

Abstract
Rationale
The function of PKN, a stress-activated protein kinase, in the heart is poorly understood.
Objective
We investigated the functional role of PKN during myocardial ischemia/reperfusion (I/R).
Methods and Results
PKN is phosphorylated at Thr774 in hearts subjected to ischemia and reperfusion. Myocardial infarction/area at risk (MI/AAR) produced by 45 min ischemia and 24 hours reperfusion was significantly smaller in transgenic mice with cardiac specific overexpression of constitutively active (CA) PKN (Tg-CAPKN) than in non-transgenic (NTg) mice (15 ± 5 vs 38 ± 5%, p<0.01). The number of TUNEL positive nuclei was significantly lower in Tg-CAPKN (0.3 ± 0.2 vs 1.0 ± 0.2%, p<0.05). Both MI/AAR (63 ± 9 vs 45 ± 8%, p<0.05) and the number of TUNEL positive cells (7.9 ± 1.0 vs 1.3 ± 0.9%, p<0.05) were greater in transgenic mice with cardiac specific overexpression of dominant negative PKN (Tg-DNPKN) than in NTg mice. Thr774 phosphorylation of PKN was also observed in response to H2O2 in cultured cardiac myocytes. Stimulation of PKN prevented, whereas inhibition of PKN aggravated cell death induced by H2O2, suggesting that the cell protective effect of PKN is cell-autonomous in cardiac myocytes. PKN induced phosphorylation of alpha B crystallin and increased cardiac proteasome activity. The infarct reducing effect in Tg-CAPKN mice was partially inhibited by epoxomicin, a proteasome inhibitor.
Conclusion
PKN is activated by I/R and inhibits apoptosis of cardiac myocytes, thereby protecting the heart from I/R injury. PKN mediates phosphorylation of alpha B crystallin and stimulation of proteasome activity, which in part mediates the protective effect of PKN in the heart.
Keywords: Ischemia/reperfusion, PKN, alpha B crystallin, proteasome
Introduction
Although myocardial ischemia and reperfusion induce lethal injury in the heart, the heart and the cardiac myocytes therein have powerful endogenous mechanisms to protect themselves from energy deficiency, oxidative stress, protein aggregation and organelle malfunction, thereby minimizing myocardial injury 1. Ischemic preconditioning is one of the strongest of these endogenous mechanisms of cardioprotection, and pharmacological intervention mimicking the ischemic preconditioning effect is considered to be a promising modality for the treatment of ischemic heart disease 1. However, other endogenous mechanisms of cardioprotection activated during ischemia and reperfusion remain to be elucidated.
PKN, also known as protein kinase C-related kinase 1 (PRK1), is a serine (Ser)/threonine (Thr) kinase, with a molecular mass of 120KDa 2. PKN consists of a catalytic domain highly homologous to that of protein kinase C (PKC) in the carboxyl-terminal region, and a regulatory domain in the amino-terminal region 2. PKN localizes primarily in the cytosolic fraction and possesses a wide variety of functions, such as cytoskeletal regulation, cell adhesion, vesicle transport, and cell cycle regulation 3, and it has been shown to be involved in the pathogenesis of Alzheimer’s disease 4 and amyotrophic lateral sclerosis 5. Interestingly, PKN is cleaved by caspase 3 6, and a truncated and kinase-active form of PKN has been found in ischemia/reperfusion (I/R) models of the rat retina 7 and brain 8, suggesting that PKN may modulate cell survival or death during I/R. Transgenic (Tg) mice overexpressing a constitutively activated (CA) form of PKN in mammary epithelium exhibit impaired tight junction sealing and increased apoptosis, resulting in precocious involution in the mammary gland 9.
PKN activates atrial natriuretic factor (ANF) gene expression in cardiac myocytes 10. However, the in vivo function of PKN is poorly understood in the heart. Thus, a major goal in this study was to elucidate the function of PKN in the heart in vivo. Our preliminary studies indicated that PKN is activated by stress, such as hypotonic stress and I/R. Therefore, we examined the role of PKN in regulating cell survival and death and myocardial injury in response to I/R, and the underlying molecular mechanisms mediating the action of PKN in cardiac myocytes. Our results suggest that activation of PKN is an endogenous mechanism of cardioprotection against I/R injury.
Materials and Methods
An expanded Methods section is available in the Online Data Supplement at http://circres.ahajournals.org. All animal protocols were approved by the review board of the Institutional Animal Care and Use Committee of the University of Medicine and Dentistry of New Jersey.
PKN kinase assay
Cardiac myocyte lysates were prepared in lysis buffer containing 20 mmol/L Tris·HCl (pH 7.5), 150 mmol/L NaCl, 1% IGEPAL CA-630, 1 mmol/LEDTA, 1 mmol/L EGTA, 1 mmol/L beta-glycerophosphate, 0.1 mmol/L Na3VO4, 1 mmol/L NaF, 50 μmol/L phenylmethylsulfonyl fluoride (PMSF), 5 μg/ml aprotinin, and 5μg/ml leupeptin. PKN was immunoprecipitated with PKN antibody (BD Transduction Laboratories) at 4°C for 120 min. Protein G Sepharose beads were added and incubated at 4°C for 50 min. After washing three times, the precipitates were incubated for 30 min at 30°C with reaction buffer containing 20 mmol/L Tris-HCl (pH7.5), 4 mmol/L MgCl2, 20 μmol/LATP, and 0.8 μCi [γ-32P]ATP, and with 40 μmol/L PKCδ peptide (AMFPTMNRRGSIKQAKI) as a peptide substrate. Incorporation of 32P into the substrate was measured by scintillation counting.
Generation of Tg-CAPKN and Tg-DNPKN mice
Transgenic mice with cardiac specific overexpression of CAPKN 6 (amino acids 561-942) and DNPKN (full length K644D 3) were generated on an FVB background using the α-myosin heavy chain promoter (courtesy, Dr. J Robbins, Children’s Hospital, Cincinnati, OH).
Construction of short hairpin RNA (shRNA) adenoviral expression vectors
The U6 RNA polymerase III promoter and the polylinker region were subcloned into the adenoviral shuttle vector pDC311 (Microbix). The hairpin-forming oligo, 5′-GATTGACATCATCCGCATGTTCAAGAGACATGCGGATGATGTCAATC-3′ from the rat PKN cDNA, and its antisense, and the hairpin-forming oligo, 5′-ACGTGGATCCTCTCACCATTATTCAAGAGATAATGGTGAGAGGATCCACGT-3′ from the rat alpha B crystallin (BC) cDNA, and its antisense with ApaI and Hind III overhangs were synthesized, annealed, and subcloned distal to the U6 promoter. The loop sequence is underlined. A recombinant adenovirus was generated as described 11.
Statistics
All data are expressed as mean ± SEM. Differences between experimental groups were evaluated for statistical significance using Student’s t-test for unpaired data or one-way ANOVA followed by Dunnett’s post-test when appropriate. P values < 0.05 were considered to be statistically significant.
Results
PKN is activated by I/R in the heart
We examined whether PKN is activated in the heart under stress conditions, using a myocardial I/R model. In hearts subjected to ischemia, Thr774 phosphorylation of PKN was observed after 15–30 min, and declined after 45 min (Fig. 1AB). After reperfusion, PKN phosphorylation was observed again, and this phosphorylation was sustained for more than 24 hours (Fig. 1AB). In the hearts of sham-operated mice, expression and phosphorylation of PKN were unchanged over the entire experimental period (Online Fig. I).
Fig. 1. PKN is activated in hearts subjected to I/R.

Effects of I/R on PKN Thr774 phosphorylation. (A) Time course of PKN Thr774 phosphorylation. (B) The level of PKN Thr774 phosphorylation was determined by densitometric analysis. The mean total PKN/actin or phosphorylated PKN/total PKN in sham operated mouse hearts was set as 1 (dotted line). n=3–4. * p<0.05 vs. sham
CAPKN transgenic mice show cardiac hypertrophy without cardiac dysfunction
To elucidate the role of PKN activation in the in vivo heart, we generated mice with cardiac specific expression of constitutively active PKN (Tg-CAPKN), using the αMHC promoter. In Tg-CAPKN mouse hearts, phosphorylation of endogenous PKN was increased 5 fold compared to in non-transgenic (NTg) hearts (Fig. 2A). Heart weight/body weight (HW/BW) and left ventricular weight/BW (LVW/BW) were significantly higher in Tg-CAPKN than in NTg mice at both 3 and 6 months old (Online Table I). Lung weight/BW was not different between Tg-CAPKN and NTg mice (Online Table I). LV cardiac myocyte cross sectional area was significantly greater in Tg-CAPKN mice than in NTg mice (Fig. 2BC). The echocardiographic parameters in Tg-CAPKN mice were normal except that the wall thickness was significantly greater in Tg-CAPKN mice than in NTg mice at both the ages of 3 and 6 months (Online Table II). The end-diastolic diameter and end-systolic diameter were similar in both Tg-CAPKN and NTg mice, and the fractional shortening and the ejection fraction were also comparable (Online Table II). Doppler analysis showed that the E/A ratio and deceleration time, indexes of diastolic function, were comparable in Tg-CAPKN and NTg mice (Online Fig. II). In Tg-CAPKN mice, the heart rate (HR) was slower by 10% compared to NTg mice (Online Table II). Taken together, these data indicate that Tg- CAPKN mice showed left ventricular hypertrophy without LV dysfunction.
Fig. 2. Development of cardiac hypertrophy in Tg-CAPKN.

Baseline characterization of Tg-CAPKN mice at 3 months of age. (A) Representative immunoblots of heart homogenates with anti-PKN and anti-phospho (Thr774) PKN antibodies. Densitometric analysis was performed to determine the relative phospho-PKN level. CAPKN is N-terminally truncated and does not exist in NTg. (B) Representative pictures of WGA staining of the heart from Tg-CAPKN and NTg mice. (C) Myocyte cross sectional area was determined from WGA staining. The mean myocyte cross sectional area in NTg mice was set as 1. * p<0.05, ** p<0.01 vs NTg.
We also generated transgenic mice with cardiac specific overexpression of dominant negative (DN) PKN (Tg-DNPKN). The baseline cardiac phenotype of Tg-DNPKN was normal (Online Tables III and IV).
I/R injury is attenuated in Tg-CAPKN mice
Using the gain of function animal model, we examined whether activation of PKN is protective against I/R injury. Tg-CAPKN and NTg mice at 3 months old were subjected to 45 min of ischemia and 24 hours of reperfusion. The area at risk (AAR) was similar in Tg-CAPKN and NTg mice. The infarct size/AAR (MI/AAR) was significantly smaller in Tg-CAPKN than in NTg mice (15.1 ± 5.2 vs 37.5± 4.9%, p<0.01, Fig. 3A–C). There were also fewer TUNEL positive cells in the AAR in Tg-CAPKN than in NTg mice (0.3 ± 0.2 vs 1.0 ± 0.2%, p<0.05, Fig. 3DE). Furthermore, in Langendorff-perfused heart preparations, LV developed pressure and LV dP/dt were higher, and LV end-diastolic pressure was lower in Tg-CAPKN mice during the recovery phase after 45 min of global ischemia (Online Fig. III). These results suggest that activation of PKN is protective against I/R injury.
Fig. 3. I/R injury was attenuated in CA-PKN Tg mice.

Tg-CAPKN and NTg mice at 3 months old were subjected to I/R. (A) Gross appearance of LV tissue sections after Alcian blue (1%) and TTC (1%) staining in Tg-CAPKN and NTg mice subjected to I/R. (B, C) The effect of I/R on the extent of LV MI in Tg-CAPKN and NTg mice. AAR/LV size (%) (B) and MI area/AAR (%) (C) are shown. (D, E) LV tissue sections were subjected to TUNEL and DAPI staining. (D) Representative staining with TUNEL and DAPI are shown. (E) The number of TUNEL-positive nuclei was expressed as a percentage of total nuclei detected by DAPI staining. *p<0.05, **p<0.01 vs NTg.
It should be noted that the heart rate was slower by 10–15% in Tg-CAPKN mice than in NTg mice. To test the possibility that the reduced heart rate alone could be the mechanism of cardioprotection in the Tg-CAPKN heart, we reduced the heart rate in the NTg mice to the same level as that in Tg-CAPKN mice using propranolol, a beta blocker, during the ischemia and early reperfusion periods. Although intraperitoneal injection of propranolol (3 mg/kg) reduced the heart rate in NTg to a similar level as that in Tg-CAPKN mice, MI/AAR was still significantly smaller in Tg-CAPKN than in NTg (Online Fig. IV), suggesting that lowering the heart rate alone in NTg mice is not sufficient to achieve the infarct reducing effect observed in Tg-CAPKN mice.
I/R injury is enhanced in Tg-DNPKN mice
To elucidate the role of endogenous PKN in mediating protection against I/R injury, we also conducted I/R in Tg-DNPKN mice. Activation of PKN in response to I/R was abolished in Tg-DNPKN (not shown, See also Fig. 6E). In the DNPKN heart, MI/AAR (62.5 ± 9.3 vs 45.2 ± 8.4%, p<0.05)and the number of TUNEL positive cells (7.9 ± 1.0 vs 1.3 ± 0.9%, p<0.05) were both greater than in NTg mice (Fig. 4A–F), indicating that activation of endogenous PKN is protective during I/R.
Fig. 6. Alpha B crystallin mediates protective effect of PKN.

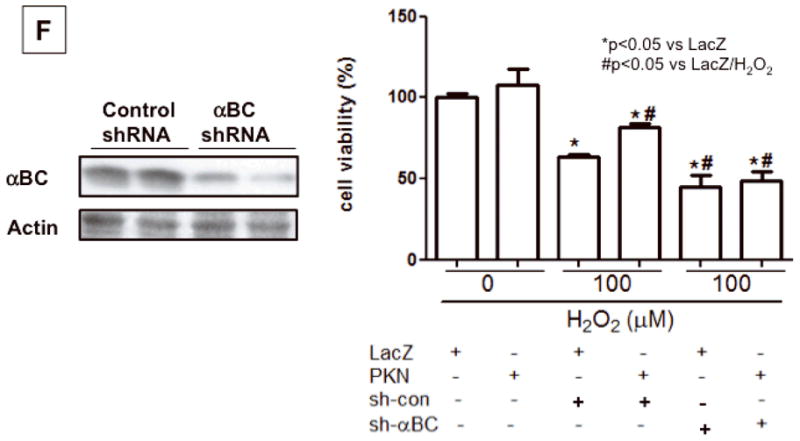
Immunoblot analyses of alpha B crystallin (αBC) and Serine 59 and Serine 45 phosphorylated alpha B crystallin (p-αBC (Ser59 or Ser45)) (A) p-αBC (Ser59) in Tg-CAPKN and NTg hearts. (B) Subcellular localization of αBC and p-αBC (Ser59) in Tg-CAPKN and NTg hearts. Cytoskeletal and cytosolic fractions were prepared from the heart in Tg-CAPKN or NTg. The purity of each fraction was analyzed by immunoblots with anti-α-sarcomeric actinin or GAPDH. (C) Immunoblot analyses of αBC and Serine 45 phosphorylated αBC (p-αBC (Ser45). (D) Subcellular localization of p-αBC (Ser45) in Tg-CAPKN and NTg hearts. (E) NTg and Tg-DNPKN mice were subjected to either sham operation or 45 min ischemia/2 hour reperfusion (I/R). Total heart homogenates were subjected to immunoblot analyses. The effect of I/R upon Ser45 and Ser59 phosphorylation of αBC and the role of PKN. * p<0.05 vs NTg. (F) Effect of αBC knockdown on the protective effect of PKN in cardiac myocytes. Immunoblot analysis of αBC expression. (Left). Myocytes were transduced with either Ad-LacZ or Ad-PKN in combination with Ad-sh-scramble or Ad-sh-αBC and then treated H2O2(Right).
Fig. 4. I/R injury was exacerbated in DN-PKN Tg mice.

Tg-DNPKN and NTg mice were subjected to I/R. (A) Expression of transgene (DNPKN, PKN K664D) is shown. (B) Gross appearance of LV tissue sections after Alcian blue and TTC staining in Tg-DNPKN and NTg mice subjected to I/R. (C, D) The quantitative analyses of AAR/LV size (%) (C) and MI area/AAR (%) (D) are shown. (E, F) LV tissue sections were subjected to TUNEL and DAPI staining. (E) Representative stainings with TUNEL and DAPI. (F) The number of TUNEL-positive nuclei was expressed as a percentage of total nuclei detected by DAPI staining. *p<0.05, **p<0.01 vs NTg.
Activation of PKN attenuates, whereas inactivation of PKN enhances death of cardiac myocytes in response to hydrogen peroxide stimulation in vitro
To evaluate whether the protective effect of PKN is cell-autonomous in cardiac myocytes, we conducted in vitro experiments, using cultured cardiac myocytes and adenovirus vectors harboring PKN (Ad-PKN), DNPKN (Ad-DNPKN) or shRNA-PKN (Ad-sh-PKN) (Online Fig. V). Oxidative stress is one of the most important causes of I/R injury in the heart. To evaluate the role of PKN in mediating cell survival and death in response to oxidative stress, cardiac myocytes were transduced with Ad-PKN, Ad-DNPKN or Ad-sh-PKN, and then treated with hydrogen peroxide (H2O2). Treatment with H2O2 induced PKN Thr774 phosphorylation in cardiac myocytes (Fig. 5A). Transduction of Ad-PKN reduced (Fig. 5B), while that of Ad-DNPKN or Ad-sh-PKN enhanced cell death (Fig. 5CD). These results suggest that activation of endogenous PKN during oxidative stress is protective, and that the cell protective effect of PKN is cell-autonomous.
Fig. 5. Transduction of PKN protects, whereas knockdown of PKN enhances cell death induced by H2O2.

Cardiac myocytes were transduced with Ad-PKN, Ad-DNPKN or Ad-sh-PKN. (A) The effect of H2O2 (100 μM) upon activity of PKN, as indicated by Thr774 phosphorylation of PKN, was evaluated. Immunoblots with anti-PKN and anti-phospho-PKN antibodies are shown. The result of densitometric analyses is shown. Phosphorylated PKN/total PKN without H2O2 was set as 1. * p<0.05 vs control. (B, C) Myocytes were treated with the indicated doses of H2O2 for 8 hours. Cell viability was determined by the Cell Titer Blue assay. Cell viability after the H2O2 treatment in cardiac myocytes transduced with Ad-PKN (B) or Ad-DNPKN (C) are shown. (D) Cell viability after H2O2 treatment in cardiac myocytes transduced with Ad-sh-PKN is shown. In B–D, cell viability in cardiac myocytes transduced with Ad-LacZ in the absence of H2O2 treatment was set as 1.
Activation of PKN causes phosphorylation and translocation of alpha BC into the cytoskeletal fraction
Alpha BC, a member of the small heat shock protein family of molecular chaperones, has a cell protective effect against stresses 12. PKN upregulates alpha BC gene expression in HeLa S3 cells 13. To address the molecular mechanism mediating the cell protective effect of PKN in cardiac myocytes, we evaluated expression of total and phosphorylated forms of alpha BC. The level of total alpha BC was not different between Tg-CAPKN and NTg hearts (Fig 6A, Online Fig. VIA). However, the level of alpha BC phosphorylated at Ser59 was significantly increased in Tg-CAPKN mice (Fig. 6A, Online Fig. VIAB). In neonatal rat cardiac myocytes, transduction with Ad-PKN increased, whereas that with Ad-sh-PKN reduced Ser59 phosphorylation of alpha BC (Online Fig. VICD). Treatment with H2O2 caused an increase in Ser59 phosphorylation of alpha BC, which was partially inhibited by shRNA PKN (Online Fig. VIEF). Alpha BC translocates to the cytoskeletal fraction and interacts with cytoskeletal proteins in response to stress 14, thereby playing a protective role in the heart 15. Translocation of alpha BC from the cytosol to the cytoskeletal fraction was also increased in Tg-CAPKN mice (Fig. 6B). In addition to Ser59 phosphorylation, Ser45 phosphorylation of alpha BC and its translocation to the cytoskeletal fraction were also increased in Tg-CAPKN mice (Fig. 6CD, Online Fig. VIG). In hearts subjected to I/R, both Ser45 and Ser59 phosphorylation of alpha BC and its translocation to the cytoskeletal fraction were increased (Online Fig. VII). The increase in alpha BC phosphorylation at both Ser45 and Ser59 in response to I/R was markedly inhibited in Tg-DNPKN mice (Fig. 6E). These results suggest that PKN plays an important role in mediating phosphorylation of alpha BC, which may in turn mediate protective effects in the heart during I/R. To address the role of alpha BC in mediating the cell protective effect of PKN, we knocked down alpha BC by shRNA, and examined the cell viability under H2O2 treatment in cultured cardiac myocytes. Ad-sh-alpha BC inhibited the cell protective effect of PKN, indicating that alpha BC mediates the cell protective effect of PKN (Fig. 6F).
PKN increases proteasome activity
Phosphorylation of alpha BC is important for its chaperone activity 16, which in turn plays an important role in regulating protein quality control via the ubiquitin-proteasome system. We therefore evaluated proteasome activity in Tg-CAPKN mice and cardiac myocytes treated with Ad-PKN. The proteasome activity, as evaluated by chymotrypsin-like activity, was significantly elevated in Tg-CAPKN hearts compared to NTg hearts (Fig. 7A). Similarly, cardiac myocytes transduced with Ad-PKN exhibited greater chymotrypsin-like activities than Ad-LacZ transduced myocytes (not shown). In hearts subjected to I/R, chymotrypsin-like activity was decreased, an effect which was significantly attenuated in Tg-CAPKN hearts (Fig. 7B). Furthermore, the decrease in chymotrypsin-like activity in response to I/R was significantly enhanced in Tg-DNPKN hearts (Fig. 7C). In order to test the functional significance of the elevated proteasome activity in Tg-CAPKN hearts, the effect of epoxomicin, a proteasome inhibitor, upon the cardioprotective effect of CAPKN was evaluated. Epoxomicin partially attenuated the infarct-reducing effect in Tg-CAPKN mice (Fig. 7D). These results are consistent with the notion that the elevated proteasome activity in Tg-CAPKN may in part mediate the protective effect against I/R.
Fig. 7. Proteasome is activated in Tg-CAPKN mouse hearts.
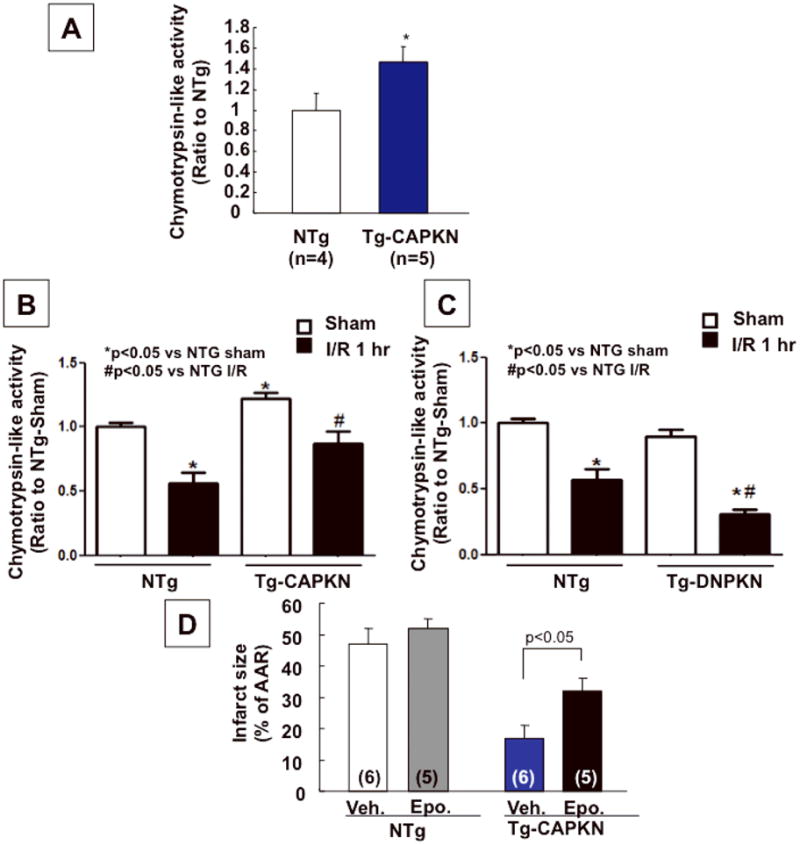
Chymotrypsin-like peptidylglutamyl-peptide hydrolase activity of proteasomes present in the cytoplasmic milieu was evaluated with the fluorogenic peptide LLVY-MCA. (A) Chymotrypsin-like activity in either Tg-CAPKN or NTg hearts was evaluated. The mean proteasome activity in NTg was set as 1. *p<0.05 vs. NTg. (B and C) Chymotrypsin-like activity in the heart subjected to I/R was measured in Tg-CAPKN (B) or Tg-DNPKN mice (C) and compared with that in NTg mice. The mean proteasome activity in NTg-Sham hearts was set as 1. N=4. (D) Epoxomicin (Epo. 0.5 mg/kg) or vehicle (Veh.) was injected intraperitoneally in Tg-CAPKN or NTg mice one day before and 1 hour before coronary occlusion. Mice were then subjected to I/R. Infarct size (% of AAR) are shown. The values of AAR were not different between all groups (not shown).
Discussion
Our main findings in this work are 1) that PKN is activated by I/R in the heart in vivo and by oxidative stress in cardiac myocytes in vitro, 2) that activation of PKN causes cardiac hypertrophy without LV dysfunction, 3) that activation of PKN plays a cell protective role in cardiac myocytes during I/R in vivo and in response to oxidative stress in vitro, and 4) that PKN mediates its protective effects against I/R in part through activation of the alpha BC/proteasome pathway.
In our study, Thr774 phosphorylation of PKN, which is essential for its activation, was observed in hearts subjected to I/R in vivo. In yeast, Pkc1, whose amino-terminal regulatory region is highly homologous to that of PKN, is activated by cell wall stress, including hypotonic stress17. Pkc1 is also important for cell viability in the adaptive response to oxidative stress 18, 19 or nitrosative stress 19. I/R causes both oxidative and osmotic stress, both of which affect survival and death of cardiac myocytes in the heart 20, 21. PKN is phosphorylated by H2O2 in cardiac myocytes in vitro, and PKN phosphorylation caused by I/R is inhibited in the presence of MPG, an antioxidant, in the heart in vivo (Online Fig. VIII), suggesting that oxidative stress plays an important role in mediating I/R-induced PKN activation. Furthermore, we found previously that hypotonic stress causes activation of PKN in vitro (manuscript in preparation by KK and JS). Thus, we speculate that osmotic stress may also mediate PKN activation in response to I/R. Interestingly, RhoA, a known activator of PKN 3, is activated by reactive oxygen species 22. Oxygen radicals can stimulate PKC directly by oxidative modification of its regulatory domain 23. The role of RhoA and oxidative posttranslational modification of PKN in mediating activation of PKN during I/R remains to be elucidated.
Our findings suggest that activation of PKN in cardiac myocytes is necessary and sufficient for cardioprotection during I/R in vivo and in response to H2O2 in vitro. Although LV function in Tg-DNPKN mice was comparable to that in NTg mice at 3 months of age, nearly complete downregulation of PKN by Ad-sh-PKN induces cell death in cardiac myocytes in vitro (Online Fig. IX), suggesting that a low level of PKN is required for survival of cardiac myocytes. Thus, all lines of experimental evidence presented in this work support the notion that PKN promotes survival of cardiac myocytes. Since PKN reduces the number of TUNEL positive myocytes in the ischemic area after I/R, we speculate that the protective effect of PKN is mediated through suppression of apoptosis, but its effect upon other forms of cell death remains to be elucidated.
Since suppression of myocardial injury during I/R was exacerbated when activation of PKN was inhibited in Tg-DNPKN mice, activation of PKN during I/R protects the heart. However, since Tg-CAPKN mice have stronger and more persistent activation of PKN, the cardioprotective effect observed in Tg-CAPKN may also be mediated in part by preconditioning effects. Our preliminary results suggest that PKN is also activated by preconditioning (data not shown). Whether or not activation of endogenous PKN can achieve protection against prolonged ischemia or late preconditioning remains to be elucidated. Neither translocation of PKCε into the membrane fraction (Online Fig. X) nor Akt phosphorylation, common mediators of ischemic preconditioning, was induced by overexpression of PKN in cardiac myocytes in vitro (Online Fig. XI). Thus, it is likely that the cardioprotective effects of CAPKN are mediated by PKCε- and Akt-independent mechanisms. Development of small molecules that specifically stimulate PKN would be relevant clinically, considering the fact that stimulators of PKCε have thus far shown promising results in their preclinical studies 24.
Tg-CAPKN mice showed mild LV hypertrophy. PKN is involved in actin reorganization in vascular smooth muscle cells 25 and stimulates ANF gene expression in cardiac myocytes 10, integral features of cardiac hypertrophy. PKN has also been shown to interact directly with alpha-actinin 26. Thus, activation of PKN by stress could stimulate cardiac hypertrophy in vivo. It should be noted that whether or not PKN is a physiological mediator of cardiac hypertrophy requires further evaluation with loss of function models of PKN. The presence of hypertrophy alone may secondarily affect the cardioprotective effect of PKN against I/R. However, since even short term activation or inactivation of PKN affects cell survival in a cell autonomous manner, the protective effect of PKN may be at least in part hypertrophy-independent.
Thus far, we have found that LV function in Tg-CAPKN mice is well maintained, even after over a year of follow up (data not shown). Together with the cardioprotective effect of PKN during I/R, we speculate that cardiac hypertrophy induced by PKN may be compensatory/physiological. However, this notion would require further testing by applying long-term hypertrophic stimulation, such as pressure overload, to Tg-CAPKN mice. In addition, the reduced heart rate could have resulted in a compensatory increase in ejection fraction, an index of LV contractility, in Tg-CAPKN mice. Thus, the effect of PKN upon the intrinsic contractility of cardiac myocytes should be evaluated.
As noted above, PKN physically interacts with RhoA 3, which positively regulates PKN. Transgenic mice with cardiac specific overexpression of constitutively active RhoA exhibit early lethality, and those with overexpression of wild type RhoA exhibit atrial enlargement, LV dilation and contractile failure, with a markedly reduced heart rate 27. The phenotype of Tg-CAPKN mice appears to be quite different from either of the above, suggesting that PKN may not be a major effector of RhoA in the heart. It should be noted, however, that Tg-CAPKN mice do show mild bradycardia. Since RhoA regulates the function of ion channels, such as Kir2.1 28 and L type calcium channel 29, PKN may participate in the regulation of ion channels by RhoA, thereby controlling heart rate.
Alpha BC, a member of the small heat shock protein and the molecular chaperone families, has protective effects against stresses 12, 30, 31. Whereas some PKC isoforms phosphorylate alpha BC 15, PKN upregulates mRNA expression of alpha BC through heat shock factor-1 in HeLa S3 cells 13. Interestingly, although total expression of alpha BC did not differ between Tg-CAPKN and NTg hearts, both alpha BC phosphorylation at Ser59 and Ser45 and its expression in the cytoskeletal fraction were increased in Tg-CAPKN mice. Furthermore, I/R-induced increases in alpha BC phosphorylation at Ser59 and Ser45 were inhibited in Tg-DNPKN mice. These results are consistent with the notion that PKN plays an important role in mediating phosphorylation of alpha BC in response to I/R in the heart. Although phosphorylation of alpha BC at Ser59 can be mediated by activation of MAPKAPK-2 32, MAPKAPK-2 is not activated in Tg-CAPKN mice (Online Fig. XII).
Although the specific role of PKN-induced Ser59 and Ser45 phosphorylation of alpha BC during I/R remains to be clarified, knockdown of alpha BC inhibited the protective effect of PKN in cardiac myocytes in response to H2O2, indicating that alpha BC mediates the cell protective effect of PKN. Phosphorylation of alpha BC at the Ser59 residue contributes to cytoprotection in the heart 12, 32, primarily due to its association with cytoskeletal elements, where the chaperone stabilizes myofilament, thereby maintaining cellular integrity 33, 34. Phosphorylation of alpha BC at either Ser59 or Ser45 stimulates its chaperone activity 16, which in turn plays an important role in mediating cellular protein quality control through modulation of the ubiquitin-proteasome system and autophagy. These mechanisms of protein degradation act as defense mechanisms against unfolded proteins, and are essential for cellular function and survival 35. It has been shown that I/R decreases the proteasome activity but ischemic preconditioning improves the proteasomal activity after I/R, and that increased proteasome activity is involved in the cardioprotective effect of preconditioning 36. Interestingly, Tg-CAPKN mice showed enhanced proteasome activity both at baseline and after I/R, and epoxomicin treatment partially reversed the cardioprotective effect of PKN against I/R injury. Furthermore, decreases in the proteasome activity during I/R were significantly enhanced in the Tg-DNPKN heart. Thus, these results suggest that the increased proteasome activity, in part, mediates the cardioprotective effect of PKN.
In conclusion, PKN is activated by I/R and activation of PKN plays a cell protective role in the heart in vivo. Furthermore, PKN mediates phosphorylation of alpha BC and stimulation of ubiquitin-proteasome activity, which in part mediates the protective effect of PKN in the heart (Online Fig. XIII). Thus, stimulation of PKN may represent a novel strategy for protecting the heart from I/R injury.
Novelty and Significance.
What Is Known?
PKN is a serine/threonine kinase with a catalytic domain homologous to protein kinase C.
PKN is activated by ischemia/reperfusion (I/R) in the retina and the brain in rats.
What New Information Does This Article Contribute?
PKN is activated by ischemia (I) and reperfusion (R) in the heart and protects the heart against I/R injury.
PKN phosphorylates alpha B crystallin (αBC) and stimulates proteasome activity, which plays an essential role in mediating the protective effect of PKN against I/R injury.
I/R in the heart causes myocardial injury and cardiac arrhythmia, which, in turn, induce left ventricular dysfunction and/or cardiac death. Identifying novel molecular mechanisms protecting the heart from I/R injury may lead to the development of new treatment for acute myocardial infarction. Here we show that PKN, a serine/threonine kinase, is activated by both ischemia and reperfusion in the mouse heart. Activation of PKN promotes phosphorylation of αBC and stimulates ubiquitin-proteasome activity in the heart, which in turn plays an essential role in mediating the protective effect of PKN against myocardial I/R injury. Our study identifies PKN as a novel endogenous mediator of cardioprotection against I/R injury. PKN is unique in that chaperone-mediated activation of the proteasome plays an important role in mediating its cardioprotective action. We propose that stimulation of endogenous PKN could be a novel therapeutic strategy for limiting myocardial injury caused by acute I/R.
Supplementary Material
Acknowledgments
We thank Rumiko Sho for technical assistance.
Sources of Funding
This work was supported in part by U.S. Public Health Service Grants HL 59139, HL67724, HL69020, HL91469, HL102738, and AG27211, and Foundation of Leducq Transatlantic Network of Excellence.
List of abbreviations
- Tg
transgenic
- NTg
non-transgenic
- CA
constitutively active
- DN
dominant negative
- LV
left ventricle
- RV
right ventricle
- BW
body weight
- shRNA
short hairpin RNA
- Ser
serine
- Thr
threonine
- I/R
ischemia/reperfusion
- AAR
area at risk
- MI
myocardial infarction
- alpha BC
alpha B crystallin
- MPG
N-(2-mercaptopropionyl)-glycine
Footnotes
Disclosures
None
References
- 1.Bolli R. Preconditioning: a paradigm shift in the biology of myocardial ischemia. Am J Physiol Heart Circ Physiol. 2007;292:H19–27. doi: 10.1152/ajpheart.00712.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mukai H, Ono Y. A novel protein kinase with leucine zipper-like sequences: its catalytic domain is highly homologous to that of protein kinase C. Biochem Biophys Res Commun. 1994;199:897–904. doi: 10.1006/bbrc.1994.1313. [DOI] [PubMed] [Google Scholar]
- 3.Mukai H. The structure and function of PKN, a protein kinase having a catalytic domain homologous to that of PKC. J Biochem. 2003;133:17–27. doi: 10.1093/jb/mvg019. [DOI] [PubMed] [Google Scholar]
- 4.Kawamata T, Taniguchi T, Mukai H, Kitagawa M, Hashimoto T, Maeda K, Ono Y, Tanaka C. A protein kinase, PKN, accumulates in Alzheimer neurofibrillary tangles and associated endoplasmic reticulum-derived vesicles and phosphorylates tau protein. J Neurosci. 1998;18:7402–7410. doi: 10.1523/JNEUROSCI.18-18-07402.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manser C, Stevenson A, Banner S, Davies J, Tudor EL, Ono Y, Leigh PN, McLoughlin DM, Shaw CE, Miller CC. Deregulation of PKN1 activity disrupts neurofilament organisation and axonal transport. FEBS Lett. 2008;582:2303–2308. doi: 10.1016/j.febslet.2008.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takahashi M, Mukai H, Toshimori M, Miyamoto M, Ono Y. Proteolytic activation of PKN by caspase-3 or related protease during apoptosis. Proc Natl Acad Sci U S A. 1998;95:11566–11571. doi: 10.1073/pnas.95.20.11566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sumioka K, Shirai Y, Sakai N, Hashimoto T, Tanaka C, Yamamoto M, Takahashi M, Ono Y, Saito N. Induction of a 55-kDa PKN cleavage product by ischemia/reperfusion model in the rat retina. Invest Ophthalmol Vis Sci. 2000;41:29–35. [PubMed] [Google Scholar]
- 8.Ueyama T, Ren Y, Sakai N, Takahashi M, Ono Y, Kondoh T, Tamaki N, Saito N. Generation of a constitutively active fragment of PKN in microglia/macrophages after middle cerebral artery occlusion in rats. J Neurochem. 2001;79:903–913. doi: 10.1046/j.1471-4159.2001.00624.x. [DOI] [PubMed] [Google Scholar]
- 9.Fischer A, Stuckas H, Gluth M, Russell TD, Rudolph MC, Beeman NE, Bachmann S, Umemura S, Ohashi Y, Neville MC, Theuring F. Impaired tight junction sealing and precocious involution in mammary glands of PKN1 transgenic mice. J Cell Sci. 2007;120:2272–2283. doi: 10.1242/jcs.03467. [DOI] [PubMed] [Google Scholar]
- 10.Morissette MR, Sah VP, Glembotski CC, Brown JH. The Rho effector, PKN, regulates ANF gene transcription in cardiomyocytes through a serum response element. Am J Physiol Heart Circ Physiol. 2000;278:H1769–1774. doi: 10.1152/ajpheart.2000.278.6.H1769. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto S, Yang G, Zablocki D, Liu J, Hong C, Kim SJ, Soler S, Odashima M, Thaisz J, Yehia G, Molina CA, Yatani A, Vatner DE, Vatner SF, Sadoshima J. Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy. J Clin Invest. 2003;111:1463–1474. doi: 10.1172/JCI17459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morrison LE, Hoover HE, Thuerauf DJ, Glembotski CC. Mimicking phosphorylation of alphaB-crystallin on serine-59 is necessary and sufficient to provide maximal protection of cardiac myocytes from apoptosis. Circ Res. 2003;92:203–211. doi: 10.1161/01.res.0000052989.83995.a5. [DOI] [PubMed] [Google Scholar]
- 13.Kitagawa M, Mukai H, Takahashi M, Ono Y. The role of PKN in the regulation of alphaB-crystallin expression via heat shock transcription factor 1. Biochem Biophys Res Commun. 1998;252:561–565. doi: 10.1006/bbrc.1998.9694. [DOI] [PubMed] [Google Scholar]
- 14.Djabali K, de Nechaud B, Landon F, Portier MM. AlphaB-crystallin interacts with intermediate filaments in response to stress. J Cell Sci. 1997;110 (Pt 21):2759–2769. doi: 10.1242/jcs.110.21.2759. [DOI] [PubMed] [Google Scholar]
- 15.Eaton P, Fuller W, Bell JR, Shattock MJ. AlphaB crystallin translocation and phosphorylation: signal transduction pathways and preconditioning in the isolated rat heart. J Mol Cell Cardiol. 2001;33:1659–1671. doi: 10.1006/jmcc.2001.1418. [DOI] [PubMed] [Google Scholar]
- 16.Koteiche HA, McHaourab HS. Mechanism of chaperone function in small heat-shock proteins. Phosphorylation-induced activation of two-mode binding in alphaB-crystallin. J Biol Chem. 2003;278:10361–10367. doi: 10.1074/jbc.M211851200. [DOI] [PubMed] [Google Scholar]
- 17.Davenport KR, Sohaskey M, Kamada Y, Levin DE, Gustin MC. A second osmosensing signal transduction pathway in yeast. Hypotonic shock activates the PKC1 protein kinase-regulated cell integrity pathway. J Biol Chem. 1995;270:30157–30161. doi: 10.1074/jbc.270.50.30157. [DOI] [PubMed] [Google Scholar]
- 18.Pujol N, Bonet C, Vilella F, Petkova MI, Mozo-Villarias A, de la Torre-Ruiz MA. Two proteins from Saccharomyces cerevisiae: Pfy1 and Pkc1, play a dual role in activating actin polymerization and in increasing cell viability in the adaptive response to oxidative stress. FEMS Yeast Res. 2009;9:1196–1207. doi: 10.1111/j.1567-1364.2009.00565.x. [DOI] [PubMed] [Google Scholar]
- 19.Gerik KJ, Bhimireddy SR, Ryerse JS, Specht CA, Lodge JK. PKC1 is essential for protection against both oxidative and nitrosative stresses, cell integrity, and normal manifestation of virulence factors in the pathogenic fungus Cryptococcus neoformans. Eukaryot Cell. 2008;7:1685–1698. doi: 10.1128/EC.00146-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vandenberg JI, Rees SA, Wright AR, Powell T. Cell swelling and ion transport pathways in cardiac myocytes. Cardiovasc Res. 1996;32:85–97. [PubMed] [Google Scholar]
- 21.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aghajanian A, Wittchen ES, Campbell SL, Burridge K. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PLoS One. 2009;4:e8045. doi: 10.1371/journal.pone.0008045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gopalakrishna R, Anderson WB. Ca2+- and phospholipid-independent activation of protein kinase C by selective oxidative modification of the regulatory domain. Proc Natl Acad Sci U S A. 1989;86:6758–6762. doi: 10.1073/pnas.86.17.6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Budas GR, Churchill EN, Mochly-Rosen D. Cardioprotective mechanisms of PKC isozyme-selective activators and inhibitors in the treatment of ischemia-reperfusion injury. Pharmacol Res. 2007;55:523–536. doi: 10.1016/j.phrs.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 25.Deaton RA, Su C, Valencia TG, Grant SR. Transforming growth factor-beta1-induced expression of smooth muscle marker genes involves activation of PKN and p38 MAPK. J Biol Chem. 2005;280:31172–31181. doi: 10.1074/jbc.M504774200. [DOI] [PubMed] [Google Scholar]
- 26.Mukai H, Toshimori M, Shibata H, Takanaga H, Kitagawa M, Miyahara M, Shimakawa M, Ono Y. Interaction of PKN with alpha-actinin. J Biolo Chem. 1997;272:4740–4746. doi: 10.1074/jbc.272.8.4740. [DOI] [PubMed] [Google Scholar]
- 27.Sah VP, Minamisawa S, Tam SP, Wu TH, Dorn GW, 2nd, Ross J, Jr, Chien KR, Brown JH. Cardiac-specific overexpression of RhoA results in sinus and atrioventricular nodal dysfunction and contractile failure. J Clin Invest. 1999;103:1627–1634. doi: 10.1172/JCI6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rossignol TM, Jones SV. Regulation of a family of inwardly rectifying potassium channels (Kir2) by the m1 muscarinic receptor and the small GTPase Rho. Pflugers Arch. 2006;452:164–174. doi: 10.1007/s00424-005-0014-9. [DOI] [PubMed] [Google Scholar]
- 29.Yatani A, Irie K, Otani T, Abdellatif M, Wei L. RhoA GTPase regulates L-type Ca2+ currents in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2005;288:H650–659. doi: 10.1152/ajpheart.00268.2004. [DOI] [PubMed] [Google Scholar]
- 30.Arrigo AP, Simon S, Gibert B, Kretz-Remy C, Nivon M, Czekalla A, Guillet D, Moulin M, Diaz-Latoud C, Vicart P. Hsp27 (HspB1) and alphaB-crystallin (HspB5) as therapeutic targets. FEBS Lett. 2007;581:3665–3674. doi: 10.1016/j.febslet.2007.04.033. [DOI] [PubMed] [Google Scholar]
- 31.Ray PS, Martin JL, Swanson EA, Otani H, Dillmann WH, Das DK. Transgene overexpression of alphaB crystallin confers simultaneous protection against cardiomyocyte apoptosis and necrosis during myocardial ischemia and reperfusion. FASEB J. 2001;15:393–402. doi: 10.1096/fj.00-0199com. [DOI] [PubMed] [Google Scholar]
- 32.Hoover HE, Thuerauf DJ, Martindale JJ, Glembotski CC. alpha B-crystallin gene induction and phosphorylation by MKK6-activated p38. A potential role for alpha B-crystallin as a target of the p38 branch of the cardiac stress response. J Biol Chem. 2000;275:23825–23833. doi: 10.1074/jbc.M003864200. [DOI] [PubMed] [Google Scholar]
- 33.Singh BN, Rao KS, Ramakrishna T, Rangaraj N, Rao Ch M. Association of alphaB-crystallin, a small heat shock protein, with actin: role in modulating actin filament dynamics in vivo. J Mol Biol. 2007;366:756–767. doi: 10.1016/j.jmb.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 34.Eaton P, Awad WI, Miller JI, Hearse DJ, Shattock MJ. Ischemic preconditioning: a potential role for constitutive low molecular weight stress protein translocation and phosphorylation? J Mol Cell Cardiol. 2000;32:961–971. doi: 10.1006/jmcc.2000.1136. [DOI] [PubMed] [Google Scholar]
- 35.Wang X, Robbins J. Heart failure and protein quality control. Circ Res. 2006;99:1315–1328. doi: 10.1161/01.RES.0000252342.61447.a2. [DOI] [PubMed] [Google Scholar]
- 36.Churchill EN, Ferreira JC, Brum PC, Szweda LI, Mochly-Rosen D. Ischaemic preconditioning improves proteasomal activity and increases the degradation of deltaPKC during reperfusion. Cardiovasc Res. 85:385–394. doi: 10.1093/cvr/cvp334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.