

Abstract
The attachment of ubiquitin (Ub) and the Ub-like (Ubl) molecule interferon-stimulated gene 15 (ISG15) to cellular proteins mediates important innate antiviral responses. Ovarian tumor (OTU) domain proteases from nairoviruses and arteriviruses were recently found to remove these molecules from host proteins, which inhibits Ub and ISG15-dependent antiviral pathways. This contrasts with the Ub-specific activity of known eukaryotic OTU-domain proteases. Here we describe crystal structures of a viral OTU domain from the highly pathogenic Crimean–Congo haemorrhagic fever virus (CCHFV) bound to Ub and to ISG15 at 2.5-Å and 2.3-Å resolution, respectively. The complexes provide a unique structural example of ISG15 bound to another protein and reveal the molecular mechanism of an ISG15 cross-reactive deubiquitinase. To accommodate structural differences between Ub and ISG15, the viral protease binds the β-grasp folds of Ub and C-terminal Ub-like domain of ISG15 in an orientation that is rotated nearly 75° with respect to that observed for Ub bound to a representative eukaryotic OTU domain from yeast. Distinct structural determinants necessary for binding either substrate were identified and allowed the reengineering of the viral OTU protease into enzymes with increased substrate specificity, either for Ub or for ISG15. Our findings now provide the basis to determine in vivo the relative contributions of deubiquitination and deISGylation to viral immune evasion tactics, and a structural template of a promiscuous deubiquitinase from a haemorrhagic fever virus that can be targeted for inhibition using small-molecule-based strategies.
Keywords: innate immunity, viral deubiquitinase, bunyavirus, Crimean–Congo haemorrhagic fever virus, viral immune evasion
The posttranslational modification of proteins by Ub and Ub-like (Ubl) molecules is a regulatory process that controls numerous biological events (1, 2). Ub is conjugated to a lysine residue of target proteins through an isopeptide bond between the terminal carboxyl group of Ub and ϵ-amino group of the target lysine (3). Additional Ub molecules can be conjugated to lysines within Ub itself to form polyubiquitin chains. Lys 48-linked polyubiquitination is the canonical signal that targets proteins for proteasomal degradation, whereas Lys 63-linked polyubiquitination can initiate proteasome-independent events (4). Both Lys 48- and Lys 63-linked polyubiquitination have been established as key signaling events that activate innate and adaptive immune responses (5).
A critical innate immune response to viral infection is the rapid production of type I interferon (IFN) and tumor necrosis factor alpha (TNFα). The induction and activity of these antiviral cytokines is controlled by, among other factors, Ub and Ubl conjugation (4). A hallmark of type I IFN stimulation is the rapid production of the Ubl molecule ISG15 (6). ISG15 is composed of two tandem Ub-like folds (7) and is known to exhibit potent antiviral activity against several important viruses (8). It conjugates to ϵ-amino groups of target lysine residues on the surface of cellular target proteins similarly to Ub (9). Although conjugation is essential (8), several possible antiviral mechanisms have recently been proposed for ISG15 (10).
Ub and ISG15 conjugation can be reversed by deubiquitinating enzymes (DUBs). Ovarian tumor (OTU) domain proteases are papain-like cysteine DUBs that have been identified in eukaryotes, bacteria, and viruses (11). We have assayed a number of eukaryotic OTU-domain-containing proteins for deubiquitinating and deISGylating activity, including human A20, Cezanne, otubain1, and otubain2 (12) and the Saccharomyces cerevisiae OTU-domain-containing protein Otu1 (see Results and Discussion). While these eukaryotic proteins exhibit deubiquitinating activity as expected, none have been found to exhibit deISGylating activity, suggesting that eukaryotic OTU domains are Ub-specific. Importantly, the deubiquitinating activities of human A20, and the OTU-domain-containing protein DUBA, have been found to down-regulate TNFα and type I IFN production, respectively, by removing Ub from signaling proteins (13, 14).
In contrast to the Ub-specific activity of known eukaryotic OTU domains, we recently found that OTU-domain-containing proteins from nairoviruses and arteriviruses, two unrelated groups of RNA viruses, act as ISG15 cross-reactive deubiquitinases, removing both Ub and ISG15 from host proteins (12). CCHFV is a nairovirus that causes severe haemorrhagic fever in humans, with a mortality rate approaching 30% (15). As for other members in the genus, its genome consists of three negative-sense RNA segments, the largest of which encodes a 448 kDa viral RNA polymerase (L) that contains an amino terminal OTU domain of approximately 18 kDa (16, 17). Arteriviruses, such as equine arteritis virus (EAV) and porcine respiratory and reproductive syndrome virus (PRRSV), are nonsegmented, positive-sense RNA viruses that contain an OTU domain within their nonstructural protein 2 (nsp2) (18). The catalytic activity of these viral OTU domains not only inhibits NF-κB-dependent signaling (12, 19), we found that expression of the OTU domain from CCHFV antagonizes the antiviral effects of ISG15, suggesting that viral OTU domains act as virulence factors to suppress host inflammatory and antiviral responses (12).
A number of eukaryotic OTU-domain structures have been determined (20–23), including a covalent complex between the OTU domain from the Saccharomyces cerevisiae protein Otu1 and Ub (24). The structure of a viral OTU domain has not been reported, however, and the mechanism of ISG15 cross-reactivity has remained unknown. Here we report crystal structures of the viral OTU-domain protease domain from CCHFV covalently bound to Ub and to ISG15 at 2.5-Å and 2.3-Å resolution, respectively. The complexes provide a unique structural example of ISG15 bound to another protein and together reveal the molecular mechanism of an ISG15 cross-reactive deubiquitinase. The expanded substrate-specificity arises from a unique topological feature of the viral OTU-domain fold, which causes the protein to bind Ub and C-terminal Ub-like domain of ISG15 in an orientation that is rotated nearly 75° with respect to that observed for Ub bound to a representative eukaryotic OTU domain from yeast. This permits the viral OTU domain to accommodate additional bulk within the C-terminal domain of ISG15 that is not present in Ub. Individual residues within the substrate-binding site of viral protease were identified as key interactions sites for Ub or ISG15, which allowed the reengineering of the protease into enzymes with increased substrate specificity, either for Ub or for ISG15. By identifying the unique structural determinants that underlie ISG15 cross-reactivity, our findings now provide the basis to determine in vivo the relative contributions of deubiquitination and deISGylation to viral immune evasion tactics and the potential to develop small-molecule based strategies that specifically inhibit viral OTU-domain proteases.
Results and Discussion
Structure of CCHFV-OTU Bound to Ub or ISG15.
Crystal structures of the CCHFV–OTU domain were obtained using recombinantly expressed CCHFV L protein residues 1–185 for the Ub complex and L residues 1–169 for the ISG15 complex. For both complexes, residues 1–162 were clearly observed in the electron density and comprise the complete viral OTU domain. It consists of two lobes that clamp around a central substrate-binding groove, which narrows into a channel to direct the conserved C-terminal LRLRGG motif of Ub or ISG15 toward the enzyme active site (Figs. 1A and 2A). The larger of the two lobes (amino acids 1–34 and 114–162) is referred to as the β-sheet lobe because it is composed predominantly of a seven-stranded β-sheet (β1↑ β2↓ β3↑ β4↓ β5↓ β6↑ β7↓) that is sandwiched between helices α1, α2, α3, and α7. The smaller lobe (residues 35–113) is exclusively α-helical (composed of helices α3, α4, α5, α6, and α7) and referred to as the α-helical lobe.
Fig. 1.

Crystal structure of CCHFV–OTU (cyan) bound to Ub (orange). (A) Ribbon diagram of the complex highlighting the three major regions of interaction between Ub and the protease. Ub is bound within a substrate-binding groove that is flanked on either side by the β-sheet lobe (left) and α-helical lobe (right) of the protease. The groove narrows into a channel that directs the C terminus of Ub toward the enzyme active site. Gly 76 of Ub is replaced with a propylamine linker (3CN) that covalently attaches Ub to the enzyme nucleophile (Cys 40) located on helix α3. Residue side chains involved in protein–protein contacts in regions 1 (B), 2 (C), and 3 (D) are shown with hydrogen bonds in black. All other side chains have been omitted for clarity. Figures were drawn using PyMol (42).
Fig. 2.

Crystal structure of the CCHFV–OTU (cyan) domain bound to ISG15 (green). (A) Ribbon diagram of the complex highlighting the three major regions of interaction between ISG15 and the protease. The C-terminal Ub-like domain of ISG15 binds the substrate-binding groove of the proteases in an analogous manner to Ub. Gly 157 of ISG15 is replaced with a propylamine linker (3CN) that is covalently attached to the enzyme nucleophile (Cys 40) on helix α3. Side chains of residues involved in protein–protein contacts in regions 1 (B), 2 (C), and 3 (D) are shown with hydrogen bonds in black. Equivalent residues of Ub are in parentheses. All other side chains have been omitted for clarity. Atoms Cϵ and Nζ of Lys 90 of ISG15 are disordered and not shown in D.
Superposing the CCHFV–OTU structure onto the crystal structure of the Ub-bound eukaryotic OTU domain from yeast Otu1 (24) (Fig. 3A) reveals the presence of a unique N-terminal β1↑-β2↓ hairpin atop the β-sheet lobe of CCHFV–OTU, a feature not present in Otu1 (Fig. 3A) or any of the other OTU-domain structures that have been reported (Fig. S1A). Relative to the OTU domains from Otu1 and human A20, Otubain1 and Otubain2, the entire N terminus of CCHFV–OTU, which includes strands β1 and β2, is relocated to the opposite side of the core β-sheet structure of the OTU-domain fold (Fig. 3 B and C). Yeast Otu1 is the only additional OTU-domain-containing protein to be determined in complex with Ub. Residues comprising the substrate-binding groove of the OTU domain of Otu1 are highly conserved in other eukaryotic OTU domains, suggesting the Otu1–Ub complex is representative of Ub complexes that are formed by other eukaryotic OTU domains (24).
Fig. 3.

Comparison of the CCHFV–OTU–Ub and yeast Otu1–Ub crystal structures. (A) Superposition of CCHFV–OTU (cyan) and the OTU domain from yeast Otu1 (yellow) (Ub omitted for clarity). Strands β1 and β2 of CCHFV–OTU are labeled. Secondary structure topologies of CCHFV–OTU (B) and yeast Otu1 (C) are shown (dashed boxes enclose the core topology that is conserved in all known OTU domain structures). (D) Superposition shown in A including Ub. (E) Superposition in D rotated 90° showing the difference in binding orientation of Ub bound to the CCHFV–OTU versus yeast Otu1. (F) Superposition of the CCHFV–OTU–Ub and CCHFV–OTU–ISG15 complexes. (G) Location of Ub Thr 9 (yellow) bound to yeast Otu1 (gray). Superposing ISG15 (green residues) onto Ub predicts that Lys90 would clash with the Otu1 binding groove. Atoms Cϵ and Nζ of Lys 90 have been modeled for clarity.
The β1↑–β2↓ hairpin comprises a significant part of the CCHFV–OTU substrate-binding groove and causes the viral protein to bind the β-grasp fold of Ub in a strikingly different manner than yeast Otu1 (Fig. 3 D and E). When bound to CCHFV–OTU, Ub is rotated about an axis defined by its extended C-terminal LRLRG-3CN motif by nearly 75° with respect to its position when bound to yeast Otu1. The C-terminal Ub-like domain of ISG15 (residues 82–157) binds CCHFV–OTU in the same position and orientation as Ub (rmsd of 0.9 Å for 75 Cα carbons) (Fig. 3F). Importantly, this permits CCHFV–OTU to accommodate bulky and polar residues on loop Arg 87–Ser 93 in the C-terminal domain of ISG15 that are not present in the analogous loop of Ub (Lys 6–Thr 12). The bulk arises primarily from Lys 90, which corresponds to Thr 9 in Ub (Fig. 3G). In the CCHFV–OTU–ISG15 complex, the Arg 87–Ser 93 loop is solvent accessible, held near the edge of the binding groove where it participates in key interactions with the viral protease (Fig. 2 A and D). In the yeast Otu1–Ub complex, the analogous loop of Ub is buried within the substrate-binding groove. Lys 90 within the Arg 87–Ser 93 loop of ISG15 would likely be too large to fit the binding groove of yeast Otu1 (which we found to be Ub-specific) (Fig. S2), or other Ub-specific eukaryotic OTU domains that bind Ub similarly to Otu1 (Fig. 3G).
Molecular Interactions of CCHFV–OTU with Ub and ISG15.
Analysis of protein interface interactions using PISA (25), revealed the binding of Ub or ISG15 buries approximately 900 Å2 of solvent accessible surface area of the CCHFV–OTU substrate-binding groove, which in turn buries roughly 20% (approximately 1,000 Å2) of the total surface area of Ub or C-terminal Ubl domain of ISG15 (Fig. 4A). Both Ub and ISG15 were found to interact with three distinct regions within this buried surface area of the enzyme (Figs. 1 and 2). Region 1 comprises the channel that guides the C-terminal LRLRGG motifs of Ub (amino acids 71–76) or ISG15 (amino acids 152–157) toward the enzyme active site (Figs. 1B and 2B). Regions 2 (Figs. 1C and 2C) and 3 (Figs. 1D and 2D) are located on the α-helical and β-sheet lobe, respectively, and form the interface that binds the β-grasp folds of Ub or C-terminal Ub-like domain of ISG15.
Fig. 4.

Regions of interaction of Ub and ISG15 with the substrate-binding groove and active site of CCHFV–OTU. (A) The Ub and ISG15 structures are separated from CCHFV–OTU and rotated 180° to expose the areas of interaction between these substrates and the binding groove of the protease. Resides marked with an asterisk were functionally characterized by mutagenesis (Table 1). Electrostatic surface potentials are drawn at -60 kT/e [red (-), blue (+)] as implemented in PyMol (42). Three distinct regions within the CCHFV–OTU binding groove [region 1 (outlined in red), region 2 (outlined in green), and region 3 (outlined in purple)] interact with patches on Ub or ISG15 that are outlined by the same color. Region 3 on CCHFV–OTU contains a notable patch of negative charge around Glu128, which plays a key role in binding the Arg 87–Ser 93 loop of ISG15. Although atoms Cϵ and Nζ of Lys 90 of ISG15 are disordered in the ISG15:CCHFV–OTU complex, they have been modeled onto the ISG15 structure shown here to reveal their position on the ISG15 structure. B and C show the CCHFV–OTU active site and propylamine linker (3CN) that covalently joins the enzyme nucleophile (Cys 40) to Ub or ISG15, respectively. Electron densities are maximum-likelihood weighted 2Fobs - Fcalc syntheses contoured at 1σ. His151 adopts two conformations in the ISG15 complex. Only one of these conformations is observed in the Ub complex and is presumed to be the catalytically competent conformation. (D) Superposition of CCHFV–OTU–Ub (cyan) and CCHFV–OTU–ISG15 (gray) with papain (PDB ID code 9PAP) (purple). The papain nucleophile (Cys 25) is oxidized. The Gln19 side chain and main chain amide of Cys 25 form the oxianion hole of papain. Gln19 is substituted by the main chain amide of Asp 37 (blue sphere) in CCHFV–OTU.
In region 1 (Figs. 1B and 2B), both lobes of CCHFV–OTU participate in forming the channel that guides the terminal carboxyl group of the LRLRGG motif of Ub and ISG15 toward the enzyme nucleophile (Cys 40) of the active site (Fig. 4 B and C). The C-terminal motifs of Ub and ISG15 adopt nearly identical conformations within this channel (Fig. 3F). It is worth noting that although the bound orientation of the β-grasp fold of Ub (and C-terminal domain ISG15) is significantly different from that observed in the yeast Otu1–Ub complex, the C-terminal tail of Ub in Otu1–Ub complex superposes with the analogous residues of Ub (or ISG15) bound to CCHFV–OTU (Fig. 3D). The carboxy termini of Ub or ISG15 enter the active site of CCHFV–OTU that contains a cataylic diad (Cys 40 and His 151), which is spatially conserved with cysteine/histidine catalytic diad found in the archetype cysteine protease papain (26) (Fig. 4D). The catalytic mechanism of cysteine proteases requires the cysteine thiol to be deprotonated by the histidine residue. This promotes a nucleophilic attack by the cysteine on the carbonyl carbon of the scissile peptide bond of the substrate and formation of a covalent acyl-enzyme intermediate, which is subsequently resolved by a water molecule (27, 28). Cys 40 of CCHFV–OTU is conserved in other OTU-domain proteases (11). Mutation of this residue to alanine was found to eliminate catalytic activity (12), and the covalent adducts we observed between Cys 40 and Ub or ISG15 confirms this cysteine as the catalytic nucleophile CCHFV–OTU (Fig. 4 B and C).
In addition to the Cys 40/His 151 catalytic dyad, the carboxyl group of Asp 153 in CCHFV–OTU likely enhances catalysis of the viral protease by hydrogen-bonding with the imidazole group of His 151 (Fig. 4 B and C). This interaction is analogous to the interaction between Asn 175 and His 159 in papain (26) and is believed to correctly orient the imidazolium ring of the histidine residue during catalysis (28). For most cysteine proteases, the main chain amide of the cysteine nucleophile and a glutamine side chain from an oxianion hole, which stabilizes the oxianion that develops along the reaction coordinate (28). While the amide of the cysteine nucleophile is present, an equivalent glutamine is not present in CCHFV–OTU and is substituted by the main chain amide of Asp 37, which appears well positioned to help stabilize the oxianion. Similar oxianion hole architectures are observed in the OTU domains of yeast Otu1 (24) and human Otubain1 (23), Otubain2 (22), and A20 (20, 21) (Fig. S1B).
Region 2 centers on helix α5 of the α-helical lobe of CCHFV–OTU. Relatively few distinct contacts are made between this region and Ub or C-terminal Ub-like domain of ISG15 with the exception of a hydrogen bond between Arg 42 of Ub and Gln 16 of strand β2 of CCHFV–OTU. The region appears to act primarily as a buttress to support the substrates against region 3 and help guide the C-terminal motifs of each substrate toward the active site (Figs. 1C and 2C).
Region 3 centers on the distinctive β1↑–β2↓ hairpin of the viral protease, where hydrophobic interactions with Ub (Fig. 1D) or electrostatic interactions with ISG15 are mediated (Fig. 2D). When bound to Ub, Val 12 and Ile 13 of strand β1, Val 18 of strand β2, and Ala 129 and Ile 131 from strand β5, comprise a hydrophobic patch that interacts with Leu 8, Ile 44, Val 70, and Leu 73 of Ub. This hydrophobic patch on Ub, in particular Ile 44, is a critical interaction site that is recognized by DUB enzymes (29), and CCHFV–OTU uses it to orient Ub within the binding groove. Interestingly, due to the difference in binding orientation of Ub to CCHFV–OTU versus yeast Otu1, a cluster of hydrophobic residues analogous to those in region 3 of CCHFV–OTU exist on the α-helical lobe of Otu1, where they serve to bind the Ile 44 patch of Ub (24). This patch of residues on Otu1 is conserved in other eukaryotic OTU domains and supports the conjecture that they bind Ub in the same orientation as Otu1.
The C-terminal Ub-like domain of ISG15 does not possess a patch of hydrophobic residues analogous to Leu 8, Ile 44, Val 70, and Leu 73 of Ub with the exception of Leu 154 (Leu 73 of Ub). Instead, this region of ISG15 is composed primarily of polar residues (equivalent Ub residues in parentheses): Asn 89 (Leu 8), Thr 125 (Ile 44), Asn 151 (Val 70). To compensate for this difference in substrate structure, region 3 contains residues Asn 20, Arg 22, Thr 120, and Glu 128, which form a network of hydrogen bonds with the Arg 87–Ser 93 loop of ISG15 (Fig. 2D). In particular, Lys 90 of the loop is fully solvent exposed and the side chain of Asn 89 appears to form critical hydrogen-bonding interactions with Glu 128 and Thr 120 of the viral protease. With the exception of region 1, this hydrogen-bonding network is the major site of interaction between CCHFV–OTU and ISG15. Thus, the Arg 87–Ser 93 loop of the C-terminal β-grasp fold of ISG15 appears to be an important site of interaction for proteins that recognize ISG15.
Functional Analysis of the CCHFV–OTU Substrate-Binding Interface.
Structural analysis revealed that Ub and ISG15 interact with discrete patches of residues within the substrate-binding groove of CCHFV–OTU (Fig. 4A). Site-directed mutagenesis was used to verify the importance of individual enzyme residues in forming the enzyme–substrate reactive complex and to determine if it was possible to selectively attenuate DUB or deISGylating activity. With this objective in mind, we produced the following mutants of CCHFV–OTU: (i) Mutations Val12Thr, Ile131Asn, and Ala129Ser of region 3 were created to disrupt the binding of region 3 of the protease to the Ile 44 patch of Ub (Figs. 1D and 4A); (ii) residue Gln16 of region 2 was mutated to serine to disrupt an apparent Ub-specific electrostatic interaction between Gln 16 in region 2 of the CCHFV–OTU and Arg 42 of Ub (Figs. 1C and 4A); and (iii) the mutation Glu128Ala was created to perturb the hydrogen-bonding network existing between region 3 of CCHFV–OTU and the Arg 87–Ser 93 loop of ISG15 (an interaction that does not occur between Ub and CCHFV–OTU) (Figs. 2D and 4A).
The Michaelis constant KM can be used as a measure of the amount of enzyme that is bound in any form to the substrate (30). However, because the commercially available Ub–AMC substrate stock was dissolved in dimethylsulfoxide (DMSO), a limit (dictated by enzyme denaturation and substrate solubility) existed on the maximum substrate concentration that could be used, making it difficult to obtain reliable Km values directly. Our kinetic analysis of protease activity toward Ub–AMC was thus limited to the linear portion of the Michaelis–Menten curve (Fig. S3), which still allowed us to quantitatively determine the specificity constant 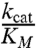 of the enzyme relative to the substrate. In order to compare the effect of each mutation, we determined the specificity constants for all mutant CCHFV–OTU enzymes toward Ub–AMC, ISG15–AMC, and the peptide RLRGG–AMC using the linear portion of the relevant Michaelis–Menten plots. These values are shown in Table 1.
of the enzyme relative to the substrate. In order to compare the effect of each mutation, we determined the specificity constants for all mutant CCHFV–OTU enzymes toward Ub–AMC, ISG15–AMC, and the peptide RLRGG–AMC using the linear portion of the relevant Michaelis–Menten plots. These values are shown in Table 1.
Table 1.
Effect of substrate binding site mutations on CCHFV–OTU activity*
| Enzyme | Substrate | |||||
| Ub–AMC | ISG15–AMC | RLRGG–AMC | ||||
| kcat/KM (× 10-5) | Ratio of specificity constants (mutant/wild type) | kcat/KM (× 10-5) | Ratio of specificity constants (mutant/wild type) | kcat/KM (× 10-5) | Ratio of specificity constants (mutant/wild type) | |
| Wild type | 3.41 ± 0.07 | 1 | 3.6 ± 0.1 | 1 | 0.60 ± 0.02 | 1 |
| Val12Thr | 0.81 ± 0.02 | 0.24 ± 0.01 | 1.40 ± 0.06 | 0.39 ± 0.02 | 0.26 ± 0.01 | 0.43 ± 0.01 |
| Ile131Asn | 0.111 ± 0.001 | 0.0326 ± 0.0006 | 1.20 ± 0.03 | 0.33 ± 0.01 | 0.008 ± 0.001 | 0.01 ± 0.001 |
| Ala129Ser | 2.47 ± 0.03 | 0.72 ± 0.01 | 2.07 ± 0.07 | 0.58 ± 0.09 | 0.22 ± 0.02 | 0.37 ± 0.04 |
| Gln16Ser | 1.81 ± 0.02 | 0.53 ± 0.01 | 1.80 ± 0.06 | 0.50 ± 0.02 | 0.63 ± 0.02 | 1.1 ± 0.03 |
| Glu128Ala | 3.8 ± 0.2 | 1.11 ± 0.06 | 0.82 ± 0.02 | 0.23 ± 0.03 | 0.65 ± 0.05 | 1.10 ± 0.09 |
*For wild-type and mutant enzymes the specificity constants were obtained from the slopes of the lines shown in Fig. S3.
In comparing the various specificity constant values in Table 1, the following points must be considered. We designed our mutations expecting that they would not strongly perturb the catalytic site of the CCHFV–OTU enzymes because they were located in regions 2 and 3 of the enzyme, which are far from the active site. We verified this expectation by using a traditional Michaelis–Menten analysis using the substrate ISG15–AMC (which could be used at concentrations that approached kinetic saturation of the enzyme) (Fig. S4). We indeed observed that the various mutations have limited effect on the kcat values: The maximum change in kcat observed between mutant enzymes and the wild type is a reduction by a factor less than 2 (Table S1). It should be noted that the value of the wild-type enzyme specificity constant reported in Table S1 is within 10% of the value reported in Table 1, and the specificity constants of the mutant enzymes reported in the Table 1 and Table S1 agree with one another within the uncertainty limits. Therefore, it is reasonable to assume that if the specificity constant of a mutant enzyme toward a given substrate is more than five times less than that of the wild type, this change is likely due to an increase in KM and thus reflects a change in the binding of substrate to enzyme rather than any drastic deformation of the catalytic site that would lead to a change in the catalytic mechanism.
Of all the mutated CCHFV–OTU residues within region 3 that form van der Waals packing interactions with the hydrophobic Ile 44 patch of Ub (Fig. 1D), Ile 131 appeared to be the most important for Ub binding. Mutating Ile 131 to an asparagine caused the specificity constant of enzyme toward Ub–AMC to drop by a factor of 30. Other residues analyzed by mutagenesis were found to contribute significantly less, with Val 12 perhaps being the next most important contributor to Ub–AMC binding and Ala129Ser having the least effect (Table 1). Residue Ile 131 also appears to significantly contribute to the binding of the peptide substrate RLRGG–AMC, because the Ile131Asn mutation caused the specificity constant of the enzyme toward the peptide to drop by two orders of magnitude. This change is consistent with our structural data, which demonstrates that the peptide extends some distance from the enzyme active site such that the Leu residue of the peptide interacts directly with Ile 131 of the enzyme (Figs. 1 B and D and 2B). The Ile131Asn mutation had less of an effect on the specificity constant of enzyme toward ISG15–AMC, dropping it by a factor of three. Interestingly, unlike Ub or the RLRGG peptide substrate, ISG15 contains polar residues within the vicinity of the Ile131Asn mutation as observed in the CCHFV–OTU:ISG15 crystal structure. In particular, the side chain of ISG15 residue Asn 89 (Ub: Leu 8) lies within 4.0 Å of Ile 131 (Fig. 2D) and may assist in stabilizing the interaction between ISG15 and the Ile131Asn mutant by hydrogen-bonding directly to the introduced Asn side chain. This interaction could not occur with Ub or the RLRGG peptide.
Based on our structural analysis, the hydrogen-bonding interaction between Gln 16 in region 2 and Arg 42 of Ub (Fig. 1C) appeared to be selective for Ub, because the corresponding residue in ISG15 is a tryptophan (Trp 123) that does not participate in an analogous interaction with the enzyme (Fig. 2C). Surprisingly, we found that this mutation had an equal effect on the specificity constants for Ub–AMC and ISG15–AMC. As expected, however, because this mutation is far from region 1, it did not have any measurable effect on the specificity constant for the RLRGG–AMC peptide.
In contrast to the above findings using Ub–AMC, our functional assessment of CCHFV–OTU residues Val12, Ile131, and Ala129 showed that they have a less important role in the binding of ISG15–AMC, with the probable exception of Ile 131 (Table 1). Instead, our structural data revealed that a hydrogen-bonding network exists between the Arg 87–Ser 93 loop of ISG15 and region 3 of CCHFV–OTU, which appeared critical for interacting with ISG15. The shortest hydrogen bond within the network occurred between Glu 128 of CCHFV–OTU and Asn 89 of ISG15 (Fig. 2D) and was thus predicted to contribute strongly to the binding of ISG15 to the enzyme. Indeed, mutating this residue reduced the specificity constant of CCHFV–OTU toward ISG15 by a factor of five, while the specificity constants of the enzyme toward Ub–AMC or the RLRGG–AMC peptide substrates were unimpaired by this mutation. Significantly decreasing the binding of ISG15 by perturbing the hydrogen-bonding network between the Arg 87–Ser 93 loop of ISG15 and region 3 of CCHFV–OTU is consistent with the conjecture that this loop is a protein interaction “hot spot” and supports a previous prediction suggesting that the loop is involved in binding the ISG15 activating enzyme UbE1L (7) and perhaps additional proteins that interact with ISG15.
OTU-Domain Proteases as Virulence Factors for Viral Immune Evasion.
The discovery of viruses that have acquired the ability to remove Ub and ISG15 from cellular targets underscores the importance of Ub and ISG15 conjugation to innate immunity. Moreover, alternative immune evasion tactics such as NS1 of influenza B virus, a protein that prevents ISGylation of proteins by binding ISG15 (31), further highlights the importance of ISG15 to host defense. The small and compact genomes of nairoviruses and arteriviruses likely selected for a unique deconjugating OTU protease with the ability to target both Ub and ISG15 at the same time, a property that appears not to be shared by eukaryotic OTU enzymes. We show here that a rearrangement in the β-sheet topology of the CCHFV OTU-domain fold yields an ISG15 cross-reactive viral DUB capable of binding Ub or ISG15, yet it retains an active site architecture that is conserved with eukaryotic OTU-domain proteases. Importantly, the ISG15 cross-reactive deubiquitinating activity of CCHFV–OTU is retained when the domain is expressed as part of the intact CCHFV RNA polymerase L protein (12). However, it remains to be determined if other structural components of the viral L protein can modulate the DUB and deISGylating activities of the CCHFV–OTU domain during viral replication. Given the low sequence conservation of ISG15 in higher eukaryotes, there is the intriguing possibility of species-specific deISGylating activity by viral OTU domains, which may correlate with host tropism. Having demonstrated that Ub and ISG15 hydrolysis can be selectively attenuated by site directed mutagenesis, it may be possible to study the relative contributions these deconjugating activities have to the immune evasion tactics of nairoviruses and arteriviruses.
In addition to viral OTU domains, the papain-like protease from severe acute respiratory syndrome (SARS) coronavirus (32, 33) and the adenoviral protease adenain (34) are also ISG15 cross-reactive DUBs, which suggests that ISG15 cross-reactive DUBs may be a widely used viral mechanism to simultaneously suppress inflammatory and IFN responses of the host. Targeting DUB viral proteases with small-molecule inhibitors might then represent an effective therapeutic approach. The CCHFV–OTU crystal structure revealed that ISG15 cross-reactivity requires a significantly altered substrate-binding groove architecture compared to Ub-specific eukaryotic OTU proteases. These structural differences may now be exploited to facilitate the development of inhibitors that are selective for viral OTU-domain-containing DUBs.
Materials and Methods
Preparation and Crystallization of CCHFV–OTU Bound to Ub or ISG15.
Residues 1–185 (CCHFV-OTU(L1-185)) or 1–169 (CCHFV-OTU(L1-169)) of the CCHFV L protein were expressed as GST fusion proteins in Escherichia coli and purified. GST tags were removed using HRV 3c protease. Ub(1–75)-3-bromopropylamine (Ub-3Br) and ISG15(Cys78Ser)(1–156)-3-bromopropylamine (ISG15-3Br) were prepared according to Messick et al. (24) and Borodovsky et al. (35). The ISG15 mutation Cys78Ser was required to reduce aggregation (7). Selenomethionyl CCHFV-OTU(L1-185) (produced according to Van Duyne et al.) (36) and native CCHFV-OTU(L1-169) were complexed with Ub–Br3 and ISG15–Br3, respectively, then purified and dialyzed into 20 mM Tris-Cl, pH 8.0, 50 mM NaCl. Crystals were grown by hanging-drop vapor diffusion. Selenomethionyl OTU(L1-185)–Ub crystallized at 10 mg/ml in 27% PEG 4000-6000, 100 mM NaOAc, pH 5.4-5.6, 210 mM (NH4)2SO4. OTU(L1-169)-ISG15 crystallized at 7 mg/ml in 100 mM MES, pH 6.3-6.5, 22–25% PEG 6000 or 8000. Crystals were flash-cooled in liquid nitrogen after the addition of 20% glycerol.
Data Collection and Structure Determination.
X-ray data were collected at the Canadian Light Source (beam line 08ID-1) and processed using MOSFLM and SCALA (37). Diffraction data from a selenomethionyl OTU(L1-185)–Ub crystal was phased by single wavelength anomalous dispersion (SAD) phasing using PHENIX AutoSol (38). Initial phases (overall FOM of 0.326) were improved by density modification using RESOLVE (final FOM of 0.66). A model was built using PHENIX autobuild and manually completed and refined using COOT (39) and phenix.refine. The structure of OTU(L1-169)–ISG15 was determined by molecular replacement using PHASER (40) and the crystal structures of CCHFV–OTU and ISG15 (7) (PDB ID code 1Z2M) as search models. Model building and refinement, which included the use of TLS partitions, was performed using COOT and phenix.refine (38, 39, 41). Crystallographic statistics are summarized in Table S2.
Enzymatic Assays.
Enzymatic assays were carried out in 50 mM Tris-HCl, pH 8.0, 2 mM DTT buffer at 25 °C in 25 ul final volume. Ub–AMC and ISG15–AMC (Boston Biochem) hydrolysis were initiated by adding enzyme to the reaction mixture having varying concentrations of substrates; the resulting final concentration of enzyme in the reaction mixture for each case was 3 nM. For RLRGG–AMC (Enzo Life Sciences) hydrolysis, identical assay conditions were used as above; however, the final concentration of enzyme in the reactive mixture was 100 nM of enzyme. The enzyme concentration for initial rate determinations was chosen so that less than 10% of the substrate was hydrolyzed. Hydrolysis at each given substrate concentration was monitored continuously by recording the fluorescence of the released AMC product as a function of time. Initial rates were calculated from the slopes of the linear portion of the time-dependent fluorescence profiles. The rate of fluorescence increase was converted to changes in concentration (nM) of product (free AMC) produced per second using the fluorescence standardization curves of the AMC standard (Boston Biochem) that were also used for calibration. All assays were performed in triplicate at minimum. The kinetic results were analyzed with Sigmaplot software.
Supplementary Material
Acknowledgments.
We thank V. Larmour and R. Cadagan for technical assistance, and J. Wilkins for providing cDNA clones of human Ub and ISG15. X-ray diffraction data were collected at the Canadian Light Source (CLS) beamline 08ID-1. The CLS is supported by the Natural Sciences and Engineering Research Council of Canada (NSERC), National Research Council, the Canadian Institutes of Health Research, and the University of Saskatchewan. The research was partly funded by NSERC and Manitoba Health Research Council (MHRC) grants to B.L.M. and by National Institute of Allergy and Infectious Disease (NIAID) Grant U54AI057158 to A.G.S. T.W.J. was supported by a studentship from the MHRC.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. J.H. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1013388108/-/DCSupplemental.
Data deposition: The coordinates and structure factors for the CCHFV–OTU–Ub and CCHFV–OTU–ISG15 protein complexes have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 3PT2 and 3PSE, respectively).
See companion article on page 2228.
References
- 1.Welchman RL, Gordon C, Mayer RJ. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat Rev Mol Cell Biol. 2005;6:599–609. doi: 10.1038/nrm1700. [DOI] [PubMed] [Google Scholar]
- 2.Hochstrasser M. Origin and function of ubiquitin-like proteins. Nature. 2009;458:422–429. doi: 10.1038/nature07958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hicke L. Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol. 2001;2:195–201. doi: 10.1038/35056583. [DOI] [PubMed] [Google Scholar]
- 4.Kirkin V, Dikic I. Role of ubiquitin- and Ubl-binding proteins in cell signaling. Curr Opin Cell Biol. 2007;19:199–205. doi: 10.1016/j.ceb.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 5.Bhoj VG, Chen ZJ. Ubiquitylation in innate and adaptive immunity. Nature. 2009;458:430–437. doi: 10.1038/nature07959. [DOI] [PubMed] [Google Scholar]
- 6.Farrell PJ, Broeze RJ, Lengyel P. Accumulation of an mRNA and protein in interferon-treated Ehrlich ascites tumour cells. Nature. 1979;279:523–525. doi: 10.1038/279523a0. [DOI] [PubMed] [Google Scholar]
- 7.Narasimhan J, et al. Crystal structure of the interferon-induced ubiquitin-like protein ISG15. J Biol Chem. 2005;280:27356–27365. doi: 10.1074/jbc.M502814200. [DOI] [PubMed] [Google Scholar]
- 8.Lenschow DJ, et al. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc Natl Acad Sci USA. 2007;104:1371–1376. doi: 10.1073/pnas.0607038104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loeb KR, Haas AL. The interferon-inducible 15-kDa ubiquitin homolog conjugates to intracellular proteins. J Biol Chem. 1992;267:7806–7813. [PubMed] [Google Scholar]
- 10.Skaug B, Chen ZJ. Emerging role of ISG15 in antiviral immunity. Cell. 2010;143:187–190. doi: 10.1016/j.cell.2010.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Makarova KS, Aravind L, Koonin EV. A novel superfamily of predicted cysteine proteases from eukaryotes, viruses and Chlamydia pneumoniae. Trends Biochem Sci. 2000;25:50–52. doi: 10.1016/s0968-0004(99)01530-3. [DOI] [PubMed] [Google Scholar]
- 12.Frias-Staheli N, et al. Ovarian tumor domain-containing viral proteases evade ubiquitin- and ISG15-dependent innate immune responses. Cell Host Microbe. 2007;2:404–416. doi: 10.1016/j.chom.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wertz IE, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-[kappa]B signalling. Nature. 2004;430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 14.Kayagaki N, et al. DUBA: A Deubiquitinase that regulates Type I interferon production. Science. 2007;318:1628–1632. doi: 10.1126/science.1145918. [DOI] [PubMed] [Google Scholar]
- 15.Whitehouse CA. Crimean–Congo hemorrhagic fever. Antiviral Res. 2004;64:145–160. doi: 10.1016/j.antiviral.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 16.Honig JE, Osborne JC, Nichol ST. Crimean–Congo hemorrhagic fever virus genome L RNA segment and encoded protein. Virology. 2004;321:29–35. doi: 10.1016/j.virol.2003.09.042. [DOI] [PubMed] [Google Scholar]
- 17.Kinsella E, et al. Sequence determination of the Crimean–Congo hemorrhagic fever virus L segment. Virology. 2004;321:23–28. doi: 10.1016/j.virol.2003.09.046. [DOI] [PubMed] [Google Scholar]
- 18.Snijder EJ, Wassenaar AL, Spaan WJ, Gorbalenya AE. The arterivirus Nsp2 protease. An unusual cysteine protease with primary structure similarities to both papain-like and chymotrypsin-like proteases. J Biol Chem. 1995;270:16671–16676. doi: 10.1074/jbc.270.28.16671. [DOI] [PubMed] [Google Scholar]
- 19.Sun Z, Chen Z, Lawson SR, Fang Y. The cysteine protease domain of porcine reproductive and respiratory syndrome virus nonstructural protein 2 possesses deubiquitinating and interferon antagonism functions. J Virol. 2010;84:7832–7846. doi: 10.1128/JVI.00217-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Komander D, Barford D. Structure of the A20 OTU domain and mechanistic insights into deubiquitination. Biochem J. 2008;409:77–85. doi: 10.1042/BJ20071399. [DOI] [PubMed] [Google Scholar]
- 21.Lin SC, et al. Molecular basis for the unique deubiquitinating activity of the NF-kappaB inhibitor A20. J Mol Biol. 2008;376:526–540. doi: 10.1016/j.jmb.2007.11.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nanao MH, et al. Crystal structure of human otubain 2. EMBO Rep. 2004;5:783–788. doi: 10.1038/sj.embor.7400201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edelmann MJ, et al. Structural basis and specificity of human otubain 1-mediated deubiquitination. Biochem J. 2009;418:379–390. doi: 10.1042/BJ20081318. [DOI] [PubMed] [Google Scholar]
- 24.Messick TE, et al. Structural basis for ubiquitin recognition by the Otu1 ovarian tumor domain protein. J Biol Chem. 2008;283:11038–11049. doi: 10.1074/jbc.M704398200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 26.Kamphuis IG, Kalk KH, Swarte MBA, Drenth J. Structure of papain refined at 1.65 ≈ resolution. J Mol Biol. 1984;179:233–256. doi: 10.1016/0022-2836(84)90467-4. [DOI] [PubMed] [Google Scholar]
- 27.Drenth j, Kalk KH, Swen HM. Binding of chloromethyl ketone substrate analogues to crystalline papain. Biochemistry. 1976;15:3731–3738. doi: 10.1021/bi00662a014. [DOI] [PubMed] [Google Scholar]
- 28.Rawlings ND, Barrett AJ, Bateman A. MEROPS: The peptidase database. Nucleic Acids Res. 2010;38(Suppl 1):D227–233. doi: 10.1093/nar/gkp971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Komander D, Clague MJ, Urbe S. Breaking the chains: Structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10:550–563. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- 30.Fersht A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. New York: WH. Freeman and Co.; 1998. [Google Scholar]
- 31.Yuan W, Krug RM. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J. 2001;20:362–371. doi: 10.1093/emboj/20.3.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lindner HA, et al. The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J Virol. 2005;79:15199–15208. doi: 10.1128/JVI.79.24.15199-15208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clementz MA, et al. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J Virol. 2010;84:4619–4629. doi: 10.1128/JVI.02406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balakirev MY, Jaquinod M, Haas AL, Chroboczek J. Deubiquitinating function of adenovirus proteinase. J Virol. 2002;76:6323–6331. doi: 10.1128/JVI.76.12.6323-6331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borodovsky A, et al. Chemistry-based functional proteomics reveals novel members of the deubiquitinating enzyme family. Chem Biol. 2002;9:1149–1159. doi: 10.1016/s1074-5521(02)00248-x. [DOI] [PubMed] [Google Scholar]
- 36.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol. 1993;229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- 37.Collaborative Computational Project Number 4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 38.Adams PD, et al. PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr D. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 39.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 40.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 41.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 42.DeLano WL. The PyMOL Molecular Graphics System. PaloAlto, CA: DeLano Scientific; 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.