error observed with this method (see below). Because the variability of ΔCt is expected to be independent of ΔCt, note that equation 1 predicts that measurement error and its relative contribution to the overall uncertainty should decrease significantly as the allele frequency is biased toward one or the other allele (Fig. 2, cf. insets).
Figure 2.

The relationship between ΔCt and allele frequency. The solid center line is a plot of equation 1 from the text. The flanking solid lines represent the expected uncertainty (1 s.d.) in estimating the allele frequency based on sampling error alone (sample size = 1000). The broken lines represent the combined uncertainty of sampling and measurement error. The measurement error is based on an average error seen amongst the measurements taken in this paper and is that expected after averaging four replicate measurements. The insets compare the impact of measurement error at the middle and at the upper extreme of allele frequencies (the lower extreme should mirror exactly the upper).
The generation of template independent primer artifact during the PCR process could confound the signal resulting from the amplification of a mismatched primer–template combination. To avoid the formation and potential interference of template independent generation of primer artifiact, we use a uracil-N-glycosylase (UNG) mediated “hot start” as well as a heat-activated polymerase enzyme (see Methods, below). An additional source of potential error for this, and any other genotyping error based on the amplification of genomic DNA, is the amplification of nonspecific regions homologous to the target sequence (e.g., pseudogenes). In the present assay the use of a version of the Stoffel fragment of Taq polymerase minimizes this problem, as it not only is highly discriminatory but is also not very processive. We amplify as small as possible specific products which are favored over larger nonspecific homologous products.
RESULTS
We demonstrate the validity of using this method to determine allele frequencies in several ways. First, to show that the method is valid over a wide range of allele frequencies, we constructed samples consisting of predetermined, different ratios of the two alleles from two DNAs that were homozygous for the respective alleles and measured their allele frequencies. The results are listed in Table 1. The ΔCt for each sample is the product of four separate measurements (a total of eight reactions), and the s.d.s of the sets of measurements are given. The experiments were conducted on two separate SNPs. As described above, we have corrected for the error introduced by unequal amplification efficiency of the two allele-specific primers for each polymorphism by measuring ΔCt on a heterozygous sample. The observed offset from zero, due to unequal amplification efficiency, is subtracted from all ΔCt measurements. The measurements confirm equation 1 and appear quite accurate.
Second, to show that the method works on an actual pool of individual samples, we determined the allele frequencies for three distinct SNPs (see Methods) in a pool made from 100 individual human DNA samples (Table 2). For the three polymorphisms, each of the 100 samples was individually genotyped either by Tm-shift genotyping (Germer and Higuchi 1999) or, for CST5, by probe strip hybridization (Saiki et al. 1988; G. Zangenberg and R. Reynolds, unpubl.). The samples were then pooled, and the allele frequency determined by kinetic PCR. Calculated allele frequencies represent the average of 12 measurements. It should be noted that an inaccuracy in any two individual genotype determinations could produce an error in the “actual” allele frequency of as much as ± 0.02. Allele frequencies determined from the pooled samples deviate from the “actual” allele frequencies by +0.02 (PON), −0.01 (B71), and −0.03 (CST5), respectively.
Table 2.
Allele Frequency Measurements on a Pool of 100 Human DNAs
| Determination | PON | B71 | CST5 |
|---|---|---|---|
| 1 | 0.45 | 0.72 | 0.41 |
| 2 | 0.49 | 0.75 | 0.37 |
| 3 | 0.41 | 0.72 | 0.39 |
| 4 | 0.43 | 0.72 | 0.43 |
| 5 | 0.46 | 0.74 | 0.41 |
| 6 | 0.45 | 0.73 | 0.42 |
| 7 | 0.44 | 0.72 | 0.42 |
| 8 | 0.47 | 0.73 | 0.42 |
| 9 | 0.49 | 0.76 | 0.42 |
| 10 | 0.44 | 0.81 | 0.40 |
| 11 | 0.44 | 0.71 | 0.42 |
| 12 | 0.44 | 0.74 | 0.41 |
| Measured allele 1 frequency | 0.45 | 0.74 | 0.41 |
| Known frequency | 0.43 | 0.75 | 0.44 |
| Standard deviation (±) | 0.024 | 0.027 | 0.018 |
Third, to test the robustness of the method, we determined the allele frequencies of five additional SNPs on a pool constructed from 10 mouse DNAs, with each sample belonging to a different inbred strain (Table 3). The 10 samples were individually genotyped for the five SNPs by kinetically monitored, allele-specific PCR amplifications. As expected for DNA samples from inbred mouse strains, all 10 samples were homozygous for every SNP. The samples were pooled, and as in Table 2, the calculated allele frequencies represent the average of 12 measurements corrected for differential amplification effeciency. Allele frequencies determined for the pool were accurate by this method.
Table 3.
Allele Frequency Measurements on a Pool of Mouse DNAs from 10 Different Inbred Strains
| Determination | TLR4 | FASL | AHR | HOX2.3 | REN1 |
|---|---|---|---|---|---|
| 1 | 0.09 | 0.13 | 0.22 | 0.30 | 0.47 |
| 2 | 0.10 | 0.14 | 0.20 | 0.28 | 0.43 |
| 3 | 0.09 | 0.11 | 0.20 | 0.27 | 0.46 |
| 4 | 0.10 | 0.13 | 0.20 | 0.28 | 0.48 |
| 5 | 0.10 | 0.14 | 0.21 | 0.29 | 0.44 |
| 6 | 0.10 | 0.16 | 0.18 | 0.27 | 0.45 |
| 7 | 0.10 | 0.16 | 0.15 | 0.27 | 0.42 |
| 8 | 0.09 | 0.13 | 0.20 | 0.29 | 0.42 |
| 9 | 0.10 | 0.15 | 0.19 | 0.31 | 0.41 |
| 10 | 0.09 | 0.16 | 0.20 | 0.30 | 0.30 |
| 11 | 0.10 | 0.19 | 0.19 | 0.29 | 0.33 |
| 12 | 0.09 | 0.18 | 0.18 | 0.24 | 0.37 |
| Measured allele 1 frequency | 0.10 | 0.15 | 0.19 | 0.28 | 0.42 |
| Known frequency | 0.10 | 0.20 | 0.20 | 0.30 | 0.40 |
| Standard deviation (±) | 0.005 | 0.023 | 0.018 | 0.017 | 0.054 |
Figure 3 is a scatter graph that summarizes all the allele frequency determinations in Tables 1, 2, and 3. The frequencies measured on pooled DNA samples are compared with the “known” frequencies determined by counting the alleles contributed by the individually genotyped samples. The pooled estimates and known values of all allele frequencies are very highly correlated (r2 = 0.997). The slope of the regression line is not significantly different from the expected value of 1.0. As predicted by equation 1 and the relative constancy of ΔCt variability, the measurement error tends to be lower at the extremes of the frequency distribution (<15% and >85%).
Figure 3.

The accuracy of allele frequency measurement by kinetic PCR. Shown is a scatter-plot of all the measurements of allele frequency made in Tables 1, 2, and 3 comparing the known frequencies (determined by DNA concentration for Table 1 and by individual genotyping and allele counting for Tables 1 and 2) with the measured frequencies. The error bars represent one s.d. in the measurement. The diagonal line is that expected for complete concordance between known and measured values.
Because for association studies the absolute accuracy of this method is ultimately less important than its ability to detect minor differences in allele frequencies between pools, we considered the impact of the observed measurement variability on this application. It is important to consider this in the context of sample size and sampling error, because sample size in most studies has an upper fixed limit that results in significant sampling error. The question then becomes, does the error in allele frequency measurement add significantly to this unavoidable sampling error? In Figure 4, sampling and measurement error is plotted for three representative SNPs from our data for sample sizes up to n = 1000.
Figure 4.

The impact of measurement error for three SNP assays. (A) Plotted as the solid line is the expected sampling error for this SNP given an allele frequency of 10% (see text) for sample sizes up to 1000. The upper broken line is the estimated combined sampling and measurement error for this assay based on Table 3 and using the average of four measurements. This measurement error alone is the lower broken line. (B) The same as A for the human CST5 locus (Table 2). (C) The same as A and B for the mouse REN1 SNP (Table 3).
In the first frame (A) are depicted these errors for the murine SNP TRL4 from Table 3 that had an actual allele frequency of 0.1 (in our pooled sample) and one of the smallest measurement errors, ± 0.0027 (lower broken line). We use for measurement error the s.e of the mean  using four measurements, which is a reasonable number by this method. Plotted as the solid line is the expected sampling error for this SNP given an allele frequency of 10% (
using four measurements, which is a reasonable number by this method. Plotted as the solid line is the expected sampling error for this SNP given an allele frequency of 10% ( P = allele frequency and n = sample size (two alleles per individual; Glantz 1997)) for sample sizes up to 1000. The upper broken line is the estimated combined sampling and measurement error for this assay
P = allele frequency and n = sample size (two alleles per individual; Glantz 1997)) for sample sizes up to 1000. The upper broken line is the estimated combined sampling and measurement error for this assay 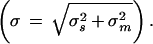 For this SNP the impactof measurement error for samples up to 1000 is negligible. In contrast, the bottom frame (C) illustrates the impact of the largest measurement error in our data set, that for the mouse SNP REN1 that had an allele frequency of 0.4 (in our pool) and a measurement error of ±0.027. At about n = 175 the measurement error begins to predominate. At n = 1000 the error is mostly measurement error.
For this SNP the impactof measurement error for samples up to 1000 is negligible. In contrast, the bottom frame (C) illustrates the impact of the largest measurement error in our data set, that for the mouse SNP REN1 that had an allele frequency of 0.4 (in our pool) and a measurement error of ±0.027. At about n = 175 the measurement error begins to predominate. At n = 1000 the error is mostly measurement error.
The middle frame (B) represents a nearly average case, in which the allele frequency is 0.44 and the measurement error is ± 0.009. At n = 1000, sampling error is still the predominant error. For an “average” assay such as this, it is instructive to consider a recent report (Barcellos et al. 1997) that performs a series of simulations to calculate the statistical power of genome scans. The hypothetical studies are designed to detect biallelic marker associations in case–control populations of varying size for markers with different association strengths (i.e., the true differences, at the population level, between marker frequencies in cases and controls). Only sampling error is taken into account. The statistical power remains high even for markers with association strengths as low as 0.05, as long as the sample sizes approach 1000 or greater, and a rate of false positives of 0.1% is acceptable (see Barcellos et al. 1997; Tables 2 and 3). From Figure 4B it can be seen that the impact of the measurement error in our assay is to increase the uncertainty at n = 1000 to about that at n = 500 without measurement error. Examination of Table 2 in Barcellos et al. (1997) shows that this should result in a significant reduction of power to detect associations of 0.05 but not of 0.1 or 0.2 . For studies with fewer markers (such as for candidate gene regions), a false positive rate of, say, 5% would be acceptable. For such studies the power to detect associations as low as 0.05 would remain high.
DISCUSSION
Clearly, as the number of markers required for a study decreases the number of potential type I errors (false-positive associations) decreases. The negative impact of both sampling and measurement errors is lessened. Logical first applications of the method proposed here would be fine mapping of candidate chromosomal regions already identified by family linkage or other studies and the testing of candidate genes chosen for their potential functional relationship to a disease (Cheng et al. 1998). The advantages of low cost and effort, once regional SNPs are identified, are attractive in this context. For the same reasons, the method may be particularly well suited to experimental genetics as a way to rapidly type large numbers of experimental intercrosses between inbred strains (G. Peltz, pers. comm.). For example, as few as 100–200 SNPs should be sufficient to cover the entire murine genome in such intercrosses. Finally, as Shaw et al. (1998) suggest, pooling strategies such as the one presented here may have a range of applications in evolutionary genetics for exploring populations' histories and the mechanisms of molecular evolution at the population level.
Once such studies have provided valuable experience with this methodology, whole genome scans might be considered. It is likely that initial genome scans will begin once a minimum number of SNPs, say 10,000 or even less, have been found. Consider an association study of 1000 case and 1000 control samples using 10,000 SNPs. Individual genotyping would require a formidable 2 × 107 typings; even without considering error rates and possible retyping, this is likely beyond the scope of current technologies. Using a pooling strategy as described here would require “only” 1.6 × 105 PCR reactions if four allele frequency determinations were performed for each SNP (for each pool). This translates into 1670 assay plates using the current commercially available 96-well format for kinetic PCR. Assuming a 96-well instrument could run six plates per day (a run takes 2.5 hr), a bank of 10 existing instruments could complete the study in less than a month. A pooling strategy also reduces the quantity of DNA required from each sample to be tested (see Methods, below).
For such a genome scan and even for smaller scale efforts, it would be impossible to completely validate each SNP assay before using it. We would propose, instead, to establish standard conditions under which most primer sets will work adequately without optimization and that only “spot-checking” of a small subset of assays be done. The two forms of assay failure, both of which are not all or none but a matter of degree, are (1) failure to discriminate alleles adequately, leading to insensitivity to actual population frequency differences, and (2) excessive assay variability leading to excess type I (false association) errors. The frequency of the first type of failure can be estimated by spot-checking. Whether it has occurred at any given SNP cannot be known, but the occurrence can be minimized. A 20% failure rate means, in essence, that a 10,000 SNP study is actually an 8000 SNP study. The occurrence of the second type of failure will be known for every SNP by the multiple measurements taken (four for each pool, or eight in all) as part of the proposed genome scan. The SNP-specific variability can be taken into account when assessing the significance of frequency differences at that SNP.
All SNP experiments reported in this paper were performed under uniform conditions. To date, we have designed 22 different primer sets (2 allele-specific and 1 common primer per set) for 10 different human SNPs, with an overall success rate of ∼80%. Based on this and our further experience, we are developing a computerized, primer design program that will automatically specify optimal SNP primers on the basis simply of the relevant sequence information and a standard set of parameters (e.g., see Beasley et al. 1999).
In conducting association studies using pools of DNA, accurate quantitation of the individual DNAs is important lest artifactual allele discrepancies between pools arise (see Methods, below). Although the routine, small errors commonly seen in DNA quantitation should increasingly cancel out as the number of samples increases, large unforeseen errors could cause problems. The simplest safeguard against errors arising from the pooling process would be to validate the pools by doing, for only one or two of the many SNPs to be screened, genotyping of the individual samples and showing concordance between allele counting and frequency measurement on the pool. Because Tm-shift genotyping (Germer and Higuchi 1999) uses the same allele-specific PCR conditions, and two of the same three primers, as the method described here and is high throughput, it should be a good choice for doing the individual genotyping.
Other kinetic PCR approaches should, in theory, allow frequency measurement in a single PCR reaction. A number of PCR-based approaches to single-tube, individual genotyping that incorporate homogeneously read, fluorescently labeled, oligonucleotide probes have been developed. 5′ Nuclease (“TaqMan”) probes (Holland et al. 1991; Lee et al. 1993) and molecular beacon probes (Tyagi and Kramer 1996) should be able to generate ΔCt's in a single tube by virtue of differentially (fluorescence wavelength) labeled probes. Molecular beacon probes have been used for individual genotyping using differences in Ct (Kostrikis et al. 1998). A disadvantage of these approaches is the much greater expense of the SNP-specific, fluorescent oligonucleotide probes (compared with unlabled primers) that, if conducting genome scans, could be prohibitive. Also, obtaining one or a few conditions under which most allele-specific probes give adequate allele discrimination or reproducible ΔCt's may be more difficult than for allele-specific priming of PCR. Differential fluorescence labeling of primers (Nazarenko et al. 1997) allows the use of allele-specific PCR but does not eliminate the objection of high cost of SNP-specific fluorescent primers. The cost objection might be overcome using approaches in which a different (for each allele, but the same for all PCRs) tag sequence is added 5′ to each of the allele-specific primers (Jeffreys et al. 1991; Neilan et al. 1997; Winn-Deen 1998). Generic, homogenously read and differentially labeled primers homologous to the two tag sequences are included in all PCRs. A similar but more complex scheme has been reported using a generic 5′ nuclease probe (Whitcombe et al. 1999).
In conclusion, we have presented in this paper a method that is a highly accurate and reproducible means of measuring the relative amounts of the two allelic variants of a SNP in pooled samples of DNA. With enough samples, this can be an accurate estimate of the frequency of the alleles in the population from which the samples were drawn. By pooling samples to measure allele frequency, a considerable savings in work over individual genotyping can be achieved when doing case/control and other study designs for the detection of associations between genes and complex diseases. This may allow for practical implementation of whole genome scans.
METHODS
DNA Samples, Pools, and Polymorphisms
Human DNA samples for testing the determination of allele frequencies were obtained from Roche Biomedical Laboratories (now, LabCorp of America) as described in a previous publication (Germer and Higuchi 1999) and were made available for this study by Gabrielle Zangenberg and Rebecca Reynolds of Roche Molecular Systems (RMS). Suzanne Cheng, Priscilla Moonsamy, and Michael Grow of RMS provided DNA samples for testing and optimizing the assay. Murine DNA samples from 10 inbred mouse strains (C57BL/6J, BALB/cJ, A/J, A/HeJ, B10.D2-H2, C3H/HeJ, DBA/2J, MRL/MpJ, NZB/BlnJ, and NZW/LacJ) were obtained from Jackson Laboratories and made available for this study by Andrew Grupe and Dee Aud at Roche Bioscience.
The samples in Table 1 were constructed by mixing two homozygous human DNAs in various proportions. An 80% allele1 sample, for instance, was made by combining 80 μl (at 2 ng/μl) of a sample homozygous for allele 1 and 20 μl (at 2 ng/μl) of a sample homozygous for allele 2. The human DNA pool (Table 2) was constructed by adding 20 μl (at 1 ng/μl) of each of the 100 individual samples for a total of 2 ml. The mouse DNA pool (Table 3) was constructed from a mixture of 10 μl each of the 10 samples, at a concentration of 10 ng/μl.
For the quantitation of individual DNA samples combined in pools, we have used both OD260 and a DNA specific fluorescent dye, PicoGreen (Molecular Probes), and, in general, have found both satisfactory on the DNAs, mostly from blood, used in this study. Both methods are available for use in high-throughput 96-well formats. PicoGreen has the advantage of greater sensitivity and specificity, although it suffers from a loss of sensitivity when DNA is degraded.
The actual quantity needed for each DNA sample obviously depends on the number of polymorphisms and samples tested and can be calculated for the conditions used in this paper as 160 ng (i.e., 20 ng/rxn × 2 pools × 4 replicate reactions) multiplied by the number of polymorphisms, divided by the number of samples included in the pool. Thus, for 10,000 SNPs and 1000 samples, 1.6 μg of each DNA is needed.
The two SNPs typed for the human samples in Table 1 (PON and B71) are as in Germer and Higuchi (1999). The third SNP in Table 2, CST5, is a polymorphism in the human cystatin D gene (exon I, a Cys to Arg amino acid substitution) (Balbin et al. 1993). The five murine SNPs were identified from a literature search (A. Grupe, Roche Bioscience), and the sequences are available in GenBank. They include polymorphisms in the Toll-like receptor 4 gene (TRL4; GenBank accession no.AF095353), the aromatic hydrocarbon receptor (AHR; accession no.L19757), the homeotic gene Hox b7 (HOX-2.3; accession no.X06762), the Renin gene (REN1; accession no.X16642), and the Fas ligand (FASL; accession no.U58995).
Reaction Optimization and PCR Amplification
PCR reaction conditions were optimized for maximum allele specificity of the amplification by the use of Stoffel fragment DNA polymerase (Lawyer et al. 1993; Tada et al. 1993), salt concentrations, amplicon length, and the use of a UNG mediated hot start (Persing and Cimino 1993). To further minimize the formation of the template-independent, artifactual product primer–dimer (Chou et al. 1992), we used a modified, “Gold” version of the Stoffel fragment polymerase (Birch 1996) to provide a simplified hot start. Additionally, enough ROX dye was added to the PCR reactions to provide a significant level of fluorescence at “baseline” reads. This helped reduce well-to-well variability in fluorescence detection and, in our hands, resulted in more reproducible Ct determinations.
All PCR reactions were performed on a 20-ng template in a total volume of 100 μl. Each reaction comprised 0.2 μm of each of the two primers; 12 units of Stoffel Gold polymerase (David Birch, RMS); 1× Stoffel buffer (10 mm Tris-HCl, 10 mm KCl at pH 8.0); an additional 30 mm KCl for a final concentration of 40 mm; 2 mm MgCl2; 50 μm each dATP, dCTP, and dGTP; 25 μm dTTP; 75 μm dUTP; 2 units of UNG; 0.2× SYBR Green I (Molecular Probes); 2 μm ROX dye (Molecular Probes); 5% DMSO; and 2.5% glycerol. The PON locus was amplified with two of the following primers: either TATTTTCTTGACCCCTACTTACA or TTTCTTGACCCCTACTTACG (forward allele-specific primers), and CCACGCTAAACCCAAATACATCTC (reverse common primer); the B71 locus with either TGAAGACCAGCCAGTGCAT or GAAGACCAGCCAGTGCAC (forward allele-specific primers), and CAAGGCTTTGCCCTCAGGGTT (reverse common primer); and CST5 with either CAATGACAAGAGTGTGCAGT or AATGACAAGAGTGTGCAGC (forward allele-specific primers), and ACCTTGTTGTACTCGCTGATGGCAAA (reverse common primer). Murine SNP primer sequences are available from the authors upon request. Of the eight total SNPs, six require for their complete typing the discrimination of a G to T mismatch that is thermodynamically the most stable mismatch (Ikuta et al. 1987).
Kinetic PCR reactions were performed on a GeneAmp 5700 Sequence Detection System (PE Applied Biosystems). A similar, CCD camera-based system has been described (Higuchi and Watson 1999; Kang and Holland 1999; Kang et al. 1999). An initial incubation step of 2 min at 50°C, to allow UNG-mediated elimination of carryover PCR product contamination (Longo et al. 1990), and an enzyme heat-activation step of 12 min at 95°C were followed by 45 two-step amplification cycles of 20 sec at 95°C for denaturation and 20 sec at 58°C for annealing and extension, and a final 20-min product extension step at 72°C.
Data Analysis
Flourescence data from kinetic PCR reactions were analyzed in a spreadsheet (MS Excel) template that performs a series of operations similar to those performed by the GeneAmp 5700 Sequence Detection System software version 1.1. to calculate Ct values for each PCR. For each allele frequency measurement, the multiple ΔCt measurements were averaged. Allele frequencies were obtained using equation 1 as described in this paper.
Acknowledgments
For advice, assistance, and/or samples, we thank Sheng-Yung Chang, Suzanne Cheng, Henry Erlich, Michael Grow, Wally Laird, Daniel Mirel, Priscilla Moonsamy, Rebecca Reynolds, Bob Watson, Gabriele Zangenberg, and especially Carita Elfstrom, of RMS, as well as Dee Aud, Andrew Grupe, and Gary Peltz of Roche Bioscience and Laura Lazzeroni of Stanford. We thank David Birch of RMS for Stoffel Gold enzyme and John Sninsky of RMS for helpful suggestions and his continuing support of this work.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL Soren.Germer@Roche.com; FAX (510) 522-1285.
REFERENCES
- Arnheim N, Strange C, Erlich H. Use of pooled DNA samples to detect linkage disequilibrium of polymorphic restriction fragments and human disease: Studies of the HLA class II loci. Proc Natl Acad Sci. 1985;82:6970–6974. doi: 10.1073/pnas.82.20.6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbin M, Freije JP, Abrahamson M, Velasco G, Grubb A, López-Otín C. A sequence variation in the human cystatin D gene resulting in an amino acid (Cys/Arg) polymorphism at the protein level. Hum Genet. 1993;90:668–669. doi: 10.1007/BF00202491. [DOI] [PubMed] [Google Scholar]
- Barcellos LF, Klitz W, Field LL, Tobias R, Bowcock AM, Wilson R, Nelson MP, Nagatomi J, Thomson G. Association mapping of disease loci, by use of pooled DNA genomic screen. Am J Hum Genet. 1997;61:734–747. doi: 10.1086/515512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beasley EM, Myers RM, Cox DR, Lazzaroni LC. Statistical refinement of primer design parameters. In: Innes MA, Gelfand DH, Sninsky JJ, editors. PCR applications: Protocols for functional genomics. San Diego, CA: Academic Press; 1999. pp. 55–73. [Google Scholar]
- Birch DE. Simplified hot start PCR. Nature. 1996;381:445–446. doi: 10.1038/381445a0. [DOI] [PubMed] [Google Scholar]
- Brookes AJ. The essence of SNPs. Gene. 1999;234:177–186. doi: 10.1016/s0378-1119(99)00219-x. [DOI] [PubMed] [Google Scholar]
- Cheng S, Pallaud C, Grow MA, Scharf SJ, Erlich HA, Klitz W, Pullinger CR, Malloy MJ, Kane JP, Siest G, Visvikis S. A multilocus genotyping assay for cardiovascular diease. Clin Chem Lab Med. 1998;36:561–566. doi: 10.1515/CCLM.1998.096. [DOI] [PubMed] [Google Scholar]
- Chou Q, Russel M, Birch DE, Raymond J, Block W. Prevention of pre-PCR mis-priming and primer dimerization improves low-copy-number amplification. Nucleic Acids Res. 1992;20:1717–1723. doi: 10.1093/nar/20.7.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS, Gruyer MS, Chakravarti A. Variations on a theme: Cataloging human DNA sequence variation. Science. 1997;278:1580–1581. doi: 10.1126/science.278.5343.1580. [DOI] [PubMed] [Google Scholar]
- Germer S, Higuchi R. Single-tube genotyping without oligonucleotide probes. Genome Res. 1999;9:72–78. [PMC free article] [PubMed] [Google Scholar]
- Glantz SA. Primer of biostatistics. 4th ed. New York, NY: McGraw-Hill; 1997. [Google Scholar]
- Higuchi R, Watson RM. Kinetic PCR analysis using a CCD-camera and without using oligonucleotide probes. In: Innis MA, Gelfand DH, Sninsky JJ, editors. PCR applications: Protocols for functional genomics. San Diego, CA: Academic Press; 1999. pp. 263–284. [Google Scholar]
- Higuchi R, Fockler C, Dollinger G, Watson R. Kinetic PCR analysis: Real-time monitoring of DNA amplification reactions. Bio/Technology. 1993;11:1026–1030. doi: 10.1038/nbt0993-1026. [DOI] [PubMed] [Google Scholar]
- Holland PM, Abramson RD, Watson R, Gelfand DH. Detection of specific polymerase chain reaction product by utilizing the 5′ → 3′ exonuclease activity of Thermus aquaticus DNA polymerase. Proc Natl Acad Sci. 1991;88:7276–7280. doi: 10.1073/pnas.88.16.7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikuta S, Takagi K, Wallace RB, Itakura K. Dissociation kinetics of 19 base paired oligonucleotide-DNA duplexes containing different single mismatched base pairs. Nucleic Acids Res. 1987;15:797–811. doi: 10.1093/nar/15.2.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys AJ, MacLeod A, Tamaki K, Neil DL, Monckton DG. Minisatellite repeat coding as a digital approach to DNA typing. Nature. 1991;354:204–209. doi: 10.1038/354204a0. [DOI] [PubMed] [Google Scholar]
- Kang JJ, Holland MJ. Cellular transcriptome analysis using a kinetic PCR assay. In: Innes MA, Gelfand DH, Sninsky JJ, editors. PCR applications: Protocols for functional genomics. San Diego, CA: Academic Press; 1999. pp. 429–445. [Google Scholar]
- Kang JJ, Watson RM, Fischer ME, Higuchi R, Gelfand DH, Holland MJ. Transcript quantitation in total yeast cellular RNA using kinetic PCR. Nucleic Acids Res. 1999;28:e2. doi: 10.1093/nar/28.2.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostrikis LG, Tyagi S, Mhlanga MM, Ho DD, Kramer FR. Spectral genotyping of human alleles. Science. 1998;279:1228–1229. doi: 10.1126/science.279.5354.1228. [DOI] [PubMed] [Google Scholar]
- Kruglyak L. Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat Genet. 1999;22:139–144. doi: 10.1038/9642. [DOI] [PubMed] [Google Scholar]
- Kwok P-Y, Carlson C, Yager TD, Ankener W, Nickerson DA. Comparative analysis of human DNA variations by fluorescence-based sequencing of PCR products. Genomics. 1994;23:138–144. doi: 10.1006/geno.1994.1469. [DOI] [PubMed] [Google Scholar]
- Lai E, Riley J, Purvis I, Roses A. A 4-Mb high-density single nucleotide polymorphism-based map around human APOE. Genomics. 1998;54:31–38. doi: 10.1006/geno.1998.5581. [DOI] [PubMed] [Google Scholar]
- Landegren U, Nilsson M, Kwok P-Y. Reading bits of genetic information: Methods for single-nucleotide polymorphism analysis. Genome Res. 1998;8:769–776. doi: 10.1101/gr.8.8.769. [DOI] [PubMed] [Google Scholar]
- Lawyer FC, Stoffel S, Saiki RK, Chang SY, Landre PA, Abramson RD, Gelfand DH. High-level expression, purification, and enzymatic characterization of full-length Thermus aquaticus DNA polymerase and a truncated form deficient in 5′ to 3′ exonuclease activity. PCR Methods Appl. 1993;2:275–287. doi: 10.1101/gr.2.4.275. [DOI] [PubMed] [Google Scholar]
- Lee LG, Connell CR, Bloch W. Allelic discrimination by nick-translation PCR with fluorogenic probes. Nucleic Acids Res. 1993;21:3761–3766. doi: 10.1093/nar/21.16.3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo MC, Berninger MS, Hartley JL. Use of uracil DNA glycosylase to control carry-over contamination in polymerase chain reactions. Gene. 1990;93:125–128. doi: 10.1016/0378-1119(90)90145-h. [DOI] [PubMed] [Google Scholar]
- Marshall E. “Playing chicken” over gene markers. Science. 1997;278:2046–2048. doi: 10.1126/science.278.5346.2046. [DOI] [PubMed] [Google Scholar]
- ————— Drug firms to create public database of genetic mutations. Science. 1999;284:406–407. doi: 10.1126/science.284.5413.406. [DOI] [PubMed] [Google Scholar]
- Nazarenko IA, Bhatnagar SK, Hohman RJ. A closed tube format for amplification and detection of DNA based on energy transfer. Nucleic Acids Res. 1997;25:2516–2521. doi: 10.1093/nar/25.12.2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neilan BA, Wilton AN, Jacobs D. A universal procedure for primer labelling of amplicons. Nucleic Acids Res. 1997;25:2938–2939. doi: 10.1093/nar/25.14.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton CR, Graham A, Heptinstall LE, Powell SJ, Summers C, Kalsheker N, Smith JC, Markham AF. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS) Nucleic Acids Res. 1989;17:2503–2516. doi: 10.1093/nar/17.7.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacek P, Sajantila A, Syvänen A-C. Determination of allele frequencies at loci with length polymorphism by quantitative analysis of DNA amplified from pooled samples. PCR Methods Applic. 1993;2:313–317. doi: 10.1101/gr.2.4.313. [DOI] [PubMed] [Google Scholar]
- Persing DH, Cimino GD. Amplification product inactivation methods. In: Persing DH, Smith TF, Tenover FC, White TJ, editors. Diagnostic molecular microbiology: Principles and applications. Washington, D.C.: American Society for Microbiology; 1993. pp. 105–121. [Google Scholar]
- Picoult-Newberg L, Ideker TE, Pohl MG, Taylor SL, Donaldson MA, Nickerson DA, Boyce-Jacino M. Mining SNPs from EST databases. Genome Res. 1999;9:167–174. [PMC free article] [PubMed] [Google Scholar]
- Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- Risch N, Teng J. The relative power of family-based and case-control designs for linkage disequilibrium studies of complex diseases I. DNA pooling. Genome Res. 1998;8:1273–1288. doi: 10.1101/gr.8.12.1273. [DOI] [PubMed] [Google Scholar]
- Saiki RK, Walsh PS, Levenson CH, Erlich HA. Genetic analysis of amplified DNA with immobilized sequence-specific oligonucleotide probes. Proc Natl Acad Sci. 1988;86:6230–6234. doi: 10.1073/pnas.86.16.6230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw SH, Carrasquillo MM, Kashuk C, Puffenberger EG, Chakravarti A. Allele frequency distributions in pooled DNA samples: Applications to mapping complex disease genes. Genome Res. 1998;8:111–123. doi: 10.1101/gr.8.2.111. [DOI] [PubMed] [Google Scholar]
- Sommer SS, Cassady JD, Sobell JL, Bottema CD. A novel method for detecting point mutations or polymorphisms and its application to population screening for carriers of phenylketonuria. Mayo Clin Proc. 1989;64:1361–1372. doi: 10.1016/s0025-6196(12)65378-6. [DOI] [PubMed] [Google Scholar]
- Syvänen A-C, Sajantila A, Lukka M. Identification of individuals by analysis of biallelic DNA markers, using PCR and solid-phase minisequencing. Am J Hum Genet. 1993;52:46–59. [PMC free article] [PubMed] [Google Scholar]
- Tada M, Omata M, Kawai S, Saisho H, Ohto M, Saiki RK, Sninsky JJ. Detection of ras gene mutations in pancreatic juice and peripheral blood of patients with pancreatic adenocarcinoma. Cancer Res. 1993;53:2472–2474. [PubMed] [Google Scholar]
- Tyagi S, Kramer FR. Molecular beacons: Probes that fluoresce upon hybridization. Nat Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- Wang DG, Fan J-B, Siao C-J, Berno A, Young P, Sapolsky R, Ghandour G, Perkins N, Winchester E, Spencer J, et al. Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science. 1998;280:1077–1082. doi: 10.1126/science.280.5366.1077. [DOI] [PubMed] [Google Scholar]
- Whitcombe D, Theaker J, Guy SP, Brown T, Little S. Detection of PCR products using self-probing amplicons and fluorescence. Nat Biotechnol. 1999;17:804–807. doi: 10.1038/11751. [DOI] [PubMed] [Google Scholar]
- Winn-Deen ES. Direct fluorescence detection of allele-specific PCR products using novel energy-transfer labeled primers. Mol Diagn. 1998;3:217–221. doi: 10.154/MODI00300217. [DOI] [PubMed] [Google Scholar]
- Wu DY, Ugozolli L, Pal BK, Wallace RB. Allele-specific enzymatic amplification of beta-globin genomic DNA for diagnosis of sickle-cell anemia. Proc Natl Acad Sci. 1989;86:2757–2560. doi: 10.1073/pnas.86.8.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]