

Abstract
T-cell-receptor (TCR) gene therapy enables for the rapid creation of antigen-specific T cells from mice of any strain and represents a valuable tool for pre-clinical immunotherapy studies. Here, we describe the superiority of gamma-retroviral vectors compared to lentiviral vectors for transduction of murine T cells and surprisingly illustrate robust gene-transfer into phenotypically naïve/memory-stem cell (CD62Lhi/CD44low) and central memory (CD62Lhi/CD44hi) CD8+ T cells using murine-stem-cell-based gamma-retroviral vectors (MSGV1). We created MSGV1 vectors for a MHC-class I restricted T-cell receptor (TCR) specific for the melanocyte-differentiation antigen, gp100 (MSGV1-pmel-1), and a MHC-class II restricted TCR specific for tyrosinase-related-protein-1 (MSGV1-TRP-1), and found that robust gene expression required codon optimization of TCR sequences for the pmel-1 TCR. To test for functionality, we adoptively transferred TCR-engineered T cells into mice bearing B16 melanomas and observed delayed growth of established tumors with pmel-1TCR engineered CD8+ T cells and significant tumor regression with TRP-1 TCR transduced CD4+ T cells. We simultaneously created lentiviral vectors encoding the pmel-1TCR, but found that these vectors mediated low TCR expression in murine T cells, but robust gene expression in other murine and human cell lines. These results indicate that preclinical murine models of adoptive immunotherapies are more practical using gamma-retroviral rather than lentiviral vectors.
INTRODUCTION
The adoptive transfer of T cells engineered to express a tumor specific T-cell receptor (TCR) represents a promising treatment for patients with metastatic cancer 1–10. To improve the results of current TCR gene therapies, preclinical models need to be established to rapidly test biological concepts that may advance existing treatment regimens. Gamma-retroviral and lentiviral vectors are capable of mediating stable gene transfer into mammalian cells 11–16. However, post-thymic murine T cells represent a unique population of cells central to a host’s adaptive immune arm and generating optimal protocols for mediating gene transfer into these cells ex vivo would be valuable for researchers seeking to rapidly create antigen-specific T cells on different genetic background stains 17.
Established murine tumor models of adoptive transfer utilize γ-retroviral mediated gene transfer of TCRs into actively dividing murine T cells 6, 18, 19. However, less differentiated CD8+ T cells may represent the ideal population of cells to mediate tumor regression and recent evidence suggests that lentiviral based vectors can mediate stable gene transfer into non-dividing naïve/memory-stem cell (TN/SCM) and central memory (TCM) T cells 20–26. Another potential advantage of lentiviral vectors is the low propensity to integrate into the promoter regions of transcribed genes, which may be associated with insertional oncogenesis 27. This has led to the rapid development of clinical grade self-inactivating lentiviral vectors for human gene therapy trials 28–31 and we therefore attempted to develop a practical preclinical model to assist in directing future clinical endeavors utilizing both γ-retroviral and lentiviral vectors.
Contrary to our initial hypothesis, we found that γ-retroviral vectors mediated efficient gene delivery into TN and TCM cells after a short-term (24 hour) anti-CD3/anti-CD28 stimulation. In comparison, lentiviral vectors mediated poor gene transfer specifically into murine T cells while robustly delivering genes into murine fibroblasts, human jurkat cells, and human CD4+ and CD8+ T cells. Developing effective models for HIV infection in murine T cells remains a challenge, but would represent a valuable tool for researchers across multiple disciplines.
MATERIALS AND METHODS
Mice and Cell Lines
Splenocytes from C57BL/6 and CD8−/− mice (Jackson Laboratories) were used to generate CD8+ and CD4+ murine T cells, respectively. Murine 3T3 and human jurkat-T3 cells (ATCC, TIB-152) were maintained in culture as previously described. Human peripheral blood lymphocytes were obtained from patients with metastatic melanoma seeking treatment at the Surgery Branch, National Cancer Institute. Murine splenocytes were stimulated with either 1μg/mL of anti-CD3 and anti-CD28 (BD Biosciences, San Jose, CA) or con A (2 μg/mL) and IL-7 (1 ng/mL; Peprotech, Rocky Hill, NJ) for 24 or 48 hours and cultured in 60 IU/mL of recombinant human IL-2 (Chiron, Emeryville, CA) IL-15 (10 ng/mL), or IL-21 (10 ng/mL; Peprotech, Rocky Hill, NJ). Complete media consisted of either DMEM high glucose for viral producer cell lines or RPMI for T cells (Invitrogen, Carlsbad, CA) mixed with 10% Fetal Bovine Serum, 1% Sodium Pyruvate, 1% MEM Non-essential amino-acids, 1% Glutamax, 1% Pen Strep, 0.1% Gentamycin, and 0.1% Fungizone (Invitrogen, Carlsbad CA). Frozen human PBL from patient pheresis samples were thawed and stimulated as previously described 22, 32.
Gamma-retroviral and Lentiviral TCR construction
MSGV1 Pmel-1 and TRP-1 TCRs
The cDNA for the native or codon optimized α and β subunits of the pmel-1 TCR (Vα1/Vβ13) and the TRP-1 TCR (Vα3.2/Vβ14) were linked either by a sequence encoding the foot and mouth disease picornavirus 2A ribosomal skip peptide or an internal ribosomal entry site, IRES and cloned into the MSGV1 vector.29, 32
Lentiviral Pmel-1 TCR
The cDNA for the native or codon optimized α and β subunits of the pmel-1 TCR (Vα1/Vβ13) were linked either by a sequence encoding the foot and mouth disease picornavirus 2A ribosomal skip peptide or an internal ribosomal entry site, IRES and cloned into a self inactivating HIV based lentiviral vector driven by a MSCV-U3 promoter.29, 32
Viral Production
Platinum Eco cell lines (Cell Biolabs, San Diego, CA) were used for gamma-retroviral production while 293T cells (Invitrogen, Carlsbad, CA) were used for lentiviral production. Producer cells were plated on 10cm poly-d-lysine coated plates (BD Discovery Labware, Bellrica, MA). Transfection with appropriate DNA plasmids were done using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) as described in Figure 1.
FIGURE 1.
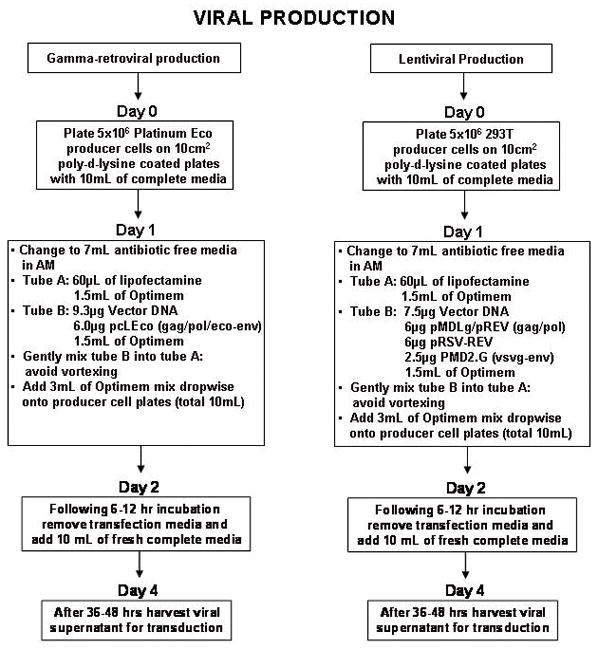
Flow diagram for gamma-retroviral and lentiviral production.
TCR Transductions
Retronectin (Takara Bio, Otsu, Japan) and protamine sulfate (Sigma, St. Louis, MO) spinoculation protocols are described in the flow diagrams presented in Figure 2.
FIGURE 2.
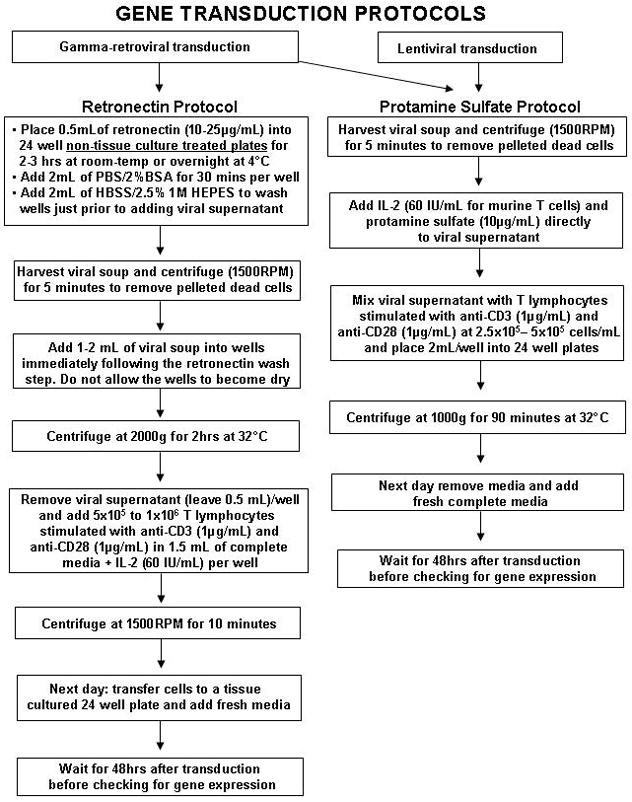
Flow diagram for gene transduction protocols
Adoptive Cell Transfer
Six to twelve week old C57BL/6 mice (n=5 for all groups) were injected subcutaneously with 5 × 105 B16 melanoma cells. Ten to fourteen days later, they were irradiated with 5 Gy total body irradiation (TBI) and given pmel-1 TCR or TRP-1 TCR transduced CD8+ or CD4+ T cells, respectively. T cells were generated from C57BL/6 splenocytes and administered in vivo by tail vein. Tumors were measured using digital calipers and the tumor area was calculated as the product of perpendicular diameter by investigators in a blinded manner. All experiments were performed independently at least twice with similar results and all tumor curve data is shown as mean +/− standard error of the mean.
Flow Cytometry
Transduced cells were stained with fluorescently labeled antibodies for mouse anti-CD8α, anti-CD4, anti-Vβ-13, anti-CD90.1 (thy1.1), anti-CD62L, anti-CD44, (BD Biosciences) and PE-labeled pmel-1 tetramer 18. Human T cells were labeled with human anti-CD8α and anti-CD4 (BD Biosciences, San Jose, CA).
Statistical Analysis
One-way ANOVA and student t-tests were used to test for significant differences. Tumor growth slopes were compared using Wilcoxon rank sum test.
RESULTS
MSGV1 vectors mediate efficient gene transfer into naïve/memory-stem cell and central memory murine T cells
To determine the ideal T cell stimulation for gene transfer, we compared anti-CD3/anti-CD28 to concalavalin A (ConA)/IL-7 for 24 and 48 hours. Transduction efficiencies between the conditions were similar (39–55%; Fig. 3A). We next measured the effects of culturing T cells in different common gamma-chain cytokines and observed optimal rates of transduction when cells were cultured in IL-2 (43%) compared to IL-15 (25%) and IL-21 (9%) following initial priming with anti-CD3/anti-CD28 (Fig. 3B). We next assessed the phenotypic characteristics of the CD8+ T cells transduced with MSGV1 vectors. Purified populations of CD8+ T cells from spleens were stimulated for 24 hours with anti-CD3/anti-CD28, transduced with a MSGV1 vector encoding thy1.1 and analyzed by flow cytometry 3 and 5 days after transductions (Fig. 3C). Surprisingly, we found that 3 days following gene transfer, the majority of CD8+ T cells expressing thy1.1 were TN/SCM (CD62Lhi CD44low; 53%) or TCM (CD62Lhi CD44hi; 24%) cells (Fig. 3C, upper panel). After prolonging cells in culture for 5 days following gene transfer, we witnessed an increase in the percentage of thy1.1+ effector memory (TEM, CD62Llow CD44hi; 30%) and TCM cells (65%) and a marked decrease in TN/SCM cells (5%; Fig. 3C, lower panel). Thus, MSGV1 based vectors mediated efficient gene transfer into short-term cultured TN/SCM and TCM cells.
FIGURE 3.
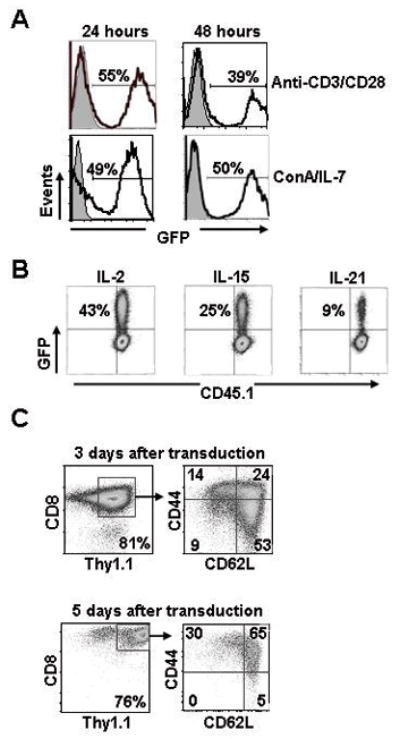
Optimization of gene transfer into T cells with gamma-retroviral vectors. A, Bulk C57BL/6 splenocytes were stimulated with either anti-CD3 and anti-CD28 or con A and IL-7 for 24 or 48 hours and transduced with the MSGV1-GFP vector. B, Congenically marked (CD45.1) CD8+ T cells isolated from C57/BL-6 spleens were stimulated with anti-CD3 and anti-CD28 and cultured in IL-2, IL-15, or IL-21 and transduced with the MSGV1-GFP vector. Transduction efficiencies for A and B were measured by flow cytometry 3 days following gene transfer (grey = non-transduced control). C, CD8+ T cells were stimulated with anti-CD3/anti-CD28 for 24 hrs, cultured in IL-2, transduced with a MSGV1-thy1.1 vector and examined by flow cytometry 3 and 5 days after transduction for CD62L and CD44 to define TN/SCM (CD62LHi CD44Low) and TCM (CD62LHi CD44Hi) populations. All results in A, B, and C are representative of at least 3 independent experiments.
MSGV1 codon optimized pmel-1TCR and TRP-1 TCR vectors efficiently redirect open-repertoire CD8+ and CD4+ T cells, respectively
To develop an effective pre-clinical gene therapy model for adoptive cell transfers against established B16 melanomas, we constructed vectors using the MSGV1 backbone33 expressing the pmel-1TCR (designated as MSGV1-pmel-1) a class I restricted receptor recognizing the melanocyte-differentiation antigen, gp10034, and the TRP-1 TCR (designated as MSGV1-TRP-1), a class II restricted TCR recognizing the melanocyte-differentiation antigen, tyrosinase-related-protein-1 (Fig. 4A).35 We used a sequence encoding the foot and mouth disease picornavirus 2A ribosomal skip peptide or an internal ribosomal entry site, IRES to separate the Vα and Vβ subunits of the TCRs. The initial vectors for the pmel-1 TCR did not yield robust levels of gene expression based on flow cytometric analysis for Vβ13 (MSGV1-pmel-1-2A, 17%; MSGV1-pmel-1-IRES, 13%; Fig. 4B, right upper panels) and gp100 tetramer (<1%; Fig. 4B, right lower panels). However, after codon optimization, we were able to achieve marked improvements in the surface expression of the pmel-1 TCR based on Vβ13 and gp100 tetramer staining (MSGV1-pmel-1-IRES, 63% Vβ13 and 39% gp100 tetramer; MSGV1-pmel-1-2A: 48% Vβ13 and 32% gp100 tetramer; Fig. 4B). We next measured the efficacy of our native TRP-1 TCR constructs and attained greater than 70% TCR expression based on Vβ14 staining with both the IRES and 2A vectors (Fig. 4C). Thus, both the codon optimized pmel-1 TCR and native-sequence TRP-1TCR vectors efficiently redirected TCR specificity in CD8+ and CD4+ T cells, respectively, although transduction efficiencies did appear to be slightly higher with the TRP-1 vectors.
FIGURE 4.
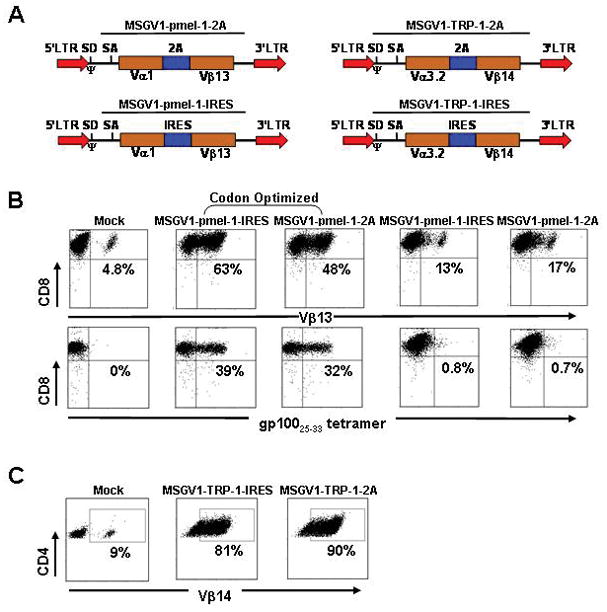
Redirection of open-repertoire CD8+ and CD4+ T cells with MSGV1 vectors encoding the codon optimized pmel-1 and native TRP-1 TCRs. A, The α and β subunits of the pmel-1 TCR (Vα1/Vβ13) and the TRP-1 TCR (Vα3.2/Vβ14) were linked either by a sequence encoding the foot and mouth disease picornavirus 2A ribosomal skip peptide (upper panels) or an internal ribosomal entry site, IRES (lower panels) and cloned into the MSGV1 vector backbone (SD, splice done; SA, splice acceptor; LTR, long terminal repeat; Ψ, packaging sequence). B, C57BL?6 splenocytes were stimulated with anti-CD3/anti-CD28 for 24 hrs, cultured in IL-2, and transduced with the MSGV-1 vectors encoding for either the native or codon optimized pmel-1 TCR sequences containing IRES or 2A. Transduction efficiencies in CD8+ T cells were measured by flow cytometry for Vβ13 (upper panel) or gp10025-33 tetramer expression (lower panel). C, C57/BL-6 splenocytes were stimulated with anti-CD3/anti-CD28 for 24 hrs, cultured in IL-2, and transduced with the MSGV1-TRP-1-IRES and MSGV1-TRP-1-2A vectors. TCR transfer efficiencies in CD4+ T cells were measured by flow cytometry for Vβ14. Mock represents cells taken through the transduction protocol with complete media instead of viral supernatant. All results A, B, and C, are representative at least 3 independent experiments.
TCR gene transduced CD8+ and CD4+ T cells mediate anti-tumor immunity
To assess the in vivo efficacy and functionality of our TCR constructs, we adoptively transferred 4 × 106 CD8+ T cells transduced with the MSGV1-GFP, MSGV-pmel-1-IRES or MSGV1-pmel-1-2A vectors into sublethally irradiated (5Gy total body irradiation) mice bearing established B16 subcutaneous tumors. We observed a significant delay in tumor progression following transfer of open-repertoire CD8+ T cells redirected to express the pmel-1 TCR (p<0.05; Fig. 5A). Both the MSGV1-pmel-1-IRES and MSGV1-pmel-1-2A vectors mediated similar degrees of anti-tumor immunity (Fig. 5A). We next measured the in vivo effects of transferring TCR redirected open-repertoire CD4+ T cells and observed a profound anti-tumor response with both the MSGV1-TRP-1-IRES and MSGV1-TRP-1-2A vectors following the adoptive transfer of 4 × 106 transduced cells (p<0.05; Fig. 5B). Similar to experiments with the pmel-1TCR, we observed no functional differences between the TRP-1 TCR vectors encoding either the IRES or 2A linker sequences (Fig. 5B). Due to the slightly higher levels of TCR expression with the TRP-1 vectors compared to the pmel-1vectors, we tested transferring up to 20 × 106 pmel-1 TCR transduced CD8+ T cells but did not observe a significant increase in anti-tumor efficacy by transferring higher number of CD8+ T cells (Supplementary Figure 1). Thus, CD4+ T cells redirected to express the TRP-1 TCR mediated robust anti-tumor immunity while pmel-1 TCR engineered CD8+ T cells delayed tumor growth.
FIGURE 5.
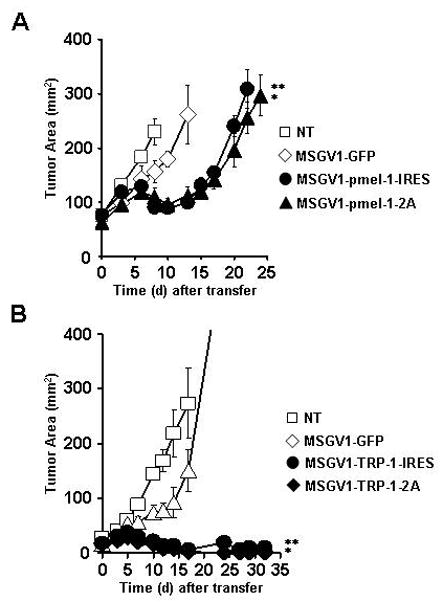
Anti-tumor immunity of CD8+ T cells engineered to express the pmel-1 TCR and CD4+ T cells transduced with the TRP-1 TCR. A, Tumor treatment following the adoptive transfer of 4×106 open repertoire CD8+ T cells transduced with the MSGV1-GFP, MSGV1-pmel-1-IRES, or MSGV1-pmel-1-2A vectors into sublethaly-irradiated (5 Gy) mice bearing B16 tumors (n=5) established for 10 days [*, ** = p<0.05 compared to no treatment (NT)] B, Anti-tumor immunity following the adoptive transfer of 4×106 open repertoire CD4+ T cells transduced with the MSGV1-GFP, MSGV1-TRP-1-IRES, or MSGV1-TRP-1-2A vectors into sublethaly-irradiated (5Gy) mice bearing B16 tumors (n=5) established for 10 days [*, ** = p<0.05 compared to no treatment (NT)]. All results for (A) and (B) are representative of at least two separate experiments.
Lentiviral vectors encoding the pmel-1 TCR mediate poor gene transfer in murine T cells
We next created two self-inactivating human-immunodeficient-virus (HIV) based lentiviral vectors encoding the codon optimized α and β subunits of the pmel-1 TCR linked either by a sequence encoding the foot and mouth disease picornavirus 2A ribosomal skip peptides (Fig. 6A, upper panel; designated LV-pmel-1-2A) or an internal ribosomal entry site, IRES (Fig. 6A, lower panel; designated LV-pmel-1-IRES). For comparative purposes with our gamma-retroviral vectors, we constructed lentiviral vectors driven by a MSCV-U3 promoter (Fig. 6A). We attempted to transduce open-repertoire murine CD8+ T cells and achieved only modest levels of gene transfer based on Vβ13 specific staining with both the LV-pmel-1-2A (11%; Fig. 6A, upper panel) and LV-pmel-1-IRES (12%; Fig. 6B, upper panel) vector. In contrast, gene transfer with the identical LV-pmel-1-2A and LV-pmel-1-IRES supernatants and similar transduction protocols led to robust Vβ13 expression of 77% and 53%, respectively, in human jurkat T cells (Fig. 6B, lower panel). To confirm these surprising findings we also measured gene transfer efficiencies by staining transduced cells with a fluorescently labeled gp10025-33 tetramer. Similar to the Vβ13 expression pattern, gene transfer with the LV-pmel-1-IRES vector mediated poor gp10025-33 tetramer staining in murine CD8+ T cells (3%, Fig. 6C, upper panel), but once again, robust expression was observed in human jurkat cells when using the identical viral supernatant and transduction protocol (84%, Fig. 6C, lower panel). We also tried several other strategies to improve lentiviral mediated TCR gene transfer into murine CD8+ T cells such as pseudo-typing the virus with an ecotropic envelope, varying the transduction protocols, and concentrating our viral supernatant, however, none of these modifications significantly improved TCR gene transfer (data not shown). Thus, lentiviral vectors mediated high levels of TCR gene transfer in human T cells, but only γ-retroviral vectors efficiently mediated TCR redirection in murine T cells.
FIGURE 6.
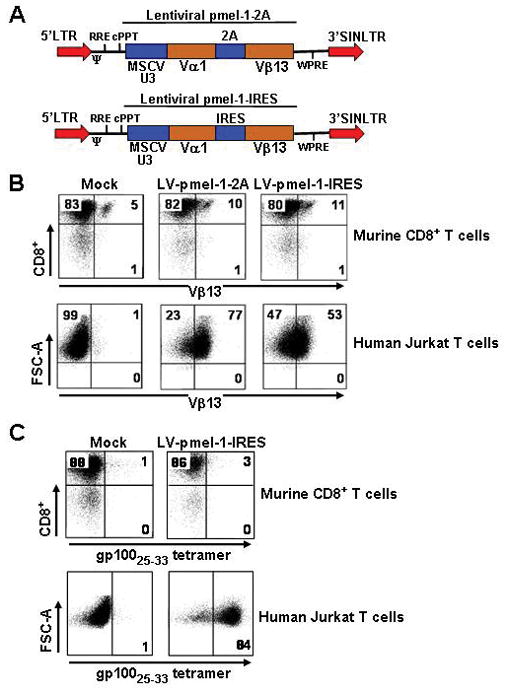
Gene transfer with HIV based self-inactivated lentiviral vectors encoding the pmel-1 TCR. A, The HIV based lentiviral vector encoding the pmel-1 TCR linked by either a 2A linker (upper panel) or an internal ribosomal entry site, IRES (lower panel) (LTR, long terminal repeat; Ψ, packaging sequence; cPPT, central polypurine tract; RRE, rev response element; MSCV-U3, murine-stem-cell-virus-based promoter). B, Open-repertoire murine CD8+ T cells from spleens were stimulated with anti-CD3 and anti-CD28 and transduced with the LV-Pmel-2A and LV-Pmel-IRES vectors. Human jurkat T cells were transduced with the same viral supernatant. Transduction efficiencies were analyzed by flow cytometry for Vβ-13 expression. C, Transduced murine CD8+ T cells and human jurkat T cells from B, were analyzed by flow cytometry for gp10025-33 tetramer expression. Mock represent cells taken through the transduction protocol with complete media instead of viral supernatant. All results for A, B, and C, are representative of at least 2 independent experiments.
Impaired lentiviral gene transfer specifically in murine T cells
We next wanted to investigate whether the poor lentiviral based transduction efficiencies in murine CD8+ T cells were a generalized phenomenon for all cell types or specific for T cells only. We used a lentiviral vector driven by MSCV-U3 promoter encoding the green fluorescent protein reporter (LV-GFP) and transduced murine 3T3 fibroblasts along with murine CD8+ and CD4+ T cells. Interestingly, the transduction efficiency in 3T3 fibroblasts was remarkably high (92%; Fig. 7A, upper left panel) compared to both murine CD8+ T cells (10%; Fig. 7A, upper middle panel) and murine CD4+ T cells (14%; Fig. 7A, upper right panel), indicating that lentiviral mediated gene transfer was impaired specifically in T cells. We next used the same LV-GFP viral supernatant and transduced human jurkat T cells along with human CD8+ and CD4+ T cells and observed excellent levels of gene transfer, 84%, 92%, and 87% respectively (Fig. 7A, lower panel). We investigated these findings quantitatively and found significantly higher GFP expression (mean fluorescence intensity; p < 0.05) in human compared to murine CD8+ and CD4+ T cells (Fig. 7B). Thus, the impairment in lentiviral mediated gene transfer appeared to be specific to murine T cells.
FIGURE 7.
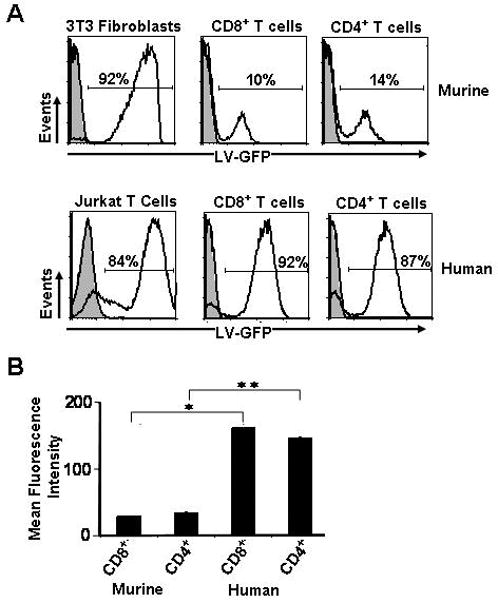
Comparative transductions between murine and human cells with lentiviral vectors. A, Murine fibroblasts, CD8+ and CD4+ T cells, along with human jurkat, CD8+ and CD4+ T cells were transduced with a HIV based lentivirus encoding the green flourescent protein (GFP). Murine and human T cells were stimulated similarily with anti-CD3 and anti-CD28 and cultured in IL-2. Transduction efficiencies in cells were analyzed by flow cytometry for GFP expression. Results are representative of at least 2 independent experiments. B, The mean flourescence intensity (MFI) was calcluated and quantified for GFP transduced murine and human CD8+ and CD4+ T cells (*, ** p <0.05; n=3). All results for A and B are representative of at least 2 independent experiments.
DISCUSSION
The advent of ex vivo gene transfer into lymphocytes followed by the infusion of engineered T cells in the autologous and allogenic setting has ushered in an exciting era for gene therapy 6, 36. The adoptive transfer of T cells redirected to express tumor-specific TCRs represents a promising option for patients with metastatic cancer refractory to standard treatment regimens 5, 37. However, clinical results from gene therapy trials have thus far been disappointing 5, 6, 19, 37, 38.
Preclinical studies often form the basis for major advancements in biomedical research. For cancer therapeutics, developing accurate animal models closely mimicking clinical endeavors allows for the rapid testing of multiple biological parameters. We therefore attempted to generate optimized mouse models for the adoptive transfer of open-repertoire CD8+ and CD4+ T cells redirected to express tumor-specific TCRs. The stable introduction of genes into rapidly dividing cells requires an established integration of desired genetic sequences into target cells 15. Both γ-retroviral and lentiviral based platforms are capable of mediating stable gene delivery into mammalian cells, but marked heterogeneity exists in regard to optimal vector backbones depending on differences in recipient species and cell types 14, 16, 39, 40. Lentiviruses are quickly becoming the standard for mediating gene transfer in human lymphocytes 20, 28, 29, 32, 41. These vectors mediate high transduction efficiencies, stable integration, and possess the ability to transfer genes into resting lymphocytes 21, 41. While much work has been conducted on human lymphocytes, the ability of lentiviral based vectors to mediate gene transfer in murine T cells has not been thoroughly explored. In contrast, γ-retroviral vectors mediate efficient delivery of genes into murine T cells but the requirement for a strong stimulus and active cell division has led to the general belief that transducing less differentiated T cells may not be possible.
We therefore developed both γ-retroviral and lentiviral vectors to try to redirect TCR specificity of murine T cells to recognize tumor antigens expressed by B16 melanomas. During optimization, we observed that 24 hour stimulation with anti-CD3 and anti-CD28 in the presence of IL-2 provided sufficient priming of T cells for efficient gene delivery. Under these culture conditions, we surprisingly found that MSGV1 based vectors effectively transduced both naïve/memory-stem cell and central memory T cells. Most likely, the short 24 hour stimulation prevented early differentiation of naïve/memory-stem cell and central memory T cells into effectors, yet still pushed T cells into active cell division, making them permissible for retroviral integration.
We previously found that the pmel-1 TCR on a PMX backbone led to modest levels of anti-tumor immunity and therefore looked to improve upon earlier results 18. We optimized gamma-retroviral transduction conditions and constructed vectors encoding for the pmel-1 and TRP-1 TCR on a MSGV1 backbone in order to redirect both CD8+ and CD4+ T cells to recognize the melanocyte differentiation antigens gp100 (class I restricted) and tyrosinase-related-protein-1 (class II restricted), respectively. We found it necessary to codon optimize the sequences for the alpha and beta subunits of the pmel-1 TCR in order to achieve robust gene expression. Interestingly, we did not need to codon optimize the TRP-1 TCR sequences and obtained the highest levels of TCR expression with these vectors. The biological reasons for these differences remain unclear, although previous studies have described the existence of ‘strong’ and ‘weak’ TCRs in regard to surface expression 42, 43.
For functional examination of the TCR vectors, we adoptively transferred CD8+ T cells engineered to express the pmel-1 TCR and CD4+ T cells engineered to express the TRP-1 TCR into mice bearing established subcutaneous B16 tumors. We observed a delay in tumor growth with pmel-1 TCR transduced CD8+ T cells and striking anti-tumor immunity with TRP-1 TCR engineered CD4+ T cells. The differences in treatment could not be explained by the increased levels of TRP-1 TCR expression because transferring higher numbers of pmel-1 TCR engineered CD8+ T cells (up to 20 X 106) did not appear to induce similar degrees of anti-tumor immunity observed after transferring TRP-1 TCR engineered CD4+ T cells. We and others have described the importance of CD4+ T cells as potent and direct mediators of tumor destruction, however, it remains unclear whether the differences in tumor treatments were due to the inherent anti-tumor abilities of CD8+ versus CD4+ T cells or intrinsic differences in the TCRs 35, 44, 45. Nevertheless, these are much needed models to perform mechanistic studies to improve the efficacy of current TCR gene therapy clinical trials46 and this study represents the first description of adoptively transferring genetically engineering CD4+ T cells expressing the TRP-1 TCR.
A recent report by Bendle et al. described toxicities after adoptively transferring TCR engineered T cells in vivo 47. However, we did not see any clear signs of toxicity with the pmel-1 TCR due to the continued growth of tumors precluding any long-term studies. We did note evidence of long term weight loss in some mice following treatment with TRP-1 TCR transduced CD4+ T cells, but need to further investigate the nature of the weight loss.
In this study, we also created HIV-based self-inactivating lentiviral vectors encoding the pmel-1 TCR and unexpectedly found low levels of TCR gene expression in murine T cells. In contrast, human T cells engineered with the lentiviral pmel-1TCR efficiently expressed high levels of the murine receptor. To test if this defect was specific for T cells, we simultaneously transduced murine fibroblasts and murine CD4+/CD8+ T cells with a LV-GFP construct and demonstrated high levels of gene transfer in only the fibroblasts, indicating that murine T cells possessed inherent characteristics preventing lentiviral infection. We tried several different transduction protocols in addition to concentrating our virus but were unable to obtain gene transfer efficiencies comparable to γ-retroviral vectors. The lentiviral vectors we constructed were driven by a MSCV-U3 promoter to allow for direct comparisons with our γ-retroviral vectors. In the future, one possible strategy to boost lentiviral transductions in murine T cells might be to use different promoters 48. However, based on our ability to efficiently mediate gene transfer into TN/SCM and TCM cells with γ-retroviral vectors, the advantages of optimizing lentiviral based gene transfer into minimally stimulated murine T cells may not be required.
In summary, HIV-based lentiviral vectors may not represent the ideal system to mediate robust TCR gene transfer in murine T cells. The most likely explanation for this phenomenon can be drawn from studies by Baumann et al.49 and Tsurutani et al.50 that describe reduced levels of HIV infectivity in murine T cells due to lower efficiencies of reverse transcription and nuclear translocation. Understanding and developing techniques to overcome these limitations would be valuable for scientists across multiple disciplines. For performing pre-clinical adoptive immunotherapy studies, utilizing lentiviral vectors is challenging due to the large number of cells needing to undergo transduction prior to cell transfer. One strategy to overcome the limitations of reduced levels of TCR gene transfer is to perform a cell sort prior to adoptive transfer experiments, but this would dramatically increase the cost of performing in vivo experiments. In conclusion, for practical reasons, we recommend using gamma-retroviral vectors for future pre-clinical adoptive immunotherapy studies requiring the genetic engineering of murine T cells.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
The authors declare that there are no potential conflicts of interests
References
- 1.Rosenberg SA. Immunotherapy and gene therapy of cancer. Cancer Res. 1991;51:5074s–9s. [PubMed] [Google Scholar]
- 2.Hwu P, Rosenberg SA. The genetic modification of T cells for cancer therapy: an overview of laboratory and clinical trials. Cancer Detect Prev. 1994;18:43–50. [PubMed] [Google Scholar]
- 3.Clay TM, Custer MC, Sachs J, Hwu P, Rosenberg SA, Nishimura MI. Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J Immunol. 1999;163:507–13. [PubMed] [Google Scholar]
- 4.Brenner MK, Heslop HE. Adoptive T cell therapy of cancer. Curr Opin Immunol. 22:251–7. doi: 10.1016/j.coi.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–9. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sadelain M. T-cell engineering for cancer immunotherapy. Cancer J. 2009;15:451–5. doi: 10.1097/PPO.0b013e3181c51f37. [DOI] [PubMed] [Google Scholar]
- 7.Kershaw MH, Westwood JA, Parker LL, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12:6106–15. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pule MA, Savoldo B, Myers GD, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–70. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coukos G, Rubin SC. Gene therapy for ovarian cancer. Oncology (Williston Park) 2001;15:1197–204. 207. discussion 207–8. [PubMed] [Google Scholar]
- 10.Koya RC, Mok S, Comin-Anduix B, et al. Kinetic phases of distribution and tumor targeting by T cell receptor engineered lymphocytes inducing robust antitumor responses. Proc Natl Acad Sci U S A. 107:14286–91. doi: 10.1073/pnas.1008300107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abordo-Adesida E, Follenzi A, Barcia C, et al. Stability of lentiviral vector-mediated transgene expression in the brain in the presence of systemic antivector immune responses. Hum Gene Ther. 2005;16:741–51. doi: 10.1089/hum.2005.16.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li M, Rossi JJ. Lentiviral vector delivery of siRNA and shRNA encoding genes into cultured and primary hematopoietic cells. Methods Mol Biol. 2008;433:287–99. doi: 10.1007/978-1-59745-237-3_18. [DOI] [PubMed] [Google Scholar]
- 13.Persons DA. Lentiviral vector gene therapy: effective and safe? Mol Ther. 18:861–2. doi: 10.1038/mt.2010.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang Y, Xie L, Tran DT, et al. Persistent expression of factor VIII in vivo following nonprimate lentiviral gene transfer. Blood. 2005;106:1552–8. doi: 10.1182/blood-2004-11-4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Modlich U, Navarro S, Zychlinski D, et al. Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors. Mol Ther. 2009;17:1919–28. doi: 10.1038/mt.2009.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mostoslavsky G, Kotton DN, Fabian AJ, Gray JT, Lee JS, Mulligan RC. Efficiency of transduction of highly purified murine hematopoietic stem cells by lentiviral and oncoretroviral vectors under conditions of minimal in vitro manipulation. Mol Ther. 2005;11:932–40. doi: 10.1016/j.ymthe.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 17.Herzog RW, Cao O, Srivastava A. Two decades of clinical gene therapy--success is finally mounting. Discov Med. 9:105–11. [PMC free article] [PubMed] [Google Scholar]
- 18.Abad JD, Wrzensinski C, Overwijk W, et al. T-cell receptor gene therapy of established tumors in a murine melanoma model. J Immunother. 2008;31:1–6. doi: 10.1097/CJI.0b013e31815c193f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bendle GM, Haanen JB, Schumacher TN. Preclinical development of T cell receptor gene therapy. Curr Opin Immunol. 2009;21:209–14. doi: 10.1016/j.coi.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 20.Zhou X, Cui Y, Huang X, et al. Lentivirus-mediated gene transfer and expression in established human tumor antigen-specific cytotoxic T cells and primary unstimulated T cells. Hum Gene Ther. 2003;14:1089–105. doi: 10.1089/104303403322124800. [DOI] [PubMed] [Google Scholar]
- 21.Unutmaz D, KewalRamani VN, Marmon S, Littman DR. Cytokine signals are sufficient for HIV-1 infection of resting human T lymphocytes. J Exp Med. 1999;189:1735–46. doi: 10.1084/jem.189.11.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gattinoni L, Klebanoff CA, Palmer DC, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. 2005;115:1616–26. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hinrichs CS, Borman ZA, Gattinoni L, et al. Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood. doi: 10.1182/blood-2010-05-286286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klebanoff CA, Gattinoni L, Torabi-Parizi P, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A. 2005;102:9571–6. doi: 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Naldini L, Blomer U, Gallay P, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–7. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 26.Berger CX, Wong CW, Forman SJ, Riddell SR, Jensen MC. Engraftment of human central memory-derived effector CD8+ T cells in immunodeficient mice. Blood. doi: 10.1182/blood-2010-10-310599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cattoglio C, Facchini G, Sartori D, et al. Hot spots of retroviral integration in human CD34+ hematopoietic cells. Blood. 2007;110:1770–8. doi: 10.1182/blood-2007-01-068759. [DOI] [PubMed] [Google Scholar]
- 28.Levine BL, Humeau LM, Boyer J, et al. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc Natl Acad Sci U S A. 2006;103:17372–7. doi: 10.1073/pnas.0608138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones S, Peng PD, Yang S, et al. Lentiviral vector design for optimal T cell receptor gene expression in the transduction of peripheral blood lymphocytes and tumor-infiltrating lymphocytes. Hum Gene Ther. 2009;20:630–40. doi: 10.1089/hum.2008.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manilla P, Rebello T, Afable C, et al. Regulatory considerations for novel gene therapy products: a review of the process leading to the first clinical lentiviral vector. Hum Gene Ther. 2005;16:17–25. doi: 10.1089/hum.2005.16.17. [DOI] [PubMed] [Google Scholar]
- 31.Varela-Rohena A, Carpenito C, Perez EE, et al. Genetic engineering of T cells for adoptive immunotherapy. Immunol Res. 2008;42:166–81. doi: 10.1007/s12026-008-8057-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang S, Rosenberg SA, Morgan RA. Clinical-scale lentiviral vector transduction of PBL for TCR gene therapy and potential for expression in less-differentiated cells. J Immunother. 2008;31:830–9. doi: 10.1097/CJI.0b013e31818817c5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hughes MS, Yu YY, Dudley ME, et al. Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T-cell effector functions. Hum Gene Ther. 2005;16:457–72. doi: 10.1089/hum.2005.16.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Overwijk WW, Theoret MR, Finkelstein SE, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–80. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muranski P, Boni A, Antony PA, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008;112:362–73. doi: 10.1182/blood-2007-11-120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosenberg SA. Gene therapy for cancer. JAMA. 1992;268:2416–9. [PubMed] [Google Scholar]
- 37.Johnson LA, Morgan RA, Dudley ME, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–46. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmitt TM, Ragnarsson GB, Greenberg PD. T cell receptor gene therapy for cancer. Hum Gene Ther. 2009;20:1240–8. doi: 10.1089/hum.2009.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ha SP, Klemen ND, Kinnebrew GH, et al. Transplantation of mouse HSCs genetically modified to express a CD4-restricted TCR results in long-term immunity that destroys tumors and initiates spontaneous autoimmunity. J Clin Invest. 120:4273–88. doi: 10.1172/JCI43274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schenkwein D, Turkki V, Karkkainen HR, Airenne K, Yla-Herttuala S. Production of HIV-1 integrase fusion protein-carrying lentiviral vectors for gene therapy and protein transduction. Hum Gene Ther. 21:589–602. doi: 10.1089/hum.2009.051. [DOI] [PubMed] [Google Scholar]
- 41.Steffens CM, Managlia EZ, Landay A, Al-Harthi L. Interleukin-7-treated naive T cells can be productively infected by T-cell-adapted and primary isolates of human immunodeficiency virus 1. Blood. 2002;99:3310–8. doi: 10.1182/blood.v99.9.3310. [DOI] [PubMed] [Google Scholar]
- 42.Hart DP, Xue SA, Thomas S, et al. Retroviral transfer of a dominant TCR prevents surface expression of a large proportion of the endogenous TCR repertoire in human T cells. Gene Ther. 2008;15:625–31. doi: 10.1038/sj.gt.3303078. [DOI] [PubMed] [Google Scholar]
- 43.Sommermeyer D, Neudorfer J, Weinhold M, et al. Designer T cells by T cell receptor replacement. Eur J Immunol. 2006;36:3052–9. doi: 10.1002/eji.200636539. [DOI] [PubMed] [Google Scholar]
- 44.Schietinger A, Philip M, Liu RB, Schreiber K, Schreiber H. Bystander killing of cancer requires the cooperation of CD4(+) and CD8(+) T cells during the effector phase. J Exp Med. 207:2469–77. doi: 10.1084/jem.20092450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frankel TL, Burns WR, Peng PD, et al. Both CD4 and CD8 T cells mediate equally effective in vivo tumor treatment when engineered with a highly avid TCR targeting tyrosinase. J Immunol. 184:5988–98. doi: 10.4049/jimmunol.1000189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kerkar SP, Muranski P, Kaiser A, et al. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res. 70:6725–34. doi: 10.1158/0008-5472.CAN-10-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bendle GM, Linnemann C, Hooijkaas AI, et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med. 16:565–70. doi: 10.1038/nm.2128. 1p following 70. [DOI] [PubMed] [Google Scholar]
- 48.Gilham DE, Lie ALM, Taylor N, Hawkins RE. Cytokine stimulation and the choice of promoter are critical factors for the efficient transduction of mouse T cells with HIV-1 vectors. J Gene Med. 12:129–36. doi: 10.1002/jgm.1421. [DOI] [PubMed] [Google Scholar]
- 49.Baumann JG, Unutmaz D, Miller MD, et al. Murine T cells potently restrict human immunodeficiency virus infection. J Virol. 2004;78:12537–47. doi: 10.1128/JVI.78.22.12537-12547.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsurutani N, Yasuda J, Yamamoto N, Choi BI, Kadoki M, Iwakura Y. Nuclear import of the preintegration complex is blocked upon infection by human immunodeficiency virus type 1 in mouse cells. J Virol. 2007;81:677–88. doi: 10.1128/JVI.00870-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.