

Abstract
Mutations in TRPV4 have been linked to three distinct axonal neuropathies. However, the pathogenic mechanism underlying these disorders remains unclear. Both gain and loss of calcium channel activity of the mutant TRPV4 have been suggested. Here, we show that the three previously reported TRPV4 mutant channels have a physiological localization and display an increased calcium channel activity, leading to increased cytotoxicity in three different cell types. Patch clamp experiments showed that cells expressing mutant TRPV4 have much larger whole-cell currents than those expressing the wild-type TRPV4 channel. Single channel recordings showed that the mutant channels have higher open probability, due to a modification of gating, and no change in single-channel conductance. These data support the hypothesis that a “gain of function” mechanism, possibly leading to increased intracellular calcium influx, underlies the pathogenesis of the TRPV4-linked axonal neuropathies, and may have immediate implications for designing rational therapies.
Keywords: Neurobiology, Neurodegeneration, Neurological Diseases, Neuroscience, TRP Channels, Axonal Neuropathy, CMT2C, Cytotoxicity, SPSMA, TRPV4
Introduction
Scapuloperoneal spinal muscle atrophy (SPSMA), congenital distal spinal muscle atrophy (CDSMA), and Charcot-Marie-Tooth disease type 2C (CMT2C, also known as hereditary motor and sensory neuropathy type 2, HMSN IIC) are phenotypically heterogeneous and dominantly inherited disorders involving topographically distinct muscles and nerves (1, 2). Genetic linkage analysis mapped the disease loci to an overlapping region at 12q24 (3–5). Recently, it has been shown that these three axonal neuropathies are allelic disorders caused by mutations in the gene encoding the transient receptor potential cation channel, subfamily V (vanilloid), member 4 (TRPV4)3 (6–8). To date, seven mutations in TRPV4 have been identified in 15 families with axonal neuropathies (6–10). Functional studies, however, yielded divergent hypotheses about the pathogenic mechanism of the diseases (6–8, 10).
In our original study, we tested two mutations, R316C and R269H, which were identified in the original SPSMA and CMT2C families, respectively (6). We found that in HEK293 cells, mutant TRPV4 had a physiological localization on the plasma membrane similar to wild-type TRPV4 (wtTRPV4). Calcium imaging showed that mutant TRPV4 had significantly increased calcium channel activity, at both basal and activated levels when compared with wtTRPV4. These findings were consistent with electrophysiological recordings, which showed a higher TRPV4 conductance in HEK293 cells expressing the R269H mutant compared with wtTRPV4. In another study, Landouré et al. (8) showed similar results in HEK293 cells and Xenopus laevis oocytes with R269H and R269C mutations. Furthermore, mutant TRPV4-mediated cytotoxicity was observed in transfected HEK293 cells and dorsal root ganglion neurons. This cytotoxicity was related to increased intracellular calcium concentrations (8). More recently, we have also shown that two other TRPV4 mutants (R232C and R316H) localize appropriately and cause reversible and toxic hypercalcemia in HEK293 and HeLa cells (10). Collectively, these data suggest a “gain of function” mechanism for mutant TRPV4-mediated axonal neuropathies. However, using tagged constructs, Auer-Grumbach et al. (7) found that the TRPV4 mutants (R269H, R315W, and R316C) accumulated exclusively in the cytoplasm in HeLa cells. They did not find any differences in basal calcium levels between wild-type and mutant TRPV4-transfected cells. Upon stimulation they found that mutant TRPV4-transfected HeLa cells responded to a lesser degree than wtTRPV4-transfected cells. No difference was observed for the cell viability in HeLa cells transfected with mutant or wild-type TRPV4 constructs. These results, therefore, suggest a “loss of function” mechanism in mutant TRPV4-linked axonal neuropathies.
The reasons for the discrepancies about the functional consequences of the TRPV4 mutations raised by these three reports remain unclear. They may be related to differences in the experimental protocols (11) and the cell lines used. Understanding the functional alterations in the mutant TRPV4 channels is essential not only for the understanding of the pathogenesis of these disorders, but also for the design of rational therapies, because both agonists and antagonists of TRPV4 are currently available.
In this study, we have focused on the three previously analyzed TRPV4 mutations (R269H, R315W, and R316C) in three different cell types (HEK293, HeLa, and Neuro2a). HEK293 and HeLa cells were used in the three previous reports and yielded divergent results. Because TRPV4-linked axonal neuropathies predominantly involve the nervous system, we also included Neuro2a cells in this study.
EXPERIMENTAL PROCEDURES
Expression Vectors
A full-length human cDNA clone (IMAGE: 40125977) was used as a template. Two primers anchored with an XhoI (TRPV4-TP1: 5′-ctgtctcgagcaggcatggcggattccagcgaag-3′) and BamHI (TRPV4-TP2: 5′-catcggatccctagagcggggcgtcatcagt-3′) were used to amplify the full-length coding sequence. The amplified fragment was cloned into plasmid vector pBluescript M13. The TRPV4 sequence was verified by direct sequencing. The respective mutation was introduced into the plasmid vector by site-directed mutagenesis using primers containing R269H, R315W, and R316C mutations. The XhoI/BamHI fragment containing wild-type TRPV4, TRPV4R269H, TRPV4R315W, or TRPV4R316C was released from the pBluescript M13 vector and cloned into the XhoI and BamHI sites of a dual expression vector pIRES2-ZsGreen1 containing a green fluorescent protein (GFP) homolog (Clontech, Mountain View, CA).
Expression of Wild-type and Mutant TRPV4
HEK293, Neuro2a, and HeLa cells were grown on collagen-coated glass coverslips in Dulbecco's modified Eagle's medium containing 10% (v/v) human serum, 2 mm l-glutamine, 2 units/ml penicillin, and 2 mg/ml streptomycin at 37 °C in a humidity-controlled incubator with 5% CO2. The cells were transiently transfected with expression vectors, wild-type TRPV4, TRPV4R269H, TRPV4R315W, or TRPV4R316C using Lipofectamine 2000 (Invitrogen).
RT-PCR
Total RNA isolated from HEK293 cells was digested by DNase 1 (RNase-free), reverse-transcribed using primers specific to human TRPV4 (hTRPV4-rtp3: 5′-cctgtcttggcagccatcatgaga-3′ located in exon 6) and SOD1 (hSOD1-rtp3: 5′-ctacagctagcaggataacagatg-3′ located in 3′UTR). Reverse-transcribed cDNA was PCR-amplified using TRPV4 primers (hTRPV4-rtp1: 5′-ggcaacatgcgtgaattcatcaa-3′ located in exon 4, and hTRPV4-rtp2: 5′-gtacatcttggtgacaaacttggt-3′ located in exon 6), or SOD1 primers (hSOD1-rtp1: 5′-gaccagtgaaggtgtggggaagc-3′ located in exon 2, hSOD1-rtp2: 5′-ccacctttgcccaagtcatctgc-3′ located in exon 5). The size of the TRPV4 PCR product is 378 bp and that of SOD1 PCR product is 310 bp.
Western Blotting
Western blotting was performed using previously published protocols (12). Briefly, transiently transfected HEK293 cells were homogenized in buffer containing 0.5% Nonidet P-40, 50 mm Tris, pH 8.0, 5 mm MgCl2, 1 mm PMSF (phenylmethylsulfonyl fluoride), and protease inhibitor mixture (Roche Applied Science, Penzberg, Germany). The samples were sonicated for 10 s, and centrifuged at 800 × g for 10 min at 4 °C to remove cell debris and nuclei. Protein concentration was measured by using a Pierce BCA Protein Assay kit. The same amount of protein was added to run the SDS/PAGE gel. Western blotting was performed using standard procedures. Rabbit anti-TRPV4 antibody (Chemicon, Temecula, CA) was used as the primary antibody. Signals were detected by using the ECL Western blotting Analysis System (Amersham Biosciences).
Confocal Microscopy
HEK293, Neuro2a, and HeLa cells were seeded on collagen-coated coverslips 24 h prior to transfection. Twenty-four hours after transfection, the cells were fixed with 3% paraformaldehyde and 0.02% glutaldehyde for 15 min. Ice-cold methanol was used to permeablize cells. Rabbit anti-TRPV4 antibody (1:50, Chemicon, Temecula, CA) and mouse anti-cadherin (1:200, Abcam, Cambridge, MA) were used as primary antibodies. Alexa Fluor 555 goat anti-mouse (1:500, Invitrogen) and Alex 633 goat anti-rabbit (1:250, Invitrogen) were used as secondary antibodies. Digital images were captured and analyzed with Carl Zeiss LSM 510 META laser-scanning confocal microscopes.
Intracellular Ca2+ Measurements
HEK293, Neuro2a, and HeLa cells were grown in 6-well plates and transfected with indicated plasmids. After 24 h of incubation with Ruthenium Red (10 μm), cells were rinsed briefly with HEPES buffer (120 mm NaCl, 5.4 mm KCl, 1.6 mm MgCl2, 1.8 mm CaCl2, 11 mm glucose, and 25 mm HEPES, pH 7.2), and loaded with 3 μm Indo1-AM (Invitrogen) at 37 °C for 45 min. Cultures were then rinsed and kept in the dark in HEPES buffer at room temperature for an additional 30 min to allow for complete dye de-esterification. Cells were harvested and resuspended in PBS containing 25 μg/ml propidium iodide (PI, Calbiochem). Calcium flux was measured as a ratio of 405/510 fluorescence using a MoFlo cell sorter before and during treatment with 2 μm 4αPDD (Sigma) and analyzed using Summit software (DakoCytomation, Fort Collins, CO). The argon ion (488 nm) and krypton (UV; 351 nm) lasers were used for excitation. The ZsGreen1 GFP and PI signals were collected using 530/40 nm and 670/20 nm bandpass filters, respectively. Indo-1 signals were collected using 405/30 nm and 510/21 nm bandpass filters, respectively. Measurements were calibrated using the Grynkiewicz equation (13). Values for Rmin and Rmax were determined in Ca2+-free solution and high Ca2+-containing solution in the presence of 5 μm ionomycin, respectively. The dissociation constant (Kd) of 250 nm for Indo-1 AM was used for calculations. Calcium flux experiments were performed at room temperature. In all experiments, data were gated on cells that were PI-negative and showed high levels of ZsGreen1 GFP expression. Gating parameters were kept constant across experiments. Two tailed unpaired Student's t test (p < 0.05) was used for statistical analysis.
Electrophysiology
Whole-cell voltage-clamp recordings were performed from HeLa cells transiently transfected with wild-type or R269H TRPV4 cDNA and cultured in the presence of 20 μm ruthenium red (Sigma) (6). Similar ZsGreen1 GFP-fluorescent cells were chosen for experiments, although the cells expressing the mutant channels seemed generally brighter. Patch pipettes were pulled from WPI (Sarasota, FL) glass (PG10–165) for whole-cell recordings (WCR) or Dagan (Minneapolis, MN) SE16 borosilicate glass (1.65 mm OD, 0.75 mm ID) for cell-attached (CAR) and outside-out recordings (O-O) using a horizontal puller (P97, Sutter, Novato, CA) and had a resistance of 2 to 4 mΩ (WCR) and 5–7 MΩ (CAR and O-O) when filled with internal solution. For WCR and O-O the intrapipette solution consisted of (in mm): CsCl 140, MgCl2 2, EGTA 10, Na2ATP 2, NaGTP 0.1, HEPES 10, pH 7.3 adjusted with CsOH. In WCR, the series resistance was 4 to 14 mΩ and was compensated between 65 and 75%. For CAR the intrapipette solution was the same as the extracellular solution and consisted of (in mm): NaCl 100, KCl 6, MgCl2 2, CaCl2 1.5, glucose 10, HEPES 10, pH 7.38 adjusted with NaOH, ∼315 mOsm adjusted with sucrose. Currents were recorded using either a Multiclamp 700A (WCR) or an Axopatch 200B (CAR, O-O) amplifier (Molecular Devices). For WCR, cells were held at −30 mV, and currents were elicited by slow voltage ramps (from −100 mV to +100 mV, 0.6 s duration), filtered at 5 KHz and sampled at 10 KHz. Ruthenium red was applied focally using a Picospritzer II (General Valve Corp.). The current density was determined by normalizing the peak current (at 100 mV) to the cell capacitance measured in voltage clamp using a −5 mV square pulse from a holding potential of −30 mV. Only cells in which at least a partial recovery from ruthenium red inhibition was detected were included in the final analysis. For CAR and O-O measurements, currents were filtered at 2 KHz (using the built-in 4-pole, low pass Bessel filter) and acquired at 5 KHz. The gating analysis was performed after off-line filtering of the signal (700 Hz, Gaussian). For CAR the voltage reported in the figures is the applied (pipette) voltage. All electrophysiological recordings were performed at 22–24 °C.
Ruthenium red was prepared in a 20 mm stock solution (in water) and stored at 2–4 °C. Working solutions were prepared freshly every day. After drug application the dish was discarded even if complete wash-out of the drug was obtained.
Currents were analyzed offline using pClamp 9 software (Molecular Devices) and IGOR (Wavemetrics). Open channels were detected using a 60% threshold. Single channel amplitude was obtained by fitting Gaussian curves to amplitude histograms (14). The open probability was defined as Po = To/NT, where To is the time spent in the open state, and N is the number of channels detectable in the patch.
Open and closed dwell times histograms were constructed from the respective data and plotted on a log-linear scale (32 bins/decade). Histograms were fit with log-transformed exponential probability density functions of the general form of Equation 1 (14),
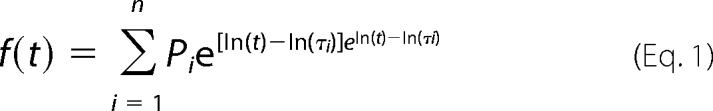 |
where τ is the state time constant, and P its fractional amplitude, N the number of exponentials and I the summation factor. Open times were fit using the sum of two functions (except two wild-type patches that showed only 1 time constant) while closed times required the sum of three functions. To verify that the observed differences in dwell times were not dependent on any particular binning, linear scale histograms of the open time distribution were also constructed. These histograms were fit with the probability density function in its usual form of Equation 2,
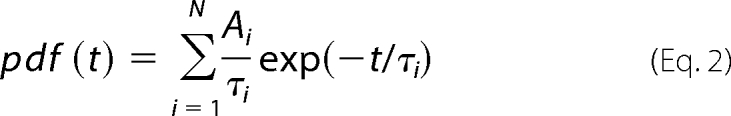 |
where Ai represents the area and t, τi, i and are the same as above. This method yielded results that were similar to those obtained with logarithmic binning and confirmed the difference between the duration of fast open states.
Cell Viability
HEK293, Neuro2a, and HeLa cells were grown in 24-well plates and transfected with indicated plasmids. After 48 h of incubation either with or without ruthenium red (20 μm), cells were harvested and resuspended in PBS containing 25 μg/ml propidium iodide (PI; Calbiochem). All flow cytometric data were collected and analyzed using a DakoCytomation Cyan flow cytometer and Summit software (DakoCytomation, Fort Collins, CO) recording at least 50,000 events. The argon ion laser (488 nm) was used for excitation. The ZsGreen1 GFP and PI signals were collected using 530/40 nm and 680/30 nm bandpass filters, respectively. In all experiments, data were gated on cells showing high levels of ZsGreen1 GFP expression. Gating parameters were kept constant across experiments. Percent cell death was the percentage of ZsGreen1 GFP-positive cells that were also PI-positive. Two tailed unpaired Student's t test (p < 0.05) was used for statistical analysis.
RESULTS
Endogenous Expression of TRPV4 in HEK293 Cells
Neuro2a cells and HeLa cells do not have detectable levels of endogenous TRPV4 expression in our experiments (data not shown) and previously published reports (15, 16). The endogenous TRPV4 expression in HEK293 cells appears variable in different batches (15, 17). In the HEK293 batch maintained in our facility, we did not observe detectable endogenous TRPV4 expression by RT-PCR and Western blot (supplemental Fig. S1), although the possibility of marginal endogenous expression of TRPV4 could not be excluded. This observation was corroborated by the observation that we did not detect any noticeable calcium flux in untransfected cells using our activation protocol (6).
Subcellular Localization of Wild-type and Mutant TRPV4
To avoid potential effects of protein tags on the subcellular trafficking and function of the TRPV4 channels, we used a dual expression vector, pIRES2ZsGreen1 (6). This vector allows expression of TRPV4 and a GFP homolog using the same mRNA transcripts simultaneously, but independently, therefore, allowing the TRPV4-expressing cells to be identified by green fluorescence. To examine the subcellular trafficking of the mutants, we transiently transfected HEK293, HeLa, and Neuro2a cells with expression vectors containing each mutation. An expression vector containing wtTRPV4 was used as a control. We observed that wild-type and the three mutant TRPV4 channels were present on the plasma membrane in all the three cell lines tested (Figs. 1, 2, and 3), suggesting that these mutations might not interfere with cellular processes of targeting mutant channels to the plasma membrane. Although the TRPV4 and ZsGreen1 GFP intensities were largely correlated in the vast majority of cells, uneven distribution of TRPV4 signal and poor correlation between TRPV4 and ZsGreen1 GFP signals were observed in some transfected cells. In addition to the plasma membrane localization, we also found cytoplasmic retention of TRPV4 in some cells (Fig. 4). Morphological changes, such as sub-plasmalemmal vesicles, have been previously described for wtTRPV4-transfected cells (18). However, the cytoplasmic retention of TRPV4 did not appear to be related to the presence of mutations or the cell type, as both wild-type and mutant TRPV4 showed the same phenomena with similar frequencies and this retention was observed in all the three cell types tested. The cytoplasmic retention seemed to predominantly occur in cells with strong ZsGreen1 GFP fluorescence, suggesting it may be related to excessive expression of the TRPV4.
FIGURE 1.

Physiological localization of wild-type and mutant TRPV4 on the plasma membrane. Confocal microscopy was performed using HeLa cells transfected with plasmids pIRES2-ZsGreen1 containing wtTRPV4 (a–d), TRPV4R269H (e–h),TRPV4R315W (i–l), and TRPV4R316C (m–p). Cells expressing exogenous TRPV4 were labeled by ZsGreen1GFP (c, g, k, and o). TRPV4 is shown by blue (a, e, i, and m) and cadherin by red (b, f, j, and n). Merged images are shown on the right panels (d, h, l, and p). Arrows indicate TRPV4 signals on the plasma membrane. Representative images are provided. For each condition, at least 50 cells were analyzed in more than two independent experiments.
FIGURE 2.

Physiological localization of wild-type and mutant TRPV4 on the plasma membrane. Confocal microscopy was performed using HEK293 cells transfected with plasmids pIRES2-ZsGreen1 containing wtTRPV4 (a–d), TRPV4R269H (e–h), TRPV4R315W (i–l), and TRPV4R316C (m–p). Cells expressing exogenous TRPV4 were labeled by ZsGreen1 GFP (c, g, k, and o). TRPV4 is shown by blue (a, e, i, and m) and cadherin by red (b, f, j, and n). Merged images are shown on the right panels (d, h, l, and p). Arrows indicate TRPV4 signals on the plasma membrane. Representative images are provided. For each condition, at least 50 cells were analyzed in more than two independent experiments.
FIGURE 3.

Physiological localization of wild-type and mutant TRPV4 on the plasma membrane. Confocal microscopy was performed using Neuro2a cells transfected with plasmids pIRES2-ZsGreen1 containing wtTRPV4 (a–d), TRPV4R269H (e–h), TRPV4R315W (i–l), and TRPV4R316C (m–p). Cells expressing exogenous TRPV4 were labeled by ZsGreen1 GFP (c, g, k, and o). TRPV4 is shown by blue (a, e, i, and m) and cadherin by red (b, f, j, and n). Merged images are shown on the right panels (d, h, l, and p). Arrows indicate TRPV4 signals on the plasma membrane. Representative images are provided. For each condition, at least 50 cells were analyzed in more than two independent experiments.
FIGURE 4.

Plasma membrane localization and cytoplasmic retention of TRPV4. Confocal microscopy was performed using HeLa cells transfected with plasmids pIRES2-ZsGreen1 containing wtTRPV4 (a–d), TRPV4R269H (e–h), TRPV4R315W (i–l), and TRPV4R316C (m–p). Cells expressing exogenous TRPV4 were labeled by ZsGreen1 GFP (c, g, k, and o). TRPV4 is shown by blue (a, e, i, and m) and cadherin by red (b, f, j, and n). Merged images are shown on the right panels (d, h, l, and p). Arrows indicate TRPV4 signals on the plasma membrane. Arrowheads indicate cytoplasmic retention of wild-type (a) and mutant TRPV4 (e, i, and m). Representative images are provided. For each condition, at least 50 cells were analyzed in more than two independent experiments.
Intracellular Ca2+ Measurements in Wild-type and Mutant TRPV4-transfected Cells
We then used flow cytometry and the calcium-sensitive probe indo-1 AM to investigate aspects of calcium homeostasis in different cell lines transfected with either wild-type or mutant TRPV4 channels. Internal Indo-1 fluorescence ratio (activity at 405 nm/activity at 510 nm) was used as an indicator of the intracellular Ca2+ levels, which depend on Ca2+ influx. In all three cell lines tested, mutant TRPV4 led to substantially higher basal calcium levels than wtTRPV4, suggesting an increased constitutive activity for the mutants (Figs. 5, 6, and 7). When the TRPV4-specific agonist 4α-phorbol 12,13-didecanoate (4αPDD) was applied, all three cell lines transfected with either wtTRPV4 or mutant channels, showed a consistent pattern of channel response. The mutant peak channel response was markedly higher compared with wtTRPV4 (Figs. 5–7). Similar to previous reports (6, 18), we observed that a subpopulation of transfected cells did not respond to agonist activation (supplemental Fig. S2). Interestingly, the percentage of cells responding to agonist stimulation appeared to be directly related to the level of ZsGreen1 GFP fluorescence. The high ZsGreen1 GFP subpopulation contained the highest proportion of 4αPDD-responding cells (supplemental Fig. S2). Hence, we used this subpopulation of cells for our further analysis. Even in this subpopulation of high ZsGreen1 GFP expressing cells, there was a much higher fraction of non-responders in HeLa cells (79.5%) compared with HEK293 (12.5%) or Neuro2a cells (11.3%). The differences in responsiveness in different cell lines used to express TRPV4 may be due to variable complement of some endogenous components, which may be necessary for the cell responsiveness toward particular stimuli. We therefore excluded these non-responding cells during analysis of the calcium flux data in HeLa cells.
FIGURE 5.

a, effect of stimulation with 2 μm 4αPDD on internal fluorescence ratio in HeLa cells transfected with either WT-, R269H-, R315W-, or R316C-TRPV4 constructs. Average basal and peak values are given. n>2000 cells for each condition. *, p < 0.0001, indicating significant differences when compared with WT-TRPV4 (two-tailed Student's t test). Error bars, means ± S.E. b, application of 4αPDD induced an increase in intracellular calcium ([Ca2+]i). Mean calcium responses before and during 4αPDD application are given. c, quantification of propidium iodide uptake in HeLa cells expressing wild-type and mutant TRPV4 indicates an increase in cytotoxicity in mutant expressing cells at 48 h that is blocked by the TRP channel blocker RR. Data are averaged from at least three independent experiments. #, p < 0.05, indicating significant differences when compared with WT-TRPV4 (two-tailed Student's t test). Error bars, means ± S.E.
FIGURE 6.

a, effect of stimulation with 2 μm 4αPDD on internal fluorescence ratio in HEK293 cells transfected with either WT-, R269H-, R315W-, or R316C-TRPV4 constructs. Average basal and peak values are given. n>5000 cells for each condition. *, p < 0.0001, indicating significant differences when compared with WT-TRPV4 (two-tailed Student's t test). Error bars, means ± S.E. b, application of 4αPDD induced an increase in intracellular calcium ([Ca2+]i). Mean calcium responses before and during 4αPDD application are given. c, quantification of propidium iodide uptake in HEK293 cells expressing wild-type and mutant TRPV4 indicates an increase in cytotoxicity in mutant expressing cells at 48 h that is blocked by the TRP channel blocker RR. Data are averaged from at least three independent experiments. #, p < 0.05, indicating significant differences when compared with WT-TRPV4 (two-tailed Student's t test). Error bars, means ± S.E.
FIGURE 7.

a, effect of stimulation with 2 μm 4αPDD on internal fluorescence ratio in Neuro2a cells transfected with either WT-, R269H-, R315W-, or R316C-TRPV4 constructs. Average basal and peak values are given. n>9000 cells for each condition. *, p < 0.0001, indicating significant differences when compared with WT-TRPV4 (two-tailed Student's t test). Error bars, means ± S.E. b, application of 4αPDD induced an increase in intracellular calcium ([Ca2+]i). Mean calcium responses before and during 4αPDD application are given. c, quantification of propidium iodide uptake in Neuro2a cells expressing wild-type and mutant TRPV4 indicates an increase in cytotoxicity in mutant expressing cells at 48 h that is blocked by the TRP channel blocker RR. Data are averaged from at least three independent experiments. #, p < 0.05, indicating significant differences when compared with WT-TRPV4 (two-tailed Student's t test). Error bars, means ± S.E.
Whole Cell Electrophysiological Recordings
Whole-cell patch-clamp recordings were obtained from acutely transfected HeLa cells. Nontransfected cells only had small background currents that did not show any rectification (data not shown). Most of the cells transfected either with the wild-type TRPV4 or the TRPV4R269H mutant channel exhibited large, outward rectifying background currents that were reversibly blocked by focal application of the TRPV blocker ruthenium red (20 μm, supplemental Fig. S3). The ruthenium red-sensitive component mediated by cells expressing wild-type TRPV4 or the TRPV4R269H mutant channel was obtained by offline digital subtraction (Fig. 8). These data show that most of the basal current in the transfected cells was actually mediated by TRPV4 channels. Consistent with the calcium flux data and our previously reported electrophysiological data in HEK293 cells (6), the ruthenium red-sensitive (TRPV4) conductance was ∼6 fold larger in HeLa cells expressing the mutant compared with wtTRPV4 (at 100 mV the normalized ruthenium red-sensitive current was 9.2 ± 4.3 and 52.4 ± 21.6 pA/pF for cells expressing the wild-type and mutated channels (n = 8 and 6, respectively, p < 0.05).
FIGURE 8.

The density of ruthenium red-sensitive current is higher in HeLa cells expressing mutant TRPV4. Patch clamp recordings were performed from HeLa cells. a and b, currents elicited by slow voltage ramps (600 ms, from −100 to +100 mV, upper traces) in control conditions (black traces) and in the presence of ruthenium red (red traces) in (a) wild-type transfected cells and (b) in cells transfected with the TRPV4R269H mutant channel. c and d, ruthenium red-sensitive currents from the traces in (a and b) obtained by digital subtraction. e, bar chart summarizing the current density (normalized to membrane capacitance) in cells transfected with either the wild-type or the mutant TRPV4 channel. No differences were detected in other properties of the ruthenium red-sensitive current such as the reversal potential (f).
Single-channel Recordings
CAR and O-O patch-clamp recordings were obtained from acutely transfected HeLa cells to investigate the mechanism(s) of the gain of TRPV4 function observed in cells expressing mutant channels. First, we used CAR recordings to compare the single channel conductance of wild-type and mutant (R269H) channels while minimizing the perturbations to the system; these measurements showed that no difference in single channel conductance between the two channel types. As expected for TRPV4 channels, both wild-type and mutant TRPV4 currents showed outward rectification: fitting the I/V curves with two straight lines (Fig. 9c) yielded two slope conductances, one for the more hyperpolarized potentials (Vpip>50 mV) and the other for the more depolarized potentials (Vpip<0 mV); no difference was observed in either one (the values were 131 ± 19 and 95 ± 25 for wt and 130 ± 13 and 93 ± 6 for mutant channels, respectively).
FIGURE 9.

Cell-attached recordings of wild-type and mutant TRPV4 channels. a, single-channel recordings form a cell expressing wild-type (black trace) and one expressing mutant (red trace) TRPV4 channels. These recordings were performed at Vpip = −100 mV (100 mV depolarization from resting potential). The amplitude histograms in b show that the mutant channels spent more time in the open state. The peak corresponding to the current in the open state was well represented in the mutant channels histogram, but it was necessary to expand the ordinate (inset) to resolve it in the wild-type histogram. c, no change was detected in single channel conductance. Recordings obtained at pipette potentials ranging from −100 to +80 mV in wild type (black traces) and mutant (red traces) TRPV4-expressing cells. The I/V curves obtained from these two cells are shown in the respective I/V plots.
Whereas no difference was detected in single channel conductance, wild-type and mutated channels clearly differed in open probability, which was largely increased in mutant channels (Fig. 9). At a pipette potential of −100 mV (i.e. 100 mV depolarization from resting potential) the open probability was 0.03 ± 0.01 for wild-type and 0.17 ± 0.04 for mutant channels (n = 4 and 8 cells, respectively, p < 0.05). Additionally, many patches from cells expressing wild-type channels did not show any TRPV4 activity at all, while this was not the case for mutant channels. A difference in channel open probability can be caused by an intrinsic change in gating, by increased channel insertion or by a combination of the two. To address this point, a more thorough kinetic analysis was performed. Because in cell-attached recordings the effective membrane potential depends on the cell resting potential and could differ between cells (although the average membrane potential did not differ between wild-type- and mutant TRPV4-expressing cells; it was −20.2 ± 5.5 (n = 9) and −22.9 ± 5 mV (n = 8), respectively) this analysis was performed in outside-out configuration, in which the membrane voltage is strictly controlled. Recordings were obtained at +50 mV in patches from wt and mutant-TRPV4 expressing cells (Fig. 10, a and b). Similar to the CAR data many patches from cells expressing wild-type channels did not show any TRPV4 activity; only 39% (7/18) of these patches showed TRPV4 currents, versus 88% (7/8) in mutant channel-expressing cells (Fig 10e). Analysis of the open probability (only patches showing TRPV4 activity were included) showed that, similar to the CAR measurements, the open probability of the mutant channels was higher (0.029 ± 0.01 versus 0.062 ± 0.01 for wt and mutant, respectively, p < 0.05, 6 patches each, Fig. 10f).
FIGURE 10.

Outside-out recordings of wild-type and mutant TRPV4 channels. a and b, single channel currents recorded at 50 mV from a patch containing wild type (a) and one containing mutant (b) TRPV4 channels. c and d, amplitude histogram of the traces shown in a and b. The vertical axis is cut at 1500 events to allow resolving the open channel currents. The overlaid dotted lines represent Gaussian functions fitted to the experimental data. The arrowheads show the value of unitary current obtained by the fitting. e, bar chart showing the fraction of outside-out patches that showed TRPV4 activity in cells expressing wild-type and mutant channels. f, bar chart showing the increased open probability of mutant channels. Only patches that showed TRPV4 activity were included in the analysis. g, bar chart of the mean open time in wild-type and mutant channels.
The mean open time (the mean duration of each open state) also differed between the two groups: it was 1.85 ± 0.24 ms in wild-type channels and 2.65 ± 0.14 ms in mutant channels (6 patches in each group, p < 0.05, Fig. 10g and Table 1). Detailed analysis of channel dwell times showed that the fast open time constant was increased by ∼60% in mutant channels (1.28 ± 0.17 ms, versus 2.02 ± 0.18 ms in wild-type, p < 0.05, Table 1), while no significant difference was found in the duration the slow open time constant (4.9 ± 0.4 ms in wild type versus 7.4 ± 1.3 ms in mutant; Table 1) or in the respective fractional contributions (Table 1). Finally, analysis of shut times showed a decreased contribution of the longer time constant in mutant channels (it decreased from 31 ± 5% for wild-type channels to 17 ± 4% for mutant channels, p = 0.05, Table 1), while no significant differences were observed in the values of the 3 shut time constants.
TABLE 1.
Gating properties of wild-type and mutant TRPV4 channels expressed in HeLa cells
| Parameter | Wild type | Mutant (R269H) | p |
|---|---|---|---|
| Mean open time (ms) | 1.85 ± 0.24 | 2.65 ± 0.14 | <0.05 |
| Open τ1 (ms) | 1.28 ± 0.17 | 2.02 ± 0.18 | <0.05 |
| Open τ1 fractional contribution (%) | 14 ± 7 | 21 ± 5 | |
| Open τ2 (ms) | 4.9 ± 0.4 | 7.4 ± 1.3 | |
| Open τ1 fractional contribution (%) | 86 ± 7 | 79 ± 5 | |
| Shut τ1 (ms) | 1.87 ± 0.27 | 1.77 ± 0.16 | |
| Shut τ1 fractional contribution (%) | 43 ± 5 | 50 ± 6 | |
| Shut τ2 (ms) | 96.8 ± 45.7 | 60.1 ± 12.5 | |
| Shut τ2 fractional contribution (%) | 27 ± 5 | 31 ± 7 | |
| Shut τ3 (ms) | 958 ± 499 | 435 ± 131 | |
| Shut τ3 fractional contribution (%) | 31 ± 5 | 17 ± 4 | 0.05 |
Cell Viability Assay in Wild-type and Mutant TRPV4-transfected Cells
We further evaluated TRPV4-mediated cytotoxicity as previously observed by Landouré et al. (8) in DRG neurons and HEK293 cells, but not by Auer-Grumbach et al. (7) who used HeLa cells. We observed that transfection with TRPV4 mutants resulted in remarkably increased cytotoxicity in all three cell lines tested at 48 h compared with wtTRPV4 (Figs. 5–7). Calcium influx and cytotoxicity could be effectively blocked by the TRP channel inhibitor ruthenium red (Figs. 5–7).
DISCUSSION
We used flow cytometry and patch clamp recordings to analyze the functional consequence of mutations in the TRPV4 channels. Flow cytometry may have advantages in some aspects. It allows acquiring data from over thousands of cells in a short time. Moreover, cells can be divided into different groups depending on the expression levels of the exogenous genes. Traditional electrophysiological techniques, on the other hand, have higher sensitivity to analyze the physiological features of a single cell or channel type. In the present study, we observed that the exogenous expression levels of TRPV4 and resultant calcium concentrations in the transfected cells could vary substantially. We and others have also observed that a significant number of TRPV4-transfected cells do not show detectable channel activity, therefore raising concerns whether data acquired from a limited number of cells can be faithfully representative. Thus, in the present study we have combined the two complementary approaches. Additionally, electrophysiological recordings were obtained from cells that showed similar amount of fluorescence at visual inspection. Thus, this study deals with a population of TRPV4-transfected cells that might be considered relatively homogeneous.
We also observed differences in responsiveness in different cell lines used. All the cells lines used in this study did not have detectable levels of endogenous TRPV4 expression. The differences in responsiveness in different cell lines used to express TRPV4 may be due to variable complement of other endogenous components, which may be necessary for the cell responsiveness toward particular stimuli.
The role of the cytoplasmic retention of TRPV4 channels and decreased calcium channel activity of the mutant TRPV4 in the pathogenesis of axonal neuropathies as described by Auer-Grumbach et al. (7) remains unclear. In their study showing a “loss of function” mechanism, the mutant TRPV4 channels appeared to have defects in transportation to plasma membrane, leading to cytoplasmic retention and loss of channel activity. These defects could be rescued by transfection of wild-type TRPV4. TRPV4-linked axonal neuropathies are dominant disorders with one allele each of wild-type and mutant on homologous chromosomes. Both wild-type and mutant TRPV4 are expressed from respective alleles. Even though mutant TRPV4 alone may not be able to be targeted to the plasma membrane in the transfected cells, this may not be a problem in the presence of the wild-type TRPV4 in neurons in the pathophysiological conditions. Therefore, it is unlikely that these defects play a critical role in the pathogenesis of TRPV4-mediated axonal neuropathies. In fact, we also observed cytoplasmic retention of mutant TRPV4 in some cells. But such retention was also observed in wtTRPV4-transfected cells with similar frequency, suggesting that cytoplasmic retention is not a specific property of mutant TRPV4, thus it is unlikely to be causal for the diseases.
“Gain of function” and “loss of function” are two disparate mechanisms underlying a large number of genetic disorders, implying opposite approaches for treatment. Understanding the disease-linked properties by a “gain of function” mechanism is a major challenge posed by most of the genetic disorders, including those related to neurodegeneration. However, for some channelopathies, such as TRPV4-linked axonal neuropathies, the gained property may be well defined, thus providing opportunities for designing rational therapies. In principle, if a mutant channel causes a disease through a “gain of function” mechanism, the use of antagonists will be beneficial, but agonists will be deleterious or vice versa.
In the present study, we used confocal microscopy, flow cytometry, and electrophysiological recordings, to test the functional alterations of three previously reported axonal neuropathy-linked TRPV4 mutants in three different cell lines, including Neuro2a, a neural cell type. These mutants displayed physiological localization and an increased calcium channel activity, leading to increased cytotoxicity in these cell lines. Additionally, the electrophysiological data on transfected HeLa cells showed that cells transfected with the TRPV4R269H mutation had ∼6-fold larger currents than the wild-type controls, which is consistent with our findings using this mutant in HEK293 cells (6). Thus, our data support the hypothesis of a “gain of function” mechanism, which is likely to lead to increased intracellular calcium influx. At least three different molecular mechanisms for this “gain of function” of the mutant TRPV4 channels can be envisaged: (i) increased single channel conductance, (ii) increased membrane insertion, and (iii) modifications of channel gating. Single channel recordings showed no change in single-channel conductance, thus ruling out hypothesis (i). These recordings showed increased open probability of the mutant channels. However, an increased open probability may be caused by either a higher number of channels or a modified gating mechanism that favors the open state. Cell surface biotinylation assays described in two of the original reports on these mutants showed that similar amounts of mutant TRPV4 incorporated into the cell membrane when compared with wtTRPV4 (6, 8). In the present study, the observation that TRPV4 activity was detectable in almost all (88%) outside-out patches from cells expressing mutant channels and only in a minority (39%) of the patches from cells expressing wild-type channels may also suggest more membrane insertion for the mutant channels. Stable cell lines with identical levels of TRPV4 expression would be more appropriate to further address this issue.
Recordings of mutant TRPV4 channels showed increased mean open time resulting from longer fast open time constant. This observation supports the idea that a gating modification that shifts the channel energy profile to favor the open state causes the “gain of function.” Additionally, we found a decreased contribution of the longer shut time constants in mutant TRPV4 channels. This parameter, however, may reflect either a difference in channels gating or the presence of more channels in the patches.
Therefore, our data show that there is a change in intrinsic gating that favors the open state; this mechanism is similar to that reported by Loukin et al. (19) for the brachyolmia-causing R616Q mutation; additionally, the R269H mutation may also cause a more effective channel insertion that may underlie the observed “gain of function” of the mutant channels. More studies, including an analysis of the intrinsic voltage dependence of the wild-type and mutant channels, will be required to provide a conclusive answer to the possibility of increased membrane insertion.
In conclusion, mutant TRPV4 channels show a “gain of function” coupled to increase calcium permeability. This could play a critical role in the pathogenesis of TRPV4-linked axonal neuropathies. Increased calcium flux is a common pathway involved in cytotoxicity and neurodegeneration (20, 21). The increased calcium channel activity and cytotoxicity can be effectively blocked by ruthenium red, a TRP channel antagonist, therefore, highlighting a rational therapy for these diseases. Ruthenium red has recently been shown to improve mitochondrial ATP levels and membrane potentials in neurons expressing mutant α-synuclein and/or PINK-1 associated with Parkinson disease (22). Furthermore, L-type calcium blockers have been suggested to reduce the chance of developing Parkinson disease (23). More recently, the selective TRPV4 channel blocker, HC-067047, has been shown to improve bladder function in experimentally induced cystitis in rodents without any major side side effects (24). Our data imply that a treatment using TRPV4 antagonists could be immediately tested as a therapeutic approach for these diseases.
Supplementary Material
Acknowledgments
We thank the staff of the Northwestern University Robert H. Lurie Comprehensive Cancer Center Flow Cytometry Core Facility.
This work was supported, in whole or in part, by NINDS Grant NS050641, NHLBI Grant HL095731, and Grants T32 AG20506 and T32 MH067564 from the National Institutes of Health. This work was also supported by the Les Turner ALS Foundation, the Vena E. Schaff ALS Research Fund, the Harold Post Research Professorship, the Herbert and Florence C. Wenske Foundation, the David C. Asselin MD Memorial Fund, the Help America Foundation, and the Les Turner ALS Foundation/Herbert C. Wenske Foundation Professorship.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
- TRPV4
- transient receptor potential cation channel, subfamily V (vanilloid), member 4
- RR
- ruthenium red.
REFERENCES
- 1. DeLong R., Siddique T. (1992) Arch. Neurol. 49, 905–908 [DOI] [PubMed] [Google Scholar]
- 2. Dyck P. J., Litchy W. J., Minnerath S., Bird T. D., Chance P. F., Schaid D. J., Aronson A. E. (1994) Ann. Neurol. 35, 608–615 [DOI] [PubMed] [Google Scholar]
- 3. Isozumi K., DeLong R., Kaplan J., Deng H. X., Iqbal Z., Hung W. Y., Wilhelmsen K. C., Hentati A., Pericak-Vance M. A., Siddique T. (1996) Hum. Mol. Genet. 5, 1377–1382 [DOI] [PubMed] [Google Scholar]
- 4. Klein C. J., Cunningham J. M., Atkinson E. J., Schaid D. J., Hebbring S. J., Anderson S. A., Klein D. M., Dyck P. J., Litchy W. J., Thibodeau S. N. (2003) Neurology 60, 1151–1156 [DOI] [PubMed] [Google Scholar]
- 5. McEntagart M. E., Reid S. L., Irrthum A., Douglas J. B., Eyre K. E., Donaghy M. J., Anderson N. E., Rahman N. (2005) Ann. Neurol. 57, 293–297 [DOI] [PubMed] [Google Scholar]
- 6. Deng H. X., Klein C. J., Yan J., Shi Y., Wu Y., Fecto F., Yau H. J., Yang Y., Zhai H., Siddique N., Hedley-Whyte E. T., Delong R., Martina M., Dyck P. J., Siddique T. (2010) Nat. Genet. 42, 165–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Auer-Grumbach M., Olschewski A., Papić L., Kremer H., McEntagart M. E., Uhrig S., Fischer C., Fröhlich E., Bálint Z., Tang B., Strohmaier H., Lochmüller H., Schlotter-Weigel B., Senderek J., Krebs A., Dick K. J., Petty R., Longman C., Anderson N. E., Padberg G. W., Schelhaas H. J., van Ravenswaaij-Arts C. M., Pieber T. R., Crosby A. H., Guelly C. (2010) Nat. Genet. 42, 160–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Landouré G., Zdebik A. A., Martinez T. L., Burnett B. G., Stanescu H. C., Inada H., Shi Y., Taye A. A., Kong L., Munns C. H., Choo S. S., Phelps C. B., Paudel R., Houlden H., Ludlow C. L., Caterina M. J., Gaudet R., Kleta R., Fischbeck K. H., Sumner C. J. (2010) Nat. Genet. 42, 170–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zimoń M., Baets J., Auer-Grumbach M., Berciano J., Garcia A., Lopez-Laso E., Merlini L., Hilton-Jones D., McEntagart M., Crosby A. H., Barisic N., Boltshauser E., Shaw C. E., Landouré G., Ludlow C. L., Gaudet R., Houlden H., Reilly M. M., Fischbeck K. H., Sumner C. J., Timmerman V., Jordanova A., Jonghe P. D. (2010) Brain 133, 1798–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Klein C. J., Shi Y., Fecto F., Donaghy M., Nicholson G., McEntagart M. E., Crosby A. H., Wu Y., Lou H., McEvoy K. M., Siddique T., Deng H. X., Dyck P. J. (2011) Neurology 76, 887–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nilius B., Owsianik G. (2010) Nat. Genet. 42, 98–100 [DOI] [PubMed] [Google Scholar]
- 12. Deng H. X., Shi Y., Furukawa Y., Zhai H., Fu R., Liu E., Gorrie G. H., Khan M. S., Hung W. Y., Bigio E. H., Lukas T., Dal Canto M. C., O'Halloran T. V., Siddique T. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 7142–7147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grynkiewicz G., Poenie M., Tsien R. Y. (1985) J. Biol. Chem. 260, 3440–3450 [PubMed] [Google Scholar]
- 14. Dempster J. (1993) Computer Analysis of Electrophysiological Signals, Academic Press, London; San Diego [Google Scholar]
- 15. Suzuki M., Hirao A., Mizuno A. (2003) J. Biol. Chem. 278, 51448–51453 [DOI] [PubMed] [Google Scholar]
- 16. Wegierski T., Lewandrowski U., Müller B., Sickmann A., Walz G. (2009) J. Biol. Chem. 284, 2923–2933 [DOI] [PubMed] [Google Scholar]
- 17. Liedtke W. (2008) Ann. N.Y. Acad. Sci. 1144, 42–52 [DOI] [PubMed] [Google Scholar]
- 18. Wissenbach U., Bödding M., Freichel M., Flockerzi V. (2000) FEBS Lett. 485, 127–134 [DOI] [PubMed] [Google Scholar]
- 19. Loukin S., Zhou X., Su Z., Saimi Y., Kung C. (2010) J. Biol. Chem. 285, 27176–27181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mattson M. P. (2007) Aging Cell 6, 337–350 [DOI] [PubMed] [Google Scholar]
- 21. Farber J. L. (1981) Life Sci. 29, 1289–1295 [DOI] [PubMed] [Google Scholar]
- 22. Marongiu R., Spencer B., Crews L., Adame A., Patrick C., Trejo M., Dallapiccola B., Valente E. M., Masliah E. (2009) J. Neurochem. 108, 1561–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ritz B., Rhodes S. L., Qian L., Schernhammer E., Olsen J. H., Friis S. (2010) Ann. Neurol. 67, 600–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Everaerts W., Zhen X., Ghosh D., Vriens J., Gevaert T., Gilbert J. P., Hayward N. J., McNamara C. R., Xue F., Moran M. M., Strassmaier T., Uykal E., Owsianik G., Vennekens R., De Ridder D., Nilius B., Fanger C. M., Voets T. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 19084–19089 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.