

Abstract
The protein-RNA interface has been regarded as “undruggable” despite its importance in many biological processes. The toll-like receptor 3 (TLR3)/double-stranded RNA (dsRNA) complex provides an exciting target for a number of infectious diseases and cancers. We describe the development of a series of small molecule probes that were shown to be competitive inhibitors of dsRNA binding to TLR3 with high affinity and specificity. In a multitude of assays, compound 4a was profiled as a potent antagonist to TLR3 signaling and also repressed the expression of downstream signaling pathways mediated by the TLR3/dsRNA complex, including TNF-α and IL-1β.
Interfering with protein-protein interactions or protein-nucleic acid interactions have been regarded as daunting goals in drug discovery.1 Major strides have been made during the last decade in developing small molecule agents to target protein-protein interactions. However, regulation of protein-RNA interactions lags behind, arguably due to the fact that RNA molecules pose a particular challenge with their high flexibility.2 RNA-binding proteins (RBPs) play key roles in post-transcriptional modifications, which, along with transcriptional regulation, is a main method of controlling gene expression during development. In the present study, we report novel molecular probes that disrupt with dsRNA binding to TLR3 as a demonstration of using specific small molecule agents to target the protein-RNA interface.
Toll-like receptors (TLRs) are highly conserved transmembrane proteins that detect pathogen-associated molecular patterns and elicit pathogen-specific immune responses.3 TLR3 signaling is activated by dsRNA released from necrotic cells during inflammation or viral infection.4 TLR3 activation induces secretion of type I interferons and proinflammatory cytokines, such as TNF-α, IL-1, and IL-6, triggering immune cell activation and recruitment that are protective during certain microbial infections.5 A dominant-negative TLR3 allele has been associated with increased susceptibility to herpes simplex encephalitis, a serious illness with significant risks of morbidity and death, upon primary infection with HSV-1 in childhood.6 In mice, TLR3 deficiency is associated with decreased survival upon coxsackie virus challenge.7 In addition, uncontrolled or sustained innate immune response via TLR3 has been shown to contribute to morbidity and mortality in certain viral infection models including the West Nile disease, phlebovirus, vaccinia, and influenza A.8–11 Therefore, modulation of TLR3 pathways offers an attractive strategy to fight a variety of diseases.
Despite the significant potential, the discovery of small molecule inhibitors of TLR3 has been slow due to the complexity associated with disrupting the protein-RNA contact: immense effort is required to design individual compounds that target specific RNA-binding domains with high binding affinity and selectivity.1 Herein, we describe the successful identification and characterization of small molecule probes for the TLR3/dsRNA complex.
In search of small molecule probes, the 1.2 million-compound Enamine database was screened against the dsRNA-binding domain of TLR3 (crystal structure PDB: 3CIY12) using the Glide 5.6 program.13 Initially, nine hits (Figure 1) were selected for cell assay screening. Interestingly, almost all of the hits identified, with an exception of T5528092, from the in silico screening generally share the common motif of a D-amino acid conjugated with an aromatic substituent, implying a novel pharmacophore to target the RNA-binding site of TLR3.
Figure 1.

Chemical structures of the nine hits from the in silico screening of a 1.2 million-compound database imply a common structural motif.
These initial hits were first evaluated using our previously established high-throughput cell assay of TLR3 activation.14 A dsRNA, polyriboinosinic:polyribocytidylic acid (Poly (I:C)), was employed to selectively activate TLR3 signaling, resulting in the activation of nitric oxide (NO) synthase and the production NO in RAW 264.7 macrophage cells.15 We monitored the NO level as an indicator of Poly (I:C)-induced TLR3 activation to evaluate the drug’s inhibitory activity.
Two compounds (T5626448 and T5260630, shown in boxes in Figure 1) demonstrated mild inhibitory activities in whole cells, with IC50 values of 154 ± 6 µM and 145 ± 4 µM, respectively. Both of these two compounds are derivatives of D-phenylalanine, suggesting the D-phenylalanine backbone as the scaffold to develop small molecule inhibitors of TLR3. Computational docking results also implied that T5626448 and T5260630 could be further optimized by varying the substituents on the benzene or thiophene rings (Supplementary Figure S1).
With the hit compounds selected, we developed concise synthetic routes for both T5626448 and T5260630 (Supplementary Scheme S1), which allows an extensive structure-activity relationship (SAR) analysis. Various substitutions with different size and electron withdrawing/donating capability were examined on the aromatic systems. To inspect the impact on the activities imposed by the stereogenic center, both R- and S-isomers were prepared.
An improvement of two orders of magnitude in inhibitory potency of T5626448 was achieved, with compound 4a (Figure 2A) showing a low µM (3.44 ± 0.41 µM) IC50 value. By contrast, no significant activity improvement for the T5260630 derivatives was observed. Therefore, we decided to focus on the development of T5626448 derivatives.
Figure 2.

Molecular model of the identified compounds docked to TLR3: (A) Global view with the murine TLR3 binding to dsRNA showing compound 4a (shown in CPK representation) compete with dsRNA for the same binding site on the TLR3 surface; (B) Close-up view comparing the binding modes of 4a (magenta) and T5626448 (green) in complex with TLR3 (surface shown), indicating that the thiophene rings in the two compounds are flipped.
SAR studies of the T5626448 derivatives lent support to the predicted binding mode of this series of TLR3 ligands (Table 1): First, substituting the 7-membered ring (T5626448) with a phenyl group decreased the inhibitory activity (1a and 1b), suggesting that perhaps the benzene ring does not fit well into the hydrophobic pocket on the TLR3 surface. Therefore we introduced some hydrophobic groups on the aromatic rings. With the replacement by smaller hydrophobic substituents, -CH3 at the R1 position and -Cl at the R2 position, the activity is increased significantly (2a, 2b). Keeping the –Cl at the R2 position and changing the R1 substituent from -CH3 to -Cl or -F (3a, 4a) resulted in an increase of activity. It is noteworthy that the fluorine at R1 and chlorine at the R2 position promoted the potency significantly by nearly 45-fold (4a) compared to that of T5626448. Our docking results suggest that the elevated potency may be due to the fact that sulfur in 4a is oriented in the opposite direction from the one in T5626448 (Figure 2B), which could facilitate hydrogen-bonding contacts with the TLR3 surface. Further, results demonstrate that an electron-withdrawing group is preferred at the R1 position (6a vs 4a). The -CF3 replacement of the -F at R1 position decreased the activity slightly (5a vs 4a), indicating that the electron density rather than the size is the dominating factor at this position. An additional hydroxyl group at the R3 position greatly decreases the activity (7a vs 4a), suggesting higher hydrophobicity is favored at the amino acid side chain. With the absence of any substitute at the R1 and the R2 positions, the activity decreased significantly or was completely abolished (9a–11a, 13a). Last, these T5626448 derivatives’ inhibitory effects are stereo-dependent, with the R- enantiomers generally demonstrating higher potency. In summary, we identified compound 4a, which shows dose-dependent inhibitory effects blocking Poly (I:C)-induced TLR3 activation with an IC50 of 3.44 ± 0.41 µM (Figure 3A). This low ~µM potency in whole cells is remarkable given that the competing dsRNA is a tight binder to TLR3 (Kd= 19 ± 0.9 nM).16
Table 1.
Structure-activity relationship analysis in RAW 264.7 cells.
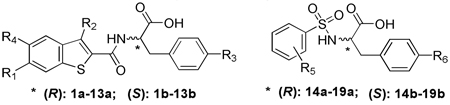 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | IC50 (µM)a |
| T5626448 | 154 ± 6 | ||||
| T5260630 | 145 ± 4 | ||||
| 1a | -H | -H | -H | -H | >155 |
| 1b | -H | -H | -H | -H | >155 |
| 2a | -CH3 | -Cl | -H | -H | 11.8 ± 1.3 |
| 2b | -CH3 | -Cl | -H | -H | 31.7 ± 0.7 |
| 3a | -Cl | -Cl | -H | -H | 5.60 ± 0.32 |
| 3b | -Cl | -Cl | -H | -H | 11.8 ± 2.5 |
| 4a | -F | -Cl | -H | -H | 3.44 ± 0.41 |
| 4b | -F | -Cl | -H | -H | 21.9 ± 0.7 |
| 5a | -CF3 | -Cl | -H | -H | 6.28 ± 1.05 |
| 5b | -CF3 | -Cl | -H | -H | 19.8 ± 3.6 |
| 6a | -OCH3 | -Cl | -H | -H | 36.7 ± 3.9 |
| 6b | -OCH3 | -Cl | -H | -H | >100 |
| 7a | -F | -Cl | -OH | -H | 56.4 ± 5.2 |
| 7b | -F | -Cl | -OH | -H | 84.7 ± 2.4 |
| 8a | -F | -Cl | -H | -F | 70.7 ± 2.1 |
| 8b | -F | -Cl | -H | -F | 79.5 ± 3.4 |
| 9a, 9b | -H | -H | -OH | -H | >100 |
| 10a, 10b | -H | -H | -H | -F | >100 |
| 11a, 11b | -H | -Cl | -H | -H | >100 |
| 12a | -H | -Cl | -H | -F | 47.3 ± 4.1 |
| 12b | -H | -Cl | -H | -F | 54.1 ± 5.7 |
| 13a, 13b | -F | -H | -H | -H | >100 |
| R5 | R6 | IC50 (µM) | |||
| 14a, 14b | 2-F | -H | >100 | ||
| 15a, 15b | 3-F | -H | >100 | ||
| 16a, 16b | 4-F | -H | >100 | ||
| 17a, 17b | 2-F | -OH | >100 | ||
| 18a, 18b | 3-F | -OH | >100 | ||
| 19a, 19b | 4-F | -OH | >100 | ||
IC50 average values and corresponding SD values (in µM) are determined from the results of at least three independent repeats.
Figure 3.

(A) Dose-dependent inhibitory response of Poly (I:C)-induced TLR3 activation by 4a and 1a (negative control). (B) Specificity test for 4a (27 µM) with TLR-specific agonists: (1) TLR4: 10 ng/mL LPS, (2) TLR3: 15 µg/mL Poly (I:C), (3) TLR2/6: 10 ng/mL FSL-1, (4) TLR1/2: 200 ng/mL Pam3CSK4, and (5) TLR7: 100 nM R848 were used to selectively activate respective TLRs. (C) Fluorescence anisotropy assay shows competitive binding between 4a and dsRNA (Kd= 19± 0.9 nM) for TLR3. Inhibition curve was fitted using a one-site competition model. (D) Normalized binding of 4a compared with the negative control, 1a.
A challenge to develop inhibitors to target TLRs is to engineer specificity and potency. There are at least 10 homologous TLRs present in murine macrophages, all sharing a ligand-binding domain with a horseshoe shape.3 We therefore tested compound 4a against a panel of homologous TLRs, including TLR1/2, TLR2/6, TLR3, TLR4, and TLR7 using TLR-specific ligands to selectively activate a particular TLR signaling pathway. We found that 4a inhibits TLR3 signaling without affecting other TLRs, showing it is highly selective in intact cells (Figure 3B). Further, compound 4a was found to have low cytotoxicity. CYP450 tests showed that 4a did not affect a panel of cytochrome enzymes (CYP3A4, 2D6, 2C19 and 1A2) (Supplementary Figure S2). The low toxicity of 4a was further confirmed in RAW 264.7 cells using the established WST-1 methodology (Supplementary Figure S3). Last, kinase profiling showed that compound 4a demonstrated negligible inhibition activity against a panel of 12 representative kinases (Supplementary Figure S4).
Biophysical tests were carried out for 4a, along with the negative control compound 1a, to demonstrate that 4a directly binds to TLR3. Fluorescence anisotropy assays showed that 4a competes with dsRNA for binding to TLR3 with a Ki of 2.96 ± 0.32 µM, which is consistent with its potency observed in the whole cell assay. The anisotropy of rhodamine labeled Poly (I:C) showed a robust increase from 0.116 to 0.171 (Figure 3C) upon addition of TLR3 (excitation= 546 nm; emission= 576 nm). This increase is consistent with the anisotropy changes seen with ligand-receptor pairs of comparable sizes.17 Increasing 4a’s concentration to 68 µM decreased the anisotropy to background levels, presumably due to release of the fluorescently labeled Poly (I:C) probe (Figure 3D). These data were then fitted to a one-site-competition model. Good fitting (R2 > 0.98) confirmed that 4a and dsRNA compete for the same binding site on the TLR3 surface. Taken together, these results support that 4a disrupts the TLR3/dsRNA association by directly targeting the RNA-binding site on TLR3.
Lastly, we used a secondary cellular assay to confirm that 4a also inhibits the downstream signaling transduction mediated by the formation of the TLR3/dsRNA complex. In addition to TLR3 signaling suppression, the release of the proinflammatory cytokines, TNF-α and IL-1β, were investigated. Results (Figure 4) showed that 4a almost completely abolishes the TLR3-mediated inflammation response at its IC90 concentration (27 µM). The inhibitory effects of TNF-α by 4a at 10 µM were also tested. Approximately 60% of inhibition was observed, agreeing with the results observed in the NO synthase assay (Supplementary Figure S5). Taken together, these results suggested that 4a suppresses the downstream signaling of TLR3 in a consistent manner in which it disrupts the TLR3/dsRNA complex formation.
Figure 4.

ELISA assay results showed that 4a, at its IC90 concentration (27 µM), abolishes the TNF-α and IL-1β production activated by 15 µg/ml Poly (I:C) in the RAW 264.7 cells.
In conclusion, we have successfully selected a series of novel small molecule inhibitors targeting the dsRNA binding region of TLR3 with high specificity and binding affinity both in vitro and in whole cells. Compound 4a provides a much needed molecular probe for studying protein-RNA interactions. In general, this effective method will shed light into the future design of potent and selective molecular probes for RNA-binding proteins, which may facilitate the studies of highly important protein-RNA complexes.
Supplementary Material
ACKNOWLEDGMENT
We thank the National Institutes of Health (DA026950, DA025740, NS067425, RR025780, and DA029119) for financial support of this work. C. K. is supported by a visiting student scholarship from the China Scholarship Council (Grant No. 2009619072). We thank Dr. Sherry A. Chavez, Dr. Peter Brown and Alexander J. Martinko for reviewing the manuscript.
Footnotes
SUPPORTING INFORMATION PARAGRAPH. Full details of the in silico virtual screening, full characterization of the individual compounds examined and complete Ref. 6. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.(a) Boger DL, Desharnais J, Capps K. Angew. Chem. Int. Ed. 2003;42:4138. doi: 10.1002/anie.200300574. [DOI] [PubMed] [Google Scholar]; (b) Yin H, Hamilton AD. Angew. Chem. Int. Ed. 2005;44:4130. doi: 10.1002/anie.200461786. [DOI] [PubMed] [Google Scholar]
- 2.(a) Jinek M, Doudna JA. Nature. 2009;457:405. doi: 10.1038/nature07755. [DOI] [PubMed] [Google Scholar]; (b) Sashital DG, Doudna JA. Curr. Opin. Struc. Biol. 2010;20:90. doi: 10.1016/j.sbi.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akira S, Uematsu S, Takeuchi O. Cell. 2006;124:783. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 4.Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T. Nat. Immunol. 2003;4:161. doi: 10.1038/ni886. [DOI] [PubMed] [Google Scholar]
- 5.Barton GM, Medzhitov R. Science. 2003;300:1524. doi: 10.1126/science.1085536. [DOI] [PubMed] [Google Scholar]
- 6.Zhang SY, et al. Science. 2007;317:1522. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- 7.Richer MJ, Lavallée DJ, Shanina I, Horwitz MS. PLoS One. 2009;4:e4127. doi: 10.1371/journal.pone.0004127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang T, Town T, Alexopoulou Lena, Anderson JF, Fikrig E, Flavell RA. Nat. Med. 2004;10:1366. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- 9.Gowen BB, Hoopes JD, Wong MH, Jung KH, Isakson KC, Alexopoulou L, Flavell RA, Sidwell RW. J. Immunol. 2006;177:6301. doi: 10.4049/jimmunol.177.9.6301. [DOI] [PubMed] [Google Scholar]
- 10.Hutchens M, Luker KE, Sottile P, Sonstein J, Lukacs NW, Núñez G, Curtis JL, Luker GD. J. Immunol. 2008;180:483. doi: 10.4049/jimmunol.180.1.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pashine A, Valiante NM, Ulmer JB. Nat. Med. 2005;11:S63. doi: 10.1038/nm1210. [DOI] [PubMed] [Google Scholar]
- 12.Liu L, Botos I, Wang Y, Leonard JN, Shiloach J, Segal DM, Davies DR. Science. 2008;320:379. doi: 10.1126/science.1155406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Glide, version 5.6. New York, NY: Schrödinger, LLC; 2010. [Google Scholar]; (b) Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. J. Med. Chem. 2004;47:1750. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 14.(a) Bevan DE, Martinko AJ, Loram LC, Stahl JA, Taylor FR, Joshee S, Watkins LR, Yin H. ACS Med. Chem. Lett. 2010;1:194. doi: 10.1021/ml100041f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Joce CM, Stahl JA, Shridhar M, Hutchinson MR, Watkins LR, Fedichev PO, Yin H. Bioorg. Med. Chem. Lett. 2010;20:5411. doi: 10.1016/j.bmcl.2010.07.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heitmeier MR, Scarim AL, Corbett JA. J. Biol. Chem. 1998;273:15301. doi: 10.1074/jbc.273.24.15301. [DOI] [PubMed] [Google Scholar]
- 16.Fukuda K, Watanabe T, Tokisue T, Tsujita T, Nishikawa S, Hasegawa T, Seya T, Matsumoto M. J. Biol. Chem. 2008;283:22787. doi: 10.1074/jbc.M802284200. [DOI] [PubMed] [Google Scholar]
- 17.Xu HQ, Zhang AH, Auclair C, Xi XG. Nucleic Acids Res. 2003;31:e70. doi: 10.1093/nar/gng070. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.