

Abstract
Neurodegenerative diseases are progressive disorders of the nervous system that affect specific cellular populations in the central and peripheral nervous systems. Although most cases are sporadic, genes associated with familial cases have been identified, thus enabling the development of animal models. Invertebrates such as Drosophila have recently emerged as model systems for studying mechanisms of neurodegeneration in several major neurodegenerative diseases. These models are also excellent in vivo systems for the testing of therapeutic compounds. Genetic studies using these animal models have provided novel insights into the disease process. We anticipate that further exploration of the animal models will further our understanding of mechanisms of neurodegeneration as well as facilitate the development of rational treatments for debilitating degenerative diseases.
Keywords: Alzheimer’s disease, Parkinson’s disease, polyglutamine disease, prion disease
INTRODUCTION
Neurodegenerative diseases represent a subgroup of human diseases with certain features in common. These diseases are often but not always of adult onset. Patients are generally asymptomatic during the development or maturation of the nervous system. Disease pathogenesis usually involves the progressive loss of specific neuronal populations characteristic of each disease type. Because of the lack of mechanism-based treatment strategies that can halt the degeneration process, most of these diseases follow an inexorably fatal course. Although neurodegenerative diseases were once considered among the most obscure and intractable of all human illnesses, this situation is changing, thanks to breakthroughs in human genetics and genomics that help pinpoint genes associated with familial forms of diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD). The identification of these disease-associated genes has allowed investigators to characterize the mechanisms of neurodegeneration at the molecular level. With an understanding of the pathogenic mechanisms of the familial disease genes, we expect to obtain insights into the pathogenesis of the more common forms of the diseases.
One powerful approach for studying disease mechanisms has been the use of animal models. Invertebrate animals, especially Drosophila, have proven to be excellent models for human neurodegenerative diseases and have attracted a great deal of attention in recent years; a number of excellent reviews have been written on this topic (1–3). The aims of this review are to introduce key features of each disease type and to summarize new developments in the methodology and analysis of disease models in genetically engineered organisms, with particular emphasis on Drosophila models. We discuss initial insights into disease mechanisms that have been gained from these models and elaborate on their relevance to mammalian systems. Finally, we present some perspectives on how further studies of these models will help us understand and ultimately treat or prevent these debilitating disorders.
BUILDING RELEVANT MODELS OF NEURODEGENERATIVE DISEASES
Despite the indisputable contribution of human genetic studies to the identification of genes associated with familial forms of neurodegenerative diseases, studies on human subjects are of limited use for elucidating the signaling pathways and cellular processes underlying the neurodegenerative process. This is because both ethical and technical constraints limit the extent to which genetic analysis can be performed in humans to determine genetic relationships among disease genes and to delineate genetic pathways. Furthermore, most human neuropathological studies use postmortem tissues that almost never reflect the earliest pathologic events at the presymptomatic stage. In these respects, animal models present excellent alternatives for studying neurodegenerative disease mechanisms from initiation to maturation.
Here we use Drosophila as an example to illustrate the methods for building relevant neurodegenerative disease models. Four approaches have been successfully applied to the study of neurodegeneration:
First, forward genetic screens have identified mutations that cause degeneration of the brain. Pioneering studies performed in the Benzer lab (4, 5) led to the identification of a number of interesting Drosophila mutants such as bubblegum, swisscheese, and drop-dead. These mutants were isolated by first screening for flies with reduced lifespan and then analyzing their brain pathology. Each mutant showed distinct patterns of degeneration in specific brain regions. The mammalian homologs of some of these genes also caused neurodegeneration phenotypes when mutated, indicating that this approach can identify genes that are essential for conserved mechanisms that maintain nervous system integrity (6). A disadvantage of this approach is that the neurodegeneration phenotypes in these mutants tend to be quite widespread, and the direct relevance of these mutants to the selective cell type vulnerability specific to individual neurodegenerative diseases is not always obvious.
Generally, mutant neurodegenerative disease genes act in a dominant manner in humans and are assumed to result in gained toxic functions. Consequently, models of these diseases can be established in Drosophila by transgenic expression of the toxic proteins. In many cases, transgenic animals overexpressing the wild-type versions of the disease-associated genes develop nearly identical disease phenotypes (7, 8). These observations, together with the fact that neurodegenerative conditions are frequently associated with mutations in proteins prone to misfolding and aggregation, lead to the notion that defects in protein homeostasis and general cellular quality control systems contribute to disease pathogenesis.
Other familial neurodegenerative diseases are transmitted recessively, suggesting that they are likely caused by a loss-of-function mechanism. The method of choice to model these diseases is to disrupt the function of endogenous genes homologous to human disease genes via a wide array of techniques available for disrupting gene function in Drosophila. Transposon-mediated mutagenesis, transgenic RNA interference (RNAi), and homologous recombination–based gene knockout can be used for this purpose (9–12).
A pharmacological approach can be used to model neurodegenerative diseases and to test candidate therapeutics in animals. This approach has been widely used in mammalian systems, and some of the toxic compounds have been successfully used in flies to recapitulate disease phenotypes (13, 14). Drugs can be mixed with food and readily delivered to the flies. One possible advantage of the fly system is the lack of a stringent blood-brain barrier, which allows compounds to easily gain access to the nervous system. Together with the smaller body size, lower cost, and shorter life cycle, this approach makes Drosophila an attractive model for the testing and screening of therapeutic compounds.
MODELING ALZHEIMER’S DISEASE AND RELATED TAUOPATHY
Pathological Features and Genes Involved in the Pathogenesis of Alzheimer’s Disease and Tauopathy
AD is the most common neurodegenerative disease and is the leading cause of dementia worldwide. Clinically, AD manifests as a gradual decline of cognitive functions such as learning and memory. Pathologically, the disease is characterized by selective atrophy of the hippocampus and the frontal cerebral cortex. Two microscopic brain lesions, neuritic or amyloid plaque and neurofibrillary tangles (NFT), are diagnostic of AD in the demented patient when present in abnormally high densities for a given age. The main components of the amyloid plaque are the Aβ-40 and Aβ-42 peptides, which are generated by endoproteolysis of the amyloid precursor protein (APP) via the sequential action of β- and γ-secretases. β-secretase activity is provided by the β-site APP-cleaving enzyme (BACE), whereas γ-secretase activity depends on a protein complex consisting of presenilin (Psn), nicastrin, aph-1, and pen-2 (15). Autosomal dominant mutations in APP, Psn-1, and Psn-2 can accelerate disease onset and progression in familial AD (FAD) cases. At the molecular level, these mutations promote the generation of amyloidogenic Aβ peptides (16). Also, defective trafficking of APP into recycling pathways may underlie the pathological accumulation of Aβ in some late-onset FAD cases (17). Taken together, these results have all but confirmed the amyloid hypothesis, which postulates that the initiating event in AD is the accumulation of Aβ peptide that may underlie synaptic failure, thereby resulting in the remarkably pure impairment of cognitive function. Excesses in Aβ accumulation trigger neuronal dysfunction, cell death, and irreversible dementia (18).
The major component of the NFT is tau protein, a highly soluble, microtubule-binding protein normally enriched in axons (19). In AD it becomes abnormally phosphorylated and is insoluble in NFT (20–22). This observation has provided circumstantial evidence that tau abnormalities may lead to neuronal dysfunction and degeneration. Direct evidence of the pathogenicity of tau was provided by the discovery that mutations in tau are associated with frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) (23–26). NFT is also a pathological hallmark of AD, where it is detectable in the abnormal neuritic processes associated with amyloid plaques (27, 28).
The mechanistic relationship between NFT and amyloid plaque in disease pathogenesis remains a matter of some controversy (18, 29). Studies in transgenic mice have shown that expression of mutant human tau (h-tau) alone can result in tangle pathology and neuronal loss without accompanying plaque pathology (30, 31). Induction of amyloid deposits in these animals can further promote NFT formation (32–34). These observations suggest that the two lesions may be causally linked, although the molecular mechanism involved is not understood. The notion that amyloid plaque and tau NFT pathologies are causally associated is further supported by recent studies demonstrating that blocking Aβ peptide production in transgenic mouse models could remove amyloid pathology as well as early-stage tau lesions (35). Together, these studies appear to lend further support to the amyloid hypothesis. Note that NFT pathology is also evident in non-AD dementias as well as in a wide variety of other conditions that do not include any amyloid plaques, suggesting that events other than Aβ peptide accumulation can also trigger the formation of NFT (36). The sequence of molecular events leading to NFT formation is poorly understood.
Modeling Alzheimer’s Disease in Drosophila
Most of the genes implicated in AD pathogenesis have clear fly homologs. There is a fly homolog of APP (known as APP-like or APPL). Flies deficient for APPL exhibit a behavioral abnormality that can be rescued by a human APP transgene, indicating functional conservation between APPL and human APP (37). However, APPL lacks the amyloidogenic Aβ peptide sequence at the C terminus, and there is no evidence that it is processed in vivo as is human APP. The γ-secretase complex components are conserved in Drosophila and are known to be involved in the processing of Notch (38, 39). Although the fly does not appear to contain a clear homolog of mammalian BACE, it is possible that other proteases may provide a similar function, as it has been reported that overexpression of human APP causes axonal transport defects and increases cell death in the larval brain in an Aβ-dependent manner (40). Furthermore, one study showed that Aβ peptide can be generated in Drosophila from a modified human APP transgene (41).
Fly models of Aβ peptide–induced amyloid formation and neurodegeneration were recently generated. In one study (42), a human BACE transgene was coexpressed with a human APP transgene and a fly Psn transgene, both of which contained FAD-linked mutations. Transgenic flies expressing all three transgenes developed β-amyloid plaques and age-dependent neurodegeneration. Genetic and pharmacological manipulation of γ-secretase activity can modulate the severity of the disease phenotype in this model (42). In studies employing another approach (43, 44), Aβ-40 and Aβ-42 peptides were directly fused to signal peptides and expressed as transgenes. Transgenic flies expressing Aβ-42 developed (a) diffuse amyloid deposits, (b) age-dependent learning defects, detected using a Pavlovian olfactory learning assay, and (c) neurodegeneration, indicated by neuronal loss and vacuole formation in various brain regions. Aβ-40-expressing flies developed only age-dependent learning defects but no amyloid formation or neurodegeneration. These studies thus emphasize the importance of Aβ-42-associated toxicity in disease pathogenesis. The development of these Aβ-toxicity models should pave the way for future studies aimed at understanding the molecular events in the pathogenic cascade of AD and at screening for drug targets and potential therapeutics.
Modeling Tauopathy in Drosophila
Tau-induced neurodegeneration has also been successfully modeled in Drosophila. The first evidence of tau-induced neurotoxicity in flies was provided by transgenic studies of a bovine tau–green fluorescent protein reporter that caused axonal degeneration in sensory neurons (45). Subsequently, fly models of tauopathy were created by expressing wild-type or FTDP-17-linked mutant forms of human tau (46, 47). The fly genome includes a fly homolog of tau (known as dtau). Overexpression studies showed that directed expression of wild-type vertebrate tau or dtau in adult mushroom body neurons, which control olfactory learning and memory in Drosophila, strongly compromised associative olfactory learning and memory in the absence of detectable neurodegeneration (48). This result suggests that behavioral plasticity decrements, possibly reflecting synaptic dysfunction, may represent the earliest neuropathological manifestation of tauopathy.
INSIGHTS GAINED FROM FLY ALZHEIMER’S DISEASE AND TAUOPATHY MODELS
Phosphorylation Control of Tau Toxicity
Tau phosphorylation and dephosphorylation have attracted considerable interest in tauopathy research, fueled by the detection of abnormally phosphorylated tau in NFT. A number of protein kinases and phosphatases have been found to regulate tau phosphorylation in vitro. These include glycogen synthase kinase (GSK)-3β, mitogen-activated protein kinase (MAPK), Cdk2/5, protein kinases A and B (PKA and PKB, respectively), calmodulin-dependent protein kinase II (CaMKII), microtubule affinity–regulating kinase (MARK), and protein phosphatases 1, 2A, 2B, and 2C (49). Despite a large body of biochemical studies, surprisingly little is known about the in vivo roles of these enzymes in regulating tau accumulation and presumed toxicity. The fact that many of these genes play essential roles during development, as well as the existence of considerable genetic redundancy, may have impeded in vivo genetic analyses of these genes in tau phosphorylation and toxicity in mammalian systems.
The compactness of the genome and the existence of an arsenal of molecular genetic tools in Drosophila make it feasible to evaluate the importance of phosphorylation in tau-mediated neurotoxicity. Through loss-of-function and overexpression genetic studies and biochemical analysis, partitioning defective–1 (PAR-1) was found to be a physiological tau kinase that plays a central role in determining tau phosphorylation and toxicity in Drosophila (50). PAR-1 is a serine/threonine kinase originally identified in Caenorhabditis elegans for its role in regulating cell polarity and asymmetric cell division (51). PAR-1 homologs have been found in eukaryotes ranging from yeast to mammals, where they possess essential cellular and developmental functions. MARKs, the mammalian homologs of PAR-1, regulate microtubule dynamics, epithelial cell polarity, and neuronal differentiation under normal conditions and are bound to NFT in AD brain (52–54). In the fly tauopathy model, overexpression of PAR-1 leads to elevated tau phosphorylation at S262 and S356 sites and to enhanced toxicity, whereas removing PAR-1 function or mutating PAR-1 phosphorylation sites in tau abolishes tau toxicity (50). Importantly, PAR-1 has been found to initiate a multisite phosphorylation process that generates pathogenic tau. In this process, phosphorylation by PAR-1 at the S262/S356 sites precedes and is obligatory for downstream phosphorylation events, including the T208 site that is phosphorylated by GSK-3β and Cdk5 (50). These results, together with the findings from a large-scale genetic screen that uncovered protein kinases and phosphatases as major modifiers of tau toxicity (55), clearly established the importance of phosphorylation in determining tau toxicity in vivo.
Although the presence of protein aggregates is a common feature of neurodegenerative diseases, these aggregates’ role in disease remains uncertain. However, studies of fly tauopathy models have shed some light on the role of pathogenic protein aggregation. In some disease models, there is a correlation between the extent of macroscopically visible protein aggregate formation and disease severity, whereas in others, disease symptoms can occur without formation of large protein aggregates. In certain cases, genetic manipulations that interfere with protein aggregate formation actually exacerbate disease symptoms (56). In fly tauopathy models, neurodegeneration can occur in the absence of NFT formation, suggesting that some soluble, nontangled form of phosphorylated tau is intrinsically toxic (46). Interestingly, when fly GSK-3β is coexpressed with tau, large NFT-like aggregates form in photoreceptor neurons, and neurodegeneration is enhanced (47). Although this observation suggests that NFT-like aggregates may contribute to tau toxicity, the evidence is also compatible with the idea that some soluble, nontangled forms of phosphorylated tau are the primary neurotoxic species and that these species also accumulate under GSK-3β-overexpression conditions. Further studies in Drosophila and vertebrate models are needed to elucidate the relationship between NFT formation and tau toxicity.
Postmortem analysis indicates that in the AD patient brain tau is hyperphosphorylated in more than 20 serine/threonine sites (57). The importance of individual phosphorylation sites in determining tau toxicity in vivo has not been clearly defined. In theory, these phosphorylation sites could act in concert to confer tau toxicity. Alternatively, a small subset of sites may play a dominant role in determining tau toxicity. Analysis of the PAR-1-triggered sequential tau phosphorylation has emphasized the importance of certain phosphorylation events in determining tau toxicity (50). The relative ease of making transgenic flies express mutant tau proteins enables us to pinpoint the key phosphorylation events that play critical roles in conferring tau toxicity. Such information will help identify the neurotoxic tau species and will reveal critical steps in the pathogenic cascade of tauopathy.
Signaling Mechanisms That Link Amyloid and Tau Lesions
The upstream signaling mechanisms that regulate tau phosphorylation under normal and disease conditions are not well understood. The identification of PAR-1 as a physiological tau kinase enables dissection of the signaling mechanisms. Recent studies show that phosphorylation of PAR-1 by the tumor-suppressor protein LKB1 is required for its activation; this, in turn, promotes tau phosphorylation in Drosophila. Diverse stress stimuli, such as high osmolarity and human APP–induced neurotoxicity (Figure 1), can promote PAR-1 activation and tau phosphorylation in an LKB1-dependent manner (58). These results reveal a previously unknown function for the tumor-suppressor protein LKB1 in a signaling cascade, through which the phosphorylation and function of tau are regulated by diverse AD-related toxic stimuli. Hyperphosphorylation of tau at the PAR-1 target site has also been observed in human AD patient brain and in APP/tau double-transgenic mouse AD models (59, 60), suggesting that aberrant activation of PAR-1-like kinases occurs in AD conditions in mammals. Together, these studies implicate the LKB1/PAR-1/tau-phosphorylation cascade as a missing molecular link between amyloid and tau lesions in AD pathogenesis (Figure 2). Because each kinase is likely to have more than one substrate, further studies are needed to identify additional substrates of LKB1 and PAR-1 and to assess their contribution to AD-related neurodegeneration.
Figure 1.
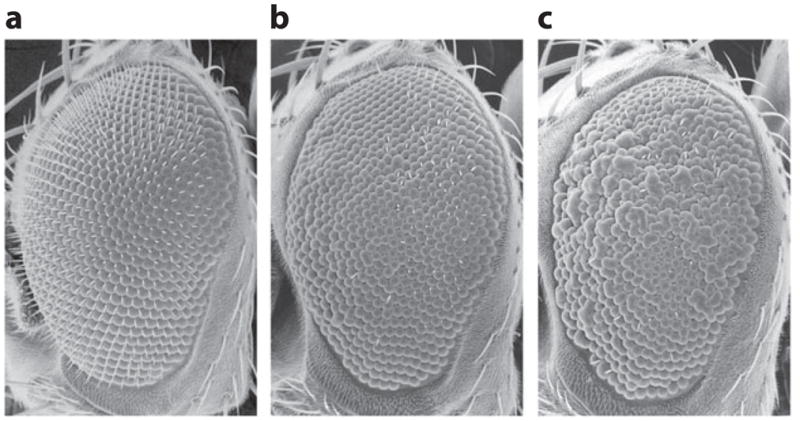
Scanning electron microscopy images of a fly eye, showing genetic interaction between amyloid precursor protein (APP) and tau. (a) Overexpression of APP in the fly eye has little effect on eye morphology, as shown by the smooth appearance of the eye surface. (b) Overexpression of tau causes mild loss of photoreceptors and a slight roughness of the eye surface. (c) Co-overexpression of APP and tau strongly enhances the rough-eye phenotype. Modified from Wang et al. (58).
Figure 2.
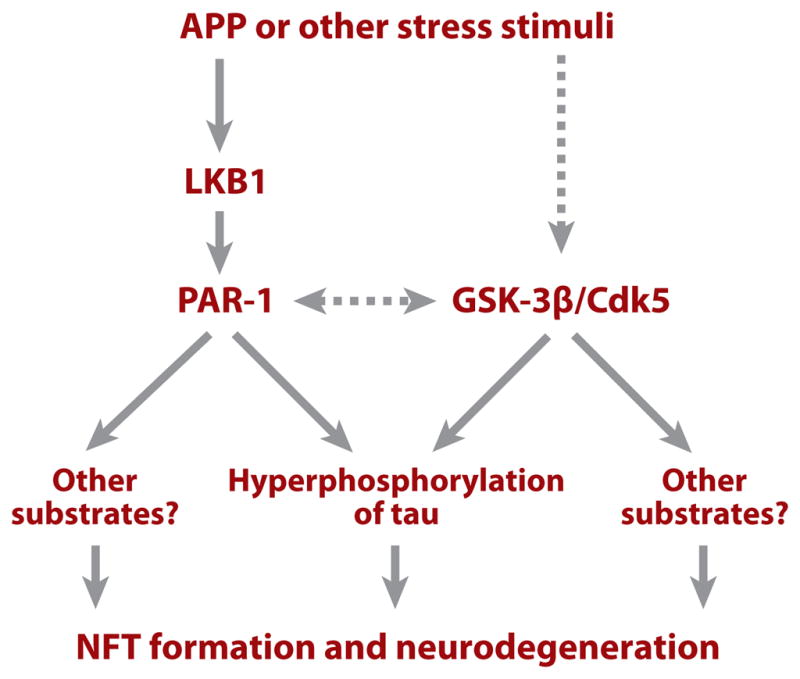
A diagram depicting putative signaling pathways linking amyloid precursor protein (APP) and tau pathology. Solid lines indicate relationships that are supported by experimental evidence; dashed lines indicate relationships for which experimental support is weak or currently missing. Abbreviations: GSK, glycogen synthase kinase; PAR-1, partitioning defective–1; NFT, neurofibrillary tangles.
Role of Synaptic Dysfunction in Disease Pathogenesis
Prior to the onset of plaque and tangle pathology and frank neuronal death in susceptible AD brain regions, mild cognitive impairment is observed. Subsequent studies have confirmed that loss of synaptic function is one of the earliest signs of the disease process in AD. This has led to the proposal that AD is primarily a synaptic failure (61). Studies in animal models have supported the role of synaptic dysfunction in disease pathogenesis. In both vertebrates and invertebrates, APP and APP-like proteins have been shown to exert normal physiological function at the synapse (37, 62). The mouse APP-transgenic model exhibits learning and memory deficits in the absence of obvious neuronal degeneration, supporting a primary role for synaptic dysfunction in APP pathogenesis (63). Interestingly, removal of endogenous tau can suppress most of the cognitive deficits in the APP model (64), indicating a critical role for tau in mediating the toxic effects of APP at the synapse. This is consistent with findings that synaptic toxicity is also observed in Drosophila and mouse transgenic tau models (65, 66).
It appears that tau is not the only synaptic protein whose abnormality may contribute to synaptic dysfunction in AD. A recent study found that overactivation of PAR-1 kinase in Drosophila causes the delocalization of Dlg, the fly homolog of the key postsynaptic scaffold protein Postsynaptic Density (PSD)-95, from the postsynaptic membrane at the neuromuscular junction (Figure 3). This effect is mediated through the direct phosphorylation of Dlg by PAR-1 (67). Dlg’s synaptic localization has been found to be decreased in AD patients and in mouse AD models (68–70). Given that aberrant activation of PAR-1-like kinases has been implicated in AD conditions, this study offers a new approach to understanding the molecular basis of synaptic dysfunction in AD.
Figure 3.
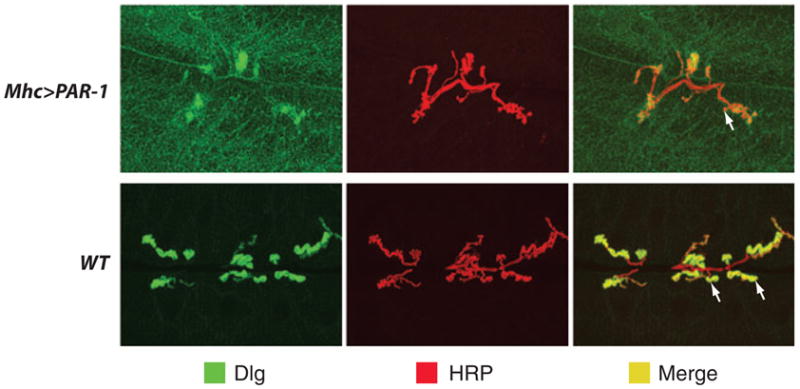
Immunostaining of the Drosophila larval neuromuscular junction (NMJ) synapse showing the effect of partitioning defective–1 (PAR-1) overactivation on Dlg localization and synaptic morphogenesis. NMJs of control (WT ) and PAR-1 overexpression (Mhc > PAR-1) animals were double immunostained for the postsynaptic marker Dlg and the presynaptic marker horseradish perxoidase (HRP). Merged images are shown on the right. Note that PAR-1 overexpression causes both the delocalization of Dlg from the synaptic membrane to the muscle cell cytoplasm and the simplification of synapse morphology and a reduction of synaptic bouton number. Synaptic boutons are swellings on motor neuron axons at sites of contact with the muscle surface. Arrows mark representative boutons. Modified from Zhang et al. (67).
MODELING PARKINSON’S DISEASE
Pathological Features and Molecular Mechanisms Involved in Parkinson’s Disease Pathogenesis
PD is the most common movement disorder and the second most common neurodegenerative disease. The classical form of the disease is clinically characterized by muscle rigidity, resting tremor, bradykinesia, and postural instability. The movement abnormality in PD is largely attributable to the deficiency of brain dopamine content, which is caused by degeneration of dopaminergic neurons in the substantia nigra. A pathological hallmark of the disease is the formation of Lewy bodies, intra-cytoplasmic inclusions that are composed of α-Synuclein (α-Syn) and ubiquitin, among other proteins. The most common forms of PD are sporadic with no known cause. However, postmortem studies of sporadic cases have identified defective mitochondrial complex I function and oxidative damage in nigrostriatal dopaminergic neurons (71, 72). The oxidative damage is thought to reflect both the increased burden of free radical production resulting from dopamine metabolism and mitochondrial dysfunction and the limited scavenging ability of dopaminergic neurons (73, 74). The identification of environmental toxins such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and rotenone, which cause PD-like symptoms by inhibiting complex I function and promoting oxidative stress, further supports the mitochondrial dysfunction and oxidative stress models of PD and provides pharmacological tools for disease studies (14, 75).
Epidemiological studies also point to the contribution of genetic factors in the pathogenesis of PD. At least 10 distinct loci (PARK1–11) have been linked to rare familial forms of PD (FPD) (76, 77). As with FAD, it is anticipated that understanding the molecular lesions associated with these FPD genes will shed light on the pathogenesis of the more common forms of the disease. To date, six FPD genes have been molecularly cloned, including α-Syn, Parkin, Ubiquitin C-terminal hydrolase–1 (Uchl-1), DJ-1, phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1), and leucine-rich repeat kinase 2 (LRRK2) (78–84). Of these six genes, α-Syn and Parkin have been the most intensively studied. Although initial biochemical and biophysical studies linked mutations of both genes to aberrant protein folding and handling, more recent in vitro cell culture studies and in vivo genetic studies have revealed their connection to mitochondrial impairment and oxidative stress (85).
DJ-1, PINK1, and LRRK2 are three recently cloned FPD genes whose molecular functions (as they pertain to PD pathogenesis) remain poorly defined. PINK1 is a putative serine/threonine kinase localized to mitochondria (82, 86). PINK1 was initially isolated as a gene induced by overexpression of the tumor suppressor PTEN (87), but its functional relationship with PTEN and whether it participates in phosphatidylinositol 3–kinase (PI3K)/PTEN signaling are not known. Nevertheless, the subcellular localization of PINK1 protein emphasizes mitochondria as its primary site of activity. DJ-1 encodes a highly conserved protein belonging to the ThiJ/PfpI superfamily. LRRK2 encodes a large protein that contains a leucine-rich repeat (LRR) domain, a kinase domain, a RAS-like GTPase domain, and a WD-40 domain. Cell culture studies have shown that overexpression of LRRK2 can cause neuronal death in a kinase activity–dependent manner and that some of the pathogenic mutations may possess enhanced kinase activity (88).
To investigate the in vivo physiological roles of the FPD genes and to model dopaminergic degeneration caused by dysfunction of these genes, transgenic or knockout mouse models for α-Syn, Parkin, DJ-1, and Pink1 have been generated (89–96). Although these models exhibit various pathological and behavioral phenotypes, the cardinal feature of PD, namely selective degeneration of dopaminergic neurons, has not been faithfully reproduced. It is possible that the short lifespan of mice may have precluded observation of the late-onset dopaminergic degeneration phenotype. Alternatively, certain fundamental differences in dopaminergic neuron physiology between mice and humans could make mice less vulnerable to the dysfunction of the individual FPD genes. Construction of double- or triple-mutant combinations or use of genetic or pharmacological manipulations to alter the aging process may help reveal a dopaminergic degeneration phenotype.
Modeling Parkinson’s Disease in Drosophila
α-Syn models
Although Drosophila does not have a clear α-Syn homolog, overexpression of wild-type- and FPD-associated mutant α-Syn in Drosophila has reproduced key features of PD, including Lewy body–like aggregate formation, selective degeneration of dopaminergic neurons, and locomotor behavior abnormality as detected by climbing ability in the negative geotaxis response (7, 97). The fact that both mutant and wild-type α-Syn form aggregates in fly neurons and cause disease phenotypes supports the notion that improper disposal of aggregation-prone abnormal proteins can result in neurodegeneration. This is also consistent with the finding that genomic triplication of α-Syn in humans can cause PD (98). The fact that Drosophila does not encode an α-Syn-like protein may have facilitated the modeling of α-Syn-related toxicity, as flies may not have evolved an effective defense mechanism against the toxic effects of α-Syn misfolding or aggregation. However, a recent study has called into question the significance of the fly α-Syn models. Pesah et al. (99) reported that no significant change of dopaminergic neurons was observed in various α-Syn models. These authors used whole mount immunofluorescence analysis of fly brain with an antibody against fly tyrosine hydroxylase (TH), a marker for dopaminergic neurons, whereas earlier studies used an antibody against vertebrate TH and applied it on paraffin brain sections. One explanation for the divergent results is that α-Syn overexpression does not lead to neuronal death but simply causes dopaminergic dysfunction and thus reduces expression of TH in certain dopaminergic neurons, rendering them undetectable by the antivertebrate TH antibody on paraffin sections due to antibody-sensitivity issues. Further studies are needed to clarify this point.
Parkin models
Three labs independently generated parkin-null mutant flies (9, 10, 100). Two prominent features of parkin-mutant flies are mitochondrial pathology and apoptotic muscle degeneration. In addition, these mutant flies exhibit sterility, reduced lifespan, reduced cell number and size, and hypersensitivity to oxidative stress. However, initial analysis reported no significant loss of dopaminergic neurons in the dorsal medial clusters (DMCs), the same clusters that were previously reported to be selectively affected in α-Syn-transgenic flies, although the sizes of these neurons appeared reduced in parkin-mutant flies. A later study using whole mount confocal analysis reported notable dopaminergic neuron loss in the protocerebral posterior lateral 1 cluster (101). Using a different approach, parkin loss-of-function phenotype was created by transgenic RNAi, which allows tissue- and cell type–specific gene knockdown (11). Targeted expression of parkin double-stranded RNA in Drosophila dopaminergic neurons also did not result in significant loss of dopaminergic neurons in the DMC, suggesting that fly Parkin is not essential for the maintenance of dopaminergic neurons in the DMC under normal physiological conditions. Targeted overexpression of human Parkin-associated endothelin-like receptor (Pael-R), a Parkin substrate protein (102), did result in a reduction of TH-positive dopaminergic neurons in the DMCs (Figure 4), and this phenotype was exacerbated in parkin RNAi background (11). Conversely, overexpression of human Parkin suppressed Pael-R-induced toxicity. Together, these in vivo genetic interaction studies confirmed the biochemical relationship between Parkin and Pael-R and indicated that accumulation of abnormal Pael-R protein in Parkin-deficient dopaminergic neurons might be one of the causes of neuronal death. Recent studies in mice showed that (a) Pael-R induces degeneration of dopaminergic neurons in the substantia nigra via endoplasmic reticulum stress and dopamine toxicity and (b) Pael-R toxicity is enhanced under Parkin-inactivation conditions (103).
Figure 4.
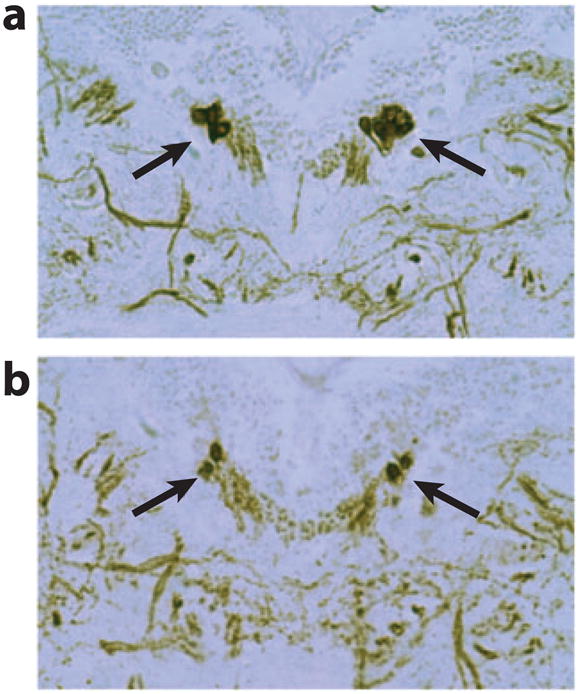
Immunostaining of dopaminergic neurons in the dorsal medial cluster (DMC) showing a reduction of neuron number in Pael-R-overexpression animals. Thin paraffin sections prepared from control (a) and Pael-R-overexpression animals (b) were immunostained with an antibody against tyrosine hydroxylase (TH), a marker specific for dopaminergic neurons. Arrows mark the DMC neurons. The expression of Pael-R was directed by the dopaminergic neuron–specific promoter from the TH-Gal4 driver. Modified from Yang et al. (11).
DJ-1 models
There are two DJ-1 homologs in Drosophila, DJ-1A and DJ-1B. Although both fly DJ-1 proteins are highly similar to human DJ-1, a putative catalytic triad characteristic of cysteine proteases is conserved in DJ-1A but not in DJ-1B, suggesting that DJ-1A is a closer homolog of human DJ-1 than DJ-1B. In one study, DJ-1A and DJ-1B double-knockout flies were generated by genomic microdeletions. These animals were viable and fertile and had a normal lifespan; however, they displayed a selective sensitivity to environmental toxins such as paraquat and rotenone. This sensitivity was thought to result primarily from loss of DJ-1B protein, which becomes modified upon oxidative stress (104). In another study, DJ-1B loss-of-function mutants were found to have an extended survival of dopaminergic neurons and resistance to paraquat stress, but they also had an acute sensitivity to hydrogen peroxide treatment. There was also a compensatory upregulation of DJ-1A expression in the brain of the DJ-1B mutant, suggesting that overexpression of DJ-1A in dopaminergic neurons may be sufficient to confer protection against paraquat (105).
The reason for the differential sensitivity to paraquat stress by the DJ-1B-mutant flies in these two studies is not clear. The nature of the different genomic deletions or the genetic backgrounds may play a role. Using a different approach, DJ-1A was knocked down by transgenic RNAi. Inhibition of DJ-1A function through cell type–specific RNAi resulted in accumulation of reactive oxygen species (ROS), organismal hypersensitivity to oxidative stress, and dysfunction and degeneration of dopaminergic and photoreceptor neurons (106). Intriguingly, DJ-1A RNAi animals exhibit apparently stronger phenotypes than do DJ-1A or DJ-1B genetic mutants; specifically, they suffer dopaminergic neuron loss, photoreceptor loss, and shortened lifespan. Although appropriate controls were included in these experiments, one cannot fully exclude the possibility that the RNAi approach may have generated “off-target” effects. Alternatively, the RNAi approach might have circumvented certain compensatory mechanisms associated with genomic deletions or that certain non–cell autonomous effects of DJ-1A inactivation may have contributed to the cell type–specific RNAi effect.
Pink1 models
Drosophila Pink1 models of PD were generated by transposon-mediated mutagenesis and RNAi approaches. Genetic removal of Drosophila Pink1 (dPink1) function results in male sterility, apoptotic muscle degeneration, defective mitochondrial morphology, and increased sensitivity to multiple stresses, including oxidative stress. Mitochondrial cristae are fragmented in dPink1 mutants (107). In addition to these phenotypes, another group found that dPink1 mutants exhibit dopaminergic neuronal degeneration accompanied by defects in locomotion (108). Furthermore, transmission electron microscopy analysis and a rescue experiment with Drosophila Bcl-2 demonstrated that mitochondrial dysfunction largely accounts for the degenerative phenotypes of dPink1 mutants (108). Using the transgenic RNAi approach, researchers have shown that inhibition of dPink1 function results in energy depletion, shortened lifespan, and degeneration of both indirect flight muscles and select dopaminergic neurons (109). The muscle pathology was preceded by mitochondrial enlargement and disintegration (Figure 5). These phenotypes could be rescued by the wild-type but not by the pathogenic C-terminal deleted form of human Pink1 (hPink1), indicating functional conservation between fly and human Pink1 (109). Another independent study (110) using the RNAi approach showed that inactivation of dPink1 results in progressive loss of dopaminergic neurons and photoreceptor neurons, which can be rescued by expression of human Pink1. Further, expression of human SOD1 and treatment with the antioxidants SOD and vitamin E can significantly inhibit photoreceptor degeneration in dPink1 RNAi flies (110). Together, these studies strongly implicate mitochondrial dysfunction and oxidative stress in Pink1 pathogenesis.
Figure 5.
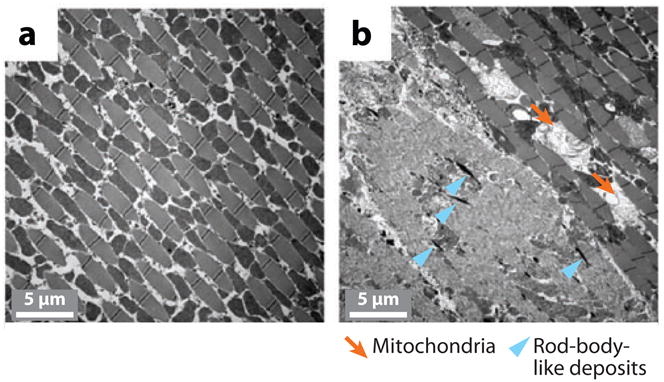
Transmission electron microscopy analysis of muscle fiber organization and mitochondria morphology in dPink1 RNA interference (RNAi) animals. (a) In wild-type animals, indirect flight muscle (IFM) fibers are regularly organized, with electron-dense mitochondria (indicated by arrows) interspersed among the muscle fibers. (b) In Pink1-knockdown animals, muscle fibers are disorganized and mitochondria are abnormally shaped. Further, electron-dense deposits appear within the area of degenerating IFMs; these are reminiscent of nemaline (rod-body) myopathy in humans. Arrowheads indicate rod-body-like deposits. Modified from Yang et al. (109).
Pharmacological models
Pharmacological PD models in mammals have been instrumental in recapitulating oxidative stress–induced dopaminergic neurodegeneration and have provided good models for sporadic forms of the disease (14). A recent study showed that pharmacological treatment could be used in Drosophila to model sporadic PD (13). Chronic exposure to rotenone in Drosophila resulted in PD-related neurodegenerative and behavioral effects. Rotenone-treated flies presented characteristic locomotor impairments as measured by the negative geotaxis response assay. At the cellular level, a selective loss of dopaminergic neurons was detected in all of the brain dopaminergic clusters. Feeding the flies with L-dopa (3,4-dihydroxy-L-phenylalanine), the precursor to dopamine in the biosynthetic pathway, rescued the behavioral deficits but not neuronal death. In contrast, the antioxidant melatonin (N-acetyl-5-methoxytryptamine) alleviated both symptomatic impairment and neuronal loss, suggesting that antioxidant agents such as melatonin may be beneficial for the treatment of PD. Pharmacological treatment in Drosophila thus provides a complementary avenue for studying mechanisms of dopaminergic degeneration.
DISEASE MECHANISMS LEARNED FROM FLY PARKINSON’S DISEASE MODELS
Role of Molecular Chaperones in Disease Pathogenesis
Using a candidate gene approach, investigators have shown that directed expression of the molecular chaperone HSP70, which was previously shown to be effective in ameliorating neurodegeneration in polyglutamine (polyQ) models (described in detail below), prevents dopaminergic neuronal loss associated with α-Syn expression in Drosophila (97) (Figure 6). Further, interference with endogenous chaperone activity exacerbates α-Syn toxicity. The authors’ observation that Lewy bodies in human postmortem tissue are immunopositive for molecular chaperones demonstrates the relevance of these findings to human diseases. These findings suggest that abnormal protein folding and aggregation may be common mechanisms in the pathogenesis of polyQ- and α-Syn-associated neurodegenerative diseases.
Figure 6.

A diagram depicting possible pathways involving familial Parkinson’s disease genes that regulate dopaminergic neuron maintenance. Solid lines indicate relationships that are supported by experimental evidence; dashed lines indicate relationships for which experimental support is weak or missing. Abbreviations: GPRK2, G protein–coupled receptor kinase 2; PI3K, phosphatidylinositol 3–kinase; Pten, phosphatase and tensin homolog; α-Syn, α-Synuclein.
A recent human genetics study indicated that polymorphisms in the human Hsp70 genes may affect susceptibility to PD (111). The potential of Hsp70 gene therapy in a mouse model of MPTP-induced PD was recently tested. Hsp70 gene transfer to dopamine neurons by a recombinant adeno-associated virus significantly protects the mouse dopaminergic system against MPTP-induced dopaminergic toxicity (112). The effectiveness of HSP70 in preventing dopaminergic degeneration in an acute pharmacological model and in a chronic genetic model suggests that increasing chaperone activity may offer a new avenue for the treatment of PD.
Regulation of α-Syn Toxicity by Phosphorylation
It was previously shown in mammalian systems that α-Syn protein found in Lewy bodies is phosphorylated at serine 129 (Ser129) (113). To assess the role of phosphorylation of Ser129 in α-Syn toxicity and inclusion formation, Chen & Feany (114) performed mutagenesis studies in the Drosophila α-Syn model of PD. Mutating Ser129 to nonphosphorylatable alanine completely suppressed dopaminergic neuronal loss caused by human α-Syn, whereas changing Ser129 to the phosphomimetic aspartate significantly enhanced α-Syn toxicity. The authors further showed that G protein–coupled receptor kinase 2 (Gprk2) phosphorylated Ser129 and enhanced α-Syn toxicity in vivo (Figure 6). Furthermore, blocking phosphorylation at Ser129 substantially increased aggregate formation. Ser129 phosphorylation thus appears to be important in regulating α-Syn neurotoxicity and inclusion formation (114). Given the broad involvement of α-Syn abnormality in familial as well as sporadic PD, additional studies investigating upstream signaling mechanisms that impinge on Gprk2 to regulate α-Syn phosphorylation and toxicity will lead to further insights into PD pathogenesis in general.
Neuroprotective Function of Parkin
The candidate gene approach was also used to study the relationship between Parkin and α-Syn in dopaminergic neuron degeneration. There are indications that these two genes may functionally interact in the disease process. Parkin colocalizes with α-Syn in Lewy bodies, and an O-glycosylated form of α-Syn is reported to be a substrate of Parkin (115). In Drosophila, overexpression of Parkin effectively suppresses α-Syn-induced degeneration of dopaminergic neurons (11). This suppression is not associated with significant changes in total cellular α-Syn protein levels, suggesting that the toxic species of α-Syn, if targeted by Parkin, may represent only a small portion of total α-Syn protein. Alternatively, Parkin suppression of α-Syn toxicity may act through a mechanism unrelated to α-Syn degradation by the ubiquitin-proteasome pathway (Figure 6). Using two other assays of α-Syn-induced toxicity, premature decline of climbing ability, and degeneration of retinal cells, another group also reported protective effects of Parkin against α-Syn toxicity in Drosophila (116).
The neuroprotective effect of Parkin has also been demonstrated in mammalian systems. In cell culture systems, overexpression of mutant α-Syn decreases proteasome function, which can be counteracted by overexpression of Parkin (117). In an in vivo study carried out in a rat lentiviral model of PD, overexpression of wild-type rat Parkin protected against the toxicity of disease-associated human α-Syn A30P. Animals overexpressing Parkin show significant reductions in α-Syn-induced neuropathology, leading to preservation of TH-positive cell bodies in the substantia nigra and to sparing of TH-positive nerve terminals in the striatum. Surprisingly, Parkin-mediated neuroprotection has been associated with an increase of hyper-phosphorylated α-Syn inclusions, supporting a role for Parkin in the genesis of Lewy bodies and implicating an intriguing neuroprotective role for α-Syn inclusion formation in the disease process (118). These studies suggest that compounds capable of upregulating Parkin activity could offer a promising treatment for PD.
DJ-1 and PI3K/AKT Signaling
Using DJ-1A RNAi-induced photoreceptor degeneration as an assay, a systematic genetic interaction study was carried out to identify genes and pathways that can modify DJ-1A-associated neurodegeneration. This search led to the isolation of components of the PI3K/Akt signaling pathway as specific genetic modifiers (106) (Figure 7). Reduction of PI3K/Akt signaling enhanced DJ-1A RNAi phenotypes, whereas activation of PI3K/Akt signaling significantly rescued DJ-1A RNAi phenotypes. The modifying effects of PI3K/Akt signaling on DJ-1A RNAi phenotypes was also manifested in dopaminergic neurons. Consistent with the genetic interaction studies, PI3K/Akt signaling was impaired in DJ-1A RNAi animals, as indicated by the reduction of p-Akt levels. An independent study (119) identified DJ-1A as a genetic modifier of PTEN-induced small-eye phenotype in Drosophila. The authors performed further biochemical studies in mammalian cells and found that DJ-1 RNAi results in decreased phosphorylation of PKB/Akt, whereas DJ-1 overexpression leads to hyperphosphorylation of PKB/Akt and increased cell survival. Furthermore, in primary breast cancer samples, DJ-1 expression correlates negatively with PTEN immunoreactivity and positively with Akt hyperphosphorylation (119). DJ-1 thus appears to be a key negative regulator of PTEN and may serve as a useful prognostic marker for cancer. These studies raise the interesting possibility that cancer and PD, two seemingly disparate diseases, may share certain underlying biochemical pathways or risk factors. Supporting a connection between cancer and PD, epidemiological studies found that incidences of tumorigenesis, especially the non-malignant tumors, were significantly lower in PD patients (120). One way to think about the connection between PD and cancer is that tumors can result from the imbalance of a common signaling pathway that controls cell proliferation and survival. Tipping the balance can result in overproliferation and cancer in one direction, and cell death and PD in the other.
Figure 7.

Scanning electron microscopy eye images showing genetic interaction between DJ-1A and genes in the phosphatidylinositol 3–kinase (PI3K) signaling pathway. Shown are eye images of a control wild-type fly (a), a DJ-1A RNA interference (RNAi) fly (b), and DJ-1A RNAi flies coexpressing PI3K (c), Akt (d ), dominant-negative PI3K (e), and phosphatase and tensin homolog (PTEN) ( f )a. Note that the eye degeneration phenotype caused by DJ-1A RNAi was suppressed by activation of PI3K signaling (panels c and d ) but was enhanced by attenuation of PI3K signaling (panels e and f ). Modified from Yang et al. (106).
Genetic Interaction Between Pink1 and Parkin and Their Physiological Roles in Mitochondria
The indirect flight muscle degeneration, male sterility, dopaminergic neuron degeneration, and mitochondrial morphological defects observed in dPink1-mutant or dPink1 RNAi flies are strikingly similar to those seen in dParkin mutants. This observation prompted genetic epistasis studies to investigate the relationship between Pink1 and Parkin. Overexpression of Parkin rescues the male sterility and mitochondrial morphology defects of dPink1 mutants, whereas double mutants lacking both Pink1 and Parkin function show phenotypes identical to those observed in either mutant alone. Overexpression of Pink1 has no effect on dParkin-mutant phenotypes. These observations suggest that Pink1 and Parkin function in the same pathway, with Pink1 acting upstream of Parkin (107–109) (Figure 6). Consistent with this notion, Parkin protein level is reduced in dPink1 RNAi animals (109). Future studies are needed to test whether Pink1 can directly modify Parkin.
The exact mitochondrial process regulated by Pink1 and Parkin is completely unknown. An understanding of this process will provide significant insight into the disease mechanism and could suggest novel therapeutic targets. Given the prominent mitochondrial morphological defects seen in dPink1 and dParkin mutants, one particular area that warrants future investigation is the mitochondrial fission/fusion pathway, a conserved mitochondria-remodeling process that controls the dynamic distribution and morphology of mitochondria in all eukaryotes (121). Mitochondrial fission/fusion has recently been shown to be important for regulating synaptic structure and plasticity (122), and imbalance of mitochondrial fission/fusion can lead to neurodegeneration (123). Future studies will ascertain whether Pink1/Parkin regulate mitochondrial morphology through interaction with components of the fission/fusion machinery.
It is intriguing that the indirect flight muscle and dopaminergic neurons are particularly vulnerable to Pink1 and Parkin mutations. It is possible that high endogenous levels of oxidative stress in these tissues may confer this vulnerability. Oxidative stress has been recognized as a major cause of aging and is considered a major pathogenic factor in PD (124). As the main cellular sites where oxygen is consumed, mitochondria produce most of the ROS in a cell. Thus it is not surprising that mitochondrial dysfunction is closely associated with oxidative stress. As one of the most metabolically active tissues, fly indirect flight muscle probably produces more ROS than any other tissue and is presumably under constant oxidative stress (125). Dopaminergic neurons are also under constant oxidative stress due to the oxidative metabolism of dopamine. These conditions may confer particular vulnerability of these tissues to additional oxidative insults caused by mitochondrial dysfunction in dPink1 and dParkin mutants.
Gene Expression Changes That Predate Neurodegeneration
Whole-genome analysis of gene expression changes represents an alternative approach to defining disease mechanism and identifying new players. To investigate the transcriptional program encoding the molecular machineries involved in α-Syn-induced neuropathology, the expression of the entire Drosophila genome at presymptomatic, early, and advanced disease stages was characterized by DNA microarray analysis (126). Signature transcripts including lipid-, energy-, and membrane-transport mRNAs were found to be closely linked with disease pathogenesis. The advantage of the DNA microarray approach is that it allows detection of gene expression changes at the presymptomatic stage. Indeed, changes in the expression of genes involved in catecholamine biosynthesis, mitochondrial function, and lipid binding were observed in histologically and behaviorally normal 1-day-old α-Syn transgenic flies that became symptomatic at 20 days. In an age-matched tauopathy fly model, the transcription of α-Syn-associated genes was normal. This result is consistent with the notion that the pathways of neurodegeneration may be highly specific for different diseases (55, 127).
MODELING OF POLYGLUTAMINEDISEASES
Genes Involved in Polyglutamine Diseases
The molecular cloning of the gene responsible for Huntington’s disease (HD) more than a decade ago revealed that the disease-causing mutation is a CAG trinucleotide repeat that encodes polyQs in the Huntingtin (Htt) protein. Later studies showed that diverse dominantly inherited neurodegenerative diseases are caused by CAG repeat expansion within disease-specific proteins. These include spinobulbar muscular atrophy (SBMA), spinocerebellar ataxia (SCA) 1, 2, 3, 6, 7, and 17, and dentatorubralpallidoluysian atrophy. These polyQ expansions confer toxic properties to the resident proteins. There is a good correlation between repeat length and disease severity (128). These polyQ repeats form intranuclear inclusions in neurons and cultured cells and are intrinsically toxic. However, when the polyQ sequence was placed in other proteins, its toxicity was altered (129). This suggested that the flanking protein sequence in the endogenous protein can modulate polyQ toxicity, leading to the suggestion that the protein context-dependent toxicity of polyQ may explain the neuronal subtype specificity of the different diseases. As is discussed below, protein cofactors binding to the nonpolyQ regions of the disease gene products could have profound effects on the toxicity of polyQ-containing proteins.
Fly Models of Polyglutamine Diseases
A number of polyQ disease models have been established in Drosophila. These include models for SCA-3, SCA-1, HD, and SBMA. In all four cases, expression of truncated, expanded polyQ-containing protein fragments leads to retinal degeneration. In the case of SCA-1, overexpression of the nonexpanded wild-type ataxin-1 protein also results in retinal degeneration, indicating that the ataxin-1 protein is intrinsically toxic at high levels (8). In the SBMA model, ingestion of androgen and its known antagonists promote neurodegeneration in flies expressing full-length mutant human androgen receptor (hAR), which is otherwise nontoxic (130). This suggests that hormone binding, subsequent structural alteration, and nuclear localization of mutant hAR protein are critical steps in the pathogenesis of SMBA. Because the fly eye is composed of ~800 eye units known as ommatidia, which are highly organized on an epithelial surface, subtle changes in gene function are amplified in this system. This feature, plus the fact that the eye is dispensable for animal viability, has enabled large-scale genetic screens to identify second-site genetic modifiers of the polyQ models. These models have also been widely used in testing the therapeutic effects of candidate compounds.
DISEASE MECHANISMS LEARNED FROM FLY POLYGLUTAMINE MODELS
Forward Genetic Screens Identify Novel Mechanisms and Pathways Involved in Spinocerebellar Ataxia–1 Pathogenesis
Significant insights into the pathogenic pathways of polyQ diseases were obtained from the SCA-1 model. In loss-of-function and overexpression screens for genes that can modify expanded ataxin-1-induced neurodegeneration, genes involved in a number of biological processes were identified (8). Not surprisingly, molecular chaperones, which have previously been implicated in regulating polyQ toxicity, were recovered in the SCA-1 screen. This screen also identified modifier genes involved in RNA processing, nuclear transport, transcriptional regulation (histone deacetylation), and cellular detoxification. Such processes had not been implicated in polyQ pathogenesis prior to this report. The identification of these new genes and mechanisms demonstrates the power of fly genetics in dissecting disease mechanisms. Further analysis of these modifier gene products in vertebrate models will help validate the findings from the fly models.
Regulation of Ataxin-1 Toxicity by Phosphorylation and 14-3-3 Binding
Using the SCA-1 model, investigators found that the 14-3-3 protein, an abundant and multifunctional protein, can bind to ataxin-1 (Atx-1), leading to the stabilization and accumulation of that toxic protein (131). 14-3-3 proteins are known to bind other proteins in a phosphorylation-dependent manner. Indeed, the interaction between Atx-1 and 14-3-3 is phosphorylation dependent. The PI3K/AKT pathway was found to regulate the association between Atx-1 and 14-3-3. AKT phosphorylates Atx-1 at a conserved Ser776 residue, and this phosphorylation event provides a binding site for 14-3-3 (Figure 8). In support of the role of AKT and 14-3-3 in SCA-1 pathogenesis, in vivo genetic manipulation of AKT and 14-3-3 levels modulates Atx-1-induced neurodegeneration. These results suggest that manipulating PI3K/AKT-pathway activity or interfering with the interaction between 14-3-3 and Atx-1 may provide new strategies for therapeutic intervention. Significantly, phosphorylation of Atx-1 also appeared to be important in regulating disease pathogenesis in a mouse SCA-1 model (132). As discussed above, activation of the PI3K/Akt pathway can suppress the DJ-1A-associated photoreceptor and dopaminergic neuron degeneration, but the same genetic manipulation enhances SCA-1 pathogenesis. This reinforces the notion that pathways of neurodegeneration are highly specific for different diseases.
Figure 8.

A diagram summarizing molecular events involved in spinocerebellar ataxia–1 (SCA-1) pathogenesis. Solid lines indicate relationships that are supported by experimental evidence; dashed lines indicate relationships for which experimental support is weak or currently missing. Abbreviations: PI3K, phosphatidylinositol 3–kinase; polyQ, polyglutamine; Pten, phosphatase and tensin homolog.
Endogenous Factors and Flanking Protein Sequences Contribute to the Selectivity of Polyglutamine Toxicity
The role of endogenous factors in influencing polyQ toxicity is best illustrated by studies of Atx-1 in fly and mouse. The Drosophila Atx-1 (dAtx-1) has a conserved AXH domain but lacks the polyQ tract found in human Atx-1 (hAtx-1). Overexpression of hAtx-1 in flies produces phenotypes similar to those of dAtx-1 but different from the polyQ peptide alone. The transcription factors Senseless and Gfi-1 have been found to interact with Atx-1’s AXH domain. In flies, overexpression of Atx-1 inhibits sensory organ development by decreasing the level of Senseless protein. Similarly, overexpression of wild-type and polyQ-expanded hAtx-1 reduces Gfi-1 levels in mouse Purkinje cells. Deletion of the AXH domain abolishes the effects of glutamine-expanded hAtx-1 on Senseless/Gfi-1. Furthermore, loss of Gfi-1 mimics SCA-1 phenotypes in Purkinje cells (133). These results suggest that endogenous factors such as Senseless/Gfi-1 contribute to the selective Purkinje cell–degeneration phenotype in SCA-1 through protein-protein interaction with the nonpolyQ portion of the Atx-1 protein (Figure 8).
The observation that the toxicity of polyQ repeats can be altered when placed in the context of other proteins provides further insights into the specificity of the different polyQ diseases (129). It has been found that polyQ peptide is intrinsically toxic in a polyQ length-dependent and progressive manner. However, when polyQ was expanded in Drosophila Dishevelled (a protein that contains a naturally occurring polyQ tract but that is not associated with disease), the toxic effect of expanded polyQ was neutralized by this new protein context. The dependence of polyQ toxicity on flanking sequence and on endogenous trans-acting factors may explain why selective neuronal populations are affected in each disease, despite the broader expression domains of the disease-gene products.
Modulation of Polyglutamine Toxicity by Sumoylation and Ubiquitination
Studies of a fly model of SBMA have emphasized the roles of the ubiquitin-proteasome pathway and the sumoylation pathway in polyQ pathogenesis. Expression of a pathogenic hAR protein with an expanded polyQ repeat results in nuclear and cytoplasmic inclusion formation and in cellular degeneration (134). The influence of ubiquitin-dependent modification and the proteasome pathway on neural degeneration was studied in this model. Compromising the ubiquitin/proteasome pathway enhanced degeneration and decreased polyQ protein solubility. Further, overexpression of a mutant form of the SUMO-1-activating enzyme Uba2 showed that polyQ-induced degeneration was intensified when the cellular SUMO-1 protein conjugation pathway was compromised, although the exact mechanism of mutant Uba2 action on polyQ was not addressed in this study. This study nevertheless implicates an interplay between the ubiquitin/proteasome and the SUMO pathways in modulating polyQ toxicity (134).
Another study of expanded Htt protein provided more conclusive evidence for the involvement of sumoylation in regulating polyQ toxicity (135). The authors found that sumoylation and ubiquitination compete for the same Lys residue in Htt. Sumoylation stabilized Htt, prevented aggregate formation, and enhanced the protein’s transcriptional repressor activity. In a fly HD model, sumoylation enhanced neurodegeneration, whereas ubiquitination had the opposite effect. Mutating the Lys residue prevented both sumoylation and ubiquitination and abrogated neurodegeneration. Sumoylation thus appears to be an essential step in polyQ pathogenesis and may represent a unique opportunity for therapeutic intervention.
Role of Specific Cell Death Regulators in Polyglutamine Pathogenesis
As in other neurodegenerative diseases, the role of programmed cell death in polyQ disease pathogenesis is a matter of debate. In fly models, expression of the antiapoptotic protein p35 has little effect on polyQ toxicity (136). Further, apoptosis-related genes have not been recovered as modifiers from the various genetic screens. However, testing on candidate cell death genes has revealed a common role for Dark in polyQ pathogenesis; Dark is the fly homolog of human Apaf-1, a key regulator of apoptosis in mammals. PolyQ-induced cell death was dramatically suppressed in flies lacking Dark. This was accompanied by suppression of caspase activation and aggregate formation. Dark colocalizes with ubiquitinated protein aggregates. Further, mammalian Apaf-1 colocalizes with Htt-containing aggregates in a murine HD model and in HD brain, suggesting a common role for Dark/Apaf-1 in polyQ pathogenesis (137). The molecular pathway mediating Dark/Apaf-1 function remains to be defined. It is possible that Dark/Apaf-1 regulates polyQ aggregation and toxicity in a cell death–independent manner.
Axonal Transport and Polyglutamine Toxicity
Aggregates of polyQ-containing protein are found in the nucleus and cytoplasm of diseased neurons. Although there is considerable evidence for transcriptional deregulation in polyQ pathogenesis, presumably caused by nuclear aggregates, it is possible that the cytoplasmic aggregates also contribute to disease pathogenesis. Several studies have indicated that defects in axonal transport may contribute to polyQ toxicity (138–140), presumably due to the physical blockage of axonal transport by the large protein aggregates. Intriguingly, inhibition of the activity of the Drosophila homolog of Htt also leads to axonal transport defects (138), suggesting that polyQ may act by interfering with endogenous Htt gene function.
OTHER DISEASE MODELS
Noncoding Trinucleotide Repeat Disease
In addition to the polyQ diseases, where trinucleotide repeat expansion leads to toxic protein production, there are other diseases caused by trinucleotide repeat expansion within noncoding regions of mRNAs. Examples include SCA-8, -10, -12, myotonic dystrophy types 1 and 2, and fragile X mental retardation (FMR). In contrast to the polyQ diseases, the mechanism by which the noncoding region trinucleotide repeats cause pathology is not clear, and there are no good mammalian models for studying these diseases.
To understand how the rCGG repeats in the 5′ UTR of the FMR1 gene cause neurodegeneration in premutation carriers, 90 rCGG repeats—a number that is intermediate between the numbers of repeats found in patients (>200) and in normal individuals (<60)—were expressed in the fly eye. These 90 noncoding rCGG repeats were sufficient to cause degeneration of photoreceptors (141). Although no mutant protein was produced from this repeat sequence, the degenerating neurons formed HSP70- and ubiquitin-positive neuronal inclusion bodies. Furthermore, overexpression of HSP70 suppressed rCGG repeat–induced neuronal death. This result demonstrates that abnormal RNAs can also cause neurodegeneration. It is possible that the expanded RNAs cause toxicity by sequestering essential RNA-binding proteins and molecular chaperones.
In another fly model of RNA-induced neurodegeneration, the human SCA-8 gene, which encodes a noncoding RNA, was expressed in fly eye (142). It induced a late-onset, progressive neurodegeneration phenotype. Interestingly, both wild-type and trinucleotide-expanded SCA-8 induced neurodegeneration phenotypes. Using these SCA-8 models, a genetic screen was carried out; it resulted in the identification of four neuronally expressed RNA-binding proteins as modifiers of SCA-8-induced neurodegeneration. Further characterization of the biochemical interaction between these RNA-binding proteins and SCA-8 RNAs, both wild type and trinucleotide expanded, and the functional significance of these interactions will help us understand the mechanism of noncoding-region trinucleotide repeats–induced neurodegeneration.
In an effort to model the pathogenesis of myotonic dystrophy using Drosophila, transgenic flies incorporating CTG repeats in the 3′ UTR of a reporter gene were generated (143). In this study, expanded CUG repeats formed discrete ribonuclear foci in muscle cells. However, flies expressing expanded CUG repeats had no detectable pathological phenotype, suggesting that in contrast to expanded polyQ-containing proteins, neither the expanded CUG-repeat RNA nor the ribonuclear foci are directly toxic in muscle cells (143).
Amyotrophic Lateral Sclerosis and Prion Diseases
Two other prominent types of neurodegenerative disease, amyotrophic lateral sclerosis and prion diseases, have been successfully modeled in mice. Prion diseases are unique among neurodegenerative diseases in that the causative agents are thought to be proteins with abnormal confirmation that can alter the conformation of normal cellular proteins. A recent study (144) suggests that Drosophila may be a useful model for studying prion diseases. Transgenic flies heterologously expressing disease-associated (P101L) mouse prion protein (PrP) in cholinergic neurons exhibited severe locomotor dysfunction and premature death as larvae and as adults. Clinical abnormalities were accompanied by age-dependent accumulation of misfolded PrP molecules, intracellular PrP aggregates, and neuronal vacuoles. These transgenic flies thus exhibited several hallmark features of human Gerstmann–Straussler–Scheinker (GSS) syndrome (144). This model should enable future genetic and pharmacologic studies of GSS.
USING FLY MODELS TO TEST AND SCREEN FOR THERAPEUTIC COMPOUNDS
Drosophila models of neurodegenerative diseases are excellent systems for in vivo testing of therapeutic effects of various compounds. The bubblegum mutant exhibits adult-onset neurodegeneration and contains elevated levels of very long chain fatty acids, as seen in the human disease adrenoleukodystrophy (ALD). In ALD, very long chain fatty acids can be lowered by dietary treatment with “Lorenzo’s oil,” a mixture of unsaturated fatty acids. In a successful application of the chemical approach to neurodegeneration in Drosophila, feeding the bub-blegum mutant with one of the components of Lorenzo’s oil, glyceryl trioleate oil, prevented neurodegeneration (145). Despite the effect of Lorenzo’s oil in lowering very long chain fatty acids, it has no therapeutic effects in ALD patients, suggesting that not all therapeutic effects seen in Drosophila may be observed in mammals.
Biochemical studies in mammalian cell culture showed that the polyQ-containing domain of Htt binds directly to the acetyl-transferase domains of certain transcriptional factors, including cAMP response element–binding (CREB)-binding protein (CBP) and p300/CBP-associated factor, resulting in inhibition of their acetyltransferase activities. Expression of Htt in cultured cells reduces the level of the acetylated histones H3 and H4, an effect that can be reversed by administering histone deacetylase (HDAC) inhibitors. In two Drosophila models of polyQ disease, HDAC inhibitors reduced cellular toxicity associated with polyQ repeat expansion and arrested neuronal degeneration (146). HDAC inhibitors thus represent lead compounds for the development of therapeutics for human polyQ diseases.
Based on the observations that mammalian target of rapamycin (mTOR) is sequestered in polyQ aggregates and that sequestration of mTOR impairs its kinase activity and induces autophagy (a key clearance pathway for mutant Htt fragments), Ravikumar et al. (147) tested the effect of the specific mTOR inhibitor rapamycin on Htt accumulation and cell death in cell models of HD. Rapamycin showed protective effects. Further experiments showed that rapamycin protects against neurodegeneration in a fly model of HD, and the rapamycin analog CCI-779 improved performance in behavioral tasks and decreased aggregate formation in a mouse model of HD. Rapamycin also appears to be effective in protecting against the neurotoxicity of other protein aggregates such as tau, suggesting that the therapeutic potential of rapamycin may go beyond polyQ diseases (148).
Fly PD and AD models have also been used to test specific therapeutic compounds. Based on the evidence that Hsp70 overexpression can prevent α-Syn-induced toxicity (97), Auluck & Bonini (149) tested the effect of feeding flies with geldanamycin, a naturally occurring benzoquinone ansamycin that (a) specifically binds and inhibits HSP90, a negative regulator of heat shock factor and (b) can induce stress response in cell culture. Geldanamycin treatment induced HSP70 expression and prevented α-Syn-induced dopaminergic neuronal loss. As mentioned above, melatonin was also effective in preventing dopaminergic degeneration in a fly rotenone model of sporadic PD (13). If geldanamycin’s and melatonin’s neuroprotective effects can be confirmed in mammalian PD models, these compounds warrant further exploration as novel therapeutics for PD.
Given the importance of protein phosphorylation in regulating neurotoxicity in a number of disease models, protein kinase inhibitors are excellent candidates for therapeutics. Two GSK-3β inhibitors, lithium and AR-A014418, can reverse both the axon transport and the locomotor defects caused by tau, suggesting that inhibition of GSK-3β may have therapeutic benefits in tauopathies (150). Lithium is currently used clinically to treat bipolar disorders. If the effects of lithium on tauopathies can be further validated in mammalian systems, lithium can be quickly moved to clinical trials to treat diseases with prominent tau pathology, such as AD, progressive supranuclear palsy, and frontotemporal dementia.
In addition to testing candidate compounds, the fly’s small size, high fecundity, and low cost make Drosophila disease models excellent systems for performing high-throughput screening of random chemical libraries for compounds with therapeutic potential. Further characterization of the effects of the compounds on the hallmark pathological features of the disease models and the identification of the cellular targets of the compounds will provide significant insights into disease mechanisms and suggest novel therapeutic strategies. Indeed, if a drug originally isolated using fly models could be used to ameliorate or cure a human neurodegenerative disease, this would arguably represent the best validation of the medical relevance of Drosophila models to human diseases.
CONCLUDING REMARKS
The development of highly relevant animal models of human diseases has ushered in a new era in neurodegenerative disease research. The power of fly models lies in the ability to perform large-scale forward genetic screens to dissect the cellular signaling pathways involved in the disease process without making any assumptions about what kind of molecules or mechanisms are involved. Once novel mechanisms or molecules are identified in fly models, their relevance to mammalian system should of course be verified. It is anticipated that ongoing and future genetic modifier screens will generate further mechanistic insights into disease processes and will help to answer some of the unresolved questions in the field, such as the identity of the neurotoxic species for each disease type, the key neuronal functions affected by the neurotoxic proteins, and the determinants of the cell type–specific vulnerability underlying each disease. The genetic modifiers identified in flies may also help reveal disease susceptibility genes in humans.
Acknowledgments
We apologize to those whose work we could not cite because of space constraints. This work was supported in part by grants from the McKnight Foundation, the Beckman Foundation, the Sloan Foundation, and the National Institutes of Health (NS043167).
- AD
Alzheimer’s disease
- PD
Parkinson’s disease
- RNA interference (RNAi)
a posttranscriptional gene-silencing process mediated by short, double-stranded RNAs that leads to the degradation of target RNA
- Tauopathy
refers to neurodegenerative diseases that are characterized by the aggregation of tau protein
- PAR-1
partitioning defective-1
- PINK1
phosphatase and tensin homolog (PTEN)-induced kinase 1
- Molecular chaperones
diverse family of prokaryotic and eukaryotic intracellular proteins involved in the assembly and transmembrane translocation of other proteins
- Polyglutamine (polyQ) diseases
neurodegenerative diseases exemplified by Huntington’s disease that are caused by the expansion of CAG repeats in the protein- coding region of certain genes, resulting in the expression of mutant proteins containing polyQ stretches
- HD
Huntington’s disease
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any biases that might be perceived as affecting the objectivity of this review.
Contributor Information
Bingwei Lu, Email: bingwei@stanford.edu.
Hannes Vogel, Email: hvogel@stanford.edu.
LITERATURE CITED
- 1.Bonini NM, Fortini ME. Human neurodegenerative disease modeling using Drosophila. Annu Rev Neurosci. 2003;26:627–56. doi: 10.1146/annurev.neuro.26.041002.131425. [DOI] [PubMed] [Google Scholar]
- 2.Muqit MM, Feany MB. Modelling neurodegenerative diseases in Drosophila: a fruitful approach? Nat Rev Neurosci. 2002;3:237–43. doi: 10.1038/nrn751. [DOI] [PubMed] [Google Scholar]
- 3.Driscoll M, Gerstbrein B. Dying for a cause: invertebrate genetics takes on human neurodegeneration. Nat Rev Genet. 2003;4:181–94. doi: 10.1038/nrg1018. [DOI] [PubMed] [Google Scholar]
- 4.Rogina B, Benzer S, Helfand SL. Drosophila drop-dead mutations accelerate the time course of age-related markers. Proc Natl Acad Sci USA. 1997;94:6303–6. doi: 10.1073/pnas.94.12.6303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kretzschmar D, Hasan G, Sharma S, Heisenberg M, Benzer S. The swisscheese mutant causes glial hyperwrapping and brain degeneration in Drosophila. J Neurosci. 1997;17:7425–32. doi: 10.1523/JNEUROSCI.17-19-07425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akassoglou K, Malester B, Xu J, Tessarollo L, Rosenbluth J, Chao MV. Brain-specific deletion of neuropathy target esterase/swisscheese results in neurodegeneration. Proc Natl Acad Sci USA. 2004;101:5075–80. doi: 10.1073/pnas.0401030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feany MB, Bender WW. A Drosophila model of Parkinson’s disease. Nature. 2000;404:394–98. doi: 10.1038/35006074. Described the first fly model of PD. [DOI] [PubMed] [Google Scholar]
- 8.Fernandez-Funez P, Nino-Rosales ML, de Gouyon B, She WC, Luchak JM, et al. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature. 2000;408:101–68. doi: 10.1038/35040584. A large-scale genetic modifier screen that identified a number of new genes and processes that are involved in Ataxin-1 pathogenesis. [DOI] [PubMed] [Google Scholar]
- 9.Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA. 2003;100:4078–83. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pesah Y, Pham T, Burgess H, Middlebrooks B, Verstreken P, et al. Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development. 2004;131:2183–94. doi: 10.1242/dev.01095. [DOI] [PubMed] [Google Scholar]
- 11.Yang Y, Nishimura I, Imai Y, Takahashi R, Lu B. Parkin suppresses dopaminergic neuron-selective neurotoxicity induced by Pael-R in Drosophila. Neuron. 2003;37:911–24. doi: 10.1016/s0896-6273(03)00143-0. [DOI] [PubMed] [Google Scholar]
- 12.Rong YS, Golic KG. Gene targeting by homologous recombination in Drosophila. Science. 2000;288:2013–18. doi: 10.1126/science.288.5473.2013. [DOI] [PubMed] [Google Scholar]
- 13.Coulom H, Birman S. Chronic exposure to rotenone models sporadic Parkinson’s disease in Drosophila melanogaster. J Neurosci. 2004;24:10993–98. doi: 10.1523/JNEUROSCI.2993-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Betarbet R, Sherer TB, Greenamyre JT. Animal models of Parkinson’s disease. Bioessays. 2002;24:308–18. doi: 10.1002/bies.10067. [DOI] [PubMed] [Google Scholar]
- 15.De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active γ-secretase complex. Neuron. 2003;38:9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- 16.Haass C, De Strooper B. The presenilins in Alzheimer’s disease—proteolysis holds the key. Science. 1999;286:916–19. doi: 10.1126/science.286.5441.916. [DOI] [PubMed] [Google Scholar]
- 17.Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39:168–77. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–56. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 19.Cleveland DW, Hwo SY, Kirschner MW. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J Mol Biol. 1977;116:207–25. doi: 10.1016/0022-2836(77)90213-3. [DOI] [PubMed] [Google Scholar]
- 20.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (Tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986;83:4913–17. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greenberg SG, Davies P. A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci USA. 1990;87:5827–31. doi: 10.1073/pnas.87.15.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee VM, Balin BJ, Otvos L, Jr, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science. 1991;251:675–78. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- 23.Foster NL, Wilhelmsen K, Sima AA, Jones MZ, D’Amato CJ, Gilman S. Frontotemporal dementia and parkinsonism linked to chromosome 17: a consensus conference. Ann Neurol. 1997;41:706–15. doi: 10.1002/ana.410410606. [DOI] [PubMed] [Google Scholar]
- 24.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–5. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 25.Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–25. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- 26.Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA. 1998;95:7737–41. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 28.Mandelkow EM, Mandelkow E. Tau in Alzheimer’s disease. Trends Cell Biol. 1998;8:425–27. doi: 10.1016/s0962-8924(98)01368-3. [DOI] [PubMed] [Google Scholar]
- 29.Davies P. A very incomplete comprehensive theory of Alzheimer’s disease. Ann N Y Acad Sci. 2000;924:8–16. doi: 10.1111/j.1749-6632.2000.tb05553.x. [DOI] [PubMed] [Google Scholar]
- 30.Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, et al. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 1999;24:751–62. doi: 10.1016/s0896-6273(00)81127-7. [DOI] [PubMed] [Google Scholar]
- 31.Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–5. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 32.Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–91. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 33.Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Aβ 42 fibrils. Science. 2001;293:1491–95. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 34.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron. 2003;39:409–21. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 35.Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Aβ immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–32. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 36.Hou CE, Carlin D, Miller BL. Non–Alzheimer’s disease dementias: anatomic, clinical, and molecular correlates. Can J Psychiatry. 2004;49:164–71. doi: 10.1177/070674370404900303. [DOI] [PubMed] [Google Scholar]
- 37.Luo L, Tully T, White K. Human amyloid precursor protein ameliorates behavioral deficit of flies deleted for Appl gene. Neuron. 1992;9:595–605. doi: 10.1016/0896-6273(92)90024-8. Described functional conservation between human APP and fly Appl in regulating animal behavior. [DOI] [PubMed] [Google Scholar]
- 38.Ye Y, Lukinova N, Fortini ME. Neurogenic phenotypes and altered Notch processing in Drosophila Presenilin mutants. Nature. 1999;398:525–29. doi: 10.1038/19096. [DOI] [PubMed] [Google Scholar]
- 39.Struhl G, Greenwald I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature. 1999;398:522–25. doi: 10.1038/19091. [DOI] [PubMed] [Google Scholar]
- 40.Gunawardena S, Goldstein LS. Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron. 2001;32:389–401. doi: 10.1016/s0896-6273(01)00496-2. [DOI] [PubMed] [Google Scholar]
- 41.Fossgreen A, Bruckner B, Czech C, Masters CL, Beyreuther K, Paro R. Transgenic Drosophila expressing human amyloid precursor protein show γ-secretase activity and a blistered-wing phenotype. Proc Natl Acad Sci USA. 1998;95:13703–8. doi: 10.1073/pnas.95.23.13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Greeve I, Kretzschmar D, Tschape JA, Beyn A, Brellinger C, et al. Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. J Neurosci. 2004;24:3899–906. doi: 10.1523/JNEUROSCI.0283-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Finelli A, Kelkar A, Song HJ, Yang H, Konsolaki M. A model for studying Alzheimer’s Aβ42-induced toxicity in Drosophila melanogaster. Mol Cell Neurosci. 2004;26:365–75. doi: 10.1016/j.mcn.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 44.Iijima K, Liu HP, Chiang AS, Hearn SA, Konsolaki M, Zhong Y. Dissecting the pathological effects of human Aβ40 and Aβ42 in Drosophila: a potential model for Alzheimer’s disease. Proc 44 Natl Acad Sci USA. 2004;101:6623–28. doi: 10.1073/pnas.0400895101. Described a fly Ab model that exhibits neurodegeneration a phenotype. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Williams DW, Tyrer M, Shepherd D. Tau and tau reporters disrupt central projections of sensory neurons in Drosophila. J Comp Neurol. 2000;428:630–40. doi: 10.1002/1096-9861(20001225)428:4<630::aid-cne4>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 46.Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, et al. Tauopathy in 46. Reported fly Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711–14. doi: 10.1126/science.1062382. Reported fly tauopathy models that exhibit neurodegeneration phenotypes without NFT pathology. [DOI] [PubMed] [Google Scholar]
- 47.Jackson GR, Wiedau-Pazos M, Sang TK, Wagle N, Brown CA, et al. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron. 2002;34:509–19. doi: 10.1016/s0896-6273(02)00706-7. [DOI] [PubMed] [Google Scholar]
- 48.Mershin A, Pavlopoulos E, Fitch O, Braden BC, Nanopoulos DV, Skoulakis EM. Learning and memory deficits upon TAU accumulation in Drosophila mushroom body neurons. Learn Mem. 2004;11:277–87. doi: 10.1101/lm.70804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–59. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 50.Nishimura I, Yang Y, Lu B. PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell. 2004;116:671–82. doi: 10.1016/s0092-8674(04)00170-9. Took advantage of the power of genetic analysis in Drosophila to implicate PAR-1 kinase in regulating tau phosphorylation and toxicity. [DOI] [PubMed] [Google Scholar]
- 51.Guo S, Kemphues KJ. par-1, a gene required for establishing polarity in C. elegans embryos, encodes a putative Ser/Thr kinase that is asymmetrically distributed. Cell. 1995;81:611–20. doi: 10.1016/0092-8674(95)90082-9. [DOI] [PubMed] [Google Scholar]
- 52.Drewes G, Ebneth A, Preuss U, Mandelkow EM, Mandelkow E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell. 1997;89:297–308. doi: 10.1016/s0092-8674(00)80208-1. [DOI] [PubMed] [Google Scholar]
- 53.Bohm H, Brinkmann V, Drab M, Henske A, Kurzchalia TV. Mammalian homologues of C. elegans PAR-1 are asymmetrically localized in epithelial cells and may influence their polarity. Curr Biol. 1997;7:603–6. doi: 10.1016/s0960-9822(06)00260-0. [DOI] [PubMed] [Google Scholar]
- 54.Biernat J, Wu YZ, Timm T, Zheng-Fischhofer Q, Mandelkow E, et al. Protein kinase MARK/PAR-1 is required for neurite outgrowth and establishment of neuronal polarity. Mol Biol Cell. 2002;13:4013–28. doi: 10.1091/mbc.02-03-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shulman JM, Feany MB. Genetic modifiers of tauopathy in Drosophila. Genetics. 2003;165:1233–42. doi: 10.1093/genetics/165.3.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cummings CJ, Reinstein E, Sun Y, Antalffy B, Jiang Y, et al. Mutation of the E6-AP ubiquitin ligase reduces nuclear inclusion frequency while accelerating polyglutamine-induced pathology in SCA1 mice. Neuron. 1999;24:879–92. doi: 10.1016/s0896-6273(00)81035-1. [DOI] [PubMed] [Google Scholar]
- 57.Hanger DP, Betts JC, Loviny TL, Blackstock WP, Anderton BH. New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer’s disease brain using nanoelectrospray mass spectrometry. J Neurochem. 1998;71:2465–76. doi: 10.1046/j.1471-4159.1998.71062465.x. [DOI] [PubMed] [Google Scholar]
- 58.Wang JW, Imai Y, Lu B. Activation of PAR-1 kinase and stimulation of tau phosphorylation by diverse signals require the tumor-suppressor protein LKB1. J Neurosci. 2007;27:574–81. doi: 10.1523/JNEUROSCI.5094-06.2007. Reported a signaling cascade involving the tumor suppressor LKB1 and the PAR-1 kinase, which links APP and tau toxicity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Augustinack JC, Schneider A, Mandelkow EM, Hyman BT. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol (Berl) 2002;103:26–35. doi: 10.1007/s004010100423. [DOI] [PubMed] [Google Scholar]
- 60.Perez M, Ribe E, Rubio A, Lim F, Moran MA, et al. Characterization of a double (amyloid precursor protein-tau) transgenic: tau phosphorylation and aggregation. Neuroscience. 2005;130:339–47. doi: 10.1016/j.neuroscience.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 61.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–91. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 62.Wang P, Yang G, Mosier DR, Chang P, Zaidi T, et al. Defective neuromuscular synapses in mice lacking amyloid precursor protein (APP) and APP-like protein 2. J Neurosci. 2005;25:1219–25. doi: 10.1523/JNEUROSCI.4660-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ashe KH. Synaptic structure and function in transgenic APP mice. Ann N Y Acad Sci. 2000;924:39–41. doi: 10.1111/j.1749-6632.2000.tb05558.x. [DOI] [PubMed] [Google Scholar]
- 64.Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, et al. Reducing endogenous tau ameliorates amyloid β–induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–54. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 65.Chee FC, Mudher A, Cuttle MF, Newman TA, MacKay D, et al. Overexpression of tau results in defective synaptic transmission in Drosophila neuromuscular junctions. Neurobiol Dis. 2005;20:918–28. doi: 10.1016/j.nbd.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 66.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–51. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 67.Zhang Y, Guo H, Kwan H, Wang JW, Kosek J, Lu B. PAR-1 kinase phosphorylates Dlg and regulates its postsynaptic targeting at the Drosophila neuromuscular junction. Neuron. 2007;53:201–15. doi: 10.1016/j.neuron.2006.12.016. Identified Dlg as a substrate protein that mediates the synaptic toxicity of PAR-1 activation; this result has implications for understanding the abnormal synaptic expression of PSD-95 in AD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, et al. β in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis. 2005;20:187–98. doi: 10.1016/j.nbd.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 69.Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM. Synaptic changes in Alzheimer’s disease: Increased amyloid-β and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am J Pathol. 2004;165:1809–17. doi: 10.1016/s0002-9440(10)63436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roselli F, Tirard M, Lu J, Hutzler P, Lamberti P, et al. Soluble β-amyloid 1–40 induces NMDA-dependent degradation of postsynaptic density–95 at glutamatergic synapses. J Neurosci. 2005;25:11061–70. doi: 10.1523/JNEUROSCI.3034-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dexter DT, Sian J, Rose S, Hindmarsh JG, Mann VM, et al. Indices of oxidative stress and mitochondrial function in individuals with incidental Lewy body disease. Ann Neurol. 1994;35:38–44. doi: 10.1002/ana.410350107. [DOI] [PubMed] [Google Scholar]
- 72.Dunnett SB, Bjorklund A. Prospects for new restorative and neuroprotective treatments in Parkin-son’s disease. Nature. 1999;399:A32–39. doi: 10.1038/399a032. [DOI] [PubMed] [Google Scholar]
- 73.Zhang Y, Dawson VL, Dawson TM. Oxidative stress and genetics in the pathogenesis of Parkinson’s disease. Neurobiol Dis. 2000;7:240–50. doi: 10.1006/nbdi.2000.0319. [DOI] [PubMed] [Google Scholar]
- 74.Jenner P. Oxidative stress in Parkinson’s disease. Ann Neurol. 2003;53(Suppl 3):S26–38. doi: 10.1002/ana.10483. [DOI] [PubMed] [Google Scholar]
- 75.Beal MF. Experimental models of Parkinson’s disease. Nat Rev Neurosci. 2001;2:325–34. doi: 10.1038/35072550. [DOI] [PubMed] [Google Scholar]
- 76.Bertoli-Avella AM, Oostra BA, Heutink P. Chasing genes in Alzheimer’s and Parkinson’s disease. Hum Genet. 2004;114:413–38. doi: 10.1007/s00439-004-1097-7. [DOI] [PubMed] [Google Scholar]
- 77.Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson’s disease. Science. 2003;302:819–22. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- 78.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, et al. Mutation in the α-Synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–47. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 79.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–8. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 80.Leroy E, Boyer R, Auburger G, Leube B, Ulm G, et al. The ubiquitin pathway in Parkinson’s disease. Nature. 1998;395:451–52. doi: 10.1038/26652. [DOI] [PubMed] [Google Scholar]
- 81.Bonifati V, Rizzu P, Squitieri F, Krieger E, Vanacore N, et al. DJ-1(PARK7), a novel gene for autosomal recessive, early-onset parkinsonism. Neurol Sci. 2003;24:159–60. doi: 10.1007/s10072-003-0108-0. [DOI] [PubMed] [Google Scholar]
- 82.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–60. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 83.Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–7. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 84.Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 85.Shen J, Cookson MR. Mitochondria and dopamine; new insights into recessive parkinsonism. Neuron. 2004;43:301–4. doi: 10.1016/j.neuron.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 86.Canet-Aviles RM, Wilson MA, Miller DW, Ahmad R, McLendon C, et al. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci USA. 2004;101:9103–8. doi: 10.1073/pnas.0402959101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Unoki M, Nakamura Y. Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene. 2001;20:4457–65. doi: 10.1038/sj.onc.1204608. [DOI] [PubMed] [Google Scholar]
- 88.Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23:329–41. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 89.Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, et al. Dopaminergic loss and inclusion body formation in α-Synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–69. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 90.Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, et al. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem. 2003;278:43628–35. doi: 10.1074/jbc.M308947200. [DOI] [PubMed] [Google Scholar]
- 91.Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, et al. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial parkinsonism-linked gene DJ-1. Neuron. 2005;45:489–96. doi: 10.1016/j.neuron.2005.01.041. [DOI] [PubMed] [Google Scholar]
- 92.Kim RH, Smith PD, Aleyasin H, Hayley S, Mount MP, et al. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc Natl Acad Sci USA. 2005;102:5215–20. doi: 10.1073/pnas.0501282102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee MK, Stirling W, Xu Y, Xu X, Qui D, et al. Human α-Synuclein-harboring familial Parkinson’s disease–linked Ala-53/Thr mutation causes neurodegenerative disease with α-Synuclein aggregation in transgenic mice. Proc Natl Acad Sci USA. 2002;99:8968–73. doi: 10.1073/pnas.132197599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM. Neuronal α-synucleinopathy with severe movement disorder in mice expressing A53T human α-Synuclein. Neuron. 2002;34:521–33. doi: 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- 95.Von Coelln R, Thomas B, Savitt JM, Lim KL, Sasaki M, et al. Loss of locus coeruleus neurons and reduced startle in parkin null mice. Proc Natl Acad Sci USA. 2004;101:10744–49. doi: 10.1073/pnas.0401297101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, et al. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci USA. 2007;104:11441–46. doi: 10.1073/pnas.0702717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of 97. Showed that HSP70 α-Synuclein toxicity in a Drosophila model for Parkinson’s disease. Science. 2002;295:865–68. doi: 10.1126/science.1067389. Showed that HSP70 overexpression can suppress α-Syn-induced a neurotoxicity in a fly PD model, implicating molecular chaperones in PD pathogenesis. [DOI] [PubMed] [Google Scholar]
- 98.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, et al. α-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 99.Pesah Y, Burgess H, Middlebrooks B, Ronningen K, Prosser J, et al. Whole-mount analysis reveals normal numbers of dopaminergic neurons following misexpression of α-Synuclein in Drosophila. Genesis. 2005;41:154–59. doi: 10.1002/gene.20106. [DOI] [PubMed] [Google Scholar]
- 100.Cha GH, Kim S, Park J, Lee E, Kim M, et al. Parkin negatively regulates JNK pathway in the dopaminergic neurons of Drosophila. Proc Natl Acad Sci USA. 2005;102:10345–50. doi: 10.1073/pnas.0500346102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Whitworth AJ, Theodore DA, Greene JC, Benes H, Wes PD, Pallanck LJ. Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson’s disease. Proc Natl Acad Sci USA. 2005;102:8024–29. doi: 10.1073/pnas.0501078102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell. 2001;105:891–902. doi: 10.1016/s0092-8674(01)00407-x. [DOI] [PubMed] [Google Scholar]
- 103.Kitao Y, Imai Y, Ozawa K, Kataoka A, Ikeda T, et al. Pael receptor induces death of dopaminergic neurons in the substantia nigra via endoplasmic reticulum stress and dopamine toxicity, which is enhanced under condition of parkin inactivation. Hum Mol Genet. 2007;16:50–60. doi: 10.1093/hmg/ddl439. [DOI] [PubMed] [Google Scholar]
- 104.Meulener M, Whitworth AJ, Armstrong-Gold CE, Rizzu P, Heutink P, et al. Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson’s disease. Curr Biol. 2005;15:1572–77. doi: 10.1016/j.cub.2005.07.064. [DOI] [PubMed] [Google Scholar]
- 105.Menzies FM, Yenisetti SC, Min KT. Roles of Drosophila DJ-1 in survival of dopaminergic neurons and oxidative stress. Curr Biol. 2005;15:1578–82. doi: 10.1016/j.cub.2005.07.036. [DOI] [PubMed] [Google Scholar]
- 106.Yang Y, Gehrke S, Haque ME, Imai Y, Kosek J, et al. Inactivation of Drosophila DJ-1 leads to impairments of oxidative stress response and phosphatidylinositol 3–kinase/Akt signaling. Proc Natl Acad Sci USA. 2005;102:13670–75. doi: 10.1073/pnas.0504610102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–66. doi: 10.1038/nature04779. Reported fly models of Pink1-induced PD that showed defective mitochondrial morphology and implicated Parkin and Pink1 in the same pathway. [DOI] [PubMed] [Google Scholar]
- 108.Park J, Lee SB, Lee S, Kim Y, Song S, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–61. doi: 10.1038/nature04788. Reported fly models of Pink1-induced PD that showed defective mitochondrial morphology and implicated Parkin and Pink1 in the same pathway. [DOI] [PubMed] [Google Scholar]
- 109.Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, et al. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci USA. 2006;103:10793–98. doi: 10.1073/pnas.0602493103. Reported fly models of Pink1-induced PD that showed defective mitochondrial morphology and implicated Parkin and Pink1 in the same pathway. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang D, Qian L, Xiong H, Liu J, Neckameyer WS, et al. Antioxidants protect PINK1- dependent dopaminergic neurons in Drosophila. Proc Natl Acad Sci USA. 2006;103:13520–25. doi: 10.1073/pnas.0604661103. Reported fly models of Pink1-induced PD that showed defective mitochondrial morphology and implicated Parkin and Pink1 in the same pathway. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wu YR, Wang CK, Chen CM, Hsu Y, Lin SJ, et al. Analysis of heat-shock protein 70 genepolymorphisms and the risk of Parkinson’s disease. Hum Genet. 2004;114:236–41. doi: 10.1007/s00439-003-1050-1. [DOI] [PubMed] [Google Scholar]
- 112.Dong Z, Wolfer DP, Lipp HP, Bueler H. Hsp70 gene transfer by adeno-associated virus inhibits MPTP-induced nigrostriatal degeneration in the mouse model of Parkinson disease. Mol Ther. 2005;11:80–88. doi: 10.1016/j.ymthe.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 113.Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, et al. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–64. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- 114.Chen L, Feany MB. α-Synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci. 2005;8:657–63. doi: 10.1038/nn1443. [DOI] [PubMed] [Google Scholar]
- 115.Shimura H, Schlossmacher MG, Hattori N, Frosch MP, Trockenbacher A, et al. Ubiquitination of a new form of α-Synuclein by parkin from human brain: implications for Parkinson’s disease. Science. 2001;293:263–69. doi: 10.1126/science.1060627. [DOI] [PubMed] [Google Scholar]
- 116.Haywood AF, Staveley BE. Parkin counteracts symptoms in a Drosophila model of Parkinson’s disease. BMC Neurosci. 2004;5:14. doi: 10.1186/1471-2202-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Petrucelli L, O’Farrell C, Lockhart PJ, Baptista M, Kehoe K, et al. Parkin protects against the toxicity associated with mutant α-Synuclein: Proteasome dysfunction selectively affects catecholaminergic neurons. Neuron. 2002;36:1007–19. doi: 10.1016/s0896-6273(02)01125-x. [DOI] [PubMed] [Google Scholar]
- 118.Lo Bianco C, Schneider BL, Bauer M, Sajadi A, Brice A, et al. Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an α-Synuclein rat model of Parkinson’s disease. Proc Natl Acad Sci USA. 2004;101:17510–15. doi: 10.1073/pnas.0405313101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kim RH, Peters M, Jang Y, Shi W, Pintilie M, et al. DJ-1, a novel regulator of the tumor suppressor PTEN. Cancer Cell. 2005;7:263–73. doi: 10.1016/j.ccr.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 120.Jansson B, Jankovic J. Low cancer rates among patients with Parkinson’s disease. Ann Neurol. 1985;17:505–9. doi: 10.1002/ana.410170514. [DOI] [PubMed] [Google Scholar]
- 121.Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–52. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 122.Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–87. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 123.Bossy-Wetzel E, Barsoum MJ, Godzik A, Schwarzenbacher R, Lipton SA. Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr Opin Cell Biol. 2003;15:706–16. doi: 10.1016/j.ceb.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 124.Beal MF. Mitochondria, oxidative damage, and inflammation in Parkinson’s disease. Ann N Y Acad Sci. 2003;991:120–31. doi: 10.1111/j.1749-6632.2003.tb07470.x. [DOI] [PubMed] [Google Scholar]
- 125.Sacktor B. Biochemical adaptations for flight in the insect. Biochem Soc Symp. 1976;41:111–31. [PubMed] [Google Scholar]
- 126.Scherzer CR, Jensen RV, Gullans SR, Feany MB. Gene expression changes presage neurodegeneration in a Drosophila model of Parkinson’s disease. Hum Mol Genet. 2003;12:2457–66. doi: 10.1093/hmg/ddg265. [DOI] [PubMed] [Google Scholar]
- 127.Ghosh S, Feany MB. Comparison of pathways controlling toxicity in the eye and brain in Drosophila models of human neurodegenerative diseases. Hum Mol Genet. 2004;13:2011–18. doi: 10.1093/hmg/ddh214. [DOI] [PubMed] [Google Scholar]
- 128.Gusella JF, MacDonald ME. Molecular genetics: unmasking polyglutamine triggers in neurodegenerative disease. Nat Rev Neurosci. 2000;1:109–15. doi: 10.1038/35039051. [DOI] [PubMed] [Google Scholar]
- 129.Marsh JL, Walker H, Theisen H, Zhu YZ, Fielder T, et al. Expanded polyglutamine peptides alone are intrinsically cytotoxic and cause neurodegeneration in Drosophila. Hum Mol Genet. 2000;9:13–25. doi: 10.1093/hmg/9.1.13. [DOI] [PubMed] [Google Scholar]
- 130.Takeyama K, Ito S, Yamamoto A, Tanimoto H, Furutani T, et al. Androgen-dependent neurodegeneration by polyglutamine-expanded human androgen receptor in Drosophila. Neuron. 2002;35:855–64. doi: 10.1016/s0896-6273(02)00875-9. [DOI] [PubMed] [Google Scholar]
- 131.Chen HK, Fernandez-Funez P, Acevedo SF, Lam YC, Kaytor MD, et al. Interaction of Akt-phosphorylated ataxin-1 with 14–3-3 mediates neurodegeneration in spinocerebellar ataxia type 1. Cell. 2003;113:457–68. doi: 10.1016/s0092-8674(03)00349-0. [DOI] [PubMed] [Google Scholar]
- 132.Emamian ES, Kaytor MD, Duvick LA, Zu T, Tousey SK, et al. Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron. 2003;38:375–87. doi: 10.1016/s0896-6273(03)00258-7. [DOI] [PubMed] [Google Scholar]
- 133.Tsuda H, Jafar-Nejad H, Patel AJ, Sun Y, Chen HK, et al. The AXH domain of Ataxin-1 mediates neurodegeneration through its interaction with Gfi-1/Senseless proteins. Cell. 2005;122:633–44. doi: 10.1016/j.cell.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 134.Chan HY, Warrick JM, Andriola I, Merry D, Bonini NM. Genetic modulation of polyglutamine toxicity by protein conjugation pathways in Drosophila. Hum Mol Genet. 2002;11:2895–904. doi: 10.1093/hmg/11.23.2895. [DOI] [PubMed] [Google Scholar]
- 135.Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, et al. SUMO modification of Huntingtin and Huntington’s disease pathology. Science. 2004;304:100–4. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- 136.Jackson GR, Salecker I, Dong X, Yao X, Arnheim N, et al. Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron. 1998;21:633–42. doi: 10.1016/s0896-6273(00)80573-5. [DOI] [PubMed] [Google Scholar]
- 137.Sang TK, Li C, Liu W, Rodriguez A, Abrams JM, et al. Inactivation of Drosophila Apaf-1-related killer suppresses formation of polyglutamine aggregates and blocks polyglutamine pathogenesis. Hum Mol Genet. 2005;14:357–72. doi: 10.1093/hmg/ddi032. [DOI] [PubMed] [Google Scholar]
- 138.Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, et al. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40:25–40. doi: 10.1016/s0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- 139.Lee WC, Yoshihara M, Littleton JT. Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington’s disease. Proc Natl Acad Sci USA. 2004;101:3224–29. doi: 10.1073/pnas.0400243101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Szebenyi G, Morfini GA, Babcock A, Gould M, Selkoe K, et al. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron. 2003;40:41–52. doi: 10.1016/s0896-6273(03)00569-5. [DOI] [PubMed] [Google Scholar]
- 141.Jin P, Zarnescu DC, Zhang F, Pearson CE, Lucchesi JC, et al. RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron. 2003;39:739–47. doi: 10.1016/s0896-6273(03)00533-6. [DOI] [PubMed] [Google Scholar]
- 142.Mutsuddi M, Marshall CM, Benzow KA, Koob MD, Rebay I. The spinocerebellar ataxia 8 noncoding RNA causes neurodegeneration and associates with staufen in Drosophila. Curr Biol. 2004;14:302–8. doi: 10.1016/j.cub.2004.01.034. [DOI] [PubMed] [Google Scholar]
- 143.Houseley JM, Wang Z, Brock GJ, Soloway J, Artero R, et al. Myotonic dystrophy associated expanded CUG repeat muscleblind positive ribonuclear foci are not toxic to Drosophila. Hum Mol Genet. 2005;14:873–83. doi: 10.1093/hmg/ddi080. [DOI] [PubMed] [Google Scholar]
- 144.Gavin BA, Dolph MJ, Deleault NR, Geoghegan JC, Khurana V, et al. Accelerated accumulation of misfolded prion protein and spongiform degeneration in a Drosophila model of Gerstmann–Straussler–Scheinker syndrome. J Neurosci. 2006;26:12408–14. doi: 10.1523/JNEUROSCI.3372-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Min KT, Benzer S. Preventing neurodegeneration in the Drosophila mutant bubblegum. Science. 1999;284:1985–88. doi: 10.1126/science.284.5422.1985. [DOI] [PubMed] [Google Scholar]
- 146.Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–43. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- 147.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 148.Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 2006;15:433–42. doi: 10.1093/hmg/ddi458. [DOI] [PubMed] [Google Scholar]
- 149.Auluck PK, Bonini NM. Pharmacological prevention of Parkinson disease in Drosophila. Nat Med. 2002;8:1185–86. doi: 10.1038/nm1102-1185. [DOI] [PubMed] [Google Scholar]
- 150.Mudher A, Shepherd D, Newman TA, Mildren P, Jukes JP, et al. GSK-3β inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol Psychiatry. 2004;9:522–30. doi: 10.1038/sj.mp.4001483. [DOI] [PubMed] [Google Scholar]