

Abstract
Type 1 (non neuronopathic) Gaucher disease was the first lysosomal storage disorder for which an effective enzyme replacement therapy was developed and it has become a prototype for treatments for related orphan diseases. There are currently four treatment options available to patients with Gaucher disease, nevertheless, almost 25% of type 1 Gaucher patients do not gain timely access to therapy because of delays in diagnosis after the onset of symptoms. Diagnosis of Gaucher disease by enzyme testing is unequivocal, but the rarity of the disease and non-specific and heterogeneous nature of Gaucher disease symptoms may impede consideration of this disease in the differential diagnosis. To help promote timely diagnosis and optimal management of the protean presentations of Gaucher disease, a consensus meeting was convened to develop algorithms for diagnosis and disease management for Gaucher disease.
Keywords: Gaucher disease, enzyme replacement therapy
Introduction
The defect in Gaucher disease is an inherited deficiency of the lysosomal enzyme acid β-glucosidase (glucocerebrosidase, GBA1), which results in the accumulation of glucocerebroside within lysosomes of macrophages. Systemic accumulation of these glycolipid-lipid engorged cells (eponymously known as Gaucher cells – Figure 1) results in variable combinations of splenomegaly with associated abdominal discomfort; anemia associated with chronic fatigue; bleeding due to thrombocytopenia and/or Gaucher disease-related coagulopathy; hepatomegaly, abnormal tests of liver function; and a diverse pattern of bone disease [1]. Increased susceptibility to infections may result from impaired neutrophil function and neutropenia [2]. Rarely, the lungs, lymphatic system, skin, eyes, kidneys and the heart are involved and, in the rare neuronopathic forms, neurodegenerative disease results [1]. Gaucher disease is traditionally classified into three broad phenotypic categories: type 1 (non-neuronopathic disease); type 2, fulminant neuronopathic disease that is fatal during infancy; and type 3, chronic neuronopathic disease, that usually results in death in childhood or early adult life [1]. Further distinct phenotypic categories may be recognized within these broad groups.
FIGURE 1.
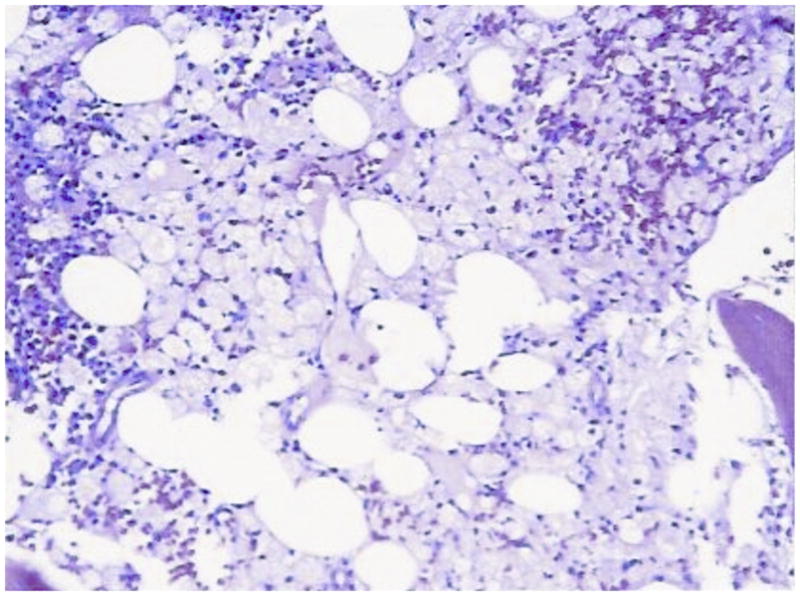
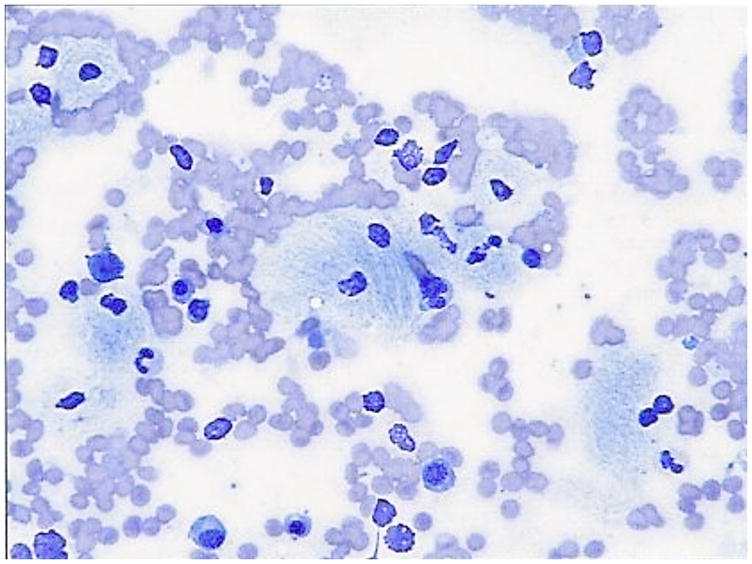
Bone marrow smear and biopsy from a patient with N370S homozygous Gaucher disease showing classical striated macrophages and marrow replacement Smear: Gaucher cells (100×)
Core: Bone marrow replacement by Gaucher cells (20×)
Type 1 (non neuronopathic) Gaucher disease accounts for more than 90% all Gaucher disease patients. Its prevalence world-wide is 1 in 50,000–100,000 but it is as high as ~1 in 850 in individuals of Ashkenazi heritage [3–6]. The broadest phenotypic spectrum in Gaucher disease with respect to age of onset, rate of progression, and organs affected occurs in type 1 Gaucher disease [7]. Homozygosity for the N370S mutation is the most common genotype in the Ashkenazim in whom it accounts for ~70% of all disease alleles. It is associated with atypical presentation in adults or the older patient and is notable for significant skeletal disease despite inconspicuous classical manifestations (splenomegaly, hepatomegaly, anemia, thrombocytopenia). However, severe disease with classic manifestations presenting in childhood may occur in a minority of N370S homozygous patients [8, 9]. The most common disease allele of GBA1 world-wide is L444P mutation, which occurs in the sequence of the closely linked pseudogene; it is believed that gene conversion events lead to L444P mutation in the active gene. It seems likely that the most frequent genotype of type 1 Gaucher disease in populations of European descent in the world is N370S/L444P. Generally this genotype leads to more severe disease compared to N370S homozygosity [1, 10, 11] (Figure 2).
FIGURE 2.
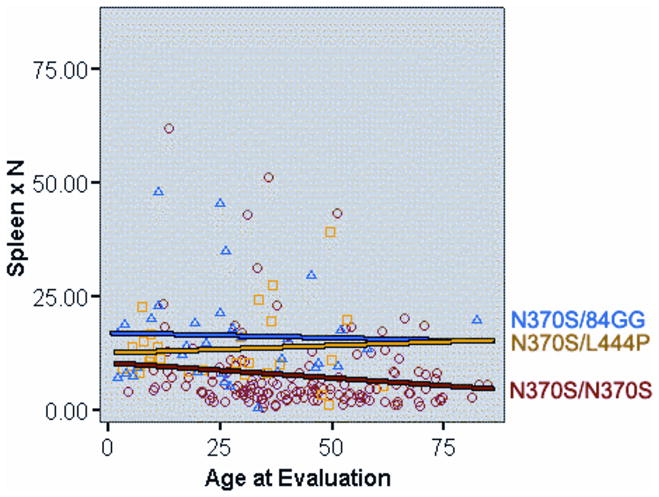
Relationship between age of presentation at extent of splenomegaly (determined by volumetric MRI with normal spleen volume at 0.2% body weight; extent of splenomegaly is indicated by multiple of normal, i.e. xN). Three most common genotypes in North American Gaucher disease patient populations are depicted: N370S/N370S, N370S/L444P and N370S/84G ins G). The results show that the majority of older N370S homozygous patients harbor no, or minimal, splenomegaly)
Treatment with macrophage-targeted mannose-terminated glucocerebrosidase enzyme replacement therapy (imiglucerase, Cerezyme®, Genzyme Corporation, Cambridge, MA) is the standard of care for type 1 Gaucher disease and of non-neuronopathic manifestations of type 3 Gaucher disease. Evidence from the ICGG Gaucher Registry, indicates that enzyme replacement with imiglucerase and its predecessor alglucerase reverses hematological and visceral manifestations of the disease [12] and reduces the bone marrow burden of Gaucher cells resulting in amelioration of osteopenia, bone pain, risk of bone crises and overall improvement in quality of life [13–16]. Several aspects of bone disease, such as osteonecrosis, osteofibrosis and lytic lesions cannot be reversed but timely initiation of enzyme therapy reduces the risk of these irreversible complications [8, 17]. Other variants of macrophage-targeted enzyme replacement therapies are undergoing clinical trials: velaglucerase, a human fibroblast-derived enzyme, was recently approved for treatment of type 1 Gaucher disease [18] and taliglucerase, a plant derived enzyme is in clinical trials [19]. Substrate reduction therapy (Zavesca® miglustat) Actelion Pharmaceuticals, Allschwil, Switzerland) is approved for patients with mild Gaucher disease who are unable to receive enzyme replacement therapy [20, 21]. A more specific and potent inhibitor of glucosylceramide synthesis, eliglustat tartrate is currently in phase 3 trials having shown impressive efficacy and safety in phase 2 trials [22, 23].
Prompt diagnosis before the occurrence of irreversible complications underpins the Gaucher disease management model [24]. The early symptoms of type 1 Gaucher disease tend to reflect the hematological aspects of the disease (splenomegaly, anemia, thrombocytopenia and bleeding tendency) [25]. Patients with Gaucher disease, therefore, are most likely to be referred to hematologists for diagnosis and management. However, only ~20% of hematologists/oncologists in one study considered Gaucher disease in differential diagnoses even in the presence of all classical symptoms [8]. Diagnosis may be achieved in high risk groups, such as individuals of Ashkenazi Jewish ancestry, by opportunistic screening (i.e., those with any of the manifestations of Gaucher disease or those with surrogate indicators of disease, i.e, severe osteoporosis, hyperferritinemia, gallstones and low HDL cholesterol) and family screening after diagnosis in a family member [26]. The aim of the consensus meeting was to develop algorithms for diagnosis and management based on current understanding of the full clinical spectrum of presentations of Gaucher disease.
Design and Methods
The project to develop diagnostic and disease management algorithms was conceived in recognition of the need for greater awareness and early diagnosis of Gaucher disease amongst hematologists [8]. Genzyme Corporation provided an unrestricted education grant to support the consensus conference. An international group of physicians experienced in hematology/oncology and the management of Gaucher disease convened in Copenhagen to review literature and personal clinical experiences with the goal of developing diagnosis and management algorithms for Gaucher disease. Each participant carried out his/her own review of relevant peer-reviewed medical literature (not restricted by language or date of publication) aided by on line tools such as PubMed to support their individual contributions to roundtable discussions. Discussions were also supported by reference to data from the International Collaborative Gaucher Group (ICGG) Registry (an observational registry of >5,000 patients world-wide). The participating physicians represented ten countries, contributing experience of Gaucher disease presentations and phenotypes in different parts of the world in an attempt to capture marked geographic differences. Members of the group completed a survey of the patterns of misdiagnosis from their respective practices (five illustrative case studies are included in appendix 1).
The consensus process
During the first meeting participants discussed their experiences of patterns of misdiagnoses in Gaucher disease, discussed presenting sign and symptoms in Gaucher patients from their clinical practice and those described in the literature. Based on these discussions, key diagnostic features were identified. These formed the basis for the development of diagnostic algorithms. The algorithms for diagnosis and management of Gaucher disease were further developed and agreed through subsequent email and telephone correspondence between authors.
Results
The authors’ combined experience of 362 patients with Gaucher disease revealed a consistent pattern of previous misdiagnoses that included leukemia, immune thrombocytopenia purpura, autoimmune disease, hepatic cirrhosis, idiopathic avascular necrosis, viral disease, idiopathic splenomegaly, and anemia of chronic disease. Misdiagnosis led to complications such as avascular necrosis, osteopenia, liver disease and bleeding complications and inappropriate procedures such as splenectomy, liver biopsy and empirical corticosteroid therapy (see appendix 1).
Malignancy is commonly the first diagnosis entertained in patients who are eventually diagnosed with Gaucher disease [8, 27]. Interestingly, the first patient ever described with Gaucher disease by Dr Philippe Gaucher in 1882 was believed to harbor malignancy affecting the spleen. In one study, the diagnosis most frequently considered was hematologic malignancy (leukemia 65%, lymphoma 36%, multiple myeloma 22%, chronic granulocytic leukemia 14%) [8]. It should be noted that the risk of hematological malignancies in Gaucher disease is increased, especially of multiple myeloma [9, 28–31]. The life-time risk of multiple myeloma in Gaucher disease is higher than 25-fold compared to the general population [9, 29–32].
Algorithms for diagnosis
In patients of Ashkenazi ancestry, the frequency of Gaucher disease is ~ 1 in 800 while hematologic malignancies are much less frequent at ~ 1 in 2,500 [33]. Therefore, in this ethnic group, it is prudent to test for Gaucher disease as a first-line investigation, in any patient presenting with splenomegaly and cytopenia. It is important to keep in mind that the most common genotype in this ethnic group, homozygosity for N370S mutation is often characterized by mild cytopenia and splenomegaly that escape initial detection. Therefore, presence of conditions associated with Gaucher disease should alert the clinician, i.e., hyperferritinemia, low HDL cholesterol, premature gallstones or osteoporosis and gammopathy.
In the non-Ashkenazi populations Gaucher disease is markedly less frequent (~1 in 40,000) compared to hematologic malignancies. In this setting, it would be appropriate to consider Gaucher disease in the differential diagnosis after malignancies have been ruled out. In this setting, bone marrow biopsy is usually performed; it should become routine to search for Gaucher cells as well as for evidence of hematological malignancies. The above considerations form the rationale for the diagnostic algorithm stratified by ethnicity (Figures 4 and 5). Portal hypertension due to advanced liver disease from well-known etiologies must be ruled out before application of the algorithm.
FIGURE 4.

Diagnosis algorithm for individuals of Ashkenazi origin. Diagnosis of Gaucher disease should be considered in any individual of Ashkenazi Jewish ancestry presenting with mild, moderate or severe splenomegaly. When splenomegaly is absent, Gaucher disease should be considered in the presence of: thrombocytopenia (even if mild), bleeding tendency, unexplained stable hyperferritinemia with normal transferrin saturation, or increased inflammatory markers. *In patients with bleeding diatheses, coagulopathies such as factor XI deficiency common in Askenazim [37] should be excluded.
FIGURE 5.

Diagnostic algorithm for individuals of non-Ashkenazi Jewish origin. Compared to Gaucher disease, malignancy is likely to be the more frequent cause of splenomegaly in this population. In this setting it is reasonable to perform bone marrow biopsy in initial investigations. The finding of Gaucher cells in bone marrow aspirate will suggest Gaucher disease although Gaucher disease and malignancy are not mutually exclusive. Pseudo-Gaucher cells have also been observed in malignant conditions in the absence of Gaucher disease [38].
In developing these diagnostic algorithms, the authors focused on splenomegaly, as a key presenting sign since it is present in the vast majority of Gaucher patients [10]. In the ICGG Registry 87% of patients have splenomegaly in excess of five times normal (median spleen volume = 15.2 x normal) [6]. However, Gaucher disease may occur without splenomegaly and the absence of splenomegaly does not exclude Gaucher disease (Figure 2). This is often the case in older adults homozygous for the N370S mutation in whom splenomegaly is mild and may not be present [9]. Splenomegaly is defined precisely by volumetric measurement of the spleen by MRI, CT or ultrasound scanning followed by conversion to weight (1 cc equals 1 gm) as multiple of the normal spleen size, i.e., 0.2% body weight [34]. Splenectomy in patients with Gaucher disease is associated with aggressive disease in the liver, skeleton and the lungs [35] (Figure 3]. Therefore, it should become mandatory to test for Gaucher disease in any patient being considered for splenectomy when the cause of splenomegaly has not been established. Assessment of splenomegaly in Gaucher disease should be combined with an examination of spleen parenchyma. There is almost 20% incidence of focal splenic defects in Gaucher disease and its incidence increases with increasing spleen size [36]. These lesions may represent focal collections of Gaucher cells or areas of infarction and have been a source of misdiagnosis as splenic neoplasms.
FIGURE 3.
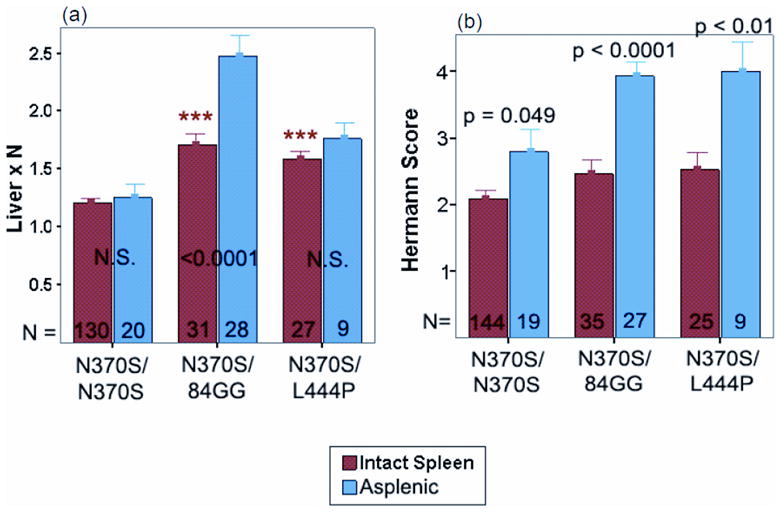
(a and b). Comparison of severity of hepatomegaly and bone disease (Herman score [9]) in patients with intact spleen and those with prior splenectomy) stratified for GBA1 gene genotype. The numbers of patients are depicted at the bottom of the bars)
Confirming diagnosis
Enzyme assay
The diagnostic test for Gaucher disease is the demonstration of low acid β-glucosidase activity in peripheral blood leukocytes (normal range 2.1–5.3 μmol/l/h) [39]. The assay is performed in blood leucocytes using a fluorescent substrate, 4-methyumbelliferone β-glucoside. The test requires 10 cc EDTA blood sample shipped at ambient temperature by overnight delivery to the lab.
Molecular diagnosis
Molecular analysis of GBA1 gene is complicated by presence of highly homologous pseudogene that harbors several mutations, which if present in the active gene leads to Gaucher disease. A number of techniques have been developed to circumvent potential problems arising from this situation in order to analyze only the active gene sequences uncontaminated by pseudogene sequences [40]. A negative screen for common GBA1 mutations does not exclude Gaucher disease (see discussion). Therefore, undertaking sequencing of the entire coding region of GBA1 gene is recommended in patients strongly suspected of harboring Gaucher disease when a screen for common mutations is negative [40]. Mutation analysis of the GBA1 gene may provide some prognostic information although there is considerable variation of disease severity among patients harboring an identical GBA1 genotype [7] ([Figure 2]). Knowledge of the GBA1 mutation in a proband also facilitates family screening for genetic counseling purposes since heterozygote carriers cannot be reliably identified by enzyme assays.
Bone marrow biopsy
Routine bone marrow examination is not necessary for diagnosis. It has been discouraged in the diagnosis of Gaucher disease, due to availability of less invasive and robust enzymatic test and the fact that it may cause bleeding complications due to thrombocytopenia and coagulopathy. Many patients report having had this procedure usually before Gaucher disease had been considered. Care should be exercised in the interpretation of bone marrow samples as false negative results are common especially upon examination of bone marrow aspirate as opposed to bone marrow biopsy. Conversely, the presence of pseudo-Gaucher cells may lead to erroneous diagnosis [38]. Pseudo-Gaucher cells have been found in diverse conditions such as multiple myeloma [41], myelodysplasia and myelodysplastic syndromes [42], chronic myeloid leukemia [43], pulmonary tuberculosis [44], mycobacterioses [45], and sickle cell disease [46]. Occasionally bone marrow biopsy may be indicated in a patient with Gaucher disease suspected of a superimposed hematologic disorder, i.e., myelofibrosis, multiple myeloma or other hematologic malignancy.
Biochemical markers
Several serum proteins are consistently elevated in Gaucher disease, with some being used as biomarkers for routine monitoring, i.e., angiotensin-converting enzyme (ACE), tartrate-resistant acid phosphatase (TRAP), chitotriosidase and a chemokine, CCL18 [47 and 48]. The finding of elevated levels of one or more of these markers is never sufficient to diagnose Gaucher disease since these markers may be elevated in other disorders. Other biochemical abnormalities are common in Gaucher disease, such as low vitamin B12, low serum lipoproteins (LDL and HDL), and elevated ferritin [9, 25, 49].
Algorithms for evaluation and management
Once a diagnosis of Gaucher disease is confirmed, the focus should be on comprehensive evaluation of all disease domains (Figure 3) to establish baseline disease characteristics, to determine candidacy for enzyme replacement therapy, and to develop personalized therapeutic goals and monitoring strategy [34, 47, 50]. Quality of life measures and a severity score calculation using one of the novel instruments that are easily applicable in the clinic are of value in making treatment decisions [51, 52]. Accurate skeletal assessment is especially important since it is the site of most disabling, irreversible complications, which can occur without warning. Evaluation of neuronopathic Gaucher disease has been described previously [53, 54]. In non-neuronopathic disease physical examination should also include a search for symptoms and signs of Parkinsonian tremor and peripheral neuropathy (see discussion below). A basic premise in the management and monitoring of Gaucher disease is that it is a chronic disease that is progressive, albeit at variable rates, and ultimately leads to reduced life-expectancy [9, 55].
Published treatment guidelines and therapeutic goals relating to the systemic manifestations of Gaucher disease are based on the experience with alglucerase and imiglucerase enzyme replacement therapy [34] which is a benchmark for all other treatment approaches. Assessment of response to treatment is derived from a large body of accumulated evidence [56]. The heterogeneous and chronic nature of type 1 Gaucher disease requires an individualized disease management model with respect to enzyme dose and therapeutic goals in each disease compartment. Moreover, maximal therapeutic gains can be achieved by pre-emptive therapy before irreversible complications occur compared to a ‘watchful waiting’ approach. It is now established that there is a dose-response relationship for imiglucerase therapy [57]. Therefore, for patients in whom immediate disease control is a priority, i.e., moderate to severe disease including those with life-threatening complications such as hepatopulmonary syndrome and pulmonary hypertension, the initial dose of imiglucerase is usually 120 U/kg body weight in a four-week period. Patients may transition to lower maintenance dosing regimens after initial disease control (Figure 6). In patients with comorbidities such as pulmonary hypertension, enzyme therapy followed by other appropriate adjuvant therapy should be planned [58]. For adult patients with less severe disease, initial dose of 60 U/kg body weight/4 weeks is a prudent approach. Some patients may not require disease-specific treatment because they have very mild or even no disease manifestations; such patients are usually of N370S homozygous genotype. These patients should be monitored regularly to detect signs of disease progression so that treatment can be initiated before new symptoms or irreversible damage occurs [47] (Figure 6). It is important to stress that patients may be asymptomatic, yet harbor significant disease manifestations such as cytopenia, splenomegaly and osteopenia. These patients should receive treatment to reverse disease manifestations.
FIGURE 6.

Baseline assessment, treatment and monitoring algorithm. Regular monitoring of patients receiving therapy ensures maximal therapeutic gains through imiglucerase dose adjustment as required. Once therapeutic goals have been met a maintenance dose may be initiated with ongoing monitoring to ensure that disease does not re-establish [50]. Treatment decisions must be personalized; no one treatment regimen will adequately treat all patients.
Discussion
The diagnosis and management algorithms presented here are likely to be applicable to the majority of patients with Gaucher disease despite there being more than 200 mutations in the GBA1 gene that contribute to vast phenotypic variability. Five mutations (N370S, L444P, 84 ins G, IVS 2+1 and R463C) account for ~90% of disease alleles in patients of Ashkenazi Jewish ancestry and ~50% in patients of non Ashkenazi descent [1]. In contrast, the most prevalent disease genotype worldwide across many ethnicities is L444P, the homozygous state being associated with severe childhood onset and a high risk of CNS involvement [1, 10,11]. There is extreme phenotypic diversity among patients with same GBA1 genotype as well as among affected sib-pairs [59] (Figure 2). Phenotype is influenced only in part by GBA1 gene mutations and the important role of genetic/epigenetic modifiers has been invoked but awaits delineation [60].
Occasionally, Gaucher disease may be associated with manifestations that escape an obvious connection with the underlying enzymatic defect and lysosomal storage of glucosylceramide. The latter underscore our incomplete understanding of the biology of GBA1 mutations and consequences of glucosylceramide accumulation in macrophages and its effects on other cell types. For example, severe Parkinsonian syndrome has been described in some patients with type 1 Gaucher disease, and this has challenged its non-neuronopathic designation [40, 61]. Parkinsonian syndrome in type 1 GD is believed to arise from synuclein aggregation within dopaminergic neurons induced by either gain of function mutations in GBA1 that lead to protein misfolding (i.e, N370S is such a mutation) or accumulation of lipids [62, 63]. Another rare but fatal complication of type 1 GD is pulmonary hypertension that typically occurs in asplenic patients, and may arise via direct involvement of endothelial cells in the disease process [58, 64]. Other examples of unusual complications include myocardial involvement [65], peripheral neuropathy [61], and hematologic malignancies [9, 25, 27, 29–31]. The increased risk of multiple myeloma and gammopathies in type 1 Gaucher disease has been attributed to chronic immune stimulation via activated macrophages [32]. Within type 3 Gaucher disease a distinct phenotype is associated with homozygosity for D409H mutation that exhibits cardiac calcification, non-atherosclerotic coronary artery disease and hydrocephalus [1], and myoclonic epilepsy [40]. Colloidion skin is the most severe manifestation of type 2 disease, a phenotype that is recapitulated in germ-line GBA1 null mice [67]. Taken together, these observations have challenged the macrophage-centric view of Gaucher disease. Indeed, recent insights into the pathophysiology of Gaucher disease have revealed important roles of cell types other than mononuclear phagocytes in development of Gaucher disease [68].
The future
During the last two decades, the management of Gaucher disease and the outlook for affected patients has undergone a remarkable transformation and it represents an excellent exemplar of personalized medicine. Herein, we attempted to harness the advances in the delineation of phenotypic diversity of Gaucher disease, genetics, the natural history and understanding of the impact of enzyme replacement therapy with imiglucerase into algorithms for individualized management. Further exciting progress is expected with advances in gene therapy [69] and small molecule pharmacological chaperone therapy tailored for mutations that result in protein folding defects [70]. A very promising approach is small molecule therapy that acts to inhibit substrate formation, akin to therapy for hypercholesterolemia with HMG CoA reductase inhibitors [22, 23, 71]; this approach is likely to represent a significant advance to current macrophage-directed therapy and promises to overcome some of the unmet needs. Advances in methods of diagnosis using dry blood spots [72, 73] will improve access to testing and timely diagnosis.
Acknowledgments
This paper derives from proceedings of a round table meeting held in Copenhagen and subsequent collaboration of international clinicians. This effort was supported by an unrestricted educational grant from Genzyme Corporation, Cambridge, MA. PKM is supported by NIH NIDDK mid-career investigator award (DK 066306). The authors are grateful to Pam Pickering (Conscience Creative LLP, Leatherhead, UK) for assistance with preparation of the manuscript. Opinions or views expressed in this paper are solely those of the authors.
Appendix 1
Case 1
An 11 year old girl suffered from back pain and had to wear a body cast for several weeks. Further investigations were not performed at this time. At the age of 26, she was diagnosed with Gaucher disease (homozygous for N370S mutation) through family screening following a diagnosis of Gaucher disease in her sister. The patient had mild splenomegaly, hemoglobin 13.0 g/dL, and platelets 135,000/μL. Skeletal involvement was not addressed. At diagnosis, the patient was reassured that she had mild asymptomatic type 1 Gaucher disease and did not require further follow-up. Four years later the patient had an acute onset of severe bone crisis in the left femur [Figure 7]. Investigations showed hemoglobin of 10.3 g/dL, platelets 121,000/μL, liver volume 1.4 x N and spleen volume 7.5 x N. X-rays showed old avascular necrosis and compression fracture of the T7 vertebra (the site of childhood back pain). MRI revealed acute avascular necrosis throughout the left femur and hip. The patient had osteoporosis (DEXA T score of −2.8 (normal range +1 to −1). At the age of 30 years, she started enzyme therapy. At the age of 35 years, she underwent left hip replacement.
FIGURE 7.
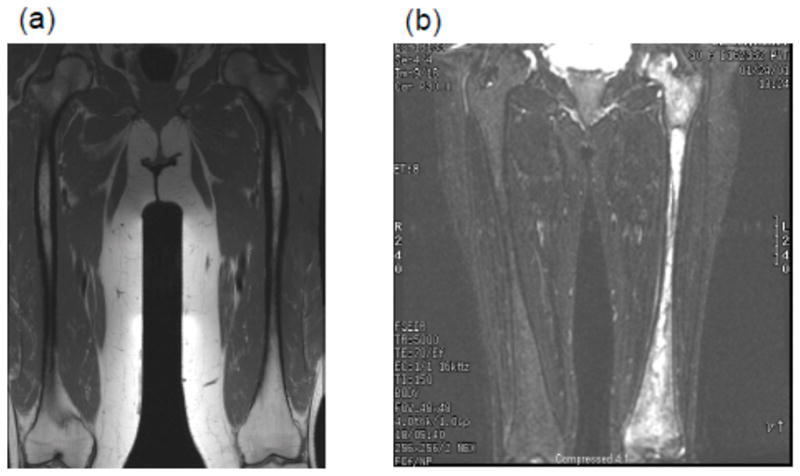
(a and b). MRI showing extensive avascular osteonecrosis in the left femur in a previously asymptomatic woman homozygous for N370S mutation with minimal hepatosplenomegaly and cytopenia (b). There is extensive medullary infarction in the left hip that eventually necessitated hip replacement. There is expansion of cellular marrow in the right femur due to marrow infiltration. For comparison, normal marrow MRI appearance is depicted in (a) that shows fatty marrow.
Comment
Back pain in childhood due to avascular necrosis was not investigated. When the patient was diagnosed with Gaucher disease as an adult through family screening, severe underlying bone disease was not identified due to undue focus on splenic/hepatic and hematologic parameters, which indicated only mild disease.
Case 2
A 20 year old woman was investigated for chronic liver disease due to the finding of hepatosplenomegaly and elevated liver enzymes. Investigations were negative and she underwent splenectomy. The patient had suffered chronic bone pain in childhood, diagnosed as osteomyelitis. At the age of 26 years, she was diagnosed with cryptogenic cirrhosis. At 58 years of age, severe anemia and hemorrhagic episodes due to thrombocytopenia developed. Bone marrow examination revealed almost complete replacement by Gaucher cells. A diagnosis of type 1 Gaucher disease was confirmed through leukocyte glucocerebrosidase assay. Investigations revealed chronic cholestatic liver disease and incipient bone marrow failure.
Comment
The patient remained undiagnosed for 38 years with progressive bone disease, liver disease and eventually bone marrow failure.
Case 3
At a routine physical examination, a 30 year old Ashkenazi male was found to have hypolipidemia (total cholesterol 89 mg/dL; LDL: 42 mg/dL; HDL: 29 mg/dL), and hyperferritinemia (1041 ng/mL). Hemoglobin was 16.1 g/dL, white blood count 7,800 μL platelets 148,000 μL, bilirubin 1.23 mg/dL. Family history was positive for an uncle with early onset Parkinson’s disease. A liver biopsy revealed Gaucher cells (Figure 8]. The diagnosis of type 1 Gaucher disease was confirmed by low acid β-glucosidase activity in blood leukocytes and homozygosity for the N370S mutation. Further evaluation revealed splenomegaly and osteopenia.
FIGURE 8.
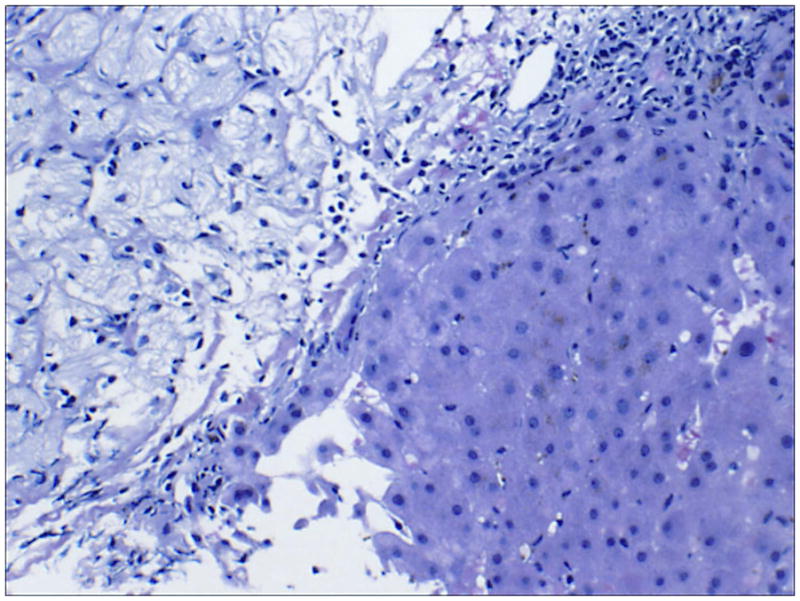
Liver biopsy showing accumulation of mass of lipid-laden Kupfer cells (hepatic Gaucher cells) in the sinusoids adjacent to normal hepatocytes.
Comment
A 30 year old man was diagnosed with Gaucher disease on liver biopsy performed for suspicion of hemochromatosis. Hyperferritinemia and hypolipidemia are common in Gaucher disease.
Case 4
A 72 year old male was followed for 12 years for severe thrombocytopenia, diagnosed as immune thrombocytopenia purpura (ITP). He was treated with steroids without response. The patient had a previous knee injury and developed hip pain, which was thought to be secondary to the knee injury. He was admitted to hospital because of gastrointestinal bleeding. Further evaluation revealed splenomegaly, advanced avascular necrosis of the hip and osteoporosis. Bone marrow biopsy revealed Gaucher cells.
Comment
Misdiagnosis as ITP led to unnecessary chronic corticosteroid therapy and eventually to avascular necrosis and osteoporosis.
Case 5
A 56 year old male patient consulted a gastroenterologist with upper gastrointestinal symptoms due to acute hepatitis A. However there was disproportionate hepatosplenomegaly. Liver and bone marrow biopsies revealed Gaucher cells. The diagnosis was confirmed by low acid β-glucosidase activity in blood leukocytes. Further evaluation confirmed marked splenomegaly, platelets 85,000 μL, elevated chitotriosidase, normal hemoglobin levels and osteopenia.
Comment
Gaucher disease may be associated with gastrointestinal symptoms. In this 56 year old man with acute hepatitis A and incidental hepatosplenomegaly, thorough abdominal examination initially and appropriate investigations could have raised the possibility of Gaucher disease obviating liver and bone marrow biopsy. Both procedures carried a high risk of bleeding in this patient.
References
- 1.Beutler E, Grabowski GA. Gaucher disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular basis of inherited disease. New York, NY: McGraw-Hill; 2001. pp. 3635–3668. [Google Scholar]
- 2.Zimran A, Altarescu G, Rudensky B, et al. Survey of hematological aspects of Gaucher disease. Hematology. 2005;10:151–156. doi: 10.1080/10245330500067181. [DOI] [PubMed] [Google Scholar]
- 3.Cox TM, Schofield JP. Gaucher’s disease: clinical features and natural history. Baillieres Clin Haematol. 1997;10:657–89. doi: 10.1016/s0950-3536(97)80033-9. [DOI] [PubMed] [Google Scholar]
- 4.Grabowski GA. Gaucher disease: gene frequencies and genotype/phenotype correlations. Genet Test. 1997;1:5–12. doi: 10.1089/gte.1997.1.5. [DOI] [PubMed] [Google Scholar]
- 5.Horowitz M, Pasmanik-Chor M, Borochowitz Z, et al. Prevalence of glucocerebrosidase mutations in the Israeli Ashkenazi Jewish population. Hum Mutat. 1998;12:240–244. doi: 10.1002/(SICI)1098-1004(1998)12:4<240::AID-HUMU4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 6.Charrow J, Andersson HC, Kaplan P, et al. The Gaucher registry: demographics and disease characteristics of 1698 patients with Gaucher disease. Arch Intern Med. 2000;160:2835–2843. doi: 10.1001/archinte.160.18.2835. [DOI] [PubMed] [Google Scholar]
- 7.Mistry P, Germain DP. Phenotype variations in Gaucher disease. Rev Med Interne. 2006;27 (Suppl 1):S3–10. doi: 10.1016/s0248-8663(06)80002-0. [DOI] [PubMed] [Google Scholar]
- 8.Mistry PK, Sadan S, Yang R, et al. Consequences of diagnostic delays in type 1 Gaucher disease: the need for greater awareness among hematologists-oncologists and an opportunity for early diagnosis and intervention. Am J Hematol. 2007;82:697–701. doi: 10.1002/ajh.20908. [DOI] [PubMed] [Google Scholar]
- 9.Taddei TH, Kacena KA, Yang M, et al. The under-recognized progressive nature of N370S Gaucher disease and high risk of cancer in 403 patients. American J Hematology. 2009;84:208–214. doi: 10.1002/ajh.21362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaplan P, Andersson HC, Kacena KA, Yee JD. The clinical and demographic characteristics of nonneuronopathic Gaucher disease in 887 children at diagnosis. Arch Pediatr Adolesc Med. 2006;160:603–8. doi: 10.1001/archpedi.160.6.603. [DOI] [PubMed] [Google Scholar]
- 11.Zimran A, Kay A, Gelbart T, et al. Gaucher disease. Clinical, laboratory, radiologic, and genetic features of 53 patients. Medicine (Baltimore) 1992;71:337–353. [PubMed] [Google Scholar]
- 12.Weinreb NJ, Charrow J, Andersson HC, et al. Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am J Med. 2002;113:112–9. doi: 10.1016/s0002-9343(02)01150-6. [DOI] [PubMed] [Google Scholar]
- 13.Weinreb N, Barranger J, Packman S, et al. Imiglucerase (Cerezyme) improves quality of life in patients with skeletal manifestations of Gaucher disease. Clin Genet. 2007;71:576–588. doi: 10.1111/j.1399-0004.2007.00811.x. [DOI] [PubMed] [Google Scholar]
- 14.Wenstrup RJ, Kacena KA, Kaplan P, et al. Effect of enzyme replacement therapy with imiglucerase on BMD in type 1 Gaucher disease. J Bone Miner Res. 2007;22:119–26. doi: 10.1359/jbmr.061004. [DOI] [PubMed] [Google Scholar]
- 15.Charrow J, Dulisse B, Grabowski GA, Weinreb NJ. The effect of enzyme replacement therapy on bone crisis and bone pain in patients with type 1 Gaucher disease. Clin Genet. 2007;71:205–211. doi: 10.1111/j.1399-0004.2007.00769.x. [DOI] [PubMed] [Google Scholar]
- 16.Sims KB, Pastores GM, Weinreb NJ, et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort study. Clin Genet. 2008;73:430–440. doi: 10.1111/j.1399-0004.2008.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mistry PK, Deegan P, Vellodi A, et al. Timing of initiation of enzyme replacement therapy after diagnosis of type 1 Gaucher disease: effect on incidence of avascular necrosis. Br J Haematol. 2009;147:561–70. doi: 10.1111/j.1365-2141.2009.07872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elstein D, Cohn GM, Wang N, et al. Early achievement and maintenance of the therapeutic goals using velaglucerase alfa in type 1 Gaucher disease. Blood Cells Mol Dis. 2010 Aug 18; doi: 10.1016/j.bcmd.2010.07.008. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 19.US National Institutes of Health. http://clinicaltrials.gov NCT01132690; NCT00712348; NCT00376168; NCT00962260.
- 20.European Agency for the Evaluation of Medicinal Products (EMEA) Zavesca prescribing data. 2002 http://www.emea.europa.eu/humandocs/PDFs/EPAR/zavesca/H-435-PI-en.pdf.
- 21.Food and Drug Adminstration (FDA) Zavesca prescribing data. 2003 http://www.fda.gov/cder/foi/label/2003/21348_zavesca_lbl.pdf.
- 22.Lukina E, Watman N, Arreguin EA, et al. A phase 2 study of eliglustat tartrate (Genz-112638), an oral substrate reduction therapy for Gaucher disease type 1. Blood. 2010;116(6):893–899. doi: 10.1182/blood-2010-03-273151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lukina E, Watman N, Avila Arreguin E, et al. Improvement in hematological, visceral, and skeletal manifestations of Gaucher disease type 1 with oral eliglustat tartrate (Genz-112638) treatment: two-year results of a Phase 2 study. Blood. 2010 Aug 16; doi: 10.1182/blood-2010-06-293902. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mistry P, Germain DP. Therapeutic goals in Gaucher disease. Rev Med Interne. 2006 Mar 27;(Suppl 1):S30–8. doi: 10.1016/s0248-8663(06)80009-3. [DOI] [PubMed] [Google Scholar]
- 25.Hughes D, Cappellini MD, Berger M, et al. Recommendations for the management of the haematological and onco-haematological aspects of Gaucher disease. Br J Haematol. 2007;138:676–86. doi: 10.1111/j.1365-2141.2007.06701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scott SA, Edelmann L, Liu L, et al. Experience with Carrier Screening and Prenatal Diagnosis for Sixteen Ashkenazi Jewish Genetic Diseases. Hum Mutat. 2010 Jul 29; doi: 10.1002/humu.21327. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee RE. The pathology of Gaucher disease. In: Desnick RJ, Grabowski GA, editors. Gaucher disease: A Century of Delineation and Research. New York, NY: Alan R Liss Inc; 1982. pp. 177–217. [Google Scholar]
- 28.Shiran A, Brenner B, Laor A, Tatarsky I. Increased risk of cancer in patients with Gaucher disease. Cancer. 1993;72:219–224. doi: 10.1002/1097-0142(19930701)72:1<219::aid-cncr2820720139>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 29.Rosenbloom BE, Weinreb NJ, Zimran A, et al. Gaucher disease and cancer incidence: a study from the Gaucher Registry. Blood. 2005;105:4569–4572. doi: 10.1182/blood-2004-12-4672. [DOI] [PubMed] [Google Scholar]
- 30.Zimran A, Liphshitz I, Barchana M, et al. Incidence of malignancies among patients with type I Gaucher disease from a single referral clinic. Blood Cells Mol Dis. 2005;34:197–200. doi: 10.1016/j.bcmd.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 31.de Fost M, Vom Dahl S, Weverling GJ, et al. Increased incidence of cancer in adult Gaucher disease in Western Europe. Blood Cells Mol Dis. 2006;36:53–58. doi: 10.1016/j.bcmd.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 32.Hughes DA. Enzyme, substrate and myeloma in Gaucher disease. Amer J Hematol. 2009;84:199–201. doi: 10.1002/ajh.21375. [DOI] [PubMed] [Google Scholar]
- 33.Israel National Cancer Registry (Israel) http://www.health.gov.il/pages/default.asp?maincat=22.
- 34.Pastores GM, Weinreb NJ, Aerts H, et al. Therapeutic goals in the treatment of Gaucher disease. Semin Hematol. 2004;41(4 Suppl 5):4–14. doi: 10.1053/j.seminhematol.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 35.Cox TM, Aerts JM, Belmatoug N, et al. Management of non-neuronopathic Gaucher disease with special reference to pregnancy, splenectomy, bisphosphonate therapy, use of biomarkers and bone disease monitoring. J Inherit Metab Dis. 2008;31:319–336. doi: 10.1007/s10545-008-0779-z. [DOI] [PubMed] [Google Scholar]
- 36.Stein P, Malhotra A, Haims A, et al. Focal splenic lesions in type I Gaucher disease are associated with poor platelet and splenic response to macrophage-targeted enzyme replacement therapy. J Inherit Metab Dis. 2010 Aug 4; doi: 10.1007/s10545-010-9175-6. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kadir RA, Kingman CE, Chi C, et al. Screening for factor XI deficiency amongst pregnant women of Ashkenazi Jewish origin. Haemophilia. 2006;12:625–628. doi: 10.1111/j.1365-2516.2006.01347.x. [DOI] [PubMed] [Google Scholar]
- 38.Costello R, O’Callaghan T, Sébahoun G. Gaucher disease and multiple myeloma. Leuk Lymphoma. 2006;47:1365–1368. doi: 10.1080/10428190600565453. [DOI] [PubMed] [Google Scholar]
- 39.Charrow J, Esplin JA, Gribble TJ, et al. Gaucher disease: recommendations on diagnosis, evaluation, and monitoring. Arch Intern Med. 1998;158:1754–1760. doi: 10.1001/archinte.158.16.1754. [DOI] [PubMed] [Google Scholar]
- 40.Sidransky E. Gaucher disease: complexity in a “simple” disorder. Mol Genet Metab. 2004;83:6–15. doi: 10.1016/j.ymgme.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 41.Shenjere P, Roy A, Eyden B, Banerjee SS. Pseudo-Gaucher cells in multiple myeloma. Int J Surg Pathol. 2008;16:176–179. doi: 10.1177/1066896907311120. [DOI] [PubMed] [Google Scholar]
- 42.Saito T, Usui N, Asai O, et al. Pseudo-Gaucher cell proliferation associated with myelodysplastic syndrome. Int J Hematol. 2007;85:350–353. doi: 10.1532/IJH97.06153. [DOI] [PubMed] [Google Scholar]
- 43.Büsche G, Majewski H, Schlué J, et al. Frequency of pseudo-Gaucher cells in diagnostic bone marrow biopsies from patients with Ph-positive chronic myeloid leukaemia. Virchows Arch. 1997;430:139–148. doi: 10.1007/BF01008035. [DOI] [PubMed] [Google Scholar]
- 44.Links TP, Karrenbeld A, Steensma JT, et al. Fatal respiratory failure caused by pulmonary infiltration by pseudo-Gaucher cells. Chest. 1992;101:265–266. doi: 10.1378/chest.101.1.265. [DOI] [PubMed] [Google Scholar]
- 45.Dunn P, Kuo MC, Sun CF. Pseudo-Gaucher cells in mycobacterial infection: a report of two cases. J Clin Pathol. 2005;58:1113–1114. doi: 10.1136/jcp.2004.018176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bain BJ, Lee L. Pseudo-Gaucher cells in sickle cell anemia. Amer J Hematol. 2010;85:435. doi: 10.1002/ajh.21647. [DOI] [PubMed] [Google Scholar]
- 47.Weinreb NJ, Aggio MC, Andersson HC, et al. Gaucher disease type 1: revised recommendations on evaluations and monitoring for adult patients. Semin Hematol. 2004;41(4 Suppl 5):15–22. doi: 10.1053/j.seminhematol.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 48.Aerts JM, Breemen MJ, Bussink AP, et al. Biomarkers for lysosomal storage disorders: identification and application as exemplified by chitotriosidase in Gaucher disease. Acta Paediatr Suppl. 2008;97:7–14. doi: 10.1111/j.1651-2227.2007.00641.x. [DOI] [PubMed] [Google Scholar]
- 49.Stein P, Yu H, Jain D, Mistry PK. Hyperferritinemia and iron overload in type 1 Gaucher disease. Amer J Hematol. 2010;85:472–476. doi: 10.1002/ajh.21721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andersson HC, Charrow J, Kaplan P, et al. International Collaborative Gaucher Group U.S. Regional Coordinators. Individualization of long-term enzyme replacement therapy for Gaucher disease. Genet Med. 2005;7:105–110. doi: 10.1097/01.gim.0000153660.88672.3c. [DOI] [PubMed] [Google Scholar]
- 51.Di Rocco M, Giona F, Carubbi F, et al. A new severity score index for phenotypic classification and evaluation of responses to treatment in type I Gaucher disease. Haematologica. 2008;93:1211–1218. doi: 10.3324/haematol.12379. [DOI] [PubMed] [Google Scholar]
- 52.Weinreb NJ, Cappellini MD, Cox TM, et al. A validated disease severity scoring system for adults with type 1 Gaucher disease. Genet Med. 2010;12:44–51. doi: 10.1097/GIM.0b013e3181c39194. [DOI] [PubMed] [Google Scholar]
- 53.Davies EH, Surtees R, DeVile C, et al. A severity scoring tool to assess the neurological features of neuronopathic Gaucher disease. J Inherit Metab Dis. 2007;30:768–782. doi: 10.1007/s10545-007-0595-x. [DOI] [PubMed] [Google Scholar]
- 54.Vellodi A, Tylki-Szymanska A, Davies EH, et al. Management of neuronopathic Gaucher disease: revised recommendations. J Inherit Metab Dis. 2009;32:660–664. doi: 10.1007/s10545-009-1164-2. [DOI] [PubMed] [Google Scholar]
- 55.Weinreb NJ, Deegan P, Kacena KA, Mistry P, Pastores GM, Velentgas P, vom Dahl S. Life expectancy in Gaucher disease type 1. Am J Hematol. 2008;83:896–900. doi: 10.1002/ajh.21305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weinreb N, Taylor J, Cox T, et al. A benchmark analysis of the achievement of therapeutic goals for type 1 Gaucher disease patients treated with imiglucerase. Amer J Hematol. 2008;83:890–895. doi: 10.1002/ajh.21280. [DOI] [PubMed] [Google Scholar]
- 57.Grabowski G, Kacena K, Hollak CE, et al. Dose-response relationships for enzyme replacement therapy with imiglucerase/alglucerase in patients with Gaucher disease type 1. Genet Med. 2009;11:92–100. doi: 10.1097/GIM.0b013e31818e2c19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mistry PK, Sirrs S, Chan A, et al. Pulmonary hypertension in type 1 Gaucher’s disease: genetic and epigenetic determinants of phenotype and response to therapy. Mol Genet Metab. 2002;77:91–98. doi: 10.1016/s1096-7192(02)00122-1. [DOI] [PubMed] [Google Scholar]
- 59.Amato D, Stachiw T, Clarke JT, Rivard GE. Gaucher disease: variability in phenotype among siblings. J Inherit Metab Dis. 2004;27:659–669. doi: 10.1023/b:boli.0000042983.60840.f3. [DOI] [PubMed] [Google Scholar]
- 60.Goker-Alpan O, Hruska KS, Orvisky E, et al. Divergent phenotypes in Gaucher disease implicate the role of modifiers. J Med Genet. 2005;42:e37. doi: 10.1136/jmg.2004.028019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bultron G, Kacena K, Pearson D, et al. The risk of Parkinson’s disease in type 1 Gaucher disease. J Inherit Metab Dis. 2010;33:167–173. doi: 10.1007/s10545-010-9055-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goker-Alpan O, Lopez G, Vithayathil J, et al. The spectrum of parkinsonian manifestations associated with glucocerebrosidase mutations. Arch Neurol. 2008;65:1353–1357. doi: 10.1001/archneur.65.10.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aharon-Peretz J, Badarny S, Rosenbaum H, et al. Mutations in the glucocerebrosidase gene and Parkinson disease: phenotype-genotype correlation. Neurology. 2005;65:1460–1461. doi: 10.1212/01.wnl.0000176987.47875.28. [DOI] [PubMed] [Google Scholar]
- 64.Elleder M. Glucosylceramide transfer from lysosomes--the missing link in molecular pathology of glucosylceramidase deficiency: a hypothesis based on existing data. J Inherit Metab Dis. 2006;29:707–715. doi: 10.1007/s10545-006-0411-z. [DOI] [PubMed] [Google Scholar]
- 65.Torloni MR, Franco K, Sass N. Gaucher’s disease with myocardial involvement in pregnancy. Sao Paulo Med J. 2002;120:90–92. doi: 10.1590/S1516-31802002000300008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Capablo JL, Saenz de Cabezón A, Fraile J, et al. Spanish Group on Gaucher disease. Neurological evaluation of patients with Gaucher disease diagnosed as type 1. J Neurol Neurosurg Psychiatry. 2008;79:219–222. doi: 10.1136/jnnp.2006.111518. [DOI] [PubMed] [Google Scholar]
- 67.Stone DL, Carey WF, Christodoulou J, et al. Type 2 Gaucher disease: the collodion baby phenotype revisited. Arch Dis Child Fetal Neonatal Ed. 2000;82:F163–6. doi: 10.1136/fn.82.2.F163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mistry PK, Liu J, Yang M, et al. GBA1 deficiency in the mouse recapitulates Gaucher disease displaying system-wide cellular and molecular dysregulation beyond the macrophage. Proc Natl Acad Sci USA. 2010 doi: 10.1073/pnas.1003308107. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McEachern KA, Nietupski JB, Chuang WL, et al. AAV8-mediated expression of glucocerebrosidase ameliorates the storage pathology in the visceral organs of a mouse model of Gaucher disease. J Gene Med. 2006;8:719–729. doi: 10.1002/jgm.901. [DOI] [PubMed] [Google Scholar]
- 70.Mu TW, Ong DS, Wang YJ, et al. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell. 2008;134:769–781. doi: 10.1016/j.cell.2008.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McEachern KA, Fung J, Komarnitsky S, et al. A specific and potent inhibitor of glucosylceramide synthase for substrate inhibition therapy of Gaucher disease. Mol Genet Metab. 2007;91:259–267. doi: 10.1016/j.ymgme.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 72.Chamoles NA, Blanco M, Gaggioli D, Casentini C. Gaucher and Niemann-Pick diseases--enzymatic diagnosis in dried blood spots on filter paper: retrospective diagnoses in newborn-screening cards. Clin Chim Acta. 2002;317:191–7. doi: 10.1016/s0009-8981(01)00798-7. [DOI] [PubMed] [Google Scholar]
- 73.Olivova P, Cullen E, Titlow M, et al. An improved high-throughput dried blood spot screening method for Gaucher disease. Clin Chim Acta. 2008;398:163–4. doi: 10.1016/j.cca.2008.08.024. [DOI] [PubMed] [Google Scholar]