

Abstract
NMDA receptor overactivation triggers intracellular Ca2+ dysregulation, which has long been thought to be critical for initiating excitotoxic cell death cascades associated with stroke and neurodegenerative disease. The inability of NMDA receptor antagonists to afford neuroprotection in clinical stroke trials has led to a re-evaluation of excitotoxic models of cell death and has focused research efforts towards identifying additional Ca2+ influx pathways. Recent studies indicate that TRPM2, a member of the TRPM subfamily of Ca2+-permeant, non-selective cation channel, plays an important role in mediating cellular responses to a wide range of stimuli that, under certain situations, can induce cell death. These include reactive oxygen and nitrogen species, tumour necrosis factor as well as soluble oli-gomers of amyloid beta. However, the molecular basis of TRPM2 channel involvement in these processes is not fully understood. In this review, we summarize recent studies about the regulation of TRPM2, its interaction with calcium and the possible implications for neurodegenerative diseases.
Keywords: TRPM2, oxidative stress, neurodegeneration, amyloid beta, stroke, NMDA receptor
Introduction
Traditional models of neuronal excitotoxicity have focused on the glutamate-mediated over-activation of receptors such as the ionotropic N-methyl-D-aspartate (NMDA) glutamate receptor subtype (NMDAR). However, glutamate receptor blockade, antioxidant or anti-inflammatory therapy has failed to provide neuroprotection in clinical stroke trials. Accordingly, the focus of recent research has shifted towards identifying neurotoxic signalling molecules positioned further downstream of NMDAR stimulation. These efforts have yielded exciting implications as to specific strategies for treating excitotoxic disorders. Specifically, several previously overlooked Ca2+ influx pathways have recently been identified. These include acid sensing and pannexin channels, as well as a member of the melas-tatin subfamily of transient receptor potential (TRP) channels [1-3]. Increasing evidence has shown that TRP channels represent an exciting new family of cation channels, several members of which are highly expressed in the brain [4-7]. The mammalian TRP family consists of six main subfamilies termed the TRPC (canonical), TRPV (vanilloid), TRPM (melastatin), TRPP (polycystin), TRPML (mucolipin), and TRPA (ankyrin) subfamilies. Collectively, the broad and varied physiological roles of TRP channels are being increasingly appreciated as is their contribution to a number of diseases [5;6;8;9]. TRPM2, one member of the TRPM subfamily, is a Ca2+-permeable, non-selective cation channel, that plays a critical role in mediating cellular responses to a wide range of physiological stimuli that can also induce cell death in response to oxidative stress [9-11].
TRPM2 channel structure
TRPM2 consists of six putative transmembrane domains, a re-entrant pore forming loop and intracellular N- and C- termini. Channels, thought to form as homotetramers, have high monovalent cation permeability with variable permeability for Ca2+, Mg2+ and other divalent cations [12-15] (Figure 1). Intriguingly, the divalent cation permeability appears to vary substantially with heat [16]. The transmembrane segments TM5 and TM6 are highly conserved with its related family member TRPM8, and the distal part of S6 is crucial for charge discrimination [17]. Furthermore, the Glu-960, Gln-981, Asp-987, and Glu-1022 residues of TRPM2 channel are found to be engaged in determining divalent cation permeation properties [13] while two conserved cysteine residues (at positions 996 and 1008), located within the putative pore region of the human TRPM2, are obligatory for TRPM2 channel function [18] (Figure 1). The C-terminus of TRPM2 can be further subdivided into an approximately 100 amino acid region of high coiled coil character, a short perhaps 30 amino acid linker region, and the approximately 270 amino acid NUDT9-homology domain (NUDT9-H) [11]. Within NUDT9-H lies the so-called Nudix box, a region of 22 amino acids that is conserved among a family of enzymes that catalyze the hydrolysis of nucleoside di-phosphates. The recognition of this motif led Scharenberg's group [19] to identify a unique mode of channel activation: TRPM2 is gated by cytosolic ADPR. Accordingly, TRPM2 has been dubbed a “chanzyme” (channel-enzyme) possessing both ion conducting and enzyme domains. However, it should be noted that the NUDT9-H enzyme activity of TRPM2 is severely restricted relative to that of NUDT9 due to specific amino acid substitutions (EF → IL) within the Nudix box [20].
Figure 1.
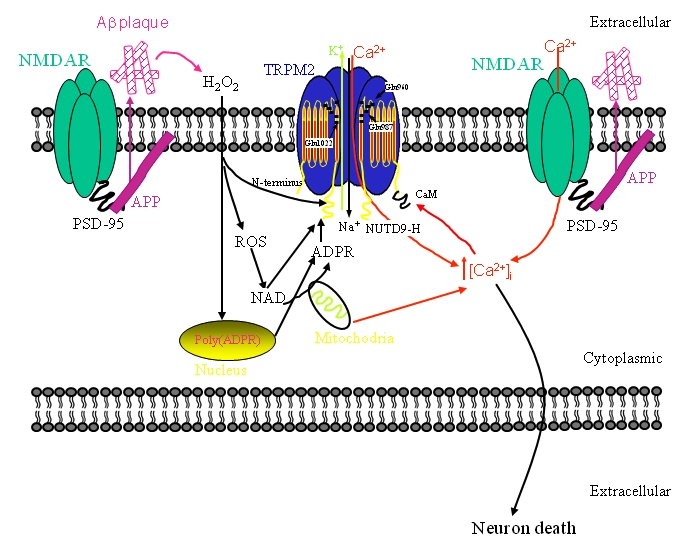
Schematic diagraph showing the signal pathway of TRPM2 and NMDAR. Abbreviations see text.
TRPM2 distribution
TRPM2 is widely distributed in the CNS including the hippocampus, cerebral cortex, thalamus, and midbrain [4, 12, 21-24]. Although initial reports suggested that the expression of TRPM2 within the CNS was restricted to non-neuronal cells, subsequent more exhaustive examination leaves little doubt that TRPM2 is also expressed within neurons [24-26]. However, that is not to say that neuronal expression is ubiquitous throughout all CNS regions and within all neuronal subtypes. Indeed, transcripts for TRPM2 could not be detected within cultured granule cells of the cerebellum [12] nor could we find functional evidence of TRPM2 expression in stratum radiatum interneurons of the hippocampus [24]. Thus, though broadly distributed, TRPM2 expression within specific cell types (e.g. neurons, astrocytes and microglia) may vary regionally which, correspondingly, likely influences the functional consequences of TRPM2 activation in each region.
At the subcellular level, limited information is available to date. In cultured cerebral cortical neurons from fetal rat, TRPM2 could be detected within cell bodies and neurites. Interestingly, molecular cloning of TRPM2 from rat brain cDNA revealed several differences in amino acid sequence within the Nudix box region as compared with those of human and mouse TRPM2 [27]. As a result, the sensitivity of rat TRPM2 to ADPR appears somewhat greater with all other properties being similar to human and mouse TRPM2. Within cultured hippocampal neurons we reported a similar expression profile within soma and neurites [24]. In addition, we found that the expression pattern appeared diffuse and did not specifically co-localize with PSD-95, a marker of excitatory synapses. Although more detailed co-localization is needed, the evidence thus far suggests that TRPM2 may preferentially be expressed within extrasynaptic regions of hippocampal pyramidal neurons.
In addition to full length transcripts, four splice variants of TRPM2 have been identified: TRPM2 -S, TRPM2-AN, TRPM2-AC and SSF-TRPM2. TRPM2-S (short) has a deletion of the entire C-terminus, including four of six C-terminal trans-membrane domains and the putative channel pore [28]. TRPM2-AN has a deletion of amino acids 538-557 in the N-terminus. TRPM2-AC has a deletion of amino acids 1292-1325 in the C-terminal CAP domain of NUDT9-H that decreases the affinity for ADPR [29]. Finally, a short form lacking the first 214 amino acids of the N-terminal was identified as being expressed uniquely within the striatum and has thus been termed the striatum short form protein (SSF-TRPM2)[30]. The functional significance of these naturally occurring variants is not presently fully appreciated nor have the relative expression levels by region been determined. It is known however, that the expression of TRPM2-S can suppress the channel activity of the full-length variant [22, 28]. Thus, the relative expression level of each variant could have important consequences for TRPM2 function.
TRPM2 gating mechanisms
In addition to ADPR, evidence has accumulated suggesting that TRPM2 may be gated by several means in a coordinated manner. Several adenine intracellular second messengers, metabolically related to ADPR, have been proposed to gate TRPM2 channels. These include nicotinamide adenine dinucleotide (NAD+), which serves as a precursor for the formation of ADPR, as well as cyclic ADPR (cADPR), a well recognized regulator of intracellular Ca2+ homeostasis. The ability of each of these adenine metabolites to gate the channels directly has been controversial [19, 31, 32]. Indeed, it is now generally agreed that activation by NAD+ at room temperature may be due to the presence of contaminating ADPR. Similarly, while two groups have reported that cADPR is incapable of gating TRPM2 directly [19, 31], another has reported that relatively high concentrations (e.g. > 100 μM) may do so [32]. Perhaps more interestingly, in the latter study the authors reported that modest concentrations of cADPR (10 μM) may facilitate TRPM2 function such that nM concentrations of ADPR may gate the channel. Previously considered to regulate Ca2+-induced Ca2+ release via ryanodine receptors, the findings suggest an expanded role for cADPR in regulating intracellular Ca2+ concentration. More recently, TRPM2 gating has been shown to be heavily influenced by temperature and is in fact directly gated by heat, the threshold for activation being 35°C [16]. Moreover, heat was shown to greatly facilitate gating by ADPR and, interestingly, by NAD+ as well as by cADPR, suggesting that TRPM2 channel activity may not be solely regulated by the intracellular levels of ADPR. A further layer of complexity is added by the finding that ADPR's breakdown product, adenosine monophosphate (AMP) specifically inhibits ADPR, but not cADPR-mediated gating of TRPM2, whereas the cADPR antagonist 8-Br-cADPR exhibits the reverse block specificity [32]. Collectively, these studies suggest that the regulation of TRPM2 channel activity is tightly regulated by the relative levels of these intracellular messengers.
TRPM2 has also been reported to be gated by oxidative and nitrosative stress (ROS/RNS). Gating by ROS/RNS requires an intact ADPR binding cleft in the C-terminal domain, suggesting that gating occurs through the intracellular production of ADPR. More definitive evidence comes from recent studies showing that ROS/ RNS induces mitochondria to produce free ADPR and release it to the cytosol where subsequent accumulation of ADPR induces TRPM2 gating [33,34] (Figure 1). Oxidative stress, may also induce the formation of ADPR in the nucleus following activation of DNA repair enzymes [34, 35] (as shown in Figure 1) poly(ADP-ribose) polymerases (PARPs) and glycohy-drolases (PARG). These enzymes first poly-ADP-ribosylates nuclear proteins and then liberate monomers of ADPR, respectively. In addition to being sensitive to ROS/RNS, more recently TRPM2 was shown to be responsive to cell stress induced by puromycin through the recruitment of Sir2 family of NAD+-dependent protein deacetylases and the production of the metabolite 2'-O-acetyl-ADP-ribose (OAADPR) [36].
Irrespective of the activating mechanism, TRPM2 currents possess a linear current-voltage relationship with a reversal potential of about 0 mV. Single channel recordings reveal a slope conductance of 60-80 pS and distinctly long open times (in the range of several hundred milliseconds to seconds). TRPM2 channels are permeable to monovalent cations as well as Ca2+ and Mg2+ and have no apparent voltage dependence. However, at high intracellular Na+ concentrations TRPM2 channels undergo a hy-perpolarization-dependent inactivation [19].
In addition to ADPR, cADPR, H2O2, intracellular ions typically used in patch-clamp experiments such as Sr2+, Ca2+, Cs+ or Na+ can alter ADPR sensitivity and voltage dependence of TRPM2 channels, complicating the evaluation of the roles of the various modulators in a physiological context. Lastly, TRPM2 currents are not suppressed by La3+, a broad spectrum blocker of non-selective cation channels, but can be inhibited by several blockers including flufenamic acid, clotrimazole, econazole and ACA in the micromolar range [7, 34].
TRPM2 and calcium
TRPM2 gating by ADPR is influenced by the intracellular concentration of Ca2+. This was initially reported by Scharenberg's group when they showed that raising intracellular Ca2+ lowered the EC50 for channel activation by ADPR [19]. The importance of Ca2+ in regulating TRPM2 channel opening is highlighted by recordings in leucocytes demonstrating that the endogenous concentration of ADPR is insufficient to activate TRPM2 when [Ca2+]i is at resting levels. However, when [Ca2+]i is elevated, these same levels of intracellular ADPR are now sufficient to permit gating [7]. Importantly, subsequent ion substitution experiments allowed the identification of one of the most distinctive features of TRPM2, namely that extracellular Ca2+ acts as a positive regulator of TRPM2 [31]. Indeed, omission of Ca2+ from external solutions prevents or rapidly abrogates TRPM2 activation. To the best of our knowledge, no other channel displays this unique property. Thus, in the absence of specific channel blockers for TRPM2, lowering of extracellular Ca2+ provides a means of distinguishing TRPM2 generated currents from other potential contributing conductances. This can be useful when studying the function of endogenously expressed TRPM2 channels; especially in neurons where numerous voltage- as well as ligand-gated conductances can contribute to cationic membrane flux [24].
Therefore, TRPM2 supports Na+ and Ca2+ influx, thereby modulating membrane potential as well as intracellular Ca2+ levels. On the other hand, the sensitivity of TRPM2 to ADPR is also facilitated by intracellular Ca2+ [37-39] via positive feedback. Rather than acting at an extracellular site, Ca2+ entering via TRPM2 acts upon an intracellular calcium sensor closely associated with the channel [31, 38]. Once opened, Ca2+ entering through the channel pore may slow channel closure by keeping a Ca2+ activating site saturated. This activating site is proposed to lie intracellularly from the gate, but in a shielded crevice near the pore entrance [40]. The activating site in question appears to correspond to a Ca2+-CAM binding site located within the N-terminus [39;41], see Figure 1. Importantly, Ca2+-CAM may do more that simply modulate the sensitivity of TRPM2 for ADPR. Indeed, a recent study, investigating the gating mechanisms in TRPM2 splice variants and mutants in which ADPR-binding has been disrupted, revealed that elevations of intracellular Ca2+ may be sufficient, in and of themselves, to gate TRPM2 channels [39, 41].
TRPM2 and Diseases
In the brain, Ca2+ is fundamental in the control of synaptic activity and memory formation, a process that leads to the activation of specific Ca2+ -dependent signal transduction pathways. The identification of several modulators of Ca2+ homeostasis as potential factors involved in the pathogenesis of Alzheimer's (AD), Parkinson's (PD), and Huntington's (HD) diseases, provides strong support for a role of Ca2+ dysregulation in neurodegeneration [42, 43]. The influx of Na+ and Ca2+ via TRPM2 channels, which promotes membrane depolarization and increases in the cytosolic Ca2+ concentration ([Ca2+]i), and the wide expression profile of TRPM2 renders it a potentially significant therapeutic target in a variety of pathological settings including cardiovascular and neurodegenerative diseases [9-11,44-48].
TRPM2 and neuronal injury and death
Activation of TRPM2 by a variety of means, including TNFα, ROS/RNS and ADPR, has been linked to cell death, suggesting that TRPM2 is likely to be a key player downstream of several signalling pathways mediating cell death in response to cerebral ischemia and reperfusion injury [49]. In the rat transient middle cerebral artery occlusion (tMCAO) stroke model, TRPM2 mRNA expression increased at 1 and 4 weeks following ischemic injury, partially as a consequence of glial activation since the profile is temporally consistent with the pattern of microglial activation seen in this model. This suggests that TRPM2, expressed in glia, may play a role in the pathophysiology produced following a transient period of ischemia [23]. However, another study indicated that oxidative stress, induced by exogenous application of H2O2, caused preferential damage of pyramidal neurons in hippocampus [25]. Consistent with this, treatment of cultured cerebral cortex neurons with small interfering RNA against rat TRPM2 efficiently suppressed TRPM2 immunoreactivity, the H2O2-induced Ca2+ influx and neuronal death, suggesting that TRPM2 plays a pivotal role in H2O2-induced neuronal death [27]. During a period of acute ischemia in vivo or oxygen-glucose deprivation (OGD) in vitro, a TRP-like channel is activated by cellular stress and contributes to ischemia-induced membrane depolarization, intracellular Ca2+ accumulation and cell swelling in CA1 neurons [4]. Moreover, clotrimazole, a TRPM2 channel blocker [7, 10], was found to block TRPM2 currents in hippo-campal pyramidal neurons [24], be neuroprotective in spinal cord clip compression injury [50] and reduce the death in cultured rat cere-bellar granule and hippocampal neurons induced by excitotoxic insults [51]. While cognizant of the fact that clotrimazole is far from being selective for TRPM2 and that other targets may have been involved in the effects described above, a collective body of work is nevertheless supportive of a role for TRPM2 as a mediator of neuronal cell death.
TRPM2 and related neurodegenerative diseases
Cognitive impairment and emotional disturbances in AD result from the degeneration of synapses and neuronal death in the limbic system and associated regions of the cerebral cortex. The accumulation of beta amyloid protein (Ab) in the brain is considered to be a key factor that causes AD. Ap can render neurons vulnerable to excitotoxicity and apoptosis by disruption of cellular Ca2+ homeostasis and membrane-associated oxidative stress [46, 52]. Recent findings, suggest that, in addition to its role in H2O2 induced-neuronal death of striatal neurons, TRPM2 may also be implicated in Ap-induced death of cultured striatal neurons [22]. Transfection with a splice variant (TRPM2-S) that acts as a dominant negative inhibitor of TRPM2 function was shown to inhibit both H2O2-and Ap-induced increases in intracellular-free Ca2+ and cell death. Furthermore, small interfering RNA targeting TRPM2 reduced TRPM2 mRNA levels and the toxicity induced by H2O2 and Ap [22]. More recently, Ap has been suggested to directly incorporate into neuronal membranes where it may form Ca2+ -permeable ion channels (amyloid channels) that cause abnormal elevations of intracellular Ca2+ [53]. This supplemental Ca2+ entry would be expected to facilitate further TRPM2 channel activation and consequent rise in intracellular Ca2+. More broadly, the specific coupling of TRPM2 activation to pathways leading to increased oxidative stress might implicate TRPM2 in other neurodegenerative diseases. Specific examples have recently been revealed by studies suggesting that TRPM2 plays important role in motor neuron death associated with Guamanian Amyotro-phic Lateral Sclerosis (ALS-G) and parkinsonism dementia (PD-G) [44, 45].
In addition, the finding that reduced mRNA levels of TRPM2 channel in B lymphoblast cell lines (BLCL) from bipolar I disorder (BD-I) patients showing elevated basal [Ca2+]i, an index of altered intracellular Ca2+ homeostasis, implicates the involvement of this gene in the Ca2+ abnormalities and the pathophysiology of bipolar disorder [54].
Potential link between Amyloid p protein, NMDA receptor and TRPM2
Acute brain injuries have been identified as a risk factor for developing AD and glutamate plays a pivotal role in these pathologies. In primary cultures of cortical neurons, sublethal NMDAR activation was found to alter the processing of the amyloid precursor protein (APP). Indeed, Ca2+ entry through NMDARs increased the expression of neuronal Kunitz protease inhibitory domain (KPI) containing APP (KPI-APP). Expression of KPI-APPs favoured p-secretase processing, instead of the normally dominant a-secretase processing, leading to increased production and secretion of Ap [55]. Interestingly, the salient features of these NMDAR-mediated changes in APP processing could be replicated in a mouse model of traumatic brain injury. A further link between NMDAR function and the production Ap comes from studies examining the localization of APP and Ap in relation to markers of excitatory synapses. For example, a pool of APP is found in the postsynaptic compartment in cortical neurons where it partially co-localizes with both NR1 and PSD-95 [56, 57], as illustrated schematically in Figure 1. In addition, immunolocalization of soluble oligomers of Ap, bound to cultured hippocampal neurons, reveals a pattern of staining suggesting that Ap associates with excitatory synapses [58, 59]. Intriguingly, the association of Ap with excitatory synapses could be prevented by pre-treatment with an antibody recognizing the extracellular N-terminus of the NMDAR subunit, NR1. Moreover, the rapid rise in intracellular Ca2+ and ROS formation, observed during acute applications of soluble oligomers of Ap to cultured neurons, could be prevented by NMDAR inhibition [60]. We have shown that hippocampal pyramidal neurons possess functional TRPM2 channels whose activation by ADPR is functionally coupled to voltage-dependent Ca2+ channels (VDCCs) and NMDARs through a rise in [Ca2+]i [24]. In light of the findings discussed above, we suggests that TRPM2, acting in concert with NMDARs, may provide the basis for a positive feedback loop in which Ca2+ influx is facilitated through a pathway involving aberrant NMDAR activation, the production of Ap and the formation of ROS all of which leads to the activation of TRPM2. A schematic figure summarizing this proposed model is shown in Figure 1.
Prospective
Neuroprotection via glutamate receptor blockade, antioxidant or anti-inflammatory therapy has not proven effective in the clinical treatment of brain damage due to narrow therapeutic windows, poor pharmacokinetics and blockade of the signalling essential for normal excitatory neurotransmission and neuronal survival. In light of the wide-spread expression and growing work indicating that TRPM2 may play important role in neuronal death that is activated by oxidative stress and downstream from excitotoxic signal pathways, useful therapeutic applications are expected for future drugs that block TRPM2 channels or inhibit their activation. This will provide an exciting new avenue for research into the pathophysiology and treatment of neuronal death and related CNS diseases.
References
- 1.Aarts M, lihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, MacDonald JF, Tymianski M. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115:863–877. doi: 10.1016/s0092-8674(03)01017-1. 26-12- [DOI] [PubMed] [Google Scholar]
- 2.Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ, Simon RP. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–698. doi: 10.1016/j.cell.2004.08.026. 17-9- [DOI] [PubMed] [Google Scholar]
- 3.Thompson RJ, Zhou N, MacVicar BA. Ischemia opens neuronal gap junction hemichannels. Science. 2006;312:924–927. doi: 10.1126/science.1126241. 12-5- [DOI] [PubMed] [Google Scholar]
- 4.Lipski J, Park TI, Li D, Lee SC, Trevarton AJ, Chung KK, Freestone PS, Bai JZ. Involvement of TRP-like channels in the acute ischemic response of hippocampal CA1 neurons in brain slices. Brain Res. 2006;1077:187–199. doi: 10.1016/j.brainres.2006.01.016. 10-3- [DOI] [PubMed] [Google Scholar]
- 5.Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem. 2007;76:387–417. doi: 10.1146/annurev.biochem.75.103004.142819. 387–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pedersen SF, Owsianik G, Nilius B. TRP channels: an overview. Cell Calcium. 2005;38:233–252. doi: 10.1016/j.ceca.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 7.Eisfeld J, Luckhoff A. TRPM2. Handb Exp Pharmacol. 2007:237–252. doi: 10.1007/978-3-540-34891-7_14. [DOI] [PubMed] [Google Scholar]
- 8.Nilius B, Voets T, Peters J. TRP channels in disease. Sci STKE. 2005 doi: 10.1126/stke.2952005re8. 2005:re8. 2-8- [DOI] [PubMed] [Google Scholar]
- 9.Jordt SE, Ehrlich BE. TRP channels in disease. Subcell Biochem. 2007;45:253–71. doi: 10.1007/978-1-4020-6191-2_9. 253-271. [DOI] [PubMed] [Google Scholar]
- 10.McNulty S, Fonfria E. The role of TRPM channels in cell death. Pflugers Arch. 2005;451:235–242. doi: 10.1007/s00424-005-1440-4. [DOI] [PubMed] [Google Scholar]
- 11.Perraud AL, Schmitz C, Scharenberg AM. TRPM2 Ca2+ permeable cation channels: from gene to biological function. Cell Calcium. 2003;33:519–531. doi: 10.1016/s0143-4160(03)00057-5. [DOI] [PubMed] [Google Scholar]
- 12.Kraft R, Grimm C, Grosse K, Hoffmann A, Sauerbruch S, Kettenmann H, Schultz G, Harteneck C. Hydrogen peroxide and ADP-ribose induce TRPM2-mediated calcium influx and cation currents in microglia. Am J Physiol Cell Physiol. 2004;286:C129–C137. doi: 10.1152/ajpcell.00331.2003. [DOI] [PubMed] [Google Scholar]
- 13.Xia R, Mei ZZ, Mao HJ, Yang W, Dong L, Bradley H, Beech DJ, Jiang LH. Identification of pore residues engaged in determining divalent cationic permeation in transient receptor potential me-lastatin subtype channel 2. J Biol Chem. 2008;283:27426–27432. doi: 10.1074/jbc.M801049200. 10-10- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sano Y, Inamura K, Miyake A, Mochizuki S, Yokoi H, Matsushime H, Furuichi K. Immunocyte Ca2+ influx system mediated by LTRPC2. Science. 2001;293:1327–1330. doi: 10.1126/science.1062473. 17-8- [DOI] [PubMed] [Google Scholar]
- 15.Schnitzler M, Waring J, Gudermann T, Chubanov V. Evolutionary determinants of divergent calcium selectivity of TRPM channels. FASEB J. 2008;22:1540–1551. doi: 10.1096/fj.07-9694com. [DOI] [PubMed] [Google Scholar]
- 16.Togashi K, Hara Y, Tominaga T, Higashi T, Konishi Y, Mori Y, Tominaga M. TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J. 2006;25:1804–1815. doi: 10.1038/sj.emboj.7601083. 6-4- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuhn FJ, Knop G, Luckhoff A. The transmem-brane segment S6 determines cation versus anion selectivity of TRPM2 and TRPM8. J Biol Chem. 2007;282:27598–27609. doi: 10.1074/jbc.M702247200. 21-9- [DOI] [PubMed] [Google Scholar]
- 18.Mei ZZ, Mao HJ, Jiang LH. Conserved cysteine residues in the pore region are obligatory for human TRPM2 channel function. Am J Physiol Cell Physiol. 2006;291:C1022–C1028. doi: 10.1152/ajpcell.00606.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, Stokes AJ, Zhu Q, Bessman MJ, Penner R, Kinet JP, Scharenberg AM. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature. 2001;411:595–599. doi: 10.1038/35079100. 31-5- [DOI] [PubMed] [Google Scholar]
- 20.Perraud AL, Shen B, Dunn CA, Rippe K, Smith MK, Bessman MJ, Stoddard BL, Scharenberg AM. NUDT9, a member of the Nudix hydrolase family, is an evolutionarily conserved mitochon-drial ADP-ribose pyrophosphatase. J Biol Chem. 2003;278:1794–1801. doi: 10.1074/jbc.M205601200. 17-l- [DOI] [PubMed] [Google Scholar]
- 21.Fonfria E, Murdock PR, Cusdin FS, Benham CD, Kelsell RE, McNulty S. Tissue distribution profiles of the human TRPM cation channel family. J Recept Signal Transduct Res. 2006;26:159–178. doi: 10.1080/10799890600637506. [DOI] [PubMed] [Google Scholar]
- 22.Fonfria E, Marshall IC, Boyfield I, Skaper SD, Hughes JP, Owen DE, Zhang W, Miller BA, Benham CD, McNulty S. Amyloid beta-peptide(1-42) and hydrogen peroxide-induced toxicity are mediated by TRPM2 in rat primary striatal cultures. J Neurochem. 2005;95:715–723. doi: 10.1111/j.1471-4159.2005.03396.x. [DOI] [PubMed] [Google Scholar]
- 23.Fonfria E, Mattei C, Hill K, Brown JT, Randall A, Benham CD, Skaper SD, Campbell CA, Crook B, Murdock PR, Wilson JM, Maurio FP, Owen DE, Tilling PL, McNulty S. TRPM2 is elevated in the tMCAO stroke model, transcriptionally regulated, and functionally expressed in C13 microglia. J Recept Signal Transduct Res. 2006;26:179–198. doi: 10.1080/10799890600637522. [DOI] [PubMed] [Google Scholar]
- 24.Olah ME, Jackson MF, Li H, Perez Y, Sun HS, Kiyonaka S, Mori Y, Tymianski M, MacDonald JF. Ca2+-dependent induction of TRPM2 currents in hippocampal neurons. J Physiol. 2009;587:965–979. doi: 10.1113/jphysiol.2008.162289. 1-3- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bai JZ, Lipski J. Differential expression of TRPM2 and TRPV4 channels and their potential role in oxidative stress-induced cell death in organo-typic hippocampal culture. Neurotoxicology. 2010 doi: 10.1016/j.neuro.2010.01.001. 12-1- [DOI] [PubMed] [Google Scholar]
- 26.Amina S, Hashii M, Ma WJ, Yokoyama S, Lopatina O, Liu HX, Islam MS, Higashida H. Intracellu-lar Calcium Elevation Induced by Extracellular Application of Cyclic-ADP-Ribose or Oxytocin is Temperature-Sensitive in Rodent NG108-15 Neuronal Cells with or without Exogenous Expression of Human Oxytocin Receptors. J Neuroendocrinol. 2010 doi: 10.1111/j.1365-2826.2010.01978.x. 12-2- [DOI] [PubMed] [Google Scholar]
- 27.Kaneko S, Kawakami S, Hara Y, Wakamori M, Itoh E, Minami T, Takada Y, Kume T, Katsuki H, Mori Y, Akaike A. A critical role of TRPM2 in neuronal cell death by hydrogen peroxide. J Pharmacol Sci. 2006;101:66–76. doi: 10.1254/jphs.fp0060128. [DOI] [PubMed] [Google Scholar]
- 28.Zhang W, Chu X, Tong Q, Cheung JY, Conrad K, Masker K, Miller BA. A novel TRPM2 isoform inhibits calcium influx and susceptibility to cell death. J Biol Chem. 2003;278:16222–16229. doi: 10.1074/jbc.M300298200. 2-5- [DOI] [PubMed] [Google Scholar]
- 29.Wehage E, Eisfeld J, Heiner I, Jungling E, Zitt C, Luckhoff A. Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. A splice variant reveals a mode of activation independent of ADP-ribose. J Biol Chem. 2002;277:23150–23156. doi: 10.1074/jbc.M112096200. 28-6- [DOI] [PubMed] [Google Scholar]
- 30.Uemura T, Kudoh J, Noda S, Kanba S, Shimizu N. Characterization of human and mouse TRPM2 genes: identification of a novel N-terminal truncated protein specifically expressed in human striatum. Biochem Biophys Res Commun. 2005;328:1232–1243. doi: 10.1016/j.bbrc.2005.01.086. 25-3- [DOI] [PubMed] [Google Scholar]
- 31.Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, Yamada H, Shimizu S, Mori E, Kudoh J, Shimizu N, Kurose H, Okada Y, Imoto K, Mori Y. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell. 2002;9:163–173. doi: 10.1016/s1097-2765(01)00438-5. [DOI] [PubMed] [Google Scholar]
- 32.Kolisek M, Beck A, Fleig A, Penner R. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol Cell. 2005;18:61–69. doi: 10.1016/j.molcel.2005.02.033. 1-4- [DOI] [PubMed] [Google Scholar]
- 33.Perraud AL, Takanishi CL, Shen B, Kang S, Smith MK, Schmitz C, Knowles HM, Ferraris D, Li W, Zhang J, Stoddard BL, Scharenberg AM. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J Biol Chem. 2005;280:6138–6148. doi: 10.1074/jbc.M411446200. 18-2- [DOI] [PubMed] [Google Scholar]
- 34.Kraft R, Harteneck C. The mammalian melas-tatin-related transient receptor potential cation channels: an overview. Pflugers Arch. 2005;451:204–211. doi: 10.1007/s00424-005-1428-0. [DOI] [PubMed] [Google Scholar]
- 35.Kuhn FJ, Heiner I, Luckhoff A. TRPM2: a calcium influx pathway regulated by oxidative stress and the novel second messenger ADP-ribose. Pflugers Arch. 2005;451:212–219. doi: 10.1007/s00424-005-1446-y. [DOI] [PubMed] [Google Scholar]
- 36.Buelow B, Song Y, Scharenberg AM. The poly (ADP-ribose) polymerase PARP-1 is required for oxidative stress-induced TRPM2 activation in lymphocytes. J Biol Chem. 2008 doi: 10.1074/jbc.M802673200. 3-7- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heiner I, Eisfeld J, Warnstedt M, Radukina N, Jungling E, Luckhoff A. Endogenous ADP-ribose enables calcium-regulated cation currents through TRPM2 channels in neutrophil granulocytes. Biochem J. 2006;398:225–232. doi: 10.1042/BJ20060183. 1-9- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McHugh D, Flemming R, Xu SZ, Perraud AL, Beech DJ. Critical intracellular Ca2+ dependence of transient receptor potential melastatin 2 (TRPM2) cation channel activation. J Biol Chem. 2003;278:11002–11006. doi: 10.1074/jbc.M210810200. 28-3- [DOI] [PubMed] [Google Scholar]
- 39.Tong Q, Zhang W, Conrad K, Mostoller K, Cheung JY, Peterson BZ, Miller BA. Regulation of the transient receptor potential channel TRPM2 by the Ca2+ sensor calmodulin. J Biol Chem. 2006;281:9076–9085. doi: 10.1074/jbc.M510422200. 7-4- [DOI] [PubMed] [Google Scholar]
- 40.Csanady L, Torocsik B. Four Ca2+ ions activate TRPM2 channels by binding in deep crevices near the pore but intracellularly of the gate. J Gen Physiol. 2009;133:189–203. doi: 10.1085/jgp.200810109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Du J, Xie J, Yue L. Intracellular calcium activates TRPM2 and its alternative spliced isoforms. Proc Natl Acad Sci U S A. 2009;106:7239–7244. doi: 10.1073/pnas.0811725106. 28-4- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marambaud P, Dreses-Werringloer U, Vingtdeux V. Calcium signaling in neurodegeneration. Mol Neurodegener. 2009;4:20. doi: 10.1186/1750-1326-4-20. 6-5-. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Small DH. Dysregulation of Calcium Homeostasis in Alzheimer's Disease. Neurochem Res. 2009 doi: 10.1007/s11064-009-9960-5. 1-4- [DOI] [PubMed] [Google Scholar]
- 44.Hermosura MC, Cui AM, Go RC, Davenport B, Shetler CM, Heizer JW, Schmitz C, Mocz G, Garruto RM, Perraud AL. Altered functional properties of a TRPM2 variant in Guamanian ALS and PD. Proc Natl Acad Sci U S A. 2008;105:18029–18034. doi: 10.1073/pnas.0808218105. 18-11- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hermosura MC, Garruto RM. TRPM7 and TRPM2 -Candidate susceptibility genes for Western Pacific ALS and PD? Biochim Biophys Acta. 2007;1772:822–835. doi: 10.1016/j.bbadis.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamamoto S, Wajima T, Hara Y, Nishida M, Mori Y. Transient receptor potential channels in Alzheimer's disease. Biochim Biophys Acta. 2007;1772:958–967. doi: 10.1016/j.bbadis.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 47.Benarroch EE. TRP channels: functions and involvement in neurologic disease. Neurology. 2008;70:648–652. doi: 10.1212/01.wnl.0000300643.95736.b4. [DOI] [PubMed] [Google Scholar]
- 48.Aarts MM, Tymianski M. TRPMs and neuronal cell death. Pflugers Arch. 2005;451:243–249. doi: 10.1007/s00424-005-1439-x. [DOI] [PubMed] [Google Scholar]
- 49.MacDonald JF, Xiong ZG, Jackson MF. Paradox of Ca2+ signaling, cell death and stroke. Trends Neurosci. 2006;29:75–81. doi: 10.1016/j.tins.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 50.Usul H, Cakir E, Arslan E, Peksoylu B, Alver A, Sayin OC, Topbas M, Baykal S. Effects of clotri-mazole on experimental spinal cord injury. Arch Med Res. 2006;37:571–575. doi: 10.1016/j.arcmed.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 51.Isaev NK, Stelmashook EV, Dirnagl U, Andreeva NA, Manuhova L, Vorobjev VS, Sharonova IN, Skrebitsky VG, Victorov IV, Katchanov J, Weih M, Zorov DB. Neuroprotective effects of the antifungal drug clotrimazole. Neuroscience. 2002;113:47–53. doi: 10.1016/s0306-4522(02)00164-1. [DOI] [PubMed] [Google Scholar]
- 52.Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 2008;31:454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kawahara M, Negishi-Kato M, Sadakane Y. Calcium dyshomeostasis and neurotoxicity of Alzheimer's beta-amyloid protein. Expert Rev Neu-rother. 2009;9:681–693. doi: 10.1586/ern.09.28. [DOI] [PubMed] [Google Scholar]
- 54.Xu C, Macciardi F, Li PP, Yoon IS, Cooke RG, Hughes B, Parikh SV, Mclntyre RS, Kennedy JL, Warsh JJ. Association of the putative susceptibility gene, transient receptor potential protein melastatin type 2, with bipolar disorder. Am J Med Genet B Neuropsychiatr Genet. 2006;B141:36–43. doi: 10.1002/ajmg.b.30239. 5-1- [DOI] [PubMed] [Google Scholar]
- 55.Lesne S, AN C, Gabriel C, Croci N, MacKenzie ET, Glabe CG, Plotkine M, Marchand-Verrecchia C, Vivien D, Buisson A. NMDA receptor activation inhibits alpha-secretase and promotes neuronal amyloid-beta production. J Neurosci. 2005;25:9367–9377. doi: 10.1523/JNEUROSCI.0849-05.2005. 12-10- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoey SE, Williams RJ, Perkinton MS. Synaptic NMDA receptor activation stimulates alpha-secretase amyloid precursor protein processing and inhibits amyloid-beta production. J Neurosci. 2009;29:4442–4460. doi: 10.1523/JNEUROSCI.6017-08.2009. 8-4- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoe HS, Fu Z, Makarova A, Lee JY, Lu C, Feng L, Pajoohesh-Ganji A, Matsuoka Y, Hyman BT, Ehlers MD, Vicini S, Pak DT, Rebeck GW. The effects of amyloid precursor protein on postsy-naptic composition and activity. J Biol Chem. 2009;284:8495–8506. doi: 10.1074/jbc.M900141200. 27-3- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer's-related amyloid beta oli-gomers. J Neurosci. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. 10-ll- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. 24-1- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–11601. doi: 10.1074/jbc.M607483200. 13-4- [DOI] [PubMed] [Google Scholar]