

Abstract
Objectives
To characterize HIV-1 env compartmentalization between cerebrospinal fluid (CSF) and peripheral blood plasma over all stages of the HIV-1 disease course, and to determine the relationship between the extent of CSF HIV-1 env compartmentalization and clinical neurologic disease status.
Design
Paired blood plasma and CSF specimens were collected from 66 HIV-infected patients cross-sectionally representing all major clinical stages relating to HIV-associated neurologic disease, including primary infection, asymptomatic chronic infection, chronic infection with minor global impairment, and immune deficiency with HIV-associated dementia.
Methods
Heteroduplex tracking assays and bulk sequence analysis targeting the V1/V2, C2-V3, and V4/V5 regions of env were performed to characterize the genetic makeup of complex HIV-1 populations in the cross-sectional blood plasma and CSF specimens. The levels of blood plasma/CSF env compartmentalization were quantified and compared across the different clinical stages of HIV-1 neurologic disease.
Results
Blood plasma/CSF env compartmentalization levels varied considerably by disease stage and were generally consistent across all three regions of env characterized. Little or no compartmentalization was observed in non-impaired individuals with primary HIV-1 infection. Compartmentalization levels were elevated in chronically infected patients, but were not significantly different between mildly impaired and non-impaired patients. Patients with HIV-associated dementia showed significantly greater blood plasma/CSF env compartmentalization relative to other groups.
Conclusion
Increased CSF compartmentalization of the HIV-1 env gene, which may reflect independent HIV-1 replication and evolution within the central nervous system, is specifically associated with HIV-associated dementia and not the less severe forms of HIV-1 neurologic disease.
Keywords: central nervous system, cerebrospinal fluid, compartmentalization, env, HIV type 1, NeuroAIDS
Introduction
HIV-1 can cause severe neurologic disease, including HIV-associated dementia (HAD), in a subset of infections [1,2]. The use of highly-active antiretroviral therapy (HAART) reduces the risk for HAD [3,4]. However, the majority of the HIV-infected population does not have access to HAART (particularly in resource poor regions), other forms of neurologic complications persist despite HAART use, and the central nervous system (CNS) may serve as an HIV-1 reservoir during HAART due to the long-lived nature of CNS-infected cells and poor penetration of certain antiretroviral therapies (ARTs) [5–9]. Thus, neurologic disease remains a serious complication of HIV-1 infection.
Studies [10,11] using autopsy tissues from infected patients have demonstrated that HIV-1 genetic populations can be distinct, or compartmentalized, between the brain and periphery. However, the inability to sample HIV-1 populations directly from the brain of living individuals poses a challenge in fully understanding the dynamics of viral CNS compartmentalization over the full disease course, and ultimately its role in HIV-1 neuropathogenesis. Although HIV-1 can be sampled from cerebrospinal fluid (CSF), the use of CSF as a surrogate CNS compartment is complicated by the fact that the anatomical and cellular sources of CSF HIV-1 remain unclear. Studies [12–15] have revealed that HIV-1 genetic populations in CSF and peripheral blood plasma are often compartmentalized, suggesting that at least a portion of the CSF viral population can be produced locally within the CNS.
Our group previously reported a small cross-sectional study [16] of 27 chronically infected patients that revealed elevated HIV-1 env V1/V2 compartmentalization between blood plasma and CSF in HAD participants. However, the small sample size of this initial study, including only four patients with HAD, prohibited a conclusive investigation into the relationships between CSF HIV-1 compartmentalization and broader neurologic disease. Furthermore, it is unclear whether the association between CSF HIV-1 env compartmentalization and neurologic disease is specific for the V1/V2 region. A better understanding of the dynamics of CSF HIV-1 compartmentalization throughout the full course of infection, including how it is reflected throughout the env gene, could provide additional insight into the mechanisms of HIV-1 CNS compartmentalization.
Here, we present the first systematic study that characterizes the extent of CSF HIV-1 env compartmentalization across all major clinical stages of disease, from primary infection to end-stage immune deficiency with HAD. To this end, we expanded our earlier characterizations of blood plasma/CSF HIV-1 V1/V2 compartmentalization [16,17], adding to the cohort for a total of 66 patients representing the major stages of HIV-1 disease and extended the genetic characterizations of env to include the V4/V5 and C2-V3 regions to enhance the measurement of compartmentalization. We found that CSF HIV-1 compartmentalization levels were comparable across all three env regions. Furthermore, the level of HIV-1 env compartmentalization between blood plasma and CSF varied considerably by disease stage. These results suggest an important role for HIV-1 env compartmentalization in the development of HAD and validate CSF HIV-1 env compartmentalization as a virus-specific biological marker of HAD that may be used to enhance future clinical studies of HIV-1 neurologic disease.
Methods
Participants
Archived human blood plasma and CSF specimens were obtained from the National NeuroAIDS Tissue Consortium and multiple clinical studies conducted at the University of North Carolina (UNC)-Chapel Hill, the HIV Neurobehavioral Research Center at the University of California, San Diego, and the University of California, San Francisco (UCSF). To limit site-specific differences in results, multiple different collection sites contributed specimens for each disease category, with the exception of primary infection (all UNC). Informed consent was obtained from all participants, and specimens were collected and used under local institutional approval. Paired blood plasma and CSF specimens were obtained at the time of neurologic diagnosis. Depending on the study site, diagnosis was based on 1991 American Academy of Neurology criteria for minor cognitive and motor disorder (MCMD) and HAD [18], AIDS dementia complex staging [19], or the Carey et al. [20] global impairment algorithm, or all. No primary infection patients had detectable impairment. Chronic infection patients without significant neurologic deficits were classified as ‘not impaired’. Because both diagnoses have significant overlap in terms of neuropsychological pathology and less severe disease symptomology relative to HAD, data from participants diagnosed as ‘Globally Impaired’ or ‘Minor Cognitive and Motor Disorder’ (MCMD) were combined (MCMD/I).
Blood plasma and CSF viral RNA specimens from approximately 85 participants were screened for reverse transcriptase (RT)-PCR robustness and heteroduplex tracking assay (HTA) reproducibility. Results were analyzed only for participants from whom adequate viral genome amplification or template sampling was obtained (see HTA methods), which yielded env compartmentalization data from 66 total participants (Table 1). Possible reasons for inadequate viral genome sampling for the remaining participants include low viral load, poor sample quality, or insufficient specimen volume.
Table 1.
Study participants.
| NeuroAIDS status | n | CD4+ cell counta | BP VLa | CSF VLa | # (%) on ART | HCV+b |
|---|---|---|---|---|---|---|
| Primary infection | 7 | 390.8 (195–542) | 5.84 (3.81–7.13) | 3.91 (3.13–5.69) | 0 | ND |
| Not impaired | 19 | 364.1 (17–1063) | 4.73 (3.74–5.89) | 3.69 (2.77–5.55) | 3 (16) | 6/17 |
| Minor cognitive and motor disorder or globally impaired | 31 | 253.5 (15–671) | 4.88 (3.07–6.00) | 3.76 (2.82–5.07) | 7 (23) | 10/26 |
| HIV-associated dementia | 9 | 105.7 (9–234)c | 5.10 (3.31–6.41) | 4.92 (3.79–6.50)c | 0 | 1/5 |
| All patients | 66 | 279.7 | 4.97 | 3.91 | 10 (15) | 17/48 |
ART, antiretroviral therapy; BP, blood plasma; CSF, cerebrospinal fluid; HAD, HIV-associated dementia; HCV, hepatitis C virus; ND, not determined; VL, viral RNA load in log copies/ml.
Values indicated are mean (range) of all patients in NeuroAIDS status category.
HCV infection based on detection of HCV RNA or antigen or both. Shown are the numbers of HCV-positive patients per total number of patients with known HCV status.
HAD patients had significantly lower CD4+ T cell counts and significantly higher CSF viral loads relative to all other patients (P <0.001, Wilcoxon rank sum test).
Heteroduplex tracking assays
Detailed methods for viral RNA isolation, RT-PCR, and HTA are described elsewhere [21,22]. A minimum of 100 genome templates based on viral RNA load were included in RT-PCR reactions to ensure adequate genome sampling. In addition, for most participants, viral RNA populations in blood plasma and CSF samples were analyzed in two independent RT-PCR and HTA analyses of the same env region to validate genome template sampling, based on HTA band pattern reproducibility. To limit the potentially misleading effects of poor quality genome sampling, HTA results (for a specific env region) that were not reproducible were not used. Therefore, for some participants, we used HTA results for only one region (13 participants with V1/V2 only and 10 with V4/V5 only) to quantify HIV-1 env compartmentalization. Only a single RT-PCR and HTA analysis was performed for four primary infection patients due to limited specimen volume.
Analysis of env compartmentalization
Two measures of blood plasma/CSF env compartmentalization were performed. The first, % Difference, measures the discordance of HTA band patterns in two different lanes:
where Lane#1i is the relative abundance of a band that migrates to a particular point on the gel, i, for all bands, n, and Lane#2i is the relative abundance of a band migrating to the same position of the gel in the other lane. For bands detected in one lane and not the other, the undetectable band was assigned a relative abundance of 0. The sum of the absolute difference for all bands was divided by 2 because an increase in abundance of one band reflects a concomitant decrease of another, and multiplied by 100 to obtain a percentage value.
A second independent measure of env compartmentalization was a bulk sequence analysis of the C2-V3 region for a subset of 54 participants. The remaining participants were not analyzed due to poor RT-PCR amplification or limited specimen volume. RNA samples were subjected to RT-PCR using primers targeting V1 (5′-TTATGGGATCAAAGCCTAAAGCCATGTGTA-3′, HXB2 nt 6555-6584) and V3 (5′-TTTTGCTCTACTAATGTTACAATGTGC-3′, HXB2 nt 7235–7209). The C2-V3 region (HXB2 nt 6891–7208) for each RT-PCR product was bulk sequenced using primers for each DNA strand. The highest electrophoregram peak was used for assigning bulk sequence for nucleotide positions with multiple signals (i.e., mixed sequence). Sequences have been deposited in GenBank (Accession numbers EU216434–EU216541).
Statistical analysis
Phosphorimaging data were analyzed using GraphPad Prism software (version 3.02) (GraphPad Software Inc., San Diego, California, USA). Box and whisker plots indicate the median, 25th/75th percentile, and maximum/minimum values. The nonparametric Wilcoxon rank-sum test (using Van der Waerden normal scores) was used for conservative two-group comparisons. Monte Carlo estimates of exact two-sided P-values were used. Nominal (unadjusted) P-values are shown. Univariable logistic regression was used to evaluate the relative strength of association of markers with dichotomized HAD (SAS, version 9.1, SAS Institute Inc., Cary, North Carolina, USA). As HAD is typically only diagnosed during chronic or late infection, these analyses excluded data from primary infection patients. An approximation to Bayes factors, known as the Schwartz Criterion, was used to assess the strength of each marker [23].
Results
Study participants
Paired blood plasma and CSF specimens collected from 66 HIV-1-infected patients were analyzed for HIV-1 env compartmentalization. At the time of blood plasma/CSF sampling, participants represented cross-sections of the major clinical stages of HIV-1 infection relating to neurologic complications, from primary infection with no neurologic disease to end-stage AIDS with accompanying HAD (Table 1). Most participants were not on ARTat the time of sampling and diagnosis, although a few in the not impaired and MCMD/I groups were on failing ART. Median plasma viral load, CSF viral load, and CD4+ cell count values within each of these groups were not significantly different between participants off ART and on failing ART (P-values of 0.10–0.98, Wilcoxon rank-sum test), and more importantly, exclusion of participants on failing ART did not affect subsequent results of blood plasma/CSF env compartmentalization (data not shown).
Characterization of HIV-1 env variants by heteroduplex tracking assay
The HTA, originally described by Delwart et al. [24,25], displays coexisting genetic variants present in a biological specimen as a series of distinct bands in a polyacrylamide gel. A major strength of utilizing HTA for characterizing complex HIV-1 genetic populations is the combined ability to sample hundreds of genome templates, reveal the extensive complexity of HIV-1 genetic populations in a simple assay, and validate genome template sampling by RT-PCR based on HTA band pattern reproducibility across multiple independent analyses. This makes it possible not only to compare qualitatively the genetic makeup of complex HIV-1 populations present in two different biological specimens, but also to quantify their level of similarity. Furthermore, this use of HTA to compare complex HIV-1 genetic populations can be performed on a scale that typically is not obtained through standard cloning, sequencing, and phylogenetic analysis. This allowed us to analyze a sufficient number of biological specimens (i.e. study participants) to derive generalizable conclusions and to validate the quality of sampling for each specimen to prevent misinterpretation of differences observed between blood plasma and CSF viral populations that arise as a result of sampling too few viral genomes.
HIV-1 env compartmentalization varies by disease state
We used RT-PCR and HTA to compare blood plasma and CSF HIV-1 env populations. Two different regions of env were analyzed, spanning either the V1/V2 or V4/V5 hypervariable regions. Representative HTA gels and % difference results illustrate qualitative differences in blood plasma/CSF env compartmentalization observed in different participants, as well as the reproducibility of HTA band patterns for individual replicates (Fig. 1). To determine the relationship between env compartmentalization and HIV-associated neurologic disease, blood plasma/CSF env compartmentalization levels were measured and compared between participants at different disease stages (Fig. 2). We observed clear group-specific differences in blood plasma/CSF env compartmentalization for both V1/V2 and V4/V5 HTA (Fig. 2a). As a control for artifactual detection of elevated env compartmentalization, RT-PCR and HTA reproducibility were shown to be similar for all groups (Fig. 2b). Also, there was a strong correlation between V1/V2 and V4/V5 HTA measurements of blood plasma/CSF env compartmentalization (Fig. 2a,c), suggesting that both regions evolve differentially between the two compartments.
Fig. 1. Analysis of HIV-1 env populations in paired blood plasma and cerebrospinal fluid.
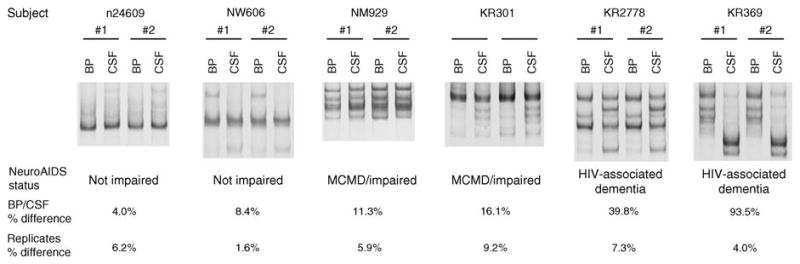
Shown are representative HTA gels for the V4/V5 region of env for six patients with varying degrees of HIV-associated neurologic disease. Two independent RT-PCR and HTA analyses were performed for each sample, and results for each replicate are shown. All bands showed run between the single-stranded probe and probe homoduplex bands, which are not shown for simplicity. The measurement of env compartmentalization (BP/CSF % difference) accounts for both the presence and absence of genetic variants in one compartment versus another, as well as differences in the relative abundance of variants present in both compartments. To monitor HTA reproducibility, two independent RT-PCR and HTA analyses were performed, and discordance between replicate HTA results was similarly quantified (replicates % difference). BP, blood plasma; CSF, cerebrospinal fluid; HTA, heteroduplex tracking assay; MCMD, minor cognitive and motor disorder; RT-PCR, reverse transcriptase-PCR.
Fig. 2. Blood plasma/cerebrospinal fluid env compartmentalization based on V1/V2 and V4/V5 heteroduplex tracking assay is associated with HIV-associated dementia.

(a) V1/V2 and V4/V5 env populations in BP and CSF of each patient were compared by HTA, and the % difference for BP/CSF HTA band patterns was determined for each patient and the data compiled for all patients in each of the different disease stages. A higher % difference indicates more discordant BP/CSF viral genetic populations for the particular region of env analyzed. (b) Discordance of band patterns for RT-PCR and HTA replicates from the same RNA sample was similarly measured by % difference to monitor reproducibility of HTA results for patients in each neurologic disease category. (c) Linear regression was performed to determine whether V1/V2 and V4/V5 BP/CSF % difference values within each patient correlate (P <0.0001, r2 = 0.4763) (d) The mean of V1/V2 and V4/V5 % difference results was determined for each patient to reflect global env compartmentalization between BP and CSF, and results were compiled for comparison between the different disease categories. Unadjusted P-values shown were determined by Wilcoxon rank-sum test (HAD versus other groups, P = 0.001, by univariable logistic regression). BP, blood plasma; CSF, cerebrospinal fluid; HAD, HIV-associated dementia; HTA, heteroduplex tracking assay; MCMD/I, minor cognitive and motor disorder/globally impaired; NI, not impaired; PI, primary infection; RT-PCR, reverse transcriptase-PCR.
Our results suggest that the ‘quality’ of HIV-1 in CSF, reflected by blood plasma/CSF env compartmentalization, is a strong indicator of HIV-1 neuropathogenesis contributing to HAD. Taking into account both V1/V2 and V4/V5 HTA measurements, blood plasma/CSF env compartmentalization was significantly greater in patients with HAD and significantly lower in primary infection patients, relative to the other groups tested (Fig. 2d). Unexpectedly, blood plasma/CSF env compartmentalization levels in chronically infected patients with less severe forms of HIV-associated neurologic disease were on average not elevated relative to non-impaired individuals. The number of HAD patients in this study (n = 9) precluded a multivariable regression analysis to determine whether CSF viral load and blood plasma/CSF env compartmentalization were independent or linked markers. Nevertheless, the CSF viral load and average % difference values for patients within individual disease groups correlated only weakly or not at all (data not shown), providing preliminary evidence that CSF viral load and blood plasma/CSF env compartmentalization are independent markers of neurologic disease.
HIV-1 env C2-V3 bulk sequencing
The C2-V3 region of env is genetically less diverse relative to V1/V2 and V4/V5, but contains determinants for receptor/coreceptor binding and epitopes for neutralizing antibodies, which might play important roles in HIV-1 neurotropism [26]. We characterized env C2-V3 sequences in blood plasma and CSF from a subset of 54 participants by determining bulk sequences of the RT-PCR products and compared the sequences by phylogenetic analysis. Similar to V1/V2 and V4/V5 HTA, C2-V3 sequence analysis revealed increased levels of blood plasma/CSF env compartmentalization in HAD patients, both at the nucleotide (Fig. 3a) and protein-coding level (data not shown). Despite this result, analysis of sequences by viral epidemiology signature pattern analysis (VESPA, Los Alamos National Laboratory HIV Database) revealed no sequence signatures for CSF relative to blood plasma in HAD or non-HAD patients within the C2-V3 region. Furthermore, characterization of V3 sequence populations by HTA and sequencing for a subset of 27 participants predicted that 24/27 participants had only chemokine receptor 5 (CCR5)-utilizing variants present (one MCMD X4 and two indeterminate) and did not reveal any biased patterns of predicted coreceptor usage (data not shown).
Fig. 3. Compartmentalization of the C2-V3 env bulk sequence between blood plasma and cerebrospinal fluid.

(a) Genetic distances estimated from alignments of the C2-V3 consensus sequences for blood plasma and CSF using the pairwise distance function in MEGA4 [28] and specifying the Kimura 2-parameter model of nucleotide substitution to allow transition–transversion rate differences. The resulting measure of genetic distance estimates the per-site number of nucleotide substitutions, after correcting for the probability of multiple substitutions at individual sites (*P <0.01 for HAD versus MCMD/I, NI, or PI). (b) A molecular clock was determined using consensus sequences obtained from longitudinal peripheral blood mononuclear cell samplings from eight chronically infected patients, as reported by Shankarappa et al. [27]. Genetic distances for the region of sequence overlap with this study (HXB2 nt positions 7023–7208) were estimated using the Kimura two-parameter model of nucleotide substitution and represent the per-site number of nucleotide substitutions that occurred in an individual patient as the initial sample in that patient. Each color corresponds to sequences sampled from a different patient. Lines represent the best-fit linear model (solid line) and the 95% confidence limits (dashed lines). On the basis of this molecular clock, we estimated the mean time of BP/CSF env divergence for patients in different neurologic disease categories. The indicated 95% confidence interval is based on the confidence limits of the molecular clock. BP, blood plasma; CSF, cerebrospinal fluid; HAD, HIV-associated dementia; MCMD/I, minor cognitive and motor disorder/globally impaired; MEGA, molecular evolutionary genetics analysis; NI, not impaired; PI, primary infection; RT-PCR, reverse transcriptase-PCR.
We performed a molecular clock analysis using a well characterized reference to assess the extent of blood plasma and CSF HIV-1 env sequence divergence. Using longitudinal HIV-1 consensus sequences obtained by Shankarappa et al. [27] from eight chronically infected patients, specifically for the region overlapping the C2-V3 region analyzed here (HXB2 nt 7023–7208), we calculated [28] the rate of HIV-1 nucleotide change in the periphery for these eight patients (Fig. 3b). This ‘molecular clock’ estimates that in the peripheral blood of chronically infected patients, this region of HIV-1 env evolves at a rate of 0.000692 substitutions per nucleotide site per month. Given this rate of sequence divergence, the measured mean genetic distance of 0.0295 substitutions per nucleotide site between blood plasma and CSF in HAD patients (Fig. 3a) corresponds to a time period of 57 months of HIV-1 evolution in the blood. If we assume that evolution is occurring independently and at an equal rate in each compartment, then the lineages were separated for one-half that time, or an equivalent of 28.5 months of HIV-1 evolution in the blood, compared with 6.9 and 8.8 months in non-impaired and MCMD/I patients, respectively (Table in Fig. 3b). However, the true time of divergence of blood plasma and CSF HIV-1 populations would vary based on the rate of evolution in each compartment, which also likely varies by disease stage. Although the assumptions oversimplify the dynamics and mechanisms of HIV-1 compartmentalization, these results illustrate the remarkable degree of divergence of HIV-1 genetic populations in blood plasma and CSF of HAD patients, comparable with the genetic divergence of HIV-1 populations in the peripheral blood after nearly 5 years of untreated chronic infection.
HIV-1 compartmentalization and immune status
Neurologic disease progression to HAD typically occurs late in HIV-1 infection, usually after the onset of immune deficiency, and CD4+ T cell counts were significantly lower in patients with HAD (Table 1). Therefore, reduced immune selective pressure throughout all compartments, independent of neurologic disease, is one possible explanation for the elevated env compartmentalization observed in HAD patients. However, we observed no correlation between CD4+ T cell count and blood plasma/CSF env compartmentalization when analyzing results for all (Fig. 4a) or only HAD patients (Fig. 4b). Furthermore, the pattern of significantly elevated blood plasma/CSF env compartmentalization in HAD patients did not change when analyzing only patients with CD4+ T cell counts of less than 250 cells/μl (Fig. 4c). These results suggest that immune deficiency alone, at least as reflected by CD4+ T cell count, does not explain the increased levels of blood plasma/CSF env compartmentalization observed in patients with HAD.
Fig. 4. Relationship between CD4+ cell count and blood plasma/cerebrospinal fluid env compartmentalization.

(a,b) Linear regression analysis was performed to determine whether CD4+ cell count levels correlate with average %difference measure of BP/CSF env compartmentalization for all patients (a) or only HAD patients (b). (c) BP/CSF env compartmentalization levels in patients with poor immune status (based on CD4+ T cell count <250 cells/μl) were compared between the different disease groups (P values based on Wilcoxon rank-sum test). Note that separate comparisons of HAD versus MCMD/I and HAD versus NI showed comparable statistical significance (P = 0.0006 and P = 0.007, respectively). CD4+ T cell counts for NI/MCMD/I and HAD patients in this subgroup analysis were not significantly different (P = 0.15, Wilcoxon rank sum test), with mean ± SD of 146 ± 66 (range 17–230) and 106 ± 71 (range 9–234), respectively. BP, blood plasma; CSF, cerebrospinal fluid; HAD, HIV-associated dementia; MCMD/I, minor cognitive and motor disorder/globally impaired; NI, not impaired, PI, primary infection.
Discussion
This first systematic, cross-sectional, and quantitative characterization of HIV-1 genetic compartmentalization between blood and CSF across the major stages of infection allowed us to make several specific observations about the nature of the CSF HIV-1 compartment. First, HIV-1 populations in blood and CSF are essentially indistinguishable during asymptomatic primary infection. Later during chronic infection, as the HIV-1 populations become more complex in both compartments, blood and CSF viral populations can remain well equilibrated in some patients, even in the presence of presumably strong systemic immune pressure. However, HIV-1 env populations in HAD patients, as well as some non-HAD patients in chronic infection, are highly compartmentalized between blood and CSF, suggesting a major shift in the nature of HIV-1 infection in the CSF/CNS compartment. Previous characterizations of CSF HIV-1 dynamics during HAART revealed that decay rates of bulk CSF HIV-1 viral load can vary by disease state, with significantly slower CSF HIV-1 viral load decay often observed in HAD patients [21,29–31]. These results suggest that the primary cellular source of CSF HIV-1 changes over the disease course, which when taken together with the present study might reflect the genetic adaptation and replication of a subset of HIV-1 populations in longer lived cells in the CNS. HTA characterizations of V4/V5 populations in the present study also revealed that this region of env, similar to V1/V2 [16], can be highly compartmentalized between blood plasma and CSF, suggesting that the distinct selective pressures in blood plasma and CSF that are responsible for divergent env evolution in these two compartments act throughout the env gene.
Results from this study are consistent with our initial smaller scale study in supporting an important relationship between CSF HIV-1 compartmentalization and neurologic disease [16]. As in the previous study, neither overall genetic complexity in any one compartment nor the simple appearance of enriched or unique HIV-1 genetic variants in CSF correlated with neurologic disease. Rather, the overall extent of env compartmentalization, quantified by blood plasma/CSF % difference in the present study, associated best with neurologic disease. In support of these observations, a recent small-scale study revealed simian immunodeficiency virus (SIVsm) env blood plasma/CSF compartmentalization throughout the course of infection in a macaque that had significant CNS inflammation at end stage, whereas two macaques with equilibrated blood and CSF SIVsm populations throughout infection had no detectable CNS inflammation [32].
The best described genetic marker for HAD is the presence, specifically in brain tissue, of an Asn residue at amino acid coding position 283 in the env gene [33]. Although slight enrichment of N283 in CSF of HAD patients was reported [33], we did not observe this sequence as either a marker of HAD or CSF env compartmentalization. However, there are differences in these two studies, including the use of viral DNA in autopsy brain tissue compared with viral RNA in CSF, with the brain-derived DNA representing a terminal disease state. It is also possible that env evolutionary pathways in CSF and brain are distinct, as the anatomical and cellular sources of compartmentalized and non-compartmentalized HIV-1 in CSF have not been well characterized. Finally, it remains unclear whether CNS HIV-1 compartmentalization, in general, is a cause or effect of HIV-1 neuropathogenesis. More comprehensive comparisons of HIV-1 genetic populations in the periphery and throughout the various regions of the CNS, although technically difficult, are an obvious next step to obtain a better understanding of the mechanisms and neuropathologic consequences of HIV-1 CNS compartmentalization.
Despite the noted limitations, our results support a dynamic model of HIV-1 CNS compartmentalization, whereby the CSF serves as an intermediate compartment between local CNS tissues and the periphery, consistent with the model by Price and Staprans [34]. In this model, infected cells from both sources can contribute to a pool of viruses in CSF, but the relative contribution of the sources varies in patients on the basis of their neurologic disease state. We speculate that the level of blood and CSF HIV-1 compartmentalization is a reflection of the amount of local CNS viral replication. Accordingly, the presence of highly equilibrated blood and CSF compartments suggests that only peripheral sources contribute to the CSF HIV-1 pool, whereas a high degree of compartmentalization suggests that the predominant source of the CSF HIV-1 pool is virus-producing cells in the CNS. The level of CNS-specific viral replication could be modulated by the migration of target cells into the CNS, reduced immune surveillance, or both, but based on this model, the detection of a CSF-compartmentalized HIV-1 variant requires the presence of an autonomously replicating CNS HIV-1 subpopulation. The observed env genetic divergence of CNS-specific populations could be due to distinct selective forces on viruses at cell entry, differences in the cellular source(s) of viral populations in the CNS and periphery, reduced immune selection in the CNS, or simply founder effects, although these mechanisms are not mutually exclusive.
Our results suggest that CSF HIV-1 env compartmentalization may serve as a novel biological marker for diagnosing neurologic disease that is specifically associated with independent replication and evolution of HIV-1 in the CNS. Although the HTA methods are not likely to provide a routine clinical application in their current form, other sensitive molecular techniques may become available in the future for routine clinical use. Importantly, HIV-1 CSF compartmentalization was a robust marker of HAD but not the less severe forms of HIV-associated neurologic disease, raising the question of whether the mechanisms of MCMD are distinct from the mechanisms of HAD? One explanation is that HIV-1 compartmentalization in the CNS plays a role in MCMD, but is either not consistently reflected in CSF or was an earlier, transient event that contributed to persistent neurologic deficits. Conversely, MCMD/I, or at least a subset of such cases, might not be attributable specifically to neuropathogenesis by HIV-1. These possible explanations illustrate the importance of utilizing novel, specific, and sensitive biological markers to improve methods of research, prevention, diagnosis, and treatment of HIV-associated neurologic complications.
Acknowledgments
We thank Dr Serena Spudich at UCSF for her collaboration and helpful discussions in writing this manuscript. A subset of blood plasma and CSF samples used in this study were provided by the National NeuroAIDS Tissue Consortium. We also thank the Duke/UNC primary infection group, particularly Drs Joseph Eron and Charles Hicks, for providing the specimens from primary infection patients and Dr Susan Fiscus and the UNC Center for AIDS Research Virology Core.
Patrick R. Harrington, Christina L. Burch, and Ronald Swanstrom designed the study; Patrick R. Harrington, Gretja Schnell, Kimberly Ritola, and Cassandra B. Jabara performed laboratory research; Scott L. Letendre, Kevin Robertson, Colin Hall, Ronald J. Ellis, and Richard W. Price recruited, diagnosed, collected CSF and blood specimens, and generated clinical data from study participants; Patrick R. Harrington, Christina L. Burch, Cassandra B. Jabara, Dominic T. Moore, and Ronald Swanstrom analyzed data; Patrick R. Harrington and Ronald Swanstrom wrote the manuscript.
This work was supported by grants from the National Institutes of Health (R01-MH67751 to Ronald Swanstrom; R01-NS37660 to Richard W. Price; UL1RR024131 to UCSF; R01-MH58076, P01-DA12065, and R01-DA16015 to Ronald J. Ellis and Scott L. Letendre), the UCSD HIV Neurobehavioral Research Center (P30-MH62512), the UNC Center for AIDS Research (P30-AI50410), the UNC Lineberger Cancer Center Core Support Grant (P30-CA16086), a postdoctoral training grant (T32-CA09156 for Patrick R. Harrington), and a predoctoral training grant (T32-AI007419 for Gretja Schnell and Kimberly Ritola).
Footnotes
The study was presented in part at: 2006 Conference on Retroviruses and Opportunistic Infections, Denver, 5–8 February 2006 (abstract #77).
References
- 1.Navia BA, Jordan BD, Price RW. The AIDS dementia complex: I. Clinical features. Ann Neurol. 1986;19:517–524. doi: 10.1002/ana.410190602. [DOI] [PubMed] [Google Scholar]
- 2.Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- 3.McArthur JC, Brew BJ, Nath A. Neurological complications of HIV infection. Lancet Neurol. 2005;4:543–555. doi: 10.1016/S1474-4422(05)70165-4. [DOI] [PubMed] [Google Scholar]
- 4.Sacktor N, McDermott MP, Marder K, Schifitto G, Selnes OA, McArthur JC, et al. HIV-associated cognitive impairment before and after the advent of combination therapy. J Neurovirol. 2002;8:136–142. doi: 10.1080/13550280290049615. [DOI] [PubMed] [Google Scholar]
- 5.Gisslen M, Hagberg L. Antiretroviral treatment of central nervous system HIV-1 infection: a review. HIV Med. 2001;2:97–104. doi: 10.1046/j.1468-1293.2001.00056.x. [DOI] [PubMed] [Google Scholar]
- 6.Lambotte O, Deiva K, Tardieu M. HIV-1 persistence, viral reservoir, and the central nervous system in the HAART era. Brain Pathol. 2003;13:95–103. doi: 10.1111/j.1750-3639.2003.tb00010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim RB, Fromm MF, Wandel C, Leake B, Wood AJ, Roden DM, Wilkinson GR. The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. J Clin Invest. 1998;101:289–294. doi: 10.1172/JCI1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sacktor N. The epidemiology of human immunodeficiency virus-associated neurological disease in the era of highly active antiretroviral therapy. J Neurovirol. 2002;8 (Suppl 2):115–121. doi: 10.1080/13550280290101094. [DOI] [PubMed] [Google Scholar]
- 9.Clifford DB. HIV-associated neurocognitive disease continues in the antiretroviral era. Top HIV Med. 2008;16:94–98. [PubMed] [Google Scholar]
- 10.Korber BT, Kunstman KJ, Patterson BK, Furtado M, McEvilly MM, Levy R, Wolinsky SM. Genetic differences between blood-and brain-derived viral sequences from human immunodeficiency virus type 1-infected patients: evidence of conserved elements in the V3 region of the envelope protein of brain-derived sequences. J Virol. 1994;68:7467–7481. doi: 10.1128/jvi.68.11.7467-7481.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong JK, Ignacio CC, Torriani F, Havlir D, Fitch NJ, Richman DD. In vivo compartmentalization of human immunodeficiency virus: evidence from the examination of pol sequences from autopsy tissues. J Virol. 1997;71:2059–2071. doi: 10.1128/jvi.71.3.2059-2071.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pillai SK, Pond SL, Liu Y, Good BM, Strain MC, Ellis RJ, et al. Genetic attributes of cerebrospinal fluid-derived HIV-1 env. Brain. 2006;129:1872–1883. doi: 10.1093/brain/awl136. [DOI] [PubMed] [Google Scholar]
- 13.Strain MC, Letendre S, Pillai SK, Russell T, Ignacio CC, Gunthard HF, et al. Genetic composition of human immunodeficiency virus type 1 in cerebrospinal fluid and blood without treatment and during failing antiretroviral therapy. J Virol. 2005;79:1772–1788. doi: 10.1128/JVI.79.3.1772-1788.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lanier ER, Sturge G, McClernon D, Brown S, Halman M, Sacktor N, et al. HIV-1 reverse transcriptase sequence in plasma and cerebrospinal fluid of patients with AIDS dementia complex treated with Abacavir. AIDS. 2001;15:747–751. doi: 10.1097/00002030-200104130-00010. [DOI] [PubMed] [Google Scholar]
- 15.Tang YW, Huong JT, Lloyd RM, Jr, Spearman P, Haas DW. Comparison of human immunodeficiency virus type 1 RNA sequence heterogeneity in cerebrospinal fluid and plasma. J Clin Microbiol. 2000;38:4637–4639. doi: 10.1128/jcm.38.12.4637-4639.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ritola K, Robertson K, Fiscus SA, Hall C, Swanstrom R. Increased human immunodeficiency virus type 1 (HIV-1) env compartmentalization in the presence of HIV-1-associated dementia. J Virol. 2005;79:10830–10834. doi: 10.1128/JVI.79.16.10830-10834.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ritola K, Pilcher CD, Fiscus SA, Hoffman NG, Nelson JA, Kitrinos KM, et al. Multiple V1/V2 env variants are frequently present during primary infection with human immunodeficiency virus type 1. J Virol. 2004;78:11208–11218. doi: 10.1128/JVI.78.20.11208-11218.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nomenclature and research case definitions for neurologic manifestations of human immunodeficiency virus-type 1 (HIV-1) infection. Report of a Working Group of the American Academy of Neurology AIDS Task Force. Neurology. 1991;41:778–785. doi: 10.1212/wnl.41.6.778. [DOI] [PubMed] [Google Scholar]
- 19.Price RW, Sidtis JJ. Evaluation of the AIDS dementia complex in clinical trials. J Acquir Immune Defic Syndr. 1990;3 (Suppl 2):S51–S60. [PubMed] [Google Scholar]
- 20.Carey CL, Woods SP, Gonzalez R, Conover E, Marcotte TD, Grant I, Heaton RK. Predictive validity of global deficit scores in detecting neuropsychological impairment in HIV infection. J Clin Exp Neuropsychol. 2004;26:307–319. doi: 10.1080/13803390490510031. [DOI] [PubMed] [Google Scholar]
- 21.Harrington PR, Haas DW, Ritola K, Swanstrom R. Compartmentalized human immunodeficiency virus type 1 present in cerebrospinal fluid is produced by short-lived cells. J Virol. 2005;79:7959–7966. doi: 10.1128/JVI.79.13.7959-7966.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kitrinos KM, Hoffman NG, Nelson JAE, Swanstrom R. Turnover of env variable region 1 and 2 genotypes in subjects with late-stage human immunodeficiency virus type 1 infection. J Virol. 2003;77:6811–6822. doi: 10.1128/JVI.77.12.6811-6822.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kass RE, Raftery A. Bayes factors. J Am Statist Soc. 1995;90:773–795. [Google Scholar]
- 24.Delwart EL, Sheppard HW, Walker BD, Goudsmit J, Mullins JI. Human immunodeficiency virus type 1 evolution in vivo tracked by DNA heteroduplex mobility assays. J Virol. 1994;68:6672–6683. doi: 10.1128/jvi.68.10.6672-6683.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Delwart EL, Shpaer EG, Louwagie J, McCutchan FE, Grez M, Rubsamen-Waigmann H, Mullins JI. Genetic relationships determined by a DNA heteroduplex mobility assay: analysis of HIV-1 env genes. Science. 1993;262:1257–1261. doi: 10.1126/science.8235655. [DOI] [PubMed] [Google Scholar]
- 26.Dunfee R, Thomas ER, Gorry PR, Wang J, Ancuta P, Gabuzda D. Mechanisms of HIV-1 neurotropism. Curr HIV Res. 2006;4:267–278. doi: 10.2174/157016206777709500. [DOI] [PubMed] [Google Scholar]
- 27.Shankarappa R, Margolick JB, Gange SJ, Rodrigo AG, Upchurch D, Farzadegan H, et al. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J Virol. 1999;73:10489–10502. doi: 10.1128/jvi.73.12.10489-10502.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 29.Eggers C, Hertogs K, Sturenburg HJ, van Lunzen J, Stellbrink HJ. Delayed central nervous system virus suppression during highly active antiretroviral therapy is associated with HIV encephalopathy, but not with viral drug resistance or poor central nervous system drug penetration. AIDS. 2003;17:1897–1906. doi: 10.1097/00002030-200309050-00008. [DOI] [PubMed] [Google Scholar]
- 30.Ellis RJ, Gamst AC, Capparelli E, Spector SA, Hsia K, Wolfson T, et al. Cerebrospinal fluid HIV RNA originates from both local CNS and systemic sources. Neurology. 2000;54:927–936. doi: 10.1212/wnl.54.4.927. [DOI] [PubMed] [Google Scholar]
- 31.Staprans S, Marlowe N, Glidden D, Novakovic-Agopian T, Grant RM, Heyes M, et al. Time course of cerebrospinal fluid responses to antiretroviral therapy: evidence for variable compartmentalization of infection. AIDS. 1999;13:1051–1061. doi: 10.1097/00002030-199906180-00008. [DOI] [PubMed] [Google Scholar]
- 32.Harrington PR, Connell MJ, Meeker RB, Johnson PR, Swanstrom R. Dynamics of simian immunodeficiency virus populations in blood and cerebrospinal fluid over the full course of infection. J Infect Dis. 2007;196:1058–1067. doi: 10.1086/520819. [DOI] [PubMed] [Google Scholar]
- 33.Dunfee RL, Thomas ER, Gorry PR, Wang J, Taylor J, Kunstman K, et al. The HIV env variant N283 enhances macrophage tropism and is associated with brain infection and dementia. Proc Natl Acad Sci U S A. 2006;103:15160–15165. doi: 10.1073/pnas.0605513103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Price RW, Staprans S. Measuring the ‘viral load’ in cerebrospinal fluid in human immunodeficiency virus infection: window into brain infection? Ann Neurol. 1997;42:675–678. doi: 10.1002/ana.410420502. [DOI] [PubMed] [Google Scholar]