

Abstract
The increasing occurrence of multi-antibiotic resistant microbes has led to the search for alternative methods of killing pathogens and treating infections. Photodynamic therapy (PDT) uses the combination of non-toxic dyes and harmless visible light to produce reactive oxygen species that can kill mammalian and microbial cells. Although the photodynamic inactivation of bacteria has been known for over a hundred years, its use to treat infections has not been much developed. This may be partly due to the difficulty of monitoring the effectiveness of PDT in animal models of infection. In order to facilitate this monitoring process, we have developed a procedure that uses bioluminescent genetically engineered bacteria and a light sensitive imaging system to allow real-time visualization of infections. When these bacteria are treated with PDT in vitro, the loss of luminescence parallels the loss of colony-forming ability. We have developed several models of infections in wounds and soft-tissue abscesses in mice that can be followed by bioluminescence imaging. The size and intensity of the infection can be sequentially monitored in a non-invasive fashion in individual mice in real-time. When photosensitizers are introduced into the infected tissue followed by illumination with red light, a light-dose dependent loss of luminescence is seen. If the bacterium is invasive, the loss of luminescence correlates with increased survival of the mice, whilst animals in control groups die of sepsis within five days. Healing of the PDT treated wounds is not impaired and may actually be improved. This approach can allow many animal models of localized infections to be accurately monitored for efficacy of treatment by PDT.
Keywords: Bioluminescence imaging, Luciferase, Photodynamic therapy, Poly-l-lysine photosensitizer conjugate, Wounds, Soft-tissue infections
1. Photodynamic therapy (PDT) for infections
PDT is a therapy for cancer and other diseases that has received regulatory approvals for several indications in many countries [1]. During PDT, the delivery of visible light of an appropriate wavelength is able to cause certain non-toxic dyes known as photosensitizers (PS) to shift to an excited singlet state. This excited state may then undergo intersystem crossing to the slightly lower energy, but longer-lived triplet state. The latter may then react further by one or both of two pathways. Type I pathway leads to radical ion production, while Type II induces the excited state singlet oxygen formation [2]. Both of these processes are oxygen dependent and lead to highly toxic reactive oxygen species (ROS). These ROS can readily react with biological molecules such as proteins, lipids and nucleic acids, and therefore can lead to the efficient killing of cells [3]. PDT has the advantage over other therapies in that it has dual selectivity: not only is the PS targeted to the diseased tissue, but the light can also be accurately delivered to the affected area. Primarily developed as a cancer therapy, PDT has the potential to be applied to various diseases, including infections [4]. The anti-microbial properties of PDT have been known for about a century and extensive knowledge on the mechanisms of its action has been gained [5,6].
In the 1990s, it was observed that fundamental differences in susceptibility to PDT exist between Gram (+) and Gram (−) bacteria. This was explained by differences in their morphology: the Gram (+) cytoplasmic membrane is surrounded by a layer of only peptidoglycan and lipoteichoic acid that is relatively porous, while Gram (−) bacteria have a somewhat more intricate, non-porous cell wall structure consisting of an inner cytoplasmic membrane and an outer membrane, which are separated by the peptidoglycan-containing periplasm. It was discovered that in general, neutral or anionic PS molecules are efficiently bound to and mediate the photodynamic inactivation (PDI) of Gram (+) bacteria whereas PDI of Gram (− ) bacteria requires the penetration of PS inside the cell [7]. The latter can be achieved by employing several different techniques. It is possible to use agents that are capable of increasing the permeability of the cell outer membrane such as polymyxin B nonapeptide [8], or EDTA [9] together with traditional PS. Alternatively one can use a PS molecule with an intrinsic positive charge [10,11], or polycationic PS conjugates formed from polymers such as polylysine [6,12-14].
Several studies have shown that antibiotic resistant bacteria are as susceptible to PDI as their naïve counterparts [15,16]. The nature of the PDI-induced damage that involves oxidative modification of vital cellular constituents, suggests that bacteria will not easily be able to develop resistance mechanisms and one study has shown that resistance to PDI does not occur [13].
The demonstration of efficient PDI of multiple classes of microorganisms, together with concern about rapidly increasing emergence of antibiotic resistance amongst pathogenic bacteria, has suggested that PDT may be a useful tool to combat infectious disease [4]. Nonetheless there are several limitations. Because the delivery of visible light is almost by definition a localized process, PDT for infections is likely to be applied exclusively to localized disease, as opposed to systemic infections such as bacteremia. The key issues to be addressed with PDT are the effectiveness of the treatment in destroying sufficient numbers of the disease causing pathogens; selectivity of the PS for the microbes, thus avoiding an unacceptable degree of PDT damage to host tissue in the area of infection; and the avoidance of regrowth of the pathogens from a few survivors in the time following the treatment. In vivo studies would provide many answers to these questions. However despite the considerable number of reports on in vitro PDI of microorganisms, in vivo studies on infection models are rare. One of the reasons for this is probably the difficulty in monitoring the development of an infection and its response to treatment. Standard microbiological techniques used to follow infections in animal models frequently involve sacrifice of the animals, removal of the infected tissue, homogenization, serial dilution, plating and colony counting. These assays use a large number of animals, are time consuming, and often are not statistically reliable.
2. Bioluminescence
Various living organisms are able to emit light [17]. The enzymes involved in this process, named luciferases, are oxygenases that utilize molecular oxygen to oxidize a substrate (luciferin), with the formation of a product molecule in an electronically excited state that emits the light. Several different luciferases and substrates from a diversity of marine and terrestrial organisms are known, many with different colors of emission [17]. Three luciferases have been cloned and their chemistries characterized to a point where they can be used routinely in the laboratory. These include the luciferases from firefly (coleoptera) [18], jellyfish and sea pansies (cnidaria) [19], and bacteria (Vibrio spp. and Photorhabdus luminescens) [20]. Representatives of each of these types of luciferases has been used for imaging in living laboratory animals.
The luciferases from firefly, click beetles and rail road worms are a single protein [21] related to the CoA ligase family of enzymes and use a benzothiazole luciferin substrate along with ATP and oxygen to generate light. The gene encoding firefly luciferase has been cloned, and the coding sequence has been optimized for mammalian expression [18]. Cells expressing this enzyme, when provided with luciferin, will emit a yellow-green light with an emission peak at 560 nm using cellular ATP as the energy source.
The luciferases from sea pansy Renilla and jellyfish Aequorea use coelenterazine as the substrate, and unlike the firefly or bacterial luciferases, neither enzyme requires a cellular energy source; the coelenterazine substrate itself provides the necessary energy [19]. The different spectra of light emission of the firefly and coelenterazine utilizing luciferases, and their mutually exclusive substrate specificity, has allowed both reporters to be used in the same cells to monitor the expression of two different tagged genes [22].
Bacterial luciferases are heterodimeric and use oxygen, long-chain fatty aldehydes (e.g., decanal) and reduced flavin mononucleotide (FMNH2) as substrates to produce a blue-green light (emission peak at 490-nm) [17]. In both marine and terrestrial bioluminescent bacteria, a five genes operon (luxCDABE) encodes the luciferase and biosynthetic enzymes (for the synthesis of the aldehyde substrate) necessary for light production. luxA and B encode the alpha and beta subunits of the luciferase, with luxC, D and E encoding proteins for aldehyde production [23]. The lux operon from P. luminescens is ideally suited for the study of pathogens in mammalian animal models as the enzyme retains significant activity at 37 °C [24].
3. Bioluminescence imaging
Optimally, the detection of light from small animals containing bioluminescent cells can be achieved using a CCD based imaging system. These systems consist of a light-tight chamber in which the animal subjects are placed, a sensitive CCD camera to detect the light emitted and a computer controller to acquire the image and allow analysis of the data. Typically, a grayscale reference image of the animal is acquired under weak illumination, and then the bioluminescent signal is captured in complete darkness. This process may take from a few seconds to several minutes, depending on the brightness of the bioluminescent signal, the depth within the tissue from which it arises, and the sensitivity of the detector. The signal intensity is then represented as a pseudocolor image and superimposed on the grayscale reference image. The magnitude of the signal can then be measured from specified regions of the animals using a ‘region of interest’ function.
Different CCD cameras may have different sensitivities for varying wavelengths of light which can be important for in vivo bioluminescence imaging (BLI) since blue and green light (shorter wavelengths) are largely absorbed by tissues, while red light (longer wavelengths) penetrates deeper. The primary absorber in the body in the visible region of the spectrum (400–600 nm) is hemoglobin. At wavelengths of 600 nm and above scattering of photons becomes a more significant attenuation factor than absorption [25]. Other pigments such as melanin in animals with dark skin and fur can also influence the efficiency of BLI. This can be avoided by using hairless or nude animals, or by breeding dark mouse strains into albino backgrounds. The thickness of the tissue is also important: the more tissue between the reporter and the detector, the more light is lost as a result of the combination of absorption and scattering of light. Therefore, the use of smaller animals (e.g., mice and rats) for BLI is preferable.
4. Bioluminescence imaging of infection
The first demonstration of detection and monitoring light emission from small mammals was performed in 1995 using a bioluminescent genetically engineered Gram-negative bacteria expressing the full bacterial lux operon [26]. Although bacterial bioluminescent reporters have been limited to use in prokaryotic systems and emit blue light, they have the advantages of being well-expressed in bacteria and not requiring the exogenous addition of a substrate. The use of BLI for imaging pathogens and infections in animal models has recently been comprehensively reviewed [27].
The luxCDABE operon from P. luminescens can be expressed successfully in a wide range of Gram-negative bacteria, including Escherichia coli and Salmonella spp. [26]. However, for expression in Gram-positive bacteria (such as Staphylococcus aureus) the operon had to be redesigned with Gram-positive ribosome binding sites (Shine–Dalgarno sites) at the start of each gene for optimal expression and bioluminescence at temperatures above 35 °C. The gene order was also changed from luxCDABE to luxABCDE in these constructs [28].
BLI can be used to either track the course of an infection or monitor the efficacy of anti-microbial therapies. Small animals are routinely used to model both human infections and the effects of antibiotics against pathogens. In such studies, groups of animals have typically been infected with the pathogenic organism and, at defined time points, subsets of these are sacrificed, and tissues are excised for determination of pathogen numbers and localization. The effects of anti-microbial drugs on the infection have been determined by measuring changes in this pathogen burden or, alternatively, by the prolonged survival of the animal after a lethal dose of the pathogen. Such approaches have traditionally required large numbers of animals and potentially time consuming processing of animal tissue to locate and quantify the infections. The use of pathogens that have been engineered to express luciferase and imaging of their location and cell number have streamlined these studies and refined these animal models as the necessity to sacrifice the animals to access the data has essentially been eliminated. Bacterial pathogenesis appeared to be unaffected by the presence of the luciferase genes, and bioluminescence can be detected throughout the study period in animals. Further, the intensity of the bioluminescence measured from the living animal correlated well with the bacterial burden subsequently determined by standard protocols [28-30]. Subsequent studies have used transposon-mediated integration of the luciferase operon into the bacterial chromosome to improve stability and to create strains that remain bioluminescent in the absence of drug selection. This means that reduction of luminescence from sites of infection in animals can be attributed to reduction of bacterial numbers rather than loss of plasmids.
5. Materials and methods
We initially used the Gram-negative species E. coli and Pseudomonas aeruginosa that had been transformed by electroporation with a plasmid, pCGSL1, containing the entire P. luminescens lux operon [20], which also confers resistance to ampicillin (carbenicillin) on the bacteria. This plasmid is a ColE1 replicon containing the luxCDABE operon from P. luminescens and an ampicillin resistance marker. For maintenance of luminescent bacteria, ampicillin (100 mg/ml) was included in the growth medium. Because the highest levels of luminescence occur in the exponential phase of growth [20], we used cells from cultures at a density of about 108 cells/ml. We subsequently used the Gram-positive Staphylococcus aureus that had been stably engineered to express bioluminescence. This was done by using a luxABCDE transposon cassette, Tn4001 luxABCDE Km(r) that allows random integration of lux genes onto the bacterial chromosome [30].
We used three systems to measure the emitted bioluminescence of the bacteria. In vitro the light from bacterial suspensions was measured on a tube luminometer (Lumat LB 9507, Berthold Technologies GmbH, Bad Wildbad, Germany) or on a 96-well plate reader (Victor 1420, Perkin–Elmer Life and Analytical Sciences, Shelton, CT). Bioluminescence was imaged using the Hamamatsu photon-counting camera controlled by Argus software. The system consisted of a ICCD camera (model C2400 Hamamatsu Photonics KK, Bridgewater, NJ) fitted with a f1.2 50 mm lens (NIKOR) and mounted in a light-tight specimen chamber (model A4178, Hamamatsu Photonics KK). The light-tight box was fitted with a light-emitting diode allowing a background grayscale image of the entire mouse to be captured with a minimum setting on the image-intensifier control module (model M4134, Hamamatsu Photonics KK). In the photon-counting mode, an image of the emitted light was captured using an integration time of 2 min at a maximum setting on the image intensifier control module. Using ARGUS software (Hamamatsu Photonics KK) the luminescence image was presented as a falsecolor image superimposed on top of the grayscale reference image. The image processing component of the software gave mean pixel values on defined areas within each wound on a 256 grayscale. The software allows the bioluminescence signal to be captured at 11 different “bit ranges” equivalent to exposure settings successively differing by a factor of 2. We captured images at each time point at several bit ranges in order to ensure that we had a set of images at the same bit range for the different time points of the whole experiment in order to make a set composite images in which the bioluminescence signals could be directly compared (even if some images had saturated pixels). However, for the Argus quantification to be accurate it is important to analyze images that contain no saturated pixels hence the need to capture many images at each time point.
For the light source for PDT, we used a 660-nm diode laser providing up to 1W of light through an SMA coupled fiber and lens that could provide a uniform spot ranging from 1 to 3 cm in diameter both in vitro and in vivo experiments.
6. PDI of bioluminescent bacteria
We first ascertained that the luminescence signal measured in a tube luminometer was linearly proportional to bacterial colony forming units (CFU) as determined by serial dilution and plating from 103 to 107 organisms (Fig. 1A). The signal saturated at large bacterial numbers, i.e., >107 . In addition to counting the colonies, it is possible to image the plates using the photon-counting camera, as shown in Fig. 1B.
Fig. 1.
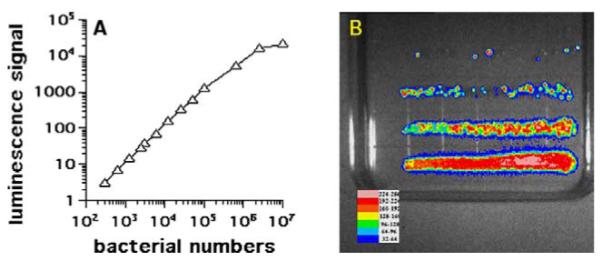
(A) Shows the relationship between luminescence and bacterial number obtained with luminescent E. coli DH5α over four logs of bacterial numbers as measured by tube luminometer. Bacterial CFU were routinely determined by streaking out a set of serial dilutions onto agar plates. (B) Shows an example of bioluminescence imaging of the agar plates.
We used a molecular construct covalently formed between poly-l-lysine chains and the PS chlorin(e6) as a microbial-targeted PS. We have previously shown [6,12,31] that these pL-ce6 conjugates are highly effective in mediating the PDI of both Gram-positive and Gram-negative bacteria. Their positive charges help them to bind to the negatively charged bacteria and their polycationic nature enables them to penetrate the outer membrane of Gram-negative cells by disturbing the structure of the lipopolysaccharide layers. The macromolecular nature of these conjugates gives a temporal selectivity for bacteria over mammalian cells as the latter take them up by the time-dependent process of endocytosis, while they bind rapidly to bacteria. The size of the pL chain was on average 160 lysine residues (range 137–173) and there was an average of 6 ce6 molecules (range 5–7) attached per polypeptide chain.
We carried out in vitro PDI experiments in order to verify that killing the bacteria, as demonstrated by loss of colony forming units, correlated with loss of luminescence. The loss of viability curves as measured by CFU and by loss of luminescence, as a function of applied light-dose, for bacteria incubated for 30-min with 3, 6, 12 and 18 μM pL-ce6 conjugate are shown in Fig. 2A and B. The CFU assay had a limit of sensitivity of six orders of magnitude in reduction of viability, while the bioluminescence assay had a limit of three orders of magnitude. Loss of luminescence showed the same dose–response curves as loss of CFU but the absolute reductions were 1–3 logs less. The reason for this is most likely to be due to the limit of sensitivity of the plate reader, which is only capable of measuring a 3 log reduction in signal, as compared to a 6 log reduction in viability measurable by plating. Nevertheless, it is important to note that luminescent measurements can be achieved in minutes, as opposed to several hours from a combination of overnight incubation followed by laborious and time consuming counting of CFU. The mechanism by which luminescence decreases after PDI is uncertain, but may be due to exhaustion of FMNH2 supplies from the bacteria (needed for the luciferase enzyme to make luminescence) and which cannot be replenished if the cells are fatally damaged.
Fig. 2.

Phototoxicity as determined by A, CFU; and B, luminescence assays. E. coli DH5a bacteria were incubated with stated concentration of pL-ce6 conjugate for 30 min, then washed and illuminated with stated fluence of 660-nm light with removal of aliquots of bacterial suspension at intervals (100, 200 and 400 s, respectively) and serial dilution and plating for CFU counting, or luminescence measurement in 96-well plates using Victor luminescence plate reader. Data points are means of triplicate determinations and two separate experiments and bars are SD.
7. PDT of E. coli infected excisional wounds in mice
All animal experiments were approved by the Sub-committee on Research Animal Care of Massachusetts General Hospital. Mice were anesthetized with a mixture of ketamine and xylazine for all procedures. We initially sought to establish the animal model of infection by inoculating bioluminescent E. coli into an excisional wound on the mouse [32]. Five million CFU from a mid-log culture in 50 μL gave a sufficiently bright luminescence signal from the wound to allow at least two logs of signal reduction to be accurately followed. When this bacterial inoculum was placed into a wound (12.5 × 8 mm) made on the back of a freshly euthanized mouse, the luminescence started to fade rapidly and was totally gone by the time the 50 μL inoculum had dried (<60 min) (data not shown). By contrast infected wounds in living mice showed only a slight loss of luminescence over a period of 4 h. We interpret these findings to mean that the living mouse wound provides nutrients and moisture to the bacteria, and thus is a reasonable model of wound infection. The next day however, control infected wounds in living mice had lost on average 90% of the original luminescence signal, but with considerable inter-animal variability (data not shown).
Since the wound infection with E. coli DH5a was found to be self-limiting, i.e., this particular strain of E. coli is non-invasive [33], it allowed the use of each mouse as its own control to follow wound healing with four wounds per mouse. The effect of topical application of pL-ce6 conjugate and successive applications of 660-nm light is presented in a series of overlaid luminescence (false-color) and grayscale reference images (Fig. 3). These data were obtained from a mouse in which bacteria were inoculated in all wounds, 30 min later conjugate was added to wounds 1 (nearest head) and 4 (nearest tail), and after another 30 min wounds 3 and 4 were illuminated with red light. Therefore, wound 1 was the dark control with conjugate, wound 2 was the absolute control, wound 3 was the light alone control, and wound 4 was PDT treated. The fluence rate was 100 mW/cm2 and mice were imaged after 7.5 min and again after 27.5 min corresponding to the delivery of 45 and 165 J/cm2, respectively. Topical application of a targeted polycationic PS conjugate followed by illumination led to a 99% reduction in luminescence as measured by the imaging software. There was a semi-logarithmic light dose-dependent reduction in luminescence from the PDT treated wound not seen with any of the control wounds, as would be expected from a standard PDT experiment (Fig. 4). There was an initial modest decrease in luminescence from wounds that received conjugate without light due to the dark toxicity of the conjugate, but the luminescence did not decrease further during the course of the experiment.
Fig. 3.

Successive overlaid luminescence false color images and monochrome LED images of a mouse with four excisional wounds infected with equal numbers of E. coli (A). Wounds 1 (nearest tail) and 4 (nearest head) received topical application of conjugate (B). Wounds 1 and 2 (two nearest tail) were then illuminated with successive fluences (45–165 J/cm2) of 660-nm light (C,D).
Fig. 4.
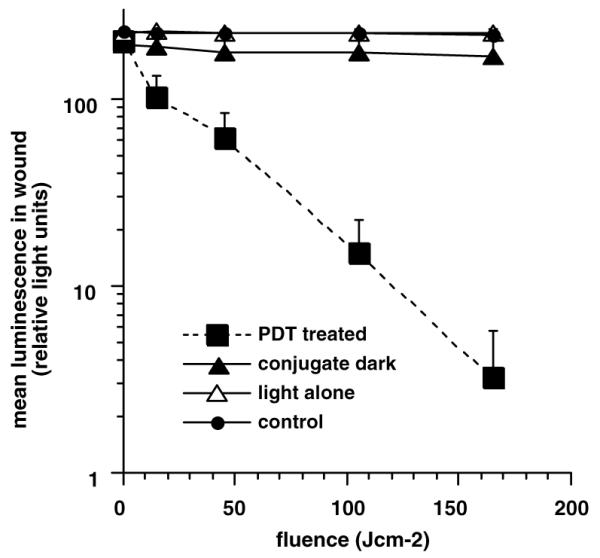
Mean pixel values of luminescence signals from defined areas of wounds measuring 1200 pixels determined by image analysis. Data points are means of values from the corresponding wound on six mice per group and bars are SD.
The assumption that considerable amounts of conjugate bound to the tissue in the wound, suggested that illumination might have caused damage to the host cells, blood vessels or extracellular matrix in the wound. However, we observed that PDT of infected wounds did not lead to any inhibition of wound healing as seen in Fig. 5. There was an indication that the PDT treated wounds actually healed somewhat faster relative to the other control wounds but this was not statistically significant. The lack of host tissue phototoxicity may have been due to the macromolecular pL-ce6 demonstrating temporal selectivity for bacteria compared to mammalian cells as discussed previously. In the present experiments, the absence of wound healing inhibition may be explained by a combination of the topical delivery method together with the large conjugate size and the relatively short incubation time. The fact that treated wounds healed as well as control wounds suggests that PDI may have advantages over topical anti-microbial products that have been reported to cause tissue damage or have other undesirable side-effects.
Fig. 5.
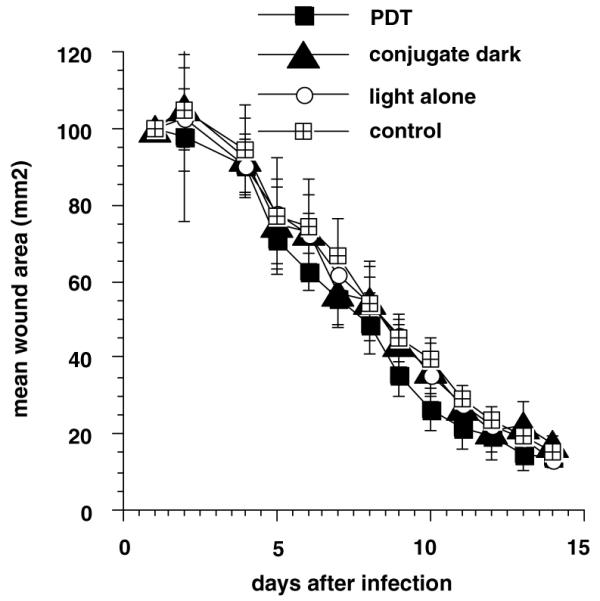
Mean areas of wounds from six mice per group treated as above. Wounds were measured daily in two dimensions and areas calculated. Bars are SD.
8. PDT of lethal P. aeruginosa wound infections in mice
The previous experiments were a proof-of principle study using a relatively non-pathogenic strain of E. coli, DH5α that lacks virulence factors necessary to cause invasive infections [33]. In order to test PDT in a more clinically relevant infection model, we used optical techniques (BLI and targeted PDT) to monitor and cure mice of an otherwise fatal P. aeruginosa wound infection [34]. We used a bioluminescent derivative of strain 180 (ATCC 19660) that has been shown to be invasive and lead to development of fatal sepsis after intraperitoneal injection in rats [35]. Mice with wounds infected with 5 × 106 CFU of bioluminescent P. aeruginosa quickly developed an illness consistent with systemic sepsis. They lost weight, had ruffled coats, and developed progressive inactivity leading to a moribund condition and death occurred between 24 and 60 h after infection. When the effect of varying the initial bacterial inoculum was studied, it was found that the LD50 was approximately 200,000 CFU. Mice infected with the lower numbers of bacteria and that did not die, suffered the same infection as the mice that died, but in a less severe form and recovered between days 6 and 8. We nevertheless decided to use a bacterial challenge for the PDT experiments that was 25 times higher than the LD50 to provide a robust test of the ability of the technology to prevent death from a fatal wound infection.
Mice were given a single dorsal excisional wound with an area of 100 mm2, infected with 5 × 106 CFU of bioluminescent P. aeruginosa suspended in 50 μL of PBS. This inoculum gave a sufficiently bright luminescence signal from the wound to allow at least two logs of signal reduction to be accurately followed. The bacteria quickly attached to the tissue surface of the panniculus carnosus as evidenced by the failure of an attempt to wash them off by irrigation with saline 30-min after infection, and as quantitated by luminescence imaging. The pL-ce6 conjugate was added as 50-μL of a 200-μM ce6 equivalent concentration as preliminary experiments had shown lower concentrations to be less effective. This volume was sufficient to spread evenly throughout the surface of the wound and was retained by the edges of the wound to prevent the liquid running off. It was necessary to give the conjugate at least 30-min to bind to, and penetrate the bacteria in order to see effective loss of luminescence after illumination with 660-nm light.
As can be seen from a set of luminescence images from a representative mouse shown in Fig. 6A–G, PDT produced a fluence-dependent loss of luminescence until only a trace remained after 240 J/cm2 had been delivered (40 min illumination). When the mouse was imaged the next day all traces of luminescence had gone (panel 6H). There was a drop in luminescence seen shortly after applying the conjugate in the dark (Fig. 6B, J), but this did not decrease further after 30 min incubation (Fig. 6C, K) or indeed after 60 min incubation (Fig. 6L), approximately equal to the time for illumination of the PDT wounds. Infected wounds left untreated or treated with illumination alone showed a rise in luminescence signal (up to twofold, Fig. 7A) presumably due to growth of the bacteria in the nutrient rich medium of the wound. There was significant luminescence present in control wounds until death occurred 2–4 days later (Fig. 6M). The mean luminescence values determined from the infections in the wounds of all the mice in the four groups were calculated using the imaging software. The resulting curves are plotted in Fig. 7A. The PDT treated group shows a semi-logarithmic relationship between bacterial luminescence and delivered fluence, until 99% of the luminescence has disappeared after 240 J/cm2. There is a significant difference between the luminescence found from the conjugate in the dark group, compared with that in the light alone and untreated control groups. This is due to two factors; firstly to a degree of dark toxicity of the conjugate towards P. aeruginosa, and secondly to the ability of the bacteria in the untreated and light alone control wounds to continue to grow.
Fig. 6.
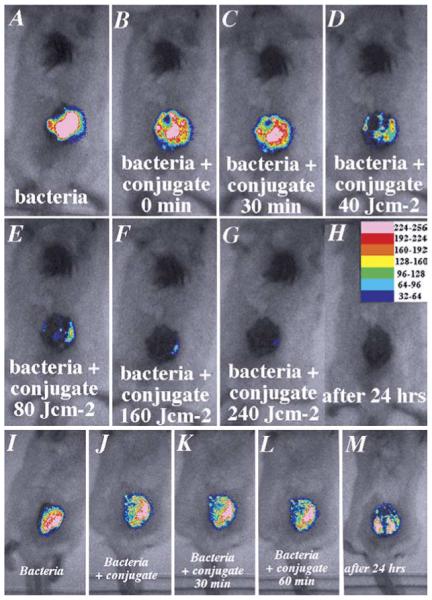
Successive overlaid luminescence false color images and monochrome LED images of mice bearing an excisional wound infected with 5 × 106 luminescent P. aeruginosa. (Panels A–H) A representative mouse treated with pL-ce6 conjugate and increasing doses of light. (Panels I–M) A representative mouse treated with pL-ce6 and kept in the dark.
Fig. 7.

(A) Mean pixel values of bioluminescence signals from defined areas measuring 1200 pixels covering infected wounds determined by image analysis. The four groups comprise untreated control, light alone control, dark conjugate control, and PDT treated. Data points are means of values from the wounds on ten mice per group and bars are SD. (B) Kaplan–Meier survival plot for the four groups of mice described in (A).
All the mice in the three control groups (untreated, light alone and dark conjugate) died within a period of five days infection. By contrast 90% of the mice treated with PDT survived as seen in Fig. 7B. These mice appeared to suffer from some symptoms of bacterial infection (weight loss, ruffled fur and inactivity) similar to those mice who received a sub-lethal dose of bacteria described above. They recovered quickly however, and by five days after infection were regaining weight and moving normally.
9. PDT of established soft-tissue infections in mice
The previous experiments were carried out on animals whose wounds were recently contaminated with relatively large numbers of CFU. It is unlikely that patients would present for treatment under these circumstances. A more realistic and clinically relevant model would consist of inoculation of a smaller number of bacteria and then allowing the infection to grow and become established in tissue over time. A second major consideration was that the previous experiments used an infection model where the bacteria were relatively near the surface of the tissue in an excisional wound. In real life the bacteria could be beneath the surface of the skin or tissue either because they had already invaded or because they were on skin or clothing that had been forcibly introduced into a penetrating wound such as those caused by gunshot. Since the penetration of visible light (even red light) into tissue is limited we asked whether PDT could be used to treat an infection where the bacteria had been allowed to multiply several 100-fold and were some distance beneath the skin surface.
For this experiment, we used a strain of stable bioluminescent S. aureus (mouse pathogenic strain 8325-4), injecting 1 million log-phase CFU into the mouse thigh muscle (2-mm deep) [36]. The mice had previously been rendered temporarily neutropenic by pre-treatment with cyclophosphamide. We used a model in which the mice developed two equivalent infections, one in each hind thigh in order to allow each mouse to have a PDT treated infection and a control untreated infection and act as its own control. Twenty-four hours after infection the bioluminescence had increased dramatically as the bacteria had multiplied within the infected tissue (Fig. 8). The pL-ce6 conjugate (50-μL) at a concentration of 1-mM ce6 equivalent was injected into the infected area resulting in a visible green coloration noticeable beneath the skin. This allowed a judgement to be made about the uniformity of the PS distribution within the infection. Although these mice were neutropenic and the infection did not accumulate the considerable quantities of pus expected from immunocompetent mice, there was still some matter present in the infection. We had previously suspected that the conjugate might diffuse relatively rapidly through the tissue, however this did not prove to be the case. The green coloration remained in place for some time (several hours) especially in unilluminated mice. Light (660-nm) was delivered to the infection as a spot on the skin about 8-mm in diameter centered on the infected area.
Fig. 8.

Series of bioluminescence images (captured at a bit range of 2–4) from a neutropenic mouse infected on day 1 in both left and right hind thighs, and treated on day 2 with injection of pL-ce6 into the right thigh, followed after 30 min by illumination of the right thigh with 660-nm light at a fluence rate of 100 mW/cm2.
There was a slight reduction in bacterial bioluminescence observed immediately after the conjugate was injected into the infection. Luminescence was further reduced after the 30-min incubation period in the dark. When illumination was commenced there was a light-dose dependent decrease in luminescence after each 40 J/cm2 increment of red light (Fig. 8). A typical mouse treated with injection of conjugate into the right infected thigh, followed by illumination of this thigh as described previously is shown in Fig. 8, together with the mean bioluminescence values from both legs of five mice in Fig. 9. After 160 J/cm2 had been delivered the bioluminescence of the treated infected legs had been reduced by >99% compared to the untreated contralateral legs. However, four out of five of these treated legs suffered a recurrence of the bioluminescence on succeeding days (data not shown).
Fig. 9.
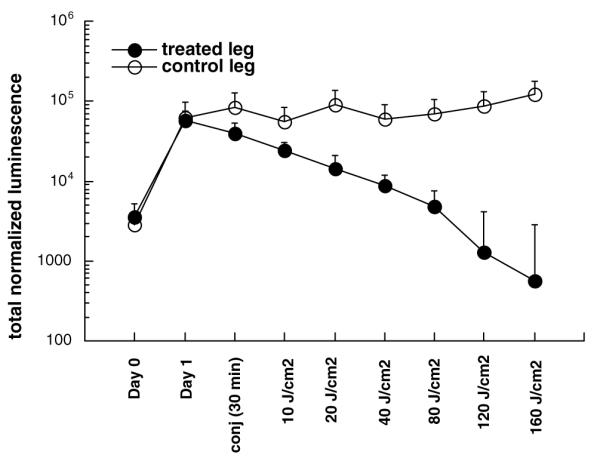
Mean total normalized bioluminescence values from left (untreated) and right (PDT treated) thighs of five mice infected in both thighs. Bars are SEM.
10. Conclusions and future work
The use of a polycationic conjugate between the PS ce6 and a poly-l-lysine chain delivers the PS in an efficient manner to bacteria in infected wounds and tissue. Moreover, the delivery of light a short time after the PS gives temporal selectivity for the bacteria over the host cells as the conjugate binds quickly to bacteria but only slowly to host cells. The use of stably transduced bioluminescent bacteria together with a low-light imaging camera provides an efficient and versatile method of monitoring the progress of the infection in experimental animals in real time and allows response to therapy to be followed in a longitudinal fashion. Due to the wide-spread occurrence of antibiotic resistance amongst pathogenic bacteria and the relatively slow response to antibiotics when bacteria are infecting traumatized tissue, alternative methods of killing bacteria in wounds are being sought. The recent development of portable and low-cost light sources such as light emitting diodes that are battery-powered may make possible the deployment of systems that can be used for wound decontamination. Our data demonstrating that PDT can also be used for established infections further extends its possible applications.
In future work, we are studying PDT for infections in thermal and chemical burns and in other medically and militarily important localized infections. These include fungal and parasitic infections of the skin (dermatophytosis and Leishmaniasis), bacterial keratitis, sinusitis, periodontitis, gastric Helicobacter pylori infection, and bacterial cystitis. The local delivery of the PS and the ability of fiber optics to deliver light anywhere within the body suggest that PDT can treat infections with many hollow organs. We are also working on second generation PS-conjugates with even higher activities and selectivities for bacteria than the pL-ce6 conjugates described.
Acknowledgments
This work was supported by the US NIH/NIAID (Grant AI050875to MRH) and by the US Air Force MFEL Program (Contract FA9550-04-1-0079). The authors are grateful to Dr. Christopher H. Contag who developed the technique of bioluminescence imaging, and Xenogen Corp. for the generous gift of bioluminescent organisms. We thank David A. O’Donnell for technical assistance and Dr. Tayyaba Hasan for support.
11. Abbreviations
- ATP
adenosine triphosphate
- BLI
bioluminescence imaging
- CCD
charge coupled device
- CFU
colony forming units
- FMNH2
reduced flavin mononucleotide
- PDI
photodynamic inactivation
- PDT
photodynamic therapy
- pL-ce6
poly-l-lysine chlorin(e6) conjugate
- PS
photosensitizer
- ROS
reactive oxygen species
References
- [1].Dougherty TJ. An update on photodynamic therapy applications. J. Clin. Laser Med. Surg. 2002;20:3–7. doi: 10.1089/104454702753474931. [DOI] [PubMed] [Google Scholar]
- [2].Ochsner M. Photophysical and photobiological processes in the photodynamic therapy of tumours. J. Photochem. Photobiol. B. 1997;39:1–18. doi: 10.1016/s1011-1344(96)07428-3. [DOI] [PubMed] [Google Scholar]
- [3].Dolmans DE, Fukumura D, Jain RK. Photodynamic therapy for cancer. Nat. Rev. Cancer. 2003;3:380–387. doi: 10.1038/nrc1071. [DOI] [PubMed] [Google Scholar]
- [4].Hamblin MR, Hasan T. Photodynamic therapy: a new antimicrobial approach to infectious disease? Photochem. Photobiol. Sci. 2004;3:436–450. doi: 10.1039/b311900a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wainwright M. Photodynamic antimicrobial chemotherapy (PACT) J. Antimicrob. Chemother. 1998;42:13–28. doi: 10.1093/jac/42.1.13. [DOI] [PubMed] [Google Scholar]
- [6].Hamblin MR, O’Donnell DA, Murthy N, Rajagopalan K, Michaud N, Sherwood ME, Hasan T. Polycationic photosensitizer conjugates: effects of chain length and Gram classification on the photodynamic inactivation of bacteria. J. Antimicrob. Chemother. 2002;49:941–951. doi: 10.1093/jac/dkf053. [DOI] [PubMed] [Google Scholar]
- [7].Malik Z, Ladan H, Nitzan Y. Photodynamic inactivation of Gram-negative bacteria: problems and possible solutions. J. Photochem. Photobiol. B. 1992;14:262–266. doi: 10.1016/1011-1344(92)85104-3. [DOI] [PubMed] [Google Scholar]
- [8].Nitzan Y, Gutterman M, Malik Z, Ehrenberg B. Inactivation of gram-negative bacteria by photosensitized porphyrins. Photochem. Photobiol. 1992;55:89–96. doi: 10.1111/j.1751-1097.1992.tb04213.x. [DOI] [PubMed] [Google Scholar]
- [9].Bertoloni G, Rossi F, Valduga G, Jori G, van Lier J. Photosensitizing activity of water- and lipid-soluble phthalocyanines on Escherichia coli. FEMS Microbiol. Lett. 1990;59:149–155. doi: 10.1111/j.1574-6968.1990.tb03814.x. [DOI] [PubMed] [Google Scholar]
- [10].Merchat M, Bertolini G, Giacomini P, Villanueva A, Jori G. Meso-substituted cationic porphyrins as efficient photosensitizers of gram-positive and gram-negative bacteria. J. Photochem. Photobiol. B. 1996;32:153–157. doi: 10.1016/1011-1344(95)07147-4. [DOI] [PubMed] [Google Scholar]
- [11].Minnock A, Vernon DI, Schofield J, Griffiths J, Parish JH, Brown SB. Mechanism of uptake of a cationic water-soluble pyridinium zinc phthalocyanine across the outer membrane of Escherichia coli. Antimicrob. Agents Chemother. 2000;44:522–527. doi: 10.1128/aac.44.3.522-527.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Soukos NS, Ximenez-Fyvie LA, Hamblin MR, Socransky SS, Hasan T. Targeted antimicrobial photochemotherapy. Antimicrob. Agents Chemother. 1998;42:2595–2601. doi: 10.1128/aac.42.10.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lauro FM, Pretto P, Covolo L, Jori G, Bertoloni G. Photoinactivation of bacterial strains involved in periodontal diseases sensitized by porphycene–polylysine conjugates. Photochem. Photobiol. Sci. 2002;1:468–470. doi: 10.1039/b200977c. [DOI] [PubMed] [Google Scholar]
- [14].Rovaldi CR, Pievsky A, Sole NA, Friden PM, Rothstein DM, Spacciapoli P. Photoactive porphyrin derivative with broad-spectrum activity against oral pathogens In vitro. Antimicrob. Agents Chemother. 2000;44:3364–3367. doi: 10.1128/aac.44.12.3364-3367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wilson M, Yianni C. Killing of methicillin-resistant Staphylococcus aureus by low-power laser light. J. Med. Microbiol. 1995;42:62–66. doi: 10.1099/00222615-42-1-62. [DOI] [PubMed] [Google Scholar]
- [16].Wainwright M, Phoenix DA, Laycock SL, Wareing DR, Wright PA. Photobactericidal activity of phenothiazinium dyes against methicillin-resistant strains of Staphylococcus aureus. FEMS Microbiol. Lett. 1998;160:177–181. doi: 10.1111/j.1574-6968.1998.tb12908.x. [DOI] [PubMed] [Google Scholar]
- [17].Hastings JW. Chemistries and colors of bioluminescent reactions: a review. Gene. 1996;173:5–11. doi: 10.1016/0378-1119(95)00676-1. [DOI] [PubMed] [Google Scholar]
- [18].de Wet JR, Wood KV, DeLuca M, Helinski DR, Subramani S. Firefly luciferase gene: structure and expression in mammalian cells. Mol. Cell. Biol. 1987;7:725–737. doi: 10.1128/mcb.7.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hart RC, Matthews JC, Hori K, Cormier MJ. Renilla reniformis bioluminescence: luciferase-catalyzed production of nonradiating excited states from luciferin analogues and elucidation of the excited state species involved in energy transfer to Renilla green fluorescent protein. Biochemistry. 1979;18:2204–2210. doi: 10.1021/bi00578a011. [DOI] [PubMed] [Google Scholar]
- [20].Frackman S, Anhalt M, Nealson KH. Cloning, organization, and expression of the bioluminescence genes of Xenorhabdus luminescens. J. Bacteriol. 1990;172:5767–5773. doi: 10.1128/jb.172.10.5767-5773.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Viviani VR. The origin, diversity, and structure function relationships of insect luciferases. Cell. Mol. Life Sci. 2002;59:1833–1850. doi: 10.1007/PL00012509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Parsons SJ, Rhodes SA, Connor HE, Rees S, Brown J, Giles H. Use of a dual firefly and Renilla luciferase reporter gene assay to simultaneously determine drug selectivity at human corticotrophin releasing hormone 1 and 2 receptors. Anal. Biochem. 2000;281:187–192. doi: 10.1006/abio.2000.4570. [DOI] [PubMed] [Google Scholar]
- [23].Meighen EA. Molecular biology of bacterial bioluminescence. Microbiol. Rev. 1991;55:123–142. doi: 10.1128/mr.55.1.123-142.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Meighen EA. Bacterial bioluminescence: organization, regulation, and application of the lux genes. FASEB J. 1993;7:1016–1022. doi: 10.1096/fasebj.7.11.8370470. [DOI] [PubMed] [Google Scholar]
- [25].Weissleder R, Ntziachristos V. Shedding light onto live molecular targets. Nat. Med. 2003;9:123–128. doi: 10.1038/nm0103-123. [DOI] [PubMed] [Google Scholar]
- [26].Contag CH, Contag PR, Mullins JI, Spilman SD, Stevenson DK, Benaron DA. Photonic detection of bacterial pathogens in living hosts. Mol. Microbiol. 1995;18:593–603. doi: 10.1111/j.1365-2958.1995.mmi_18040593.x. [DOI] [PubMed] [Google Scholar]
- [27].Doyle TC, Burns SM, Contag CH. In vivo bioluminescence imaging for integrated studies of infection. Cell. Microbiol. 2004;6:303–317. doi: 10.1111/j.1462-5822.2004.00378.x. [DOI] [PubMed] [Google Scholar]
- [28].Francis KP, Joh D, Bellinger-Kawahara C, Hawkinson MJ, Purchio TF, Contag PR. Monitoring bioluminescent Staphylococcus aureus infections in living mice using a novel luxABCDE construct. Infect. Immun. 2000;68:3594–3600. doi: 10.1128/iai.68.6.3594-3600.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Rocchetta HL, Boylan CJ, Foley JW, Iversen PW, LeTourneau DL, McMillian CL, Contag PR, Jenkins DE, Parr TR., Jr. Validation of a noninvasive, real-time imaging technology using bioluminescent Escherichia coli in the neutropenic mouse thigh model of infection. Antimicrob. Agents Chemother. 2001;45:129–137. doi: 10.1128/AAC.45.1.129-137.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Francis KP, Yu J, Bellinger-Kawahara C, Joh D, Hawkinson MJ, Xiao G, Purchio TF, Caparon MG, Lipsitch M, Contag PR. Visualizing pneumococcal infections in the lungs of live mice using bioluminescent Streptococcus pneumoniae transformed with a novel gram-positive lux transposon. Infect. Immun. 2001;69:3350–3358. doi: 10.1128/IAI.69.5.3350-3358.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gad F, Zahra T, Hasan T, Hamblin MR. Effects of growth phase and extracellular slime on photodynamic inactivation of gram-positive pathogenic bacteria. Antimicrob. Agents Chemother. 2004;48:2173–2178. doi: 10.1128/AAC.48.6.2173-2178.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hamblin MR, O’Donnell DA, Murthy N, Contag CH, Hasan T. Rapid control of wound infections by targeted photodynamic therapy monitored by in vivo bioluminescence imaging. Photochem. Photobiol. 2002;75:51–57. doi: 10.1562/0031-8655(2002)075<0051:rcowib>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- [33].Busch NA, Zanzot EM, Loiselle PM, Carter EA, Allaire JE, Yarmush ML, Warren HS. A model of infected burn wounds using Escherichia coli O18:K1:H7 for the study of gram-negative bacteremia and sepsis. Infect. Immun. 2000;68:3349–3351. doi: 10.1128/iai.68.6.3349-3351.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hamblin MR, Zahra T, Contag CH, McManus AT, Hasan T. Optical monitoring and treatment of potentially lethal wound infections in vivo. J. Infect. Dis. 2003;187:1717–1726. doi: 10.1086/375244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Milligan RC, Rust J, Rosenthal SM. Gamma globulin factors protective against infections from Pseudomonas and other organisms. Science. 1957;126:509–511. doi: 10.1126/science.126.3272.509-a. [DOI] [PubMed] [Google Scholar]
- [36].Gad F, Zahra T, Francis KP, Hasan T, Hamblin MR. Targeted photodynamic therapy of established soft-tissue infections in mice. Photochem. Photobiol. Sci. 2004;3:451–458. doi: 10.1039/b311901g. [DOI] [PMC free article] [PubMed] [Google Scholar]