

Abstract
Coronary artery disease in patients with hypertension is increasing worldwide and leads to severe cardiovascular complications. The cellular and molecular mechanisms that underlie this pathologic condition are not well understood. Experimental and clinical research indicates that immune cells and inflammation play a central role in the pathogenesis of cardiovascular diseases. Recently, it has been reported that CD4+CD25+ regulatory T cells (Tregs) regulate heart fibrosis in hypertension. In this study, we determined the role of Tregs in coronary arteriolar endothelial dysfunction in angiotensin II–dependent hypertensive mice. Mice infused with angiotensin II had significantly increased blood pressure, as determined using telemetry, and apoptotic Treg numbers, as measured using flow cytometry. The mice displayed inflammation, assessed by macrophage activation/infiltration into coronary arterioles and the heart, and increased local tumor necrosis factor-α release, which participates in reduced coronary arteriolar endothelial-dependent relaxation in response to acetylcholine using an arteriograph. Hypertensive mice injected with Tregs isolated from control mice had significantly reduced macrophage activation and infiltration, reduced tumor necrosis factor-α release, and improved coronary arteriolar endothelium-dependent relaxation. Our novel data indicate that Tregs are important in the development of coronary arteriolar endothelial dysfunction in hypertension. These results suggest a new direction in the investigation of vascular disease in hypertension and could lead to a therapeutic strategy that involves immune system modulation using Tregs.
Hypertension is still a major cause of morbidity and mortality in the world. Endothelial dysfunction is considered an early indicator of cardiovascular risk factors, such as hypercholesterolemia, diabetes mellitus, smoking, and hypertension. We and other investigators demonstrated endothelial dysfunction in mesenteric resistance arteries1–4 and coronary arterioles in hypertensive (HT) animal models5 caused by a reduction in the relaxing factors nitric oxide, prostaglandin I2, and endothelial-derived hyperpolarizing factor. Recent evidence highlights the involvement of inflammation as a critical pathologic factor in cardiovascular diseases.6–8 In addition, an increasing body of data suggest that an imbalance in the immune system triggers inflammation, and this could represent an important and highly promising new direction for modifying cardiovascular pathophysiologic effects.9–12 Therefore, it is likely that microvascular endothelial dysfunction in hypertension could be the result of perturbation in the immune system and inflammation.
Regulatory T cells (Tregs), generally characterized by the expression of CD4, CD25, and Foxp3, develop in the thymus, identify specific self-antigen, and play a critical role in maintaining self-tolerance and protection from autoimmune diseases13–16 and in affecting immune responses to different pathogens. This leads to new insights into mechanisms of tolerance breakdown in human diseases, including those resulting from allergic, autoimmune, or infectious causes.17,18 Experimental studies and successful clinical trials advanced the concept that Tregs are important as a therapy for type 1 diabetes mellitus.
It has been shown that stimulation of lymphocytes by angiotensin II (Ang II) may contribute to the pathogenesis of hypertensive kidney disease,19,20 suggesting a critical role for the immune system in the pathogenesis of hypertension. A recent study indicates that Tregs play a key role in the regulation of arterial wall disease, especially atherosclerosis. Thus, increased Treg numbers have been shown to be powerful inhibitors of atherosclerosis in several mouse models.12 Recently, it has been reported that T cells play an important role in the genesis of hypertension.21,22 Mice lacking T and B cells (Rag1−/− mice) do not develop hypertension and abnormalities of vascular function during Ang II infusion.21 Note that there are fundamental differences in function and structure between large and small arteries.23 Recently, it has been reported that Tregs reverse hypertension-induced heart damage.24 Thus, the overall goal of our study is to elucidate a new pathway for the pathogenesis of hypertension and coronary arteriolar endothelial dysfunction involving Tregs and inflammatory processes that are macrophage dependent.
Materials and Methods
Animals and Telemetry
All the protocols were approved by the Tulane University Health Sciences Center Animal Care and Use Committee. Mice (C57BL/6J, 8-week-old males) were purchased from The Jackson Laboratory (Bar Harbor, ME) and were maintained in a temperature-controlled room on a 12:12-hour light-dark cycle with free access to food (sodium ion content, 0.4%) and water.
Mice were subjected to isoflurane anesthesia, and a catheter connected to a radiotelemetry device was inserted into the left carotid artery to monitor blood pressure using telemetry in conscious, unrestrained mice (model PA-C10; Data Sciences International, St. Paul, MN).25,26 After a 5-day recovery phase, baseline mean arterial pressure levels were measured, and the animals were again subjected to general anesthesia for osmotic mini-pump implantation. Data were collected, stored, and analyzed using Dataquest A.R.T 4.0 software (Data Sciences International). Mice were infused with Ang II (400 ng/kg/min) through subcutaneous mini-osmotic pumps and then received, through i.p. injection, isolated and purified Tregs (200,000 cells) from control mice three times a week for 2 weeks. On day 14, the animals were given isoflurane, and the spleen, macrophages, and heart were collected.
Coronary Arteriolar Function
Septal arterioles (60 to 80 μm in diameter) were isolated, and endothelial function was measured as previously described.27,28 Coronary arteriolar endothelium-dependent relaxation was investigated in sham mice ± Tregs and in HT mice ± Tregs or CD4− T cells. Endothelium-dependent relaxation was also determined after the inhibition of endothelial nitric oxide synthase (eNOS) with 100 μmol/L NG-nitro-L-arginine-methyl ester (L-NAME).
Isolated and Cultured Tregs
In Vivo Studies
Six C57Bl/6 mice were sacrificed, and the spleens were isolated, dissected, and washed with cold and sterile PBS. The spleens were cut into small pieces and were strained using a 40-μm cell strainer (BD Falcon, Fisher Scientific, Pittsburgh, PA). A single cell suspension from all the mice was prepared and then incubated with 0.85% ammonium chloride–Tris-hydrochloride buffer to destroy red blood cells. The cells were washed and then incubated for 30 minutes with antimouse CD4/L3T4–fluorescein isothiocyanate, antimouse CD25/interleukin-2 (IL-2) receptor α–phosphatidylethanolamine and using rat IgG2b–phosphatidylethanolamine as a positive clone (all from Beckman Coulter Inc., Brea, CA). Treg-stained cells were washed with sterile PBS and were passed through a Becton Dickinson (Mountain View, CA) FACScan flow cytometer. Data were analyzed using Cell Quest software (Becton Dickinson). Suspensions of 200,000 Tregs from eight C57/Bl6 mice were injected three times into HT mice in the first week. In the second week, Tregs were isolated from another group of eight C57/Bl6 mice and then were injected into HT mice (three times a week).
In Vitro Studies
Tregs were isolated from cell suspensions of spleens from eight C57Bl/6 mice in two-step procedures. Briefly, CD4+ T cells were pre-enriched by depletion of unwanted cells, and then CD25+ cells were positively selected from the enriched CD4+ T-cell fraction using a magnetic separation column fitted between a MACS magnet (Miltenyi Biotech, Sunnyvale, CA). The purity of CD4+CD25+ Treg subsets was determined using a fluorescence-activated cell sorter (FACS) and was found to be greater than 96%. Six-well plates were precoated with anti-mCD3 (clone 2C11; R&D Systems, Minneapolis, MN), and the cells were grown in RPMI 1640 medium with 10% fetal bovine serum, 2 mmol/L glutamine, 10 mmol/L HEPES, 100 U/ml of penicillin, 100 μg/ml of streptomycin (all from Gibco, Invitrogen, Carlsbad, CA), 50 μg of 2-mercaptoethanol (Sigma-Aldrich, St. Louis, MO), and 10 ng/ml of human recombinant IL-2 (R&D Systems). Cells were starved overnight and then were treated for 24 hours with Ang II and Ang II + candesartan (all from Sigma-Aldrich). The cells were then harvested and subjected to FACS analysis to determine the apoptotic number of Tregs. Apoptotic Treg levels were determined using antibodies against fluorochrome-labeled annexin V to detect exposed phosphatidylserine.
IL-10 Level Measurement
IL-10 measurement was determined in plasma from all groups according to the manufacturer's mouse cytokines (Bio-Plex Assay; Bio-Rad Laboratories, Hercules, CA).
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick-End Labeling Assay
Briefly, Tregs were stimulated with or without Ang II in the presence or absence of candesartan. DNA fragmentation was measured using the DeadEnd colorimetric apoptosis detection system according to the manufacturer's instructions (Promega Corp., Madison, WI).
Macrophages
Macrophages were isolated from the peritoneal cavity of mice using a syringe-gauge device and then were subjected to Western blot analysis.
Immunostaining
The staining protocol was performed using an Autostainer Plus (Dako, Carpenteria, CA). Immunostaining was performed as previously described.27 The antibodies used were Foxp3-specific antibodies (a marker of Tregs) and CD68-specific antibodies (a marker of macrophages).
Western Blot Analysis
Western blot analysis was performed as previously described.27,29 Isolated coronary arterioles were sonicated and then subjected to centrifugation for 10 minutes (10,000 × g). The supernatant was collected and subjected to a standard procedure for Western blot analysis.
Statistical Analysis
Results are expressed as mean ± SEM, where n is the number of arterial segments and cultured cells studied. The significance of the differences between groups was determined using one-repeated or two-factor analysis of variance, where appropriate. Differences were considered significant at P < 0.05.
Results
Eight- to 10-week-old male C57/bl6 mice were randomized into three groups: i) normotensive (sham) mice, ii) HT mice infused with HT doses of Ang II (400 ng/kg/min) for 2 weeks, and iii) HT mice injected with CD4+CD25+ Tregs. To assess hypertension, blood pressure was measured using telemetry (Figure 1). Animals infused with Ang II displayed elevated blood pressure compared with the sham group (n = 5 to 7). Blood pressure was significantly reduced in mice receiving Ang II + Tregs (Figure 1).
Figure 1.
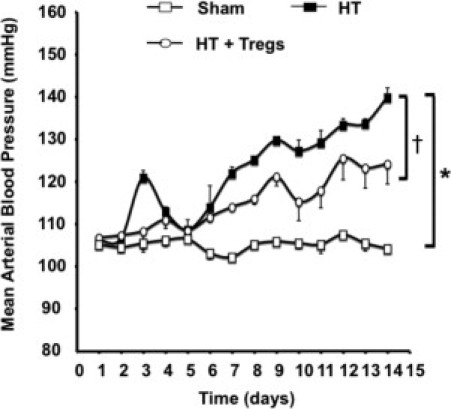
Effect of long-term Ang II (400 ng/kg/min) infusion with or without Tregs (200,000 cells) on mean arterial blood pressure as determined by means of telemetry. *P < 0.05, sham versus HT mice; †P < 0.05, sham versus HT + Tregs mice.
To determine the reduction in the number of Tregs in hypertension, we counted isolated and purified Tregs from the spleens of sham and HT mice using flow cytometry. Our data showed a reduction in the number of Tregs apoptosis-dependent mechanism in HT compared with sham mice (Figure 2A). The number of Tregs was similar in sham mice and in mice receiving Ang II + Tregs (Figure 2A). To expand our data, we isolated, purified, and cultured Tregs from the spleen of sham mice and then stimulated the cells with Ang II (100 nmol/L) for 24 hours. The results revealed an increase in apoptotic Tregs stimulated with Ang II compared with nonstimulated cells assessed using flow cytometry (Figure 2B). The increase in the number of apoptotic Tregs in response to Ang II was prevented when Tregs were pretreated with the AT-1 receptor antagonist (candesartan, 10 μmol/L). Figure 2C illustrates the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling assay and quantitative data for Tregs in response to Ang II with or without candesartan. The data revealed an increase in apoptotic Treg numbers in response to Ang II, which was normalized when the cells were pretreated with candesartan (Figure 2C).
Figure 2.
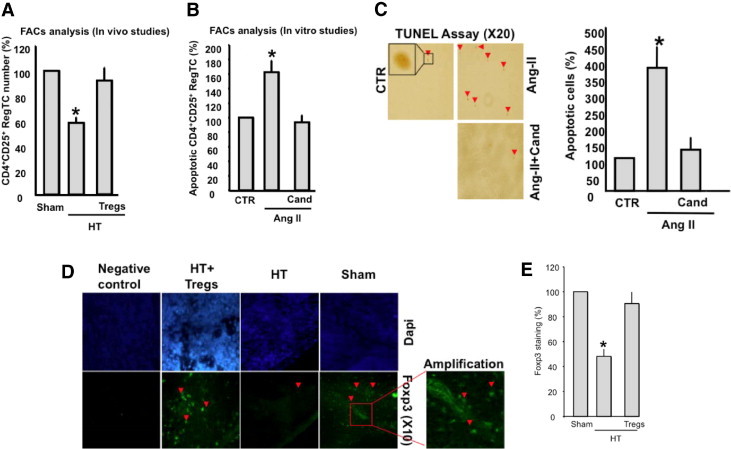
A:In vivo number of Tregs in sham, HT, and HT + Tregs mice as determined using FACS analysis (n = 5). *P < 0.05, sham versus HT or HT + Tregs mice. B:In vitro apoptotic Tregs as determined by means of FACS analysis. Isolated, purified, and cultured Tregs from sham mice were then stimulated with Ang II for 24 hours with or without candesartan (Cand; 10 μmol/L) (n = 6). *P < 0.05, Ang II versus CTR or Ang II + Cand. C:In vitro terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling assay and cumulative data indicating apoptotic Tregs under CTR, Ang II, or Cand (10 μmol/L) conditions. *P < 0.05, Ang II versus CTR or Ang II + Cand. D, E: Illustrates in vivo Foxp3 staining, and cumulative data, in green with DAPI nuclear counterstaining in blue of the spleen from sham, HT, and HT + Tregs mice (n = 5); negative control represents staining using only secondary antibody (n = 3). CTR, control.
Figure 2D illustrates Foxp3 staining in green with 4-6-diamidino-2-phenylindole nuclear counterstaining in blue in the spleen from sham, HT, and HT + Tregs animals. Foxp3 (a marker of Treg positive) staining was significantly reduced in the spleen from HT mice compared with sham mice. HT mice injected with Tregs had significantly enhanced Foxp3 staining compared with HT animals (Figure 2, D and E). The staining was absent in the negative control, in which we used only secondary antibody, indicating the specificity of the staining for Foxp3 (Figure 2D). Figure 3A shows apoptotic Tregs in the spleen assessed by the ratio of active caspase-3 to Foxp3 expression. The data revealed an increase in apoptotic Treg numbers in HT mice, which was significantly reduced in sham mice and in HT mice injected with Tregs (Figure 3A). Secreted anti-inflammatory IL-10 from Tregs was measured in the plasma from all groups. The data revealed a significant reduction in IL-10 release in HT mice compared with sham mice and HT mice injected with Tregs (Figure 3B). Coronary arterioles from HT mice displayed a significant increase in the expression of adhesion molecules (intercellular adhesion molecule-1 [ICAM-1] and vascular cell adhesion molecule-1 [VCAM-1]) compared with sham and HT + Tregs mice (Figure 3, C and D).
Figure 3.
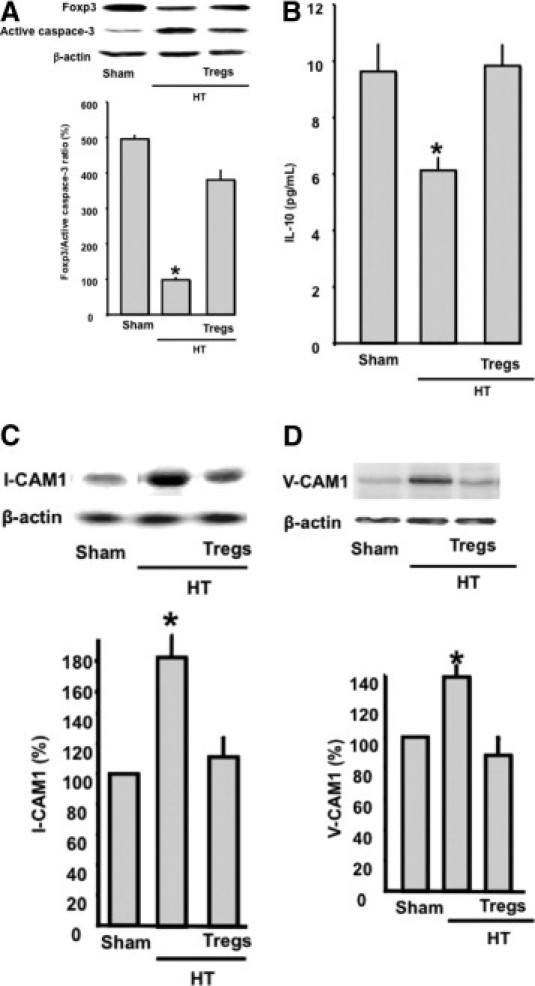
A: Western blot analysis and cumulative data of the ratio of Foxp3 to active caspase-3 expression in spleens from sham, HT, and HT + Tregs mice. β-Actin indicates the equal loading between samples (n = 6). *P < 0.05, HT versus sham or HT + Tregs mice. B: IL-10 measurement in the plasma from sham, HT, and HT + Tregs mice (n = 6). *P < 0.05, HT versus sham or HT + Tregs mice. C, D: Western blot analysis and quantitative data of ICAM-1 and VCAM-1 in coronary arterioles from sham, HT, and HT + Tregs mice. β-Actin indicates the equal loading between samples (n = 6). *P < 0.05, HT versus sham or HT + Tregs mice.
To determine the involvement of macrophages, we isolated macrophages from sham, HT, and HT + Tregs mice and then subjected them to Western blot analysis for adhesion molecule induction (ICAM-1 and VCAM-1). The data revealed an increase in ICAM-1 and VCAM-1 expression in macrophages from HT mice compared with sham and HT + Tregs mice (Figure 4, A and B). Immunostaining of the heart from sham, HT, and HT + Tregs mice revealed an increase in macrophage infiltration into the coronary arterioles and the heart of HT mice compared with sham and HT + Tregs mice (Figure 4C). A negative control (without primary antibody) was performed, and no staining was detected (data not shown). We measured tumor necrosis factor (TNF) α in the heart using Western blot analysis, and the data revealed an increase in TNF-α in HT mice compared with sham and HT + Tregs mice (Figure 4D). Expression of the chemokine receptor CCR5 in coronary arterioles was significantly increased in HT mice compared with sham and HT + Tregs mice (Figure 4E).
Figure 4.
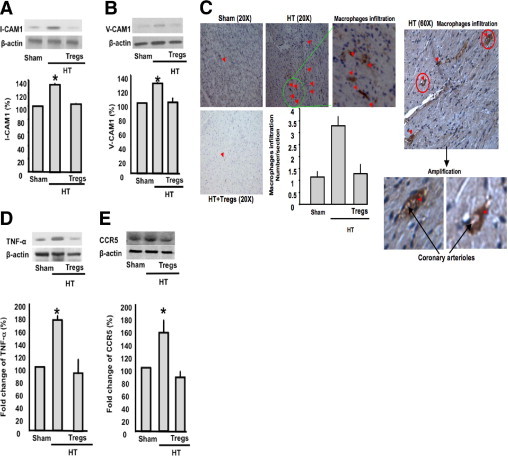
A: Western blot analysis and cumulative data of ICAM-1 in macrophages from sham, HT, and HT + Tregs mice. β-Actin indicates the equal loading between samples (n = 6). *P < 0.05, HT versus sham or HT + Tregs mice. B: Western blot analysis and quantitative data of VCAM-1 in macrophages from sham, HT, and HT + Tregs mice. β-Actin indicates the equal loading between samples (n = 5). *P < 0.05, HT versus sham or HT + Tregs mice. C: Staining showing macrophage (using CD68 antibody) infiltration into coronary arterioles and the heart in sham, HT, and HT + Tregs mice. All red points are macrophage infiltration. D: Western blot analysis and cumulative data of TNF-α in the heart from sham, HT, and HT + Tregs mice. β-Actin indicates the equal loading between samples (n = 6). *P < 0.05, HT versus sham or HT + Tregs mice. E: Western blot analysis and cumulative data indicating the expression of the chemokine receptor CCR5 in coronary arterioles from sham, HT, and HT + Tregs mice. β-Actin indicates the equal loading between samples (n = 4). *P < 0.05, HT versus sham or HT + Tregs mice.
Endothelial-dependent relaxation in response to acetylcholine was significantly reduced in coronary arterioles from HT mice compared with sham mice (Figure 5A). Inhibition of nitric oxide synthesis significantly reduced the relaxation response to acetylcholine in coronary arterioles from sham mice, and no effect on the response to acetylcholine was observed in HT mice (Figure 5, A and B), indicating that the nitric oxide pathway is compromised in HT mice. To extend our hypothesis, we isolated, purified, and cultured Tregs from sham mice and then injected the cells into HT mice (200,000 cells three times a week for 2 weeks). Our data revealed a significant improvement in coronary arteriolar endothelial function (Figure 5C). The inhibition of eNOS significantly reduced endothelium-dependent relaxation in HT mice injected with Tregs (Figure 5C). As controls, shams were injected with Tregs, and no effect was observed on endothelium-dependent relaxation with or without eNOS inhibition (Figure 5D). Similarly, in HT mice, injection with CD4− T cells did not affect endothelium-dependent relaxation with or without eNOS inhibition (Figure 5E). Phosphorylated eNOS was significantly reduced in coronary arterioles from HT mice compared with sham mice (Figure 5D). eNOS phosphorylation was normalized in coronary arterioles from HT mice injected with Tregs (Figure 5D). As a control, we infused HT mice with vehicle by i.p. injection, and this did not affect endothelial function (data not shown).
Figure 5.
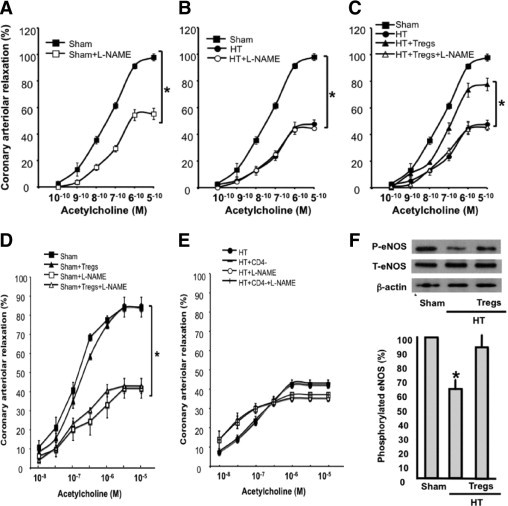
A: Changes in diameter in response to dose response (10−10 to 10−5 mol/L) of acetylcholine in coronary arterioles from sham mice with or without L-NAME (100 μmol/L) (n = 6). *P < 0.05, sham versus sham + L-NAME mice. B: Changes in diameter in response to dose response (10−10 to 10−5 mol/L) of acetylcholine in coronary arterioles from sham, HT, and HT + L-NAME mice (n = 6). *P < 0.05, sham versus HT mice. C: Changes in diameter in response to dose response (10−10 to 10−5 mol/L) of acetylcholine in coronary arterioles from sham, HT, and HT + Tregs mice (n = 6). *P < 0.05, sham versus HT + Tregs mice; *P < 0.05, HT + Tregs versus HT mice. D: Endothelium-dependent relaxation of coronary arterioles from sham mice with or without L-NAME injected with Tregs (n = 6). *P < 0.05, sham/sham + Tregs versus sham + L-NAME or sham + Tregs + L-NAME mice. E: Endothelium-dependent relaxation of coronary arterioles from HT mice with or without L-NAME injected with CD4− T cells (n = 7). F: Western blot analysis and cumulative data of phosphorylated and total eNOS in coronary arterioles from sham, HT, and HT + Tregs mice. β-Actin indicates the equal loading between samples (n = 5). *P < 0.05, HT mice versus sham or HT + Tregs mice.
Discussion
The critical function of coronary arterioles is to supply nutrients and oxygen to myocardial tissue; these vessels also have a significant role in regulating coronary blood flow. Alterations in coronary arteriolar function are mainly responsible for heart disease in hypertension. In this study, we showed that hypertension and coronary arteriolar endothelial dysfunction are associated with increased apoptotic Treg numbers, a reduction in IL-10 content, and the induction of inflammation. The major finding of the present study is that transfer of Tregs to HT animals reduced blood pressure and improved coronary arteriolar endothelial function.
The Treg population is widely accepted as an important component of the immune system. Tregs have recently emerged as a key element in the maintenance of a normal peripheral immune system. These cells play an indispensable role in maintaining self-tolerance and protection from autoimmune diseases13–16 and in influencing the immune response to different pathogens.17 It has been shown that lymphocytes stimulated by Ang II may contribute to the pathogenesis of HT kidney injury.19,20 These data indicate the involvement of the immune system in the regulation of blood pressure. A recent study indicated a critical role of Tregs in the inhibition of atherosclerosis in several mouse models.12 Another recent study showed that a single injection of Tregs ameliorates hypertension-induced heart damage.24
Hypertension is a “silent” condition and is a major cause of morbidity and mortality in the world. Microvascular dysfunction plays important roles in the pathogenesis of hypertension. The combination of systemic, local hemodynamic, and humoral factors is responsible for microvascular homeostasis. Despite the current knowledge of the association between hypertension and vascular complications, cellular and molecular mechanisms involved in altered coronary arteriolar endothelial function have yet to be determined. We and other investigators demonstrated a reduction in mesenteric resistance arterial1–4 and coronary arteriolar endothelial-dependent relaxation in HT animal models5 resulting from the reduction in the relaxing factors nitric oxide, prostaglandin I2, and endothelial-derived hyperpolarizing factor. Evidence from human and animal models suggests a link between immune system and cardiovascular pathophysiologic conditions.30,31 Thus, it is likely that microvascular dysfunction could be the result of perturbation of the immune system combined with inflammation. Recent evidence suggests the involvement of inflammation as a critical pathologic factor in cardiovascular diseases.6–8
In humans, it has been shown that coronary arteriolar dysfunction is related to smoking behavior, hyperlipidemia, diabetes mellitus, microvascular dysfunction, and hypertension.32–34 Coronary arteriolar dysfunction predisposes to heart disease. Our study showed dysfunction of coronary arterioles in Ang II–dependent HT mice. Our data are in agreement with the concept that Tregs play a critical role in hypertension-induced tissue damage. The divergence between the two studies is related to the injection of Tregs.24 In our case, we used injection three times per week for 2 weeks, and the previous study used a single injection for 2 weeks.24 Vascular impairment was assessed by the reduction in endothelial-dependent relaxation in coronary arterioles in response to acetylcholine compared with the response in sham mice. The incubation of coronary arterioles with nitric oxide synthesis inhibitor (L-NAME) significantly reduced the relaxation of coronary arterioles in response to dose response of acetylcholine in sham mice, and no effect was observed in coronary arterioles from HT mice. These data indicate that the eNOS pathway is compromised, which is in agreement with previous studies.1,32 The inhibition of eNOS significantly reduced endothelium-dependent relaxation in HT mice injected with Tregs, indicating the effect of Tregs on the eNOS pathway. As controls, shams were injected with Tregs, and no effect was observed on endothelium-dependent relaxation with or without eNOS inhibition. In addition, in HT mice, injection with CD4− T cells did not affect endothelium-dependent relaxation with or without eNOS inhibition. These data indicate the specific effect of Tregs on endothelium-dependent relaxation through the eNOS pathway. To extend our data, we performed Western blot analysis on coronary arterioles from sham and HT mice. The data revealed a decrease in eNOS phosphorylation in coronary arterioles from HT mice compared with sham mice.
Our data showed that Ang II–dependent hypertension is associated with significantly reduced numbers of Tregs compared with sham. The reduction in Treg numbers was due to apoptosis as determined by FACS analysis. To extend our study, we performed Western blot analysis, and the data revealed that the ratio of Foxp3 to active caspase-3 was significantly reduced in the spleen from HT mice compared with sham and HT + Tregs mice. These data provide evidence of an important association between the reduction in Treg numbers and the impairment of coronary arteriolar endothelial function in Ang II–dependent hypertension. To extend our study, we isolated, purified, and cultured Tregs from sham animals and then stimulated the cells with Ang II for 24 hours. Cultured Tregs stimulated with Ang II for 24 hours displayed significantly increased apoptotic Treg numbers as assessed by FACS analysis. The increased apoptotic Treg numbers in response to Ang II stimulation were significantly reduced when Tregs were pretreated with candesartan. These data suggest that increased apoptotic Treg numbers in hypertension could be the trigger for the onset of hypertension and vascular complications. To determine the link between the reduction in the number of Tregs and hypertension and coronary arteriolar endothelial dysfunction, we injected HT mice with Tregs isolated from sham animals. Mice receiving Ang II + Tregs displayed a significant reduction in blood pressure (not reaching the control level) and an improvement in coronary arteriolar endothelial function. These data are not in agreement with those of a previous study showing the involvement of T cells in blood pressure regulation and aorta smooth muscle function in Rag1−/− mice.21 These divergence results could be related to the animal model used because Rag1−/− mice are susceptible to type 1 diabetes mellitus, loss of weight, and compromised physiologic function related to T and B cells. On the other hand, our data are in agreement with the concept that cardiovascular injury is linked to Tregs. Thus, the single injection of Tregs reduces hypertension-induced cardiac damage independently of the blood pressure–lowering effect.24 One explanation regarding discrepancies in blood pressure would be the fact that mice received Tregs three times a weeks for 2 weeks rather than a single injection.
To further delineate the mechanism of how a reduction in Treg numbers triggers coronary arteriolar endothelial dysfunction in hypertension, we examined the role of inflammation. Recent studies reported that inflammation is critical in the pathogenesis of cardiovascular diseases.6,8 Thus, in our study, we suggested that the reduction in IL-10 release is associated with increased macrophage activity assessed by the up-regulation of adhesion molecule (ICAM-1 and VCAM-1) expression in HT animals. HT mice injected with Tregs displayed significantly increased IL-10 content associated with the reduction in ICAM-1 and VCAM-1 expression on macrophages. We also found that coronary arterioles from HT mice display an increase in ICAM-1 and VCAM-1 expression, which facilitates macrophage infiltration into coronary arterioles and the heart. It has been reported that chemokine receptors are also important in the infiltration of macrophages.35 In this study, we found an increase in the expression of CCR5 in coronary arterioles from HT mice compared with sham and HT + Tregs mice. These data indicate that macrophage infiltration into coronary arterioles and the heart could be facilitated by the increase in CCR5 expression in hypertension.
We also found a large number of infiltrated macrophages associated with increased levels of TNF-α in the heart of HT mice compared with sham mice. HT mice infused with purified Tregs display significant reductions in macrophage activity and infiltration and TNF-α levels compared with HT mice. These data indicate that the reduction in Treg numbers leads to inflammation, which, in turn, is responsible for endothelium-dependent relaxation dysfunction and sustained hypertension. It is well-known that an increased TNF-α level contributes to endothelial dysfunction.36 TNF-α knockout mice infused with Ang II do not develop high blood pressure, indicating the critical role of TNF-α in the regulation of blood pressure.37 It is well-known that Ang II has a broad effect on different organs, such as the heart, kidneys, brain, and vasculature, to develop blood pressure. Based on our data, Tregs significantly participate in the effect of Ang II on coronary function and blood pressure, but still we do not know the in vivo link between Ang II effects, Tregs, and other organs for the development of blood pressure.
In conclusion, we showed that the Treg population, termed natural regulatory cells and known to be crucial in the maintenance of peripheral tolerance, has an important role in coronary arteriolar endothelial dysfunction and hypertension. These results identify a new target for the modulation of hypertension and coronary arteriolar dysfunction and may provide new insight into potential mechanisms relating autoimmune and coronary arteriolar disease and hypertension.
Footnotes
Supported by grants 1R01HL095566 (K.M.) and P20RR017659 from the National Institutes of Health.
References
- 1.Matrougui K., Maclouf J., Levy B.I., Henrion D. Impaired nitric oxide- and prostaglandin-mediated responses to flow in resistance arteries of hypertensive rats. Hypertension. 1997;30:942–947. doi: 10.1161/01.hyp.30.4.942. [DOI] [PubMed] [Google Scholar]
- 2.Matrougui K., Loufrani L., Levy B.I., Henrion D. High NaCl intake decreases both flow-induced dilation and pressure-induced myogenic tone in resistance arteries from normotensive rats: involvement of cyclooxygenase-2. Pharmacol Toxicol. 2001;89:183–187. doi: 10.1111/j.0901-9928.2001.890407.x. [DOI] [PubMed] [Google Scholar]
- 3.Matrougui K., Levy B.I., Schiavi P., Guez D., Henrion D. Indapamide improves flow-induced dilation in hypertensive rats with a high salt intake. J Hypertens. 1998;16:1485–1490. doi: 10.1097/00004872-199816100-00013. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki H., DeLano F.A., Parks D.A., Jamshidi N., Granger D.N., Ishii H., Suematsu M., Zweifach B.W., Schmid-Schonbein G.W. Xanthine oxidase activity associated with arterial blood pressure in spontaneously hypertensive rats. Proc Natl Acad Sci U S A. 1998;95:4754–4759. doi: 10.1073/pnas.95.8.4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lominadze D., Joshua I.G., Schuschke D.A. Blood flow shear rates in arterioles of spontaneously hypertensive rats at early and established stages of hypertension. Clin Exp Hypertens. 2001;23:317–328. doi: 10.1081/ceh-100102670. [DOI] [PubMed] [Google Scholar]
- 6.Scott J. Pathophysiology and biochemistry of cardiovascular disease. Curr Opin Genet Dev. 2004;14:271–279. doi: 10.1016/j.gde.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 7.Gallego J., Martinez Vila E., Munoz R. Patients at high risk for ischemic stroke: identification and actions. Cerebrovasc Dis. 2007;24(Suppl 1):49–63. doi: 10.1159/000107379. [DOI] [PubMed] [Google Scholar]
- 8.Schiffrin E.L., Lipman M.L., Mann J.F. Chronic kidney disease: effects on the cardiovascular system. Circulation. 2007;116:85–97. doi: 10.1161/CIRCULATIONAHA.106.678342. [DOI] [PubMed] [Google Scholar]
- 9.Meier P., Meier R., Blanc E. Influence of CD4+/CD25+ regulatory T cells on atherogenesis in patients with end-stage kidney disease. Expert Rev Cardiovasc Ther. 2008;6:987–997. doi: 10.1586/14779072.6.7.987. [DOI] [PubMed] [Google Scholar]
- 10.Wei L., Wei-Min L., Cheng G., Bao-Guo Z. Upregulation of CD4+CD25+ T lymphocyte by adenovirus-mediated gene transfer of CTLA4Ig fusion protein in experimental autoimmune myocarditis. Autoimmunity. 2006;39:289–298. doi: 10.1080/08916930600758035. [DOI] [PubMed] [Google Scholar]
- 11.George J. Mechanisms of disease: the evolving role of regulatory T cells in atherosclerosis. Nat Clin Pract Cardiovasc Med. 2008;5:531–540. doi: 10.1038/ncpcardio1279. [DOI] [PubMed] [Google Scholar]
- 12.Ait-Oufella H., Salomon B.L., Potteaux S., Robertson A.K., Gourdy P., Zoll J., Merval R., Esposito B., Cohen J.L., Fisson S., Flavell R.A., Hansson G.K., Klatzmann D., Tedgui A., Mallat Z. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med. 2006;12:178–180. doi: 10.1038/nm1343. [DOI] [PubMed] [Google Scholar]
- 13.Maloy K.J., Powrie F. Regulatory T cells in the control of immune pathology. Nat Immunol. 2001;2:816–822. doi: 10.1038/ni0901-816. [DOI] [PubMed] [Google Scholar]
- 14.Maloy K.J., Salaun L., Cahill R., Dougan G., Saunders N.J., Powrie F. CD4+CD25+ T(R) cells suppress innate immune pathology through cytokine-dependent mechanisms. J Exp Med. 2003;197:111–119. doi: 10.1084/jem.20021345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shevach E.M. CD4+ CD25+ suppressor T cells: more questions than answers. Nat Rev Immunol. 2002;2:389–400. doi: 10.1038/nri821. [DOI] [PubMed] [Google Scholar]
- 16.Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–352. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- 17.Zwar T.D., Read S., van Driel I.R., Gleeson P.A. CD4+CD25+ regulatory T cells inhibit the antigen-dependent expansion of self-reactive T cells in vivo. J Immunol. 2006;176:1609–1617. doi: 10.4049/jimmunol.176.3.1609. [DOI] [PubMed] [Google Scholar]
- 18.Chatila T.A. Role of regulatory T cells in human diseases. J Allergy Clin Immunol. 2005;116:949–959. doi: 10.1016/j.jaci.2005.08.047. quiz 960. [DOI] [PubMed] [Google Scholar]
- 19.Crowley S.D., Coffman T.M. In hypertension, the kidney breaks your heart. Curr Cardiol Rep. 2008;10:470–476. doi: 10.1007/s11886-008-0074-5. [DOI] [PubMed] [Google Scholar]
- 20.Crowley S.D., Frey C.W., Gould S.K., Griffiths R., Ruiz P., Burchette J.L., Howell D.N., Makhanova N., Yan M., Kim H.S., Tharaux P.L., Coffman T.M. Stimulation of lymphocyte responses by angiotensin II promotes kidney injury in hypertension. Am J Physiol Renal Physiol. 2008;295:F515–F524. doi: 10.1152/ajprenal.00527.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guzik T.J., Hoch N.E., Brown K.A., McCann L.A., Rahman A., Dikalov S., Goronzy J., Weyand C., Harrison D.G. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harrison D.G., Guzik T.J., Goronzy J., Weyand C. Is hypertension an immunologic disease. Curr Cardiol Rep. 2008;10:464–469. doi: 10.1007/s11886-008-0073-6. [DOI] [PubMed] [Google Scholar]
- 23.Tanko L.B., Matrougui K. Can we apply results from large to small arteries. Circ Res. 2002;90:e68. doi: 10.1161/01.res.0000013737.02527.15. [DOI] [PubMed] [Google Scholar]
- 24.Kvakan H., Kleinewietfeld M., Qadri F., Park J.K., Fischer R., Schwarz I., Rahn H.P., Plehm R., Wellner M., Elitok S., Gratze P., Dechend R., Luft F.C., Muller D.N. Regulatory T cells ameliorate angiotensin II-induced cardiac damage. Circulation. 2009;119:2904–2912. doi: 10.1161/CIRCULATIONAHA.108.832782. [DOI] [PubMed] [Google Scholar]
- 25.Carlson S.H., Wyss J.M. Long-term telemetric recording of arterial pressure and heart rate in mice fed basal and high NaCl diets. Hypertension. 2000;35:E1–E5. doi: 10.1161/01.hyp.35.2.e1. [DOI] [PubMed] [Google Scholar]
- 26.Mills P.A., Huetteman D.A., Brockway B.P., Zwiers L.M., Gelsema A.J., Schwartz R.S., Kramer K. A new method for measurement of blood pressure, heart rate, and activity in the mouse by radiotelemetry. J Appl Physiol. 2000;88:1537–1544. doi: 10.1152/jappl.2000.88.5.1537. [DOI] [PubMed] [Google Scholar]
- 27.Belmadani S., Palen D.I., Gonzalez-Villalobos R.A., Boulares H.A., Matrougui K. Elevated epidermal growth factor receptor phosphorylation induces resistance artery dysfunction in diabetic db/db mice. Diabetes. 2008;57:1629–1637. doi: 10.2337/db07-0739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lucchesi P.A., Sabri A., Belmadani S., Matrougui K. Involvement of metalloproteinases 2/9 in epidermal growth factor receptor transactivation in pressure-induced myogenic tone in mouse mesenteric resistance arteries. Circulation. 2004;110:3587–3593. doi: 10.1161/01.CIR.0000148780.36121.47. [DOI] [PubMed] [Google Scholar]
- 29.Belmadani S., Zerfaoui M., Boulares H.A., Palen D.I., Matrougui K. Microvessel vascular smooth muscle cells contribute to collagen type I deposition through ERK1/2 MAP kinase, alphavbeta3-integrin, and TGF-beta1 in response to ANG II and high glucose. Am J Physiol Heart Circ Physiol. 2008;295:H69–H76. doi: 10.1152/ajpheart.00341.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nijm J., Jonasson L. Inflammation and cortisol response in coronary artery disease. Ann Med. 2009;41:224–233. doi: 10.1080/07853890802508934. [DOI] [PubMed] [Google Scholar]
- 31.van Gils J.M., Zwaginga J.J., Hordijk P.L. Molecular and functional interactions among monocytes, platelets, and endothelial cells and their relevance for cardiovascular diseases. J Leukoc Biol. 2009;85:195–204. doi: 10.1189/jlb.0708400. [DOI] [PubMed] [Google Scholar]
- 32.Camici P.G., Crea F. Coronary microvascular dysfunction. N Engl J Med. 2007;356:830–840. doi: 10.1056/NEJMra061889. [DOI] [PubMed] [Google Scholar]
- 33.Knaapen P., Germans T., Camici P.G., Rimoldi O.E., ten Cate F.J., ten Berg J.M., Dijkmans P.A., Boellaard R., van Dockum W.G., Gotte M.J., Twisk J.W., van Rossum A.C., Lammertsma A.A., Visser F.C. Determinants of coronary microvascular dysfunction in symptomatic hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2008;294:H986–H993. doi: 10.1152/ajpheart.00233.2007. [DOI] [PubMed] [Google Scholar]
- 34.McIntyre C.W., Burton J.O., Selby N.M., Leccisotti L., Korsheed S., Baker C.S., Camici P.G. Hemodialysis-induced cardiac dysfunction is associated with an acute reduction in global and segmental myocardial blood flow. Clin J Am Soc Nephrol. 2008;3:19–26. doi: 10.2215/CJN.03170707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glass W.G., Liu M.T., Kuziel W.A., Lane T.E. Reduced macrophage infiltration and demyelination in mice lacking the chemokine receptor CCR5 following infection with a neurotropic coronavirus. Virology. 2001;288:8–17. doi: 10.1006/viro.2001.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao X., Xu X., Belmadani S., Park Y., Tang Z., Feldman A.M., Chilian W.M., Zhang C. TNF-alpha contributes to endothelial dysfunction by upregulating arginase in ischemia/reperfusion injury. Arterioscler Thromb Vasc Biol. 2007;27:1269–1275. doi: 10.1161/ATVBAHA.107.142521. [DOI] [PubMed] [Google Scholar]
- 37.Sriramula S., Haque M., Majid D.S., Francis J. Involvement of tumor necrosis factor-alpha in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension. 2008;51:1345–1351. doi: 10.1161/HYPERTENSIONAHA.107.102152. [DOI] [PMC free article] [PubMed] [Google Scholar]