

Abstract
Hepatitis B virus (HBV)–induced hepatitis and carcinogen-induced hepatocellular carcinoma (HCC) are associated with serum androgen concentration. However, how androgen or the androgen receptor (AR) contributes to HBV-induced hepatocarcinogenesis remains unclear. We found that hepatic AR promotes HBV-induced hepatocarcinogenesis in HBV transgenic mice that lack AR only in the liver hepatocytes (HBV-L-AR−/y). HBV-L-AR−/y mice that received a low dose of the carcinogen N′-N′-diethylnitrosamine (DEN) have a lower incidence of HCC and present with smaller tumor sizes, fewer foci formations, and less α-fetoprotein HCC marker than do their wild-type HBV-AR+/y littermates. We found that hepatic AR increases the HBV viral titer by enhancing HBV RNA transcription through direct binding to the androgen response element near the viral core promoter. This activity forms a positive feedback mechanism with cooperation with its downstream target gene HBx protein to promote hepatocarcinogenesis. Administration of a chemical compound that selectively degrades AR, ASC-J9, was able to suppress HCC tumor size in DEN-HBV-AR+/y mice. These results demonstrate that targeting the AR, rather than the androgen, could be developed as a new therapy to battle HBV-induced HCC.
Introduction
Hepatitis B virus (HBV) infection is a major epidemiological factor for the development of hepatocellular carcinoma (HCC) (1), as evidenced by a high incidence of HCC in association with an increase in serum HBV DNA concentrations (2). Higher serum androgen concentrations (3, 4) or an androgen receptor (AR) gene containing shorter CAG repeats (which leads to higher AR activities) has also been linked to higher risks in HBV-mediated HCC (3, 4). Furthermore, there is a gender disparity in HBV-mediated HCC, where males are five to seven times as likely as females to contract disease and two to three times as likely if infected with hepatitis C virus (5, 6). Large cohort studies also demonstrated that the combination of male gender and HBV infection had a significant synergistic effect on HCC progression (7). These epidemiological data suggest that androgen-AR signals may play key roles in the etiology of HBV-mediated HCC cancer.
However, a reliable animal model that can be used to tease apart the mechanisms by which HBV contributes to hepatocarcinogenesis is still lacking. We recently generated a transgenic HBV mouse model that developed HCC upon exposure to a low dose (2 mg per kilogram of body weight) of N′-N′-diethylnitrosamine (DEN) (8). This animal model closely resembles human HBV-induced HCC because HBV carriers who were exposed to aflatoxins had a higher incidence of HCC (9–12). This phenomenon was prevalent in sub-Saharan Africa, east Asia, and southeast Asia, where food and feed crops were more easily contaminated with aflatoxins, DEN, and alkylbenzene derivatives (13).
Using this newly generated HBV transgenic mouse model, we report here that loss of hepatic AR suppresses HBV-induced hepatocarcinogenesis.
Results
Loss of hepatic AR results in suppression of HBV-induced HCC in a transgenic HBV mouse model
To examine the role of hepatic AR in HBV-mediated HCC, we generated HBV transgenic mice (Fig. 1A) that specifically lack hepatic AR (HBV-L-AR−/y). HBV-AR+/y mice treated with a low dose (2 mg/kg body weight) of DEN developed HCC at the age of 22 to 32 weeks (Fig. 1, C to E). These results mimic previous findings that showed that HCC could be induced in most HBV transgenic mice at the age of 28 to 32 weeks, but the wild-type AR (AR+/y) DEN-treated mice that lack HBV transgene remain tumor-free (8).
Fig. 1.
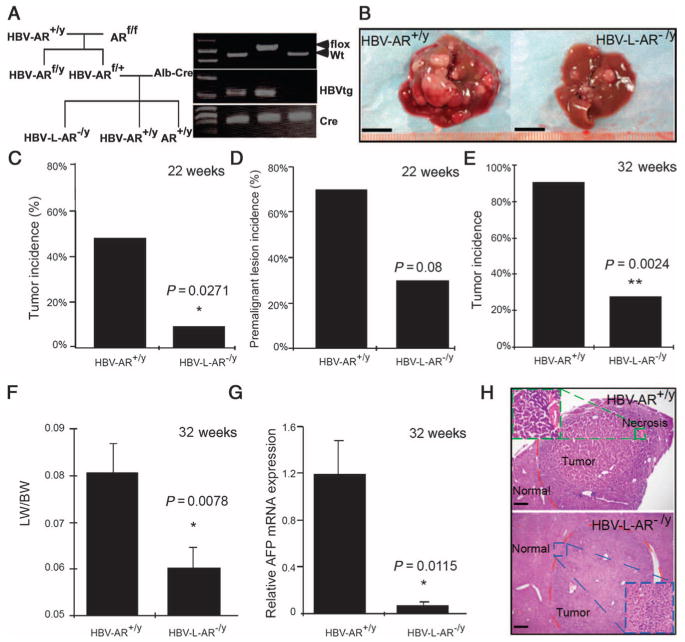
HBV-enhanced hepatocarcinogenesis is suppressed in mice with loss of hepatic AR. (A) Mating scheme of generating AR+/y, HBV-AR+/y, and HBV-L-AR−/y mice. (B) Gross observation of 32-week-old DEN-injected HBV-AR+/y and HBV-L-AR−/y mouse livers. Scale bar, 1.0 cm. (C) Tumor incidence [defined as observable tumors >2 mm in diameter (27)] of 22-week-old DEN-injected HBV-AR+/y (n = 13) and HBV-L-AR−/y (n = 13) mice. (D) Percentage of mice with premalignant liver lesions in 22-week-old DEN-injected HBV-AR+/y (n = 13) and HBV-L-AR−/y (n = 13) mice. (E) Percentage of mice with >20 liver tumor foci in 32-week-old HBV-AR+/y (n = 11) and HBV-L-AR−/y (n = 11) mice injected with DEN. (F) Ratio of liver weight (LW) to whole-body weight (BW) in 32-week-old HBV-L-AR−/y and HBV-AR+/y mice injected with DEN. (G) Liver tumor marker α-AFP mRNA expression in HBV-L-AR−/y mice compared to HBV-AR+/y mice. *P < 0.05, **P < 0.005. Relative α-AFP mRNA expression is normalized by the α-AFP mRNA expression of HBV-AR+/y. (H) Hematoxylin and eosin staining showing representative tumors in the two groups of mice. The tumor from the HBV-AR+/y shows a higher grade with an area of necrosis. Scale bars, 100 μm.
We found that liver tumor volumes were reduced in HBV-L-AR−/y mice as compared to wild-type HBV-AR (HBV-AR+/y) littermates (Fig. 1B). At an early stage (22 weeks), we found that 7.7% of HBV-L-AR−/y mice developed HCC compared to 46.2% of HBV-AR+/y mice (P = 0.0271, Fig. 1C), and none of the wild-type AR+/y mice developed HCC. At 22 weeks, we also found that 23.1% of HBV-L-AR−/y mice developed premalignant lesions (including benign lesions such as adenomas or dysplasia), whereas 69.2% of HBV-AR+/y mice developed premalignant lesions (P = 0.08, Fig. 1D). Furthermore, we found that 27.3% of HBV-L-AR−/y mice developed tumors with >20 foci, whereas >90% of HBV-AR+/y mice have >20 tumor foci (P = 0.002, Fig. 1E). The ratio of liver weight to body weight, an indicator of the tumor mass (14), was significantly less in HBV-L-AR−/y mice compared to HBV-AR+/y littermates (P = 0.0078, Fig. 1F). Also, the messenger RNA (mRNA) expression of α-fetoprotein (α-AFP), a HCC tumor marker, was much reduced in HBV-L-AR−/y mice compared to HBV-AR+/y littermates (P = 0.0115, Fig. 1G), and HCC tumor histology examination revealed fewer preneoplastic lesions (Fig. 1H) in HBV-L-AR−/y mice compared to those found in HBV-AR+/y littermates. Together, these results demonstrate that loss of hepatic AR results in suppression of HBV-induced HCC.
Hepatic AR increases HBV viral titer by enhancing HBV RNA transcription by direct binding to ARE near viral core promoter
Because higher AR activity with shorter CAG repeats has been linked to higher risks in HBV-mediated HCC (3, 4) and a high incidence of HCC is associated with an increase in serum HBV DNA concentrations (2), we hypothesized that hepatic AR may influence HBV-mediated HCC by modulating HBV viral titer. To test this, we measured HBV viral DNA from liver samples by PCR (15) and found that the amount of liver HBV DNA in HBV-L-AR−/y mice is less than in HBV-AR+/y littermates by a factor of 5 at the age of 22 weeks (Fig. 2A), suggesting that hepatic AR might promote HBV DNA replication. AR has been shown to function as a transcription factor to enhance RNA transcription of its target genes (16), so we examined the effects of hepatic AR on HBV RNA transcription and observed a decrease in HBV mRNA expression in HBV-L-AR−/y mice compared to HBV-AR+/y littermates (Fig. 2B). We then determined AR effects at the transcriptional level and found that AR could increase HBV core promoter activity in a dose-dependent manner in human HepG2 cells (Fig. 2C). We identified two potential androgen response elements (AREs) (Fig. 2D, top panel) on the HBV genome and found that deletion of the ARE1 site led to minor changes in core promoter activity (Fig. 2D, bottom panel, lanes 2 and 3), whereas deletion of both ARE1 and ARE2 sites resulted in a significant suppression of core promoter activity (Fig. 2D, lanes 2 and 4). These results suggest that the ARE2, but not the ARE1, site is essential for AR to regulate transactivation of the HBV core promoter (Fig. 2D). Chromatin immunoprecipitation (ChIP) assay confirmed that AR binds directly to the ARE2 site in vivo (Fig. 2E). AR-promoted HBV core promoter activity could be further enhanced by addition of its downstream target gene, HBx protein (HBx) (Fig. 2F). This suggests that the hepatic AR might be able to increase HBV RNA transcription in the liver by a positive feedback mechanism in cooperation with its downstream target gene HBx.
Fig. 2.
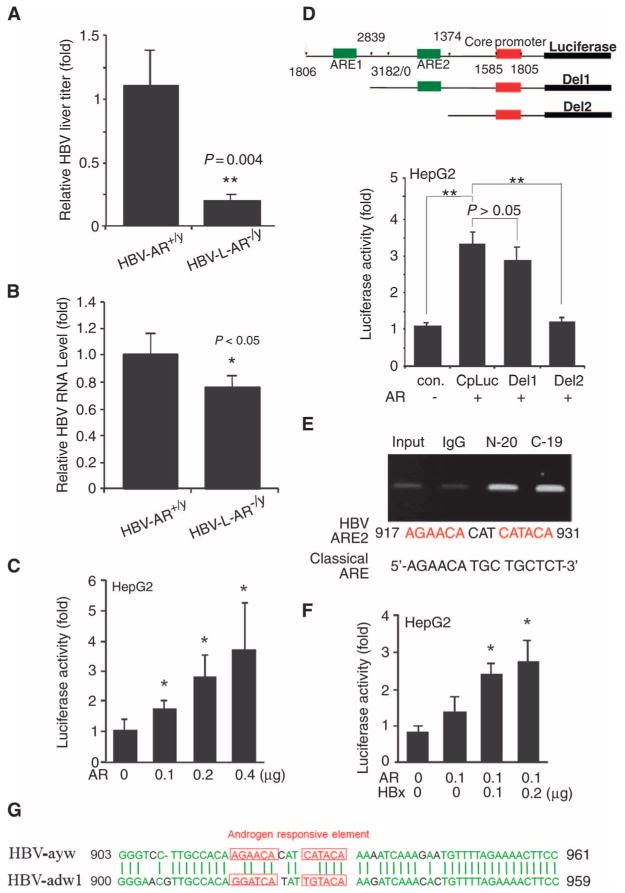
AR enhances HBV replication through increase of core promoter activity. (A) HBV DNA was decreased in 22-week-old HBV-L-AR−/y mice (n = 8) compared to HBV-AR+/y mice (n = 11). The relative HBV DNA titer was normalized by the HBV DNA titer of HBV-AR+/y. The P value indicates the comparison between HBV-AR+/y and HBV-L-AR−/y mice. **P < 0.005. (B) HBV mRNA in liver decreased in 32-week-old HBV-L-AR−/y mice as compared to HBV-AR+/y mice. The relative HBV RNA expression was normalized by the HBV RNA of HBV-AR+/y. The P value indicates the comparison between HBV-AR+/y (n = 5) and HBV-L-AR−/y (n = 5) mice. *P < 0.05. (C) AR increases HBV core promoter activity in a dose-dependent manner. *P < 0.05, compared to cells transfected with the vector. Luciferase activities are normalized by Renilla luciferase activity and are shown as mean ± SEM. (D) Top panel: Structure of 5′-deletion constructs of the core promoter. The numbers represent the nucleotide positions in the HBV genome. Green box, putative ARE; red box, HBV pregenome–core promoter. Bottom panel: AR-induced core promoter activity was abolished after deletion of ARE2. Different core promoter constructs were transfected into HepG2 vector or HepG2-AR stable expression cells. Lu-ciferase activity was measured 24 hours after transfection. **P < 0.005. (E) ChIP assay confirms that AR can bind to the ARE2 site of HBV genome. The putative ARE on HBV and classical ARE are compared in the bottom panel. (F) Enhancement of AR-induced core promoter activity by HBx. pSG5, pSG5-AR, core promoter-driven luciferase reporter (CpLuc), pRC/CMV, pCMV-HBx, and pRenilla-TK plasmids were transfected into HepG2 cells in different combinations. Luciferase activities are normalized by Renilla luciferase activity and are shown as mean ± SEM. *P < 0.05, **P < 0.005, compared to cells transfected with vectors. (G) HBV-ayw and HBV-adw1 AREs are of low similarity. Green letters are the aligned conserved sequences, whereas red letters are read as putative ARE on the HBV.
Recent data from a different HBV strain revealed that AR could increase HBV RNA transcription by binding to an ARE located on the HBV genome (17). The ARE sequence in the alternative HBV strain is quite different from the ARE sequence we found in the HBV strain used here (Fig. 2G). Yet, AR was able to bind both AREs and enhance HBV RNA transcription.
Together, the results suggest that hepatic AR may be able to increase HBV viral titer by promoting HBV RNA transcription through direct binding to the ARE in the HBV genome.
Hepatic AR increases hepatocarcinogenesis by facilitating the transformation of normal liver cells to liver tumor cells in HBV mice
To determine whether the hepatic AR-mediated increase in HBV viral titer leads to an acceleration in hepatocarcinogenesis by transforming normal liver cells to liver tumors, we analyzed hepatocyte primary cultures obtained from HBV-AR+/y and HBV-L-AR−/y livers with low DEN injection and HBV-AR+/y mice livers without low DEN injection. We found that hepatocytes obtained from HBV-AR+/y mice with low DEN injection survive well in culture compared to those from HBV-L-ART −/y mice with low DEN injection. However, hepatocytes from HBV-AR+/y mice without low DEN injection could not be maintained in culture (one passage) (Fig. 3A). To determine the role of hepatic AR in hepatocyte transformation, we stably transfected normal hepatocyte BNL CL.2 cells with functional AR (Fig. 3, B and C) and showed that they were more easily transformed by SV40 large T antigen (Fig. 3D, left top panel) and N-methyl-N-nitrosourea (NMU; Fig. 3D, left bottom panel) compared to BNL CL.2 cells stably transfected with vector control. Together, these results demonstrate that the increase in hepatic AR-mediated HBV viral titer may lead to an increase in hepatocarcinogenesis by promoting the transformation from normal liver cells to liver tumors.
Fig. 3.
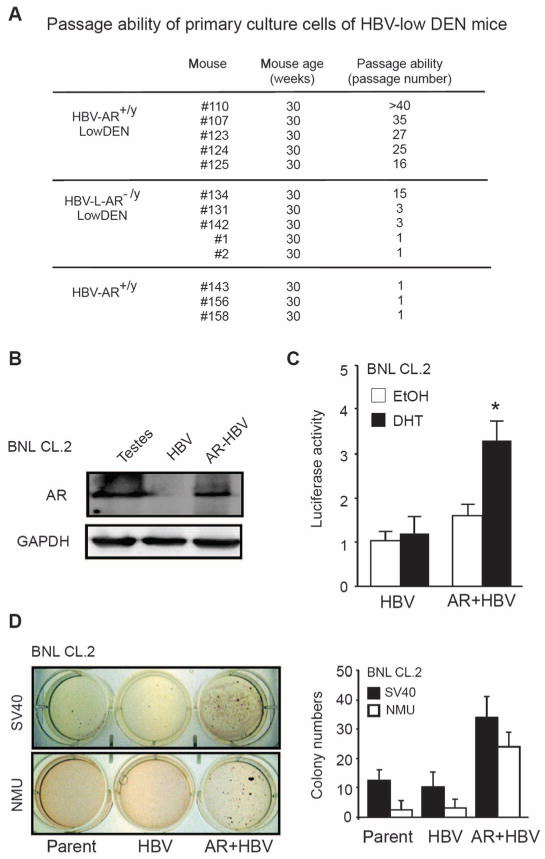
AR enhances HBV-promoted hepatocyte transformation. (A) Primary hepatocytes from HBV-AR+/y mice livers have a better passage ability and viability than those from HBV-L-AR−/y mice when cultured in vitro. The hepatocytes were isolated from HBV-AR and HBV-L-AR−/y mice, and cells were continuously propagated until these cells no longer can be passaged. (B) AR stably transfected into BNL CL.2 cells. Mouse testes protein extract served as positive control of AR expression, whereas glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as loading controls. (C) AR transactivation function was tested by cotransfection of mouse mammary tumor virus (MMTV) luciferase and pRenilla-TK constructs. The luciferase activity was examined after 24 hours of treatment [ethanol (EtOH) or 10 nM dihydrotestosterone (DHT)]. The luciferase activities are normalized by Renilla luciferase activity and are shown as means ± SEM. *P < 0.05, compared with transfected cells treated with ethanol. (D) AR enhances the transformation of HBV-containing hepatocytes. The left top panel shows representative photos of BNL CL.2 cells transformed by SV40 large T antigen, whereas the left bottom panel shows the cells transformed by NMU. The right panel shows the quantitation of colony numbers.
Hepatic AR promotes HBV-mediated HCC tumorigenicity
We generated four new HepG2 cell lines via stable transfection of control, AR, HBV, and AR+HBV, and demonstrated that the functional AR was stably transfected in the HepG2-AR and HepG2-HBV(+) AR cells, and addition of HBV further induces AR transactivation in HBV(+)AR cells (Fig. 4A). Furthermore, HepG2-HBV(+)AR cells exhibited the strongest colony-forming ability among the four stable HepG2 cell lines using anchorage-independent growth assays (Fig. 4B) and have the least number of apoptotic cells (Fig. 4C and fig. S1). These data clearly demonstrate that AR can cooperate with HBV to promote HCC tumorigenicity. This conclusion is further strengthened by evidence showing that HCC tumors in HBV-L-AR−/y mice have less proliferating cell nuclear antigen (PCNA) staining, an indicator of proliferating cells, as compared to HBV-AR+/y littermates (Fig. 4, D and E).
Fig. 4.
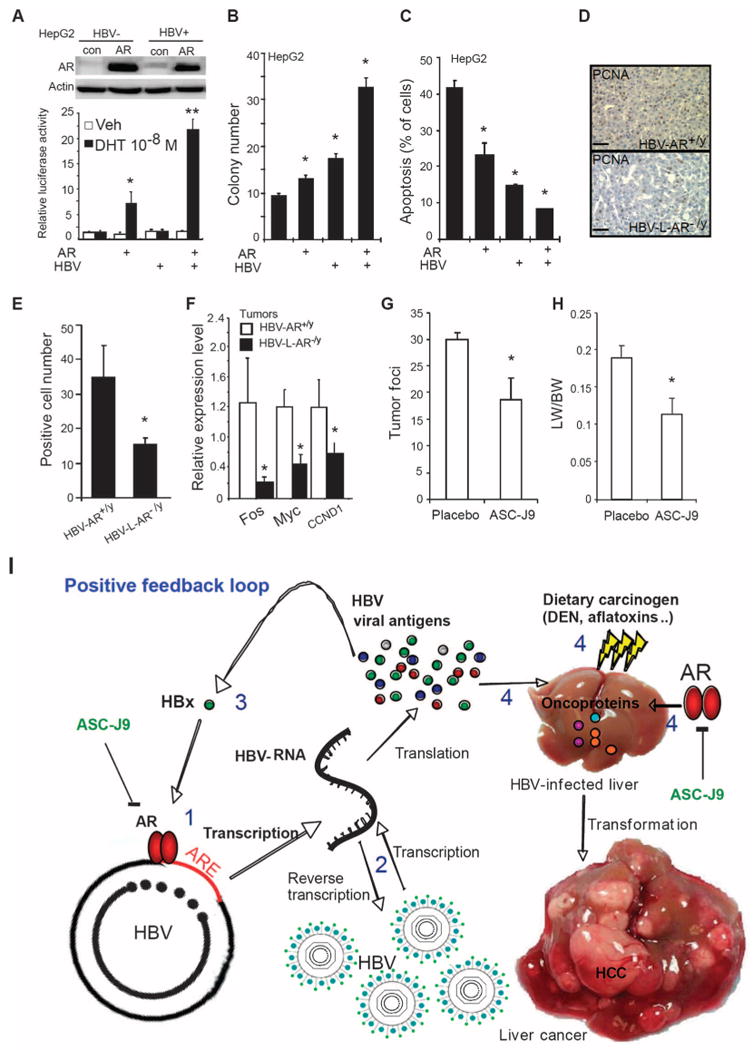
AR enhances HBV-mediated tumorigenicity and ASC-J9 suppresses HBV-mediated tumor growth in mice. (A) Top panel: Characterizations of AR expression in HepG2 cells with or without containing the HBV whole genome. AR expression was determined by Western blot. Bottom panel: AR transactivation activity was enhanced by HBV. Cells were cotransfected with pMMTV luciferase reporter and pRenilla-TK plasmid DNA. Luciferase activity was measured 24 hours after treatments (ethanol or 10 nM DHT). *P < 0.05, **P < 0.005, compared with control cells treated with ethanol. (B) AR and HBV cooperatively increased the anchorage-independent growth in HepG2 cells. Colony formation assay was performed in four stable cell lines as described in Materials and Methods. Colonies were stained and counted after 2 weeks of incubation. *P < 0.05, compared with HepG2 control cells. (C) AR and HBV cooperatively decrease stress-induced apoptosis. *P < 0.05, compared with HepG2 control cells. (D) Histological staining of cell proliferation marker PCNA in liver tissues of HBV-AR+/y and HBV-L-AR−/y mice. PCNA signals were measured by immunostaining and representative pictures are shown. Magnification, ×100. Scale bars, 80 μm. (E) Quantitation results of the PCNA-positive staining. Proliferating cells in 32-week-old DEN-injected HBV-AR+/y mice liver tumor are more than HBV-L-AR−/y mice. We counted PCNA-positive cells in three areas (100×) of liver tumor sections of HBV-AR+/y (n = 4) and HBV-L-AR−/y (n = 4) mice. Data are shown as mean ± SEM. The P value indicates the comparison between HBV-AR+/y and HBV-L-AR−/y mice. *P < 0.05. (F) Expression of HCC-associated oncogenes was suppressed in mice with loss of hepatic AR. The 32-week-old mouse liver RNA was extracted and assayed. All the results were shown as mean ± SEM. The P value indicates the comparison between HBV-AR+/y and HBV-L-AR−/y mice. *P < 0.05. (G) ASC-J9 suppresses HBV-induced liver tumor foci in mice. (H) The ratio of liver weight to body weight was reduced in the low-dose, DEN-injected, 36-week-old HBV-AR+/y mice fed with ASC-J9 (50 mg/kg per day) for 45 days. The P value indicates the comparison between placebo and ASC-J9. *P < 0.05. (I) AR promotes HBV-induced hepatocarcinogenesis in a positive feedback manner. The model illustrates that AR induces a positive feed-back response on HBV viral production to mediate hepatocarcinogenesis. (1) Activated AR enhances HBV RNA transcription via binding to ARE on HBV genome. (2) Increased HBV RNA produces more HBV viral antigen and HBV DNA. (3) Increased HBx from AR-enhanced HBV RNA transcription can consequently promote AR-enhanced HBV RNA transcription in a feedback manner. (4) Increased viral antigens in the presence of subminimal dosage of dietary carcinogens can cooperate with AR to promote hepatocyte transformation.
To further characterize the AR effects on the HBV-induced liver tumors, we then measured several HCC tumorigenicity-related oncogene expressions by three different assays [complementary DNA (cDNA) micro-array, oncogene polymerase chain reaction (PCR) array, and quantitative real-time PCR (qRT-PCR)] (table S1). We found that Fos, Myc, and CCND1 were consistently suppressed by three different assays in tumors from HBV-L-AR−/y mice compared to those from HBV-AR+/y (Fig. 4F and table S1), suggesting that AR may be able to enhance HBV-induced HCC tumorigenesis by modulating the expression of multiple HCC tumorigenicity-related oncogenes. These results are consistent with earlier studies showing links between c-Fos and c-Myc to HBV-mediated carcinogenesis (18, 19) as well as AR regulation of Fos mRNA expression (20). However, we failed to find any mutations in p53 and CTNNB1, which were reported frequently in tumors from HCC patients (21).
Targeting AR rather than androgen is a better therapy for HBV-related HCC
The gender disparity in HBV DNA and hepatitis B surface antigen (HBsAg) amounts (17, 22) suggests that androgen ablation therapy may be a potential therapy for HBV carriers. However, targeting androgen via androgen ablation therapy yielded poor results in a small clinical study of HBV carriers: Treatment with triptorelin in chronic HBV-infected patients led to little influence on HBV DNA abundance and serum HBsAg concentration (23). Here, we demonstrate that there is a marginal effect of androgen in HBV-induced HCC. Similar serum testosterone concentrations in HBV-L-AR−/y and HBV-AR+/y mice were observed despite the reduced HCC incidence and HBV DNA abundance in HBV-L-AR−/y mice (fig. S2). These results strongly suggested that AR, rather than androgen, is the key influence on HBV-induced HCC, and AR might be a better therapeutic target in HBV-induced HCC than androgen itself. To further confirm this conclusion, we gavage-fed HBV-AR+/y mice with ASC-J9, a small chemical compound that could degrade AR in selective cells with little toxicity (24), and found that tumor foci and volume were significantly reduced (Fig. 4, G and H), demonstrating that targeting AR with ASC-J9 [or AR-siRNA (small interfering RNA)] might represent a new therapeutic approach to battle the HBV-induced HCC.
Discussion
The vast majority of the evidence that indicates a role for androgen or the AR in HBV-induced HCC has been derived from indirect epidemiological studies (3, 4). Our animal data represent in vivo evidence that hepatic AR can directly influence HBV-induced HCC formation. We demonstrate that hepatic AR can positively regulate HBV-induced hepatocarcinogenesis in a newly generated animal model that mimics clinical HBV-related HCC, which is in agreement with early studies showing that although HCC and cirrhosis share similar risk factors and the male gender is one of the dominant risks (25, 26), there were still HBV carriers who developed HCC without cirrhosis (13). This suggests that male gender, upon exposure to environmental toxins such as aflatoxins, may play a significant role in HBV-induced hepatocarcinogenesis. Using another approach, Ma et al. (27) demonstrated that mice lacking hepatic AR had less hepatocarcinogenesis after exposure to a higher dose (20 mg/kg body weight) of DEN. Thus, AR participates in HCC development, whether it is induced by a high dose of DEN or by a lower dose of DEN administered together with HBV.
The current study demonstrates that AR can promote hepatocarcinogenesis as well as HCC tumorigenicity at an early stage. In prostate cancer, studies have demonstrated that suppression of prostate AR at an early stage resulted in suppression of prostate tumor progression, yet suppression of prostate AR at a much later stage led to increased prostate cancer metastasis (28, 29). These dual yet opposite roles of prostate AR could be due to differential AR signals in different cells, serving as a stimulator in prostate stromal cells, a prosurvival signal in prostate epithelial luminal cells, and a suppressor in prostate epithelial basal intermediate cells (28–30). Suppression of androgen or AR signals also led to increased infiltrating cells, such as macrophages as well as B or T cells (31, 32). The HCC tumor micro-environment is also rich in various cells, including hepatocytes, Kupffer cells (specialized macrophages in liver), and infiltrating T or B cells, which show distinct and important roles for the promotion of HCC tumorigenicity (30, 33–35). It will be interesting to see whether targeting AR at different stages or in different liver cancer cell components may also lead to differential effects during HCC progression.
Regardless of which role AR may play in individual HCC cells, the results from the current study clearly demonstrate that targeting AR, by either ASC-J9 or AR-siRNA, and not by targeting androgens, as used in current clinical trials (36), may lead to effective results to suppress hepatocarcinogenesis and/or HCC tumorigenicity at early stages.
In summary, our data demonstrate that hepatic AR can enhance HBV-induced HCC operating via up-regulating HBV DNA replication-RNA transcription through direct binding to ARE near the viral core promoter. The consequences of this influence result in promoting the transformation from normal liver cells to liver tumors and subsequent tumor proliferation in a positive feedback manner (Fig. 4I). These results illuminate a new pathway for the development of enhanced therapeutics to battle the HBV-induced hepatocarcinogenesis by specifically targeting both AR and HBx.
Materials and Methods
Tumor induction and ASC-J9 treatment in mice
All of the animal experiments followed the Guidance of the Care and Use of Laboratory Animals of the National Institutes of Health, with approval from the Department of Laboratory Animal Medicine, University of Rochester. The mating strategy is illustrated in Fig. 1A. The female floxed AR homozygous transgenic mice (ARflox/flox) mice (37) were first crossed with male HBV transgenic (HBV) mice (8) to generate HBV-ARflox/x female mice, and then the HBV-ARflox/x female mice were mated with male albumin promoter-driven Cre (Alb-Cre) transgenic mice (B6.Cg-Tg[Alb-Cre]21Mgn/J; Jackson Laboratories). After genotyping, the male AR+/y, HBV-AR+/y, and HBV-L-AR−/y mice were used for experiments. HCC was induced by intraperitoneal injection of low dose of DEN (2 mg/kg body weight) in 16-day-old pups as previously described (8). For the ASC-J9 (gift from AndroScience) treatment, the 36-week-old HBV-AR+/y mice treated with low dose of DEN (2 mg/kg body weight) were gavage-fed with ASC-J9 (50 mg/kg per mice; daily) for 45 days. The 4× stock solutions were prepared by dissolving ASC-J9 in solvent (cremophor CL/dimethyl sulfoxide, 1:1) and stored at −20°C The stock ASC-J9 was diluted in distilled water just before administration. After 45 days of feeding, the mice were killed. The tumor foci, liver weight, and body weight were evaluated at the time of killing.
HBV RNA and DNA measurement
Total RNA and DNA were extracted from mice liver tissues, and the HBV DNA amount was measured by qRT-PCR as previously described (15). The following primers were used: 5′-CAAGGTATG-TTGCCCGTTTG-3′ and 5′-AAAGCCCTRCGAACCACTGA-3′. The total RNA was reverse-transcribed, and HBV RNA amount was measured by qRT-PCR The relative HBV DNA and RNA amount was compared between livers of HBV-AR+/y and HBV-L-AR−/y mice.
Luciferase assay
HepG2 cells were seeded at 2 × 105 per well in 24-well dishes (Falcon) and transfected with 1.0 g of DNA per well by using Lipofectamine according to the manufacturer's protocol (Invitrogen). The plasmid pRenilla-TK (Promega) was cotransfected in all transfection experiments as internal control. At 24 hours after transfection, the cells were harvested and luciferase activity was measured by using the Dual-Luciferase kit (Promega). All of the luciferase activities were normalized by Renilla luciferase activity.
Quantitative real-time PCR
Total RNA was extracted using TRIzol reagent according to the manufacturer's instructions (Invitrogen). Total RNA (4 μg) was reverse-transcripted to cDNA by ImProm-II Reverse Transcriptase (Promega). Expression of different mRNAs was analyzed by qRT-PCR as previously described (38). For the HCC-related oncogene analysis, from cDNA microarray and oncogene PCR array analysis, Fos, Myc, and CCND1 were found consistently down-regulated in HBV-L-AR−/y mice compared to HBV-AR+/y mice (table S1). Therefore, these gene expressions were further confirmed via qRT-PCR in HBV-AR+/y (n = 4) and HBV-L-AR−/y (n = 4) mice tumors.
Immunohistochemical staining of paraffin-embedded sections
Serial 5-μm histologic sections were deparaffinized and rehydrated. Endogenous peroxidase activity was blocked by incubation with 3% hydrogen peroxide, and antigen retrieval was done by boiling for 20 min in citrate buffer (pH 6.0). The slides were incubated with the antibodies overnight at 4°C. After washing twice with phosphate-buffered saline (PBS), the signals were detected by applying streptavidin-biotin-peroxidase complex (Vector Laboratories) and revealed by 3,3′-diaminobenzidine (Vector Laboratories). The slides were counterstained with hematoxylin (Vector Laboratories) and mounted with mounting solution (Vector Laboratories).
Chromatin immunoprecipitation
AR was overexpressed in HepG2 cells containing HBV whole genome and cultured in 10% fetal bovine serum–Dulbecco's modified Eagle's medium. The cells were subjected to ChIP assay as described previously (17). Finally, the eluted products were detected by PCR The primer sequences flanking both ends of ARE2 in HBV are 5′-GTC-ATTGGAAGTTATGGGTCCTTGC-3′ and 5′-ACACTTGGCACA-GACCTGGC-3′. The amplified products were detected by running 2% agarose gel and visualized by ethidium bromide staining. The AR-binding DNA was pulled down by two AR antibodies (C-19 for C-terminus and N-20 for N-terminus of AR from Santa Cruz Biotechnology).
Cell transformation assay
BNL CL.2 cells (1 × 105) were seeded in 6-cm2 dishes and infected with retrovirus carrying SV40 large T antigen or treated with NMU for 24 hours. After washing three times with PBS, the retrovirus-infected cells were cultured in the growth medium containing puromycin (1 (μg/ml) for 2 weeks. The drug-resistant colonies were mixed and passaged at the density 1 × 105 in 6-cm dishes. After five passages, the cells were subjected to soft agar assay as previously described (39).
Primary hepatocyte culture from mice and cell passage ability assay
The primary culture was modified from Sawada et al. (40) and Invitrogen protocol. In brief, the mice were anesthetized and the livers were perfused from hepatic portal vein by Liver Perfusion Medium (Gibco), and then perfusion was continued by collagenase-containing Liver Digest Medium (Gibco) for 5 min. After perfusion, the livers were soaked under Liver Digestion Medium, chopped into small pieces, placed into sterile 50-ml tube, and incubated by shaking at 37°C for 20 min. After livers were digested, erythrocytes were lysed by ammonium chloride solution (StemCell Technologies) for 1 min, then washed using hepatocyte wash medium (Gibco), and centrifuged at 1500 rpm (Beckman GS6-R; rotor: G.H.3.8) for 5 min to discard tissue debris. After washing, the cells were placed onto 75-cm2 culture flask in 107 cells per flask and maintained in Williams Medium E containing 15% fetal calf serum. After 24 hours, the cells were then washed by PBS to remove unattached cells. After the cells reached 90% confluence, they were subcultured to new flask at 106 cells per flask concentration.
Serum testosterone concentration and tissue preservation
We killed mice at the indicated time points, drew 1 ml of blood by cardiocentesis, and immediately assayed for serum testosterone concentration (27) using the Coat-A-Count Total Testosterone radio-immunoassay (Diagnostic Products). We flash-froze fresh tissues in liquid nitrogen for preservation at −80°C for gene expression assay. We subjected the hepatic major lobe to 10% neutralized buffered formalin (Sigma) for histological analysis.
Apoptosis assay
Cells were subjected to one exposure of ultraviolet (50 J/m2), harvested 24 hours later, and fixed in 70% ethanol. The cells were stained with propidium iodide (40 mg/ml in PBS). Apoptotic cells were quantified by FACScan with CellQuest software (Becton-Dickinson) and presented as the percentage of hypodiploid cells.
Statistical analysis
The cancer incidence was analyzed by using the χ2 test and the Fisher exact test in GraphPad software. The other experiments were assessed by unpaired Student's t test and shown as mean ± SEM. P values <0.05 were considered to be statistically significant.
Supplementary Material
Acknowledgments
Funding: George Whipple Professorship Endowment, NIH grant CA122295, and Taiwan Department of Health Clinical Trial and Research Center of Excellence grant DOH99-TD-B-111-004 (China Medical University, Taichung, Taiwan).
Footnotes
Author contributions: C.C. provided funding and wrote the paper; M.-H.W., W.-L.M., and C.-L.H. designed and performed all experiments; Y.-L.C., C.K.R., Y.-C.H., and S.Y. provided technical support and contributed to writing; J.-H.J.O. provided mice and technical support.
Competing interests: ASC-J9 was patented by the University of Rochester, the University of North Carolina, and AndroScience, and then licensed to AndroScience. Both the University of Rochester and C.C. own royalties and equity in AndroScience.
References and Notes
- 1.Chen CJ, Yu MW, Liaw YF. Epidemiological characteristics and risk factors of hepatocellular carcinoma. J Gastroenterol Hepatol. 1997;12:S294–S308. doi: 10.1111/j.1440-1746.1997.tb00513.x. [DOI] [PubMed] [Google Scholar]
- 2.Chen CJ, Yang HI, Iloeje UH. REVEAL-HBV Study Group, Hepatitis B virus DNA levels and outcomes in chronic hepatitis B. Hepatology. 2009;49:S72–S84. doi: 10.1002/hep.22884. [DOI] [PubMed] [Google Scholar]
- 3.Yu MW, Yang YC, Yang SY, Cheng SW, Liaw YF, Lin SM, Chen CJ. Hormonal markers and hepatitis B virus-related hepatocellular carcinoma risk: A nested case–control study among men. J Natl Cancer Inst. 2001;93:1644–1651. doi: 10.1093/jnci/93.21.1644. [DOI] [PubMed] [Google Scholar]
- 4.Yu MW, Cheng SW, Lin MW, Yang SY, Liaw YF, Chang HC, Hsiao TJ, Lin SM, Lee SD, Chen PJ, Liu CJ, Chen CJ. Androgen-receptor gene CAG repeats, plasma testosterone levels, and risk of hepatitis B-related hepatocellular carcinoma. J Natl Cancer Inst. 2000;92:2023–2028. doi: 10.1093/jnci/92.24.2023. [DOI] [PubMed] [Google Scholar]
- 5.Lee CM, Lu SN, Changchien CS, Yeh CT, Hsu TT, Tang JH, Wang JH, Lin DY, Chen CL, Chen WJ. Age, gender, and local geographic variations of viral etiology of hepatocellular carcinoma in a hyperendemic area for hepatitis B virus infection. Cancer. 1999;86:1143–1150. doi: 10.1002/(sici)1097-0142(19991001)86:7<1143::aid-cncr7>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 6.Shiratori Y, Shiina S, Imamura M, Kato N, Kanai F, Okudaira T, Teratani T, Tohgo G, Toda N, Ohashi M, Ogura K, Niwa Y, Kawabe T, Omata M. Characteristic difference of hepatocellular carcinoma between hepatitis B- and C- viral infection in Japan. Hepatology. 1995;22:1027–1033. doi: 10.1016/0270-9139(95)90605-3. [DOI] [PubMed] [Google Scholar]
- 7.Wang N, Zheng Y, Yu X, Lin W, Chen Y, Jiang Q. Sex-modified effect of hepatitis B virus infection on mortality from primary liver cancer. Am J Epidemiol. 2009;169:990–995. doi: 10.1093/aje/kwn418. [DOI] [PubMed] [Google Scholar]
- 8.Zheng Y, Chen WL, Louie SG, Yen TS, Ou JH. Hepatitis B virus promotes hepatocarcinogenesis in transgenic mice. Hepatology. 2007;45:16–21. doi: 10.1002/hep.21445. [DOI] [PubMed] [Google Scholar]
- 9.Ross RK, Yuan JM, Yu MC, Wogan GN, Qian GS, Tu JT, Groopman JD, Gao YT, Henderson BE. Urinary aflatoxin biomarkers and risk of hepatocellular carcinoma. Lancet. 1992;339:943–946. doi: 10.1016/0140-6736(92)91528-g. [DOI] [PubMed] [Google Scholar]
- 10.Ming L, Thorgeirsson SS, Gail MH, Lu P, Harris CC, Wang N, Shao Y, Wu Z, Liu G, Wang X, Sun Z. Dominant role of hepatitis B virus and cofactor role of aflatoxin in hepatocarcinogenesis in Qidong, China. Hepatology. 2002;36:1214–1220. doi: 10.1053/jhep.2002.36366. [DOI] [PubMed] [Google Scholar]
- 11.Wang LY, Hatch M, Chen CJ, Levin B, You SL, Lu SN, Wu MH, Wu WP, Wang LW, Wang Q, Huang GT, Yang PM, Lee HS, Santella RM. Aflatoxin exposure and risk of hepatocellular carcinoma in Taiwan. Int J Cancer. 1996;67:620–625. doi: 10.1002/(SICI)1097-0215(19960904)67:5<620::AID-IJC5>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 12.McGlynn KA, Rosvold EA, Lustbader ED, Hu Y, Clapper ML, Zhou T, Wild CP, Xia XL, Baffoe-Bonnie A, Ofori-Adjei D, Chen GC, London WT, Shen FM, Buetow KH. Susceptibility to hepatocellular carcinoma is associated with genetic variation in the enzymatic detoxification of aflatoxin B1. Proc Natl Acad Sci USA. 1995;92:2384–2387. doi: 10.1073/pnas.92.6.2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wogan GN. Impacts of chemicals on liver cancer risk. Semin Cancer Biol. 2000;10:201–210. doi: 10.1006/scbi.2000.0320. [DOI] [PubMed] [Google Scholar]
- 14.Earl TM, Nicoud IB, Pierce JM, Wright JP, Majoras NE, Rubin JE, Pierre KP, Gorden DL, Chari RS. Silencing of TLR4 decreases liver tumor burden in a murine model of colorectal metastasis and hepatic steatosis. Ann Surg Oncol. 2009;16:1043–1050. doi: 10.1245/s10434-009-0325-8. [DOI] [PubMed] [Google Scholar]
- 15.Wong DK, Yuen MF, Tse E, Yuan H, Sum SS, Hui CK, Lai CL. Detection of intrahepatic hepatitis B virus DNA and correlation with hepatic necroinflammation and fibrosis. J Clin Microbiol. 2004;42:3920–3924. doi: 10.1128/JCM.42.9.3920-3924.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yeh S, Lin HK, Kang HY, Thin TH, Lin MF, Chang C. From Her2/Neu signal cascade to androgen receptor and its coactivators: A novel pathway by induction of androgen target genes through MAP kinase in prostate cancer cells. Proc Natl Acad Sci USA. 1999;96:5458–5463. doi: 10.1073/pnas.96.10.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang SH, Yeh SH, Lin WH, Wang HY, Chen DS, Chen PJ. Identification of androgen response elements in the enhancer I of hepatitis B virus: A mechanism for sex disparity in chronic hepatitis B. Hepatology. 2009;50:1392–1402. doi: 10.1002/hep.23163. [DOI] [PubMed] [Google Scholar]
- 18.Singh M, Kumar V. Transgenic mouse models of hepatitis B virus-associated hepatocellular carcinoma. Rev Med Virol. 2003;13:243–253. doi: 10.1002/rmv.392. [DOI] [PubMed] [Google Scholar]
- 19.Nijhara R, Jana SS, Goswami SK, Rana A, Majumdar SS, Kumar V, Sarkar DP. Sustained activation of mitogen-activated protein kinases and activator protein 1 by the hepatitis B virus X protein in mouse hepatocytes in vivo. J Virol. 2001;75:10348–10358. doi: 10.1128/JVI.75.21.10348-10358.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kerr JE, Beck SG, Handa RJ. Androgens selectively modulate c-Fos messenger RNA induction in the rat hippocampus following novelty. Neuroscience. 1996;74:757–766. doi: 10.1016/0306-4522(96)00219-9. [DOI] [PubMed] [Google Scholar]
- 21.Laurent-Puig P, Zucman-Rossi J. Genetics of hepatocellular tumors. Oncogene. 2006;25:3778–3786. doi: 10.1038/sj.onc.1209547. [DOI] [PubMed] [Google Scholar]
- 22.DeLoia JA, Burk RD, Gearhart JD. Developmental regulation of hepatitis B surface antigen expression in two lines of hepatitis B virus transgenic mice. J Virol. 1989;63:4069–4073. doi: 10.1128/jvi.63.9.4069-4073.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jilma B, Eichler HG, Köppl C, Weber B, Pidlich JP, Ferenci P, Müller C. Effects of testosterone suppression on serum levels of hepatitis B surface antigen and HBV-DNA in men. Liver. 1998;18:162–165. doi: 10.1111/j.1600-0676.1998.tb00144.x. [DOI] [PubMed] [Google Scholar]
- 24.Yang Z, Chang YJ, Yu IC, Yeh S, Wu CC, Miyamoto H, Merry DE, Sobue G, Chen LM, Chang SS, Chang C. ASC-J9 ameliorates spinal and bulbar muscular atrophy phenotype via degradation of androgen receptor. Nat Med. 2007;13:348–353. doi: 10.1038/nm1547. [DOI] [PubMed] [Google Scholar]
- 25.Liaw YF, Chu CM. Hepatitis B virus infection. Lancet. 2009;373:582–592. doi: 10.1016/S0140-6736(09)60207-5. [DOI] [PubMed] [Google Scholar]
- 26.Chen JD, Yang HI, Iloeje UH, You SL, Lu SN, Wang LY, Su J, Sun CA, Liaw YF, Chen CJ. Risk Evaluation of Viral Load Elevation and Associated Liver Disease/Cancer in HBV (REVEAL-HBV) Study Group, Carriers of inactive hepatitis B virus are still at risk for hepatocellular carcinoma and liver-related death. Gastroenterology. 2010;138:1747–1754. doi: 10.1053/j.gastro.2010.01.042. [DOI] [PubMed] [Google Scholar]
- 27.Ma WL, Hsu CL, Wu MH, Wu CT, Wu CC, Lai JJ, Jou YS, Chen CW, Yeh S, Chang C. Androgen receptor is a new potential therapeutic target for the treatment of hepatocellular carcinoma. Gastroenterology. 2008;135:947–955. doi: 10.1053/j.gastro.2008.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niu Y, Altuwaijri S, Yeh S, Lai KP, Yu S, Chuang KH, Huang SP, Lardy H, Chang C. Targeting the stromal androgen receptor in primary prostate tumors at earlier stages. Proc Natl Acad Sci USA. 2008;105:12188–12193. doi: 10.1073/pnas.0804701105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niu Y, Altuwaijri S, Lai KP, Wu CT, Ricke WA, Messing EM, Yao J, Yeh S, Chang C. Androgen receptor is a tumor suppressor and proliferator in prostate cancer. Proc Natl Acad Sci USA. 2008;105:12182–12187. doi: 10.1073/pnas.0804700105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niu Y, Chang TM, Yeh S, Ma WL, Wang Y, Chang C. Differential androgen receptor signals in different cells explain why androgen-deprivation therapy of prostate cancer fails. Oncogene. 2010 doi: 10.1038/onc.2010.121. [DOI] [PubMed] [Google Scholar]
- 31.Mercader M, Bodner BK, Moser MT, Kwon PS, Park ES, Manecke RG, Ellis TM, Wojcik EM, Yang D, Flanigan RC, Waters WB, Kast WM, Kwon ED. T cell infiltration of the prostate induced by androgen withdrawal in patients with prostate cancer. Proc Natl Acad Sci USA. 2001;98:14565–14570. doi: 10.1073/pnas.251140998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ammirante M, Luo JL, Grivennikov S, Nedospasov S, Karin M. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature. 2010;464:302–305. doi: 10.1038/nature08782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berasain C, Castillo J, Perugorria MJ, Latasa MU, Prieto J, Avila MA. Inflammation and liver cancer: New molecular links. Ann N Y Acad Sci. 2009;1155:206–221. doi: 10.1111/j.1749-6632.2009.03704.x. [DOI] [PubMed] [Google Scholar]
- 34.Guidotti LG, Chisari FV. Immunobiology and pathogenesis of viral hepatitis. Annu Rev Pathol. 2006;1:23–61. doi: 10.1146/annurev.pathol.1.110304.100230. [DOI] [PubMed] [Google Scholar]
- 35.Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro-environment in tumor progression: The role of tumor-associated macrophages. Crit Rev Oncol Hematol. 2008;66:1–9. doi: 10.1016/j.critrevonc.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 36.Groupe d'Etude et de Traitement du Carcinome Hépatocellulaire Randomized trial of leuprorelin and flutamide in male patients with hepatocellular carcinoma treated with tamoxifen. Hepatology. 2004;40:1361–1369. doi: 10.1002/hep.20474. [DOI] [PubMed] [Google Scholar]
- 37.Yeh S, Tsai MY, Xu Q, Mu XM, Lardy H, Huang KE, Lin H, Yeh SD, Altuwaijri S, Zhou X, Xing L, Boyce BF, Hung MC, Zhang S, Gan L, Chang C. Generation and characterization of androgen receptor knockout (ARKO) mice: An in vivo model for the study of androgen functions in selective tissues. Proc Natl Acad Sci USA. 2002;99:13498–13503. doi: 10.1073/pnas.212474399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Z, Chang YJ, Miyamoto H, Yeh S, Yao JL, di Sant'Agnese PA, Tsai MY, Chang C. Suppression of androgen receptor transactivation and prostate cancer cell growth by heterogeneous nuclear ribonucleoprotein A1 via interaction with androgen receptor coregulator ARA54. Endocrinology. 2007;148:1340–1349. doi: 10.1210/en.2006-0716. [DOI] [PubMed] [Google Scholar]
- 39.Huang RP, Peng A, Hossain MZ, Fan Y, Jagdale A, Boynton AL. Tumor promotion by hydrogen peroxide in rat liver epithelial cells. Carcinogenesis. 1999;20:485–492. doi: 10.1093/carcin/20.3.485. [DOI] [PubMed] [Google Scholar]
- 40.Sawada N, Lee GH, Mochizuki Y, Ishikawa T. Active proliferation of mouse hepatocytes in primary culture under defined conditions as compared to rat hepatocytes. Jpn J Cancer Res. 1988;79:983–988. doi: 10.1111/j.1349-7006.1988.tb00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.