

Abstract
mRNA turnover is a critical step in the control of gene expression. In mammalian cells, a subset of mRNAs regulated at the level of mRNA turnover contain destabilizing AU-rich elements (AREs) in their 3′ untranslated regions. These transcripts are bound by a suite of ARE-binding proteins (AUBPs) that receive information from cell signaling events to modulate rates of ARE mRNA decay. Here we show that a key destabilizing AUBP, tristetraprolin (TTP), is repressed by the p38 mitogen-activated protein kinase (MAPK)-activated kinase MK2 due to the inability of phospho-TTP to recruit deadenylases to target mRNAs. TTP is tightly associated with cytoplasmic deadenylases and promotes rapid deadenylation of target mRNAs both in vitro and in cells. TTP can direct the deadenylation of substrate mRNAs when tethered to a heterologous mRNA, yet its ability to do so is inhibited upon phosphorylation by MK2. Phospho-TTP is not impaired in mRNA binding but does fail to recruit the major cytoplasmic deadenylases. These observations suggest that phosphorylation of TTP by MK2 primarily affects mRNA decay downstream of RNA binding by preventing recruitment of the deadenylation machinery. Thus, TTP may remain poised to rapidly reactivate deadenylation of bound transcripts to downregulate gene expression once the p38 MAPK pathway is deactivated.
Human cells can fine-tune gene expression in response to a rapidly changing cellular environment by regulating the rates of mRNA decay (28). Although a variety of RNases in eukaryotic cells participate in mRNA turnover, the shortening and removal of the poly(A) tail by the process of deadenylation are frequently the first steps in this process (4, 52, 70). They are carried out by two major cytoplasmic deadenylase complexes, the Pan2-Pan3 and Ccr4-Caf1-Not complexes (52, 59, 70). Deadenylation generally leads to translational silencing of the mRNA, after which the mRNA can either be stored for later use or subjected to rapid decay (23, 25, 63). In the case of deadenylated mRNAs targeted for decay, the decapping complex removes the cap structure from the 5′ end of the transcript to allow access to the mRNA body by the 5′-to-3′ exonuclease, Xrn1 (28). Alternatively, the exosome, a 3′-to-5′ exonuclease complex, degrades the deadenylated mRNA from the 3′ end (65).
mRNA-binding proteins can influence the activity of degradative enzymes on a given message by recognizing cis elements within the transcript. One such sequence is the AU-rich element (ARE) located in the 3′ untranslated region (UTR) of many short-lived mRNAs encoding cytokines, growth factors, proto-oncogenes, and other transiently expressed proteins (11, 15, 36, 72). A number of ARE-binding proteins (AUBPs) that activate or suppress decay have been identified. The protein tristetraprolin (TTP) belongs to a family of CCCH zinc finger AUBPs that trigger rapid mRNA decay by recruiting enzymes involved in mRNA turnover (32, 39, 44). The importance of TTP as a physiological regulator of gene expression is underscored by TTP knockout mice in which chronic inflammation due to elevated levels of tumor necrosis factor alpha (TNF-α) protein has been directly attributed to the increased stability of TNF-α mRNA in TTP−/− macrophages (10).
Several cell signaling pathways are known to modulate mRNA decay, but it is poorly understood how these signals are communicated to the mRNA decay machinery. Activation of the p38 mitogen-activated protein kinase (MAPK) pathway by a number of external stresses has been shown to impair the deadenylation of ARE mRNA in vivo (18, 67, 68), although the mechanism by which deadenylation is regulated remains unknown. It has been proposed that AUBPs such as TTP can act as adaptor proteins to communicate signaling events to the mRNA decay machinery (8, 45, 54). Phosphorylation of TTP by the p38 MAPK-activated kinase MK2 has been associated with increased stability of ARE-containing transcripts (45, 58, 60). However, as several AUBPs can bind the ARE and exert synergistic or opposing effects in response to the activation of various signaling pathways, it can be difficult to determine the precise role of phosphorylation of TTP by MK2 in ARE mRNA decay. Phosphorylation of TTP by MK2 creates a substrate for the phospho-serine/threonine binding protein 14-3-3 (14, 55). This has been shown to localize TTP to the cytoplasm (35) and to protect TTP from dephosphorylation by protein phosphatase 2A (PP2A) (57). It remains unclear whether 14-3-3 recruitment plays a direct role in inhibiting the activity of phosphorylated TTP.
Here we show that TTP in cells can trigger rapid deadenylation and decay of an mRNA to which it is tethered in isolation from other AUBPs. Using in vitro deadenylation assays, we show that TTP copurifies with a deadenylase activity which specifically deadenylates ARE-containing RNA. Phosphorylation of TTP by the p38-activated kinase MK2 promotes 14-3-3 association and inhibits the ability of TTP to trigger the deadenylation of tethered mRNA in cells by preventing the recruitment of cytoplasmic deadenylases. In contrast, phosphorylation does not affect the ability of TTP to bind mRNA. Taken together, these results suggest that phosphorylated TTP may remain poised to rapidly reactivate mRNA deadenylation and decay upon dephosphorylation.
MATERIALS AND METHODS
Plasmids.
The open reading frames encoding human Pan2, 14-3-3ɛ, and poly(A)-specific RNase (PARN) were cloned into pcDNA3-FLAG expression vectors, which are derivatives of pcDNA3 (Invitrogen), by using BglII and NotI or BamHI and NotI sites, respectively. Expression plasmids for hnRNP-A1, TTP-F126N, TTP1-100, hCcr4a, hCcr4b, hCaf1a, hCaf1b, PABPC1, and Dcp2-E148Q, as well as the N-terminal MS2 coat protein fusion and β-globin mRNA decay reporter transcripts, have been described previously (41-44, 62). Similarly, expression plasmids for MK2EE, MK2KR, and TTP-AA were described earlier (55). pcNMS2-TTP-EE, expressing TTP-EE fused a FLAG tag and the MS2 coat protein, was prepared using site-specific mutagenesis via the QuikChange protocol (Stratagene) to change the AGC codons at positions S52 and S178 to GAA codons, encoding glutamate residues. pcDNA3-FLAG-hCaf1b-D40,44A, expressing catalytically inactive, FLAG-tagged hCaf1b-D40,44A mutant protein, was generated from pcDNA3-FLAG-hCaf1b by mutating the catalytic aspartate codons at positions 40 and 44 to alanine codons using the QuikChange protocol.
Pulse-chase mRNA decay assays.
Pulse-chase mRNA decay assays were performed as described previously (16, 43). Briefly, HeLa Tet-off cells (Clontech; derived from cervical cancer cells from cancer patient Henrietta Lacks in 1951 [53]) were grown in Dulbecco modified Eagle medium (DMEM; Invitrogen) containing 10% fetal bovine serum (FBS; Invitrogen) in 3.5-cm plates and transiently transfected in the presence of 50 ng/ml tetracycline using TransIt HeLaMonster transfection reagent (Mirus) in accordance with the manufacturer's protocol. A total of 2 μg of DNA was used per well, including 25 ng of the internal control expression plasmid producing β-GAP mRNA, 0.5 μg of mRNA reporter plasmid containing six binding sites for the MS2 coat protein (pPCβwt-6BS), together with 0.2 μg pcNMS2-FLAG, pcNMS2-mTTPwt, pcNMS2-mTTP-AA, or pcNMS2-mTTP-EE and 0.5 μg pcDNA3-FLAG-MK2EE or pcDNA3-FLAG-MK2KR. In addition, some assay mixtures (see Fig. 1B and 4B) contained 0.5 μg of the expression vector for Dcp2-E148Q while others (see Fig. 1C) contained 0.5 μg of Caf1b D40,44A expression plasmid. Empty vector pcDNA3 was added to 2 μg total plasmid. Two days posttransfection, transcription was pulsed for 6 h by removal of tetracycline and then shut off by addition of 1 μg/ml tetracycline. Cells were harvested at time points thereafter in Trizol (Invitrogen). RNA was analyzed by Northern blotting as described previously (16, 43). Deadenylated samples (see Fig. 1 and 4B) were generated by hybridization of extracted RNA samples with 1 μM oligo(dT)24 in 10 μl of TKE (10 mM Tris-HCl [pH 7.5], 40 mM KCl, 0.1 mM EDTA) at 80°C for 2 min, annealing at room temperature for 5 min, addition of 10 μl of 2× RNase H buffer (80 mM Tris-HCl [pH 7.0], 20 mM MgCl2, 1 mM dithiothreitol [DTT], 1.5 U of RNase H; Invitrogen), and 30 min of incubation at 37°C.
FIG. 1.
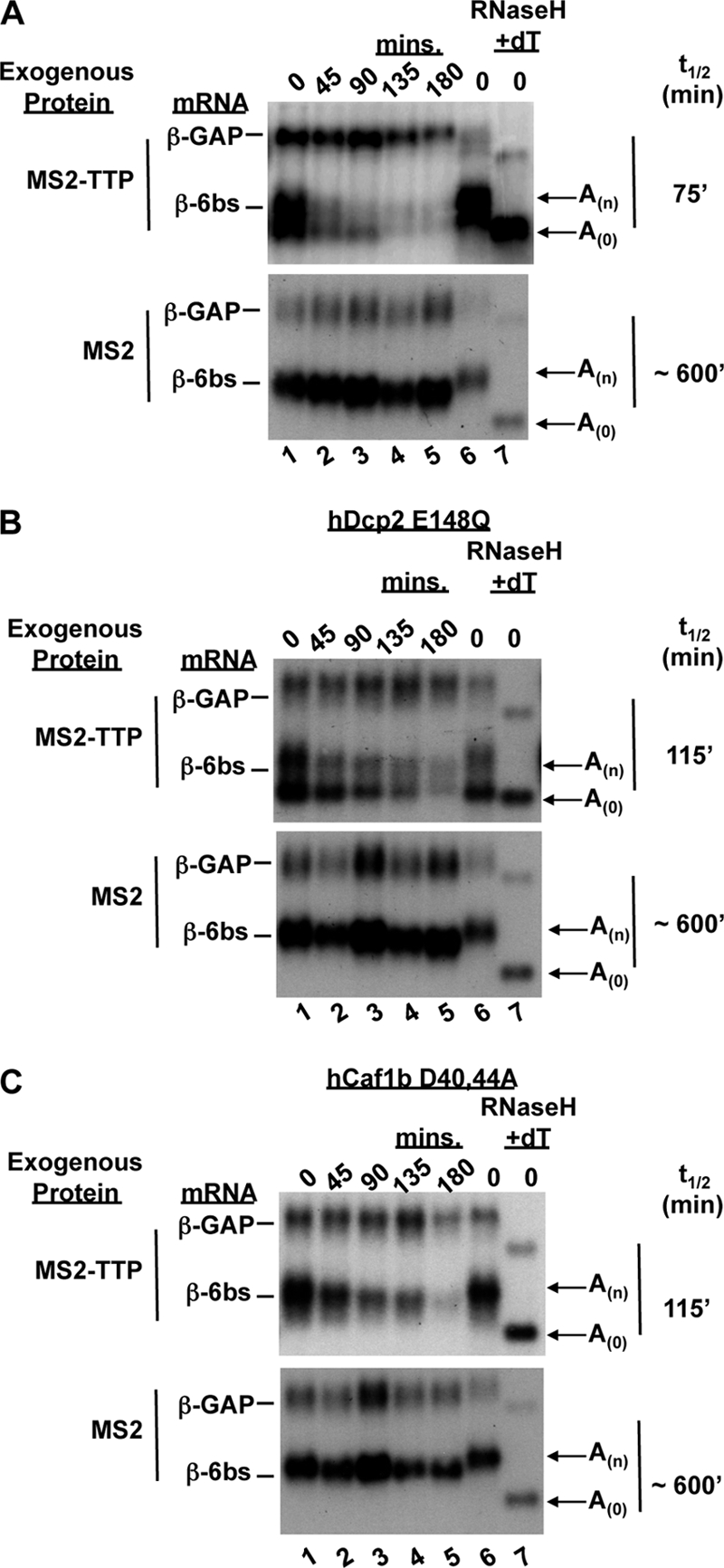
Tethered TTP promotes mRNA deadenylation. (A) Northern blot assays of transcriptional pulse-chase mRNA decay assays in human HeLa (53) Tet-off cells of a reporter mRNA bearing six MS2 coat protein binding sites in the 3′ UTR (β-6bs) in the presence of exogenous MS2-TTP or MS2 protein alone, as indicated at the left. The time points at the top are minutes after transcriptional repression by tetracycline addition. β-6bs mRNA t1/2s are indicated at the right and were calculated by normalizing to the constitutively expressed internal control, β-GAP mRNA. Arrows indicate the two distinct bands that reflect poly(A) (An) and deadenylated (A0) mRNAs, as determined by comigration with deadenylated β-6bs mRNA generated by RNase H digestion in the presence of oligo(dT) (lane 7). (B) Northern blot assays as in panel A showing the decay of β-6bs in the presence of an exogenously expressed dominant negative form of the decapping enzyme hDcp2-E148Q together with MS2-TTP or MS2. (C) Northern blot assays showing the decay of β-6bs in the presence of an exogenously expressed dominant negative form of the deadenylase hCaf1b-D40,44A together with MS2-TTP or MS2.
Protein purification.
FLAG-tagged TTP or PABPC1 was expressed in transiently transfected human embryonic kidney 293T (HEK293T) cells grown in DMEM-10% FBS. Transfection was performed with TransIT-293 reagent in accordance with the manufacturer's protocol (Mirus) using 1 μg pcDNA3-FLAG-TTP or 5 μg pcDNA3-FLAG-PABPC1 per 10-cm plate. Empty vector pcDNA3 was added to a total of 10 μg. Cells were lysed in 1.0 ml of hypotonic lysis buffer containing 20 mM HEPES-KOH (pH 7.4), 10 mM NaCl, 2 mM MgCl2, 0.1% IGEPAL CA-630, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin, and 2 μg/ml leupeptin. Cell extracts were brought to 150 mM NaCl and incubated with a 40-μl bead volume of anti-FLAG M2 agarose (Sigma) at 4°C for 2 h. Unbound proteins were removed by washing twice with ice-cold isotonic wash buffer (20 mM HEPES-KOH [pH 7.4], 150 mM NaCl, 0.01% IGEPAL CA-620, 10% glycerol), followed by six times with a high-salt wash buffer identical to the isotonic wash buffer but containing 500 mM NaCl. FLAG-TTP was eluted with 0.2 mg/ml FLAG peptide (Sigma) in 50 μl of low-salt buffer (20 mM HEPES-KOH [pH 7.4], 0.5 mM NaCl, 0.01% IGEPAL, 20% glycerol) with gentle shaking at 4°C for 1 h, and eluates were stored at −80°C.
Preparation of deadenylation substrates.
The ARE-A60 substrate containing the 62-nucleotide (nt) granulocyte-macrophage colony-stimulating factor (GM-CSF) ARE (44) was transcribed using the Uhlenbeck single-stranded T7 transcription protocol (47). Briefly, a template for transcription by T7 RNA polymerase (Ambion) was created by hybridizing a 181-nt DNA oligonucleotide, ARE-T60 (IDT) (5′-TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT CCC GGG AAT TTA AAT AAA TAA ATA AAT AAA TAT TTT AAA AAT ATA AAT ATA TAA ATA TTA AGC TTT AGA TCT TCT TCT GAA ATC AGC TTT TGT TCC ATT CTC CCT ATA GTG AGT CGT ATT A-3′), with a 23-nt T7 promoter DNA oligonucleotide (5′-TAATACGACTCACTATAGGGAGA-3′) to create a double-stranded T7 promoter. The mutant ARE-A60 substrate was transcribed in a similar manner using the 181-nt ultramer oligonucleotide ARE-MUT-T60 (5′-TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT TTT CCC GGG AAT TTC GAC TAA GCT TAT CAA TCA ATA TTC CAG ACA TCA GAT GTA TTC ATA TTA AGC TTT AGA TCT TCT TCT GAA ATC AGC TTT TGT TCC ATT CTC CCT ATA GTG AGT CGT ATT A-3′). The internal control nonadenylated RNA was transcribed from plasmid pcDNA3-FLAG-NMS2 digested with BglII. Twenty-microliter in vitro transcription reaction mixtures were prepared using 20 pmol annealed oligonucleotides or 2.0 μg linearized plasmid using the Ambion Megascript system. RNAs were precipitated with ethanol and separated in 6% polyacrylamide-6 M urea denaturing gels, visualized by UV shadowing, excised, and eluted overnight at room temperature in 400 μl of 0.5% sodium dodecyl sulfate (SDS)-300 mM sodium acetate (pH 5.2)-5 mM EDTA-400 μl phenol-chloroform-isoamyl alcohol. Following elution, RNAs were ethanol precipitated and resuspended in water. The substrates were subsequently cap labeled with [α-32P]GTP using bacterially expressed vaccinia virus capping enzyme (construct kindly provided by Stewart Shuman) as described by Zhang et al. (64).
In vitro deadenylation assays.
Deadenylation assays were performed in 30 μl of 20 mM HEPES-KOH (pH 7.4)-2 mM MgCl2-0.1 mg/ml bovine serum albumin-1 mM spermidine-0.1% IGEPAL CA-630-0.5 U/μl RNaseOut (Invitrogen) at 30°C for 1 h, including 10,000 cpm of both control and substrate RNAs and 5 to 200 ng of FLAG-TTP protein as indicated in the figures. RNAs were ethanol precipitated, separated in 6% polyacrylamide-6 M urea denaturing gels, and visualized by autoradiography. The deadenylated control RNA was created by removal of the A tail of the deadenylation substrate by RNase H treatment in the presence of oligo(dT) as described above.
Coimmunoprecipitation assays.
Four hundred microliters of extracts of HEK293T cells derived from ∼2 × 106 transiently transfected cells (corresponding to one 3.5-cm plate) was prepared as described earlier (41), in the presence or absence of RNase A (125 μg/ml), and incubated with 20 μl (bead volume) anti-FLAG M2 agarose (Sigma) at 4°C for 2 h. Complexes were washed eight times with Net-2 (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.1% IGEPAL-CA630) and eluted in 40 μl of 2× SDS-polyacrylamide gel electrophoresis (PAGE) buffer (100 mM Tris-HCl [pH 6.8], 200 mM DTT, 4% SDS, 0.2% bromophenol blue, 20% glycerol) and stored at −20°C. In some experiments, cells were treated for 2 h with 1 μM okadaic acid (Calbiochem) prior to extract preparation. Proteins were detected after SDS-PAGE by immunoblotting with rabbit polyclonal antibodies against Myc or FLAG epitopes in accordance with the manufacturer's recommendation (Sigma).
RESULTS
TTP promotes deadenylation of a tethered mRNA substrate.
TTP is an important regulator of the decay of a subset of ARE-containing mRNAs (17, 21, 34, 48, 56, 58). However, since multiple cellular AUBPs other than TTP can recognize the ARE sequence and exert synergistic or opposing effects (40, 68), it can be difficult to discern the exact role of TTP in the context of the ARE in cells. Consequently, we wished to study the function of TTP in isolation from other AUBPs. To do this, we used a pulse-chase mRNA decay assay (12, 69) combined with a tethering approach described previously (16, 42, 44) in which TTP fused to the MS2 coat protein was tested for the ability to activate the decay of a β-globin reporter mRNA that contains six MS2 coat protein-binding sites in the 3′ UTR (β-6bs). As shown in the mRNA decay assays in Fig. 1A, in which transcription of the β-6bs mRNA was pulsed for 6 h in human HeLa Tet-off cells in the absence of tetracycline and subsequently shut off by the addition of tetracycline, expression of MS2-TTP protein results in destabilization of the reporter transcript, with a half-life (t1/2) of approximately 75 min (top panel), compared to a t1/2 of ∼600 min for cells expressing MS2 protein alone (bottom panel).
In these assays, we also observed the accumulation of a faster-migrating species of β-6bs mRNA in cells expressing the MS2-TTP fusion protein (Fig. 1A, top), which was absent in cells expressing unfused MS2 (bottom). This band corresponds to deadenylated reporter mRNA, as determined by comigration with the same mRNA treated with oligo(dT) and RNase H to remove the poly(A) tail (Fig. 1, lane 7). The deadenylated mRNA is more evident upon the coexpression of a catalytically inactive mutant form of human decapping enzyme (hDcp2-E148Q) (41), which impairs removal of the 5′ cap and subsequent 5′-to-3′ exonucleolytic decay and results in the strong accumulation of deadenylated mRNA in the presence of MS2-TTP (Fig. 1B, top panel). The deadenylated mRNA does not accumulate in cells lacking MS2-TTP (Fig. 1B, bottom panel), demonstrating that tethered TTP activates rapid deadenylation. Furthermore, impairing deadenylation by exogenous expression of a catalytically inactive component of the Ccr4-Caf1-Not deadenylase complex (hCaf1b-D40,44A) prevents the accumulation of deadenylated mRNA (Fig. 1C). However, it is important to note that even in the presence of mutant hDcp2 or hCaf1b, MS2-TTP retains its ability to activate the decay of β-6bs mRNA (Fig. 1B and C), likely due its known ability to associate with multiple other mRNA decay enzymes (44).
TTP copurifies with deadenylase activity.
Deadenylation of ARE-containing transcripts is impaired in TTP knockout mice (37), suggesting that TTP is responsible for activating deadenylation on these substrates. Further investigation of the mechanism behind the activation of deadenylation of ARE mRNA by TTP indicated that TTP could activate deadenylation by PARN in human cell extracts (39). However, no interaction between TTP and PARN was observed (39, 44). This could suggest that limiting accessory factors are required for TTP to activate deadenylation via PARN or that TTP normally activates deadenylation through other deadenylases. To determine whether TTP copurifies with deadenylase activity, we immunopurified exogenously expressed FLAG-tagged TTP from HEK293T cells and tested its ability to deadenylate in vitro a radiolabeled substrate RNA containing the ARE from GM-CSF mRNA and bearing a 60-nt poly(A) tail. As shown in Fig. 2A, incubation of this substrate with increasing amounts of TTP results in the accumulation of a deadenylated product (lanes 5 to 8, ARE-A0) corresponding in length to an oligo(dT)/RNase H-treated control (lane 3). This activity is specific for poly(A) RNA, as the larger, nonadenylated control RNA remains intact (Ctrl, lanes 5 to 8 and 9 to 12).
FIG. 2.

TTP copurifies with deadenylase activity. (A) In vitro deadenylase assay with FLAG-tagged TTP purified from human cells. 5′-32P-m7G-labeled deadenylation substrate (ARE-A60) (lanes 4 to 8) and/or nonadenylated control (Ctrl) RNA (lanes 4 to 12) were incubated for 1 h at 30°C with increasing amounts of FLAG-TTP immunopurified from HEK293T cells (lanes 5 to 12). The control (lane C), ARE-A60 (A60), and deadenylated ARE-A60, [A0; generated by RNase H-oligo(dT) cleavage] RNAs, are shown in lanes 1 to 3. (B) ARE-A60 substrate incubated with increasing amounts of ARE binding-deficient TTP protein, TTP-F126N (lanes 4 to 7), and a mutated ARE substrate (ARE-MUT-A60) incubated with increasing amounts of wild-type TTP (lanes 11 to 14).
The ability of TTP to stimulate deadenylation of the reporter mRNA is dependent upon mRNA binding, as TTP does not stimulate deadenylation of a reporter mRNA with a mutated ARE sequence (ARE-MUT-A60, Fig. 2B, lanes 11 to 14). Likewise, a TTP point mutant (F126N) which fails to bind mRNA (38) fails to stimulate deadenylation of the wild-type ARE-RNA substrate (Fig. 2B, lanes 4 to 7). The deadenylation observed with FLAG-tagged TTP is the result of a copurifying activity and not due to intrinsic deadenylase activity of TTP itself, as accumulation of the deadenylated band does not occur when the ARE-A60 substrate is incubated with recombinant His-tagged TTP purified from Escherichia coli (C.S., unpublished observations). These observations suggest that TTP copurifies with endogenous deadenylase activity and that the TTP-deadenylase complex specifically deadenylates ARE mRNA.
TTP associates with multiple deadenylases.
Deadenylation in the cytoplasm of eukaryotic cells is thought to be executed by two major deadenylase complexes: the Pan2-Pan3 complex and the Ccr4-Caf1-Not complex (70). We have previously observed that TTP copurifies with the human Ccr4 protein hCcr4b (44). To test whether TTP exists in complex with other deadenylases, transiently expressed Myc-tagged deadenylases were assayed for the ability to coprecipitate with coexpressed FLAG-tagged TTP from RNase-treated HEK293T cell extracts. Figure 3 shows that TTP exists in complex with components of both cytoplasmic deadenylase complexes, including the hCcr4a and hCcr4b paralogs, as well as the hCaf1a and hCaf1b paralogs, of the human Ccr4-Caf1-Not complex, and hPan2 of the human Pan2-Pan3 complex (compare lane 4 with lane 2). Importantly, hnRNP A1, which serves as a negative control, did not copurify with TTP (lane 4) and none of the proteins tested were found in the precipitates in the absence of TTP (lane 2). Similar to previous reports (39, 44), we were unable to detect an association in these assays between the deadenylase PARN and TTP. These observations suggest that TTP exists in complex with multiple deadenylases in cell extracts. While any of these enzymes may be responsible for the deadenylating activity observed from immunopurified TTP (Fig. 2A), the deadenylation assays shown in Fig. 3B and C suggest that the major activity is most consistent with that of Ccr4-Caf1-Not complexes, because in contrast to the Pan2-Pan3 complex (2, 70), the activity is inhibited by PABP (Fig. 3B), and in contrast to PARN (19), the complex is active on an RNA lacking a 5′ cap (Fig. 3C).
FIG. 3.

TTP copurifies with multiple deadenylases. Shown are coimmunoprecipitation assays of Myc-tagged deadenylase proteins with FLAG-tagged TTP (lanes 3 and 4) coexpressed in HEK293T cells. RNase A-treated cell extracts were subjected to anti-FLAG immunopurification (IP), and precipitates were detected via immunoblotting with anti-Myc antibody. Negative immunoprecipitation controls (lanes 1 and 2) were derived from cells expressing no FLAG-tagged proteins. Lanes 1 and 3, 5% of total input fractions (T); lanes 2 and 4, pellet fractions (P). Myc-hnRNP A1, an RNA-binding protein, served as a negative coimmunoprecipitation control. (B) Deadenylase assay performed as described in the legend to Fig. 2 but with the addition of increasing amounts of FLAG-PABPC1 purified from RNase A-treated HEK293T cells. (C) Deadenylase assay using a 5′-32P-monophosphorylated ARE-A60 substrate instead of a cap-labeled substrate. A nonspecific 3′-truncated product derived from the ARE-A60 substrate is indicated by the asterisk. wt, wild type.
Phosphorylation of TTP by MK2 impairs deadenylation in cells.
The activation of the p38 MAPK pathway has been shown to result in the phosphorylation of TTP on two serine residues (S52 and S178) by the kinase MK2, resulting in increased stability of ARE mRNAs (14, 55). However, p38 MAPK and MK2 can affect other AUBPs as well (3, 26, 68). It can therefore be difficult to distinguish the effect of TTP phosphorylation from the phosphorylation of other AUBPs in the cell. In order to determine the consequence of phosphorylation specifically of TTP for its ability to activate deadenylation and decay, we performed tethered pulse-chase mRNA decay assays with cells coexpressing a constitutively active mutant version of the MK2 kinase (MK2EE) or catalytically dead MK2 (MK2KR) (22).
As shown in Fig. 4A, the t1/2 of the β-6bs reporter mRNA tethered to TTP increases 3- to 4-fold when TTP is phosphorylated by constitutively active MK2EE (compare upper two panels). In contrast, kinase-dead MK2KR did not mediate an increase in the mRNA t1/2 (third panel from top). The slower decay rate is due to the phosphorylation of TTP, as a mutant form of TTP in which serines 52 and 178 have been changed to alanines (TTP-AA) is more active than wild-type TTP and is minimally affected by the coexpression of MK2EE (Fig. 4A, lower three panels). These results show that phosphorylation of TTP by MK2 directly impairs its ability to activate the decay of a tethered mRNA.
FIG. 4.

Phosphorylation of TTP by MK2 inhibits TTP-mediated mRNA deadenylation in cells. (A) Northern blot assays showing decay rates of β-6bs in the presence of tethered wild-type TTP (WT) or TTP-AA, in which serines 52 and 178 have been mutated to alanines. Cells were transfected with TTP expression vectors in the presence of no exogenous kinase (−), a constitutively active mutant kinase, MK2EE, or a kinase-dead mutant kinase, MK2KR, as indicated on the left. Calculated β-6bs mRNA t1/2s and average n-fold changes (average of three experiments) in the presence of phosphorylated TTP relative to nonphosphorylated TTP are given on the right. (B) Northern blot assays of mRNA decay assays as in panel A but without (lanes 1 to 7) or with (lanes 8 to 12) coexpression of the catalytically inactive form of the human decapping enzyme hDcp2-E148Q in the presence of MS2-TTP and MK2EE (top panel) or MK2KR (bottom panel).
In addition to an increase in reporter t1/2, we also observed that the deadenylated β-6bs mRNA species fails to accumulate when TTP is phosphorylated by coexpressed constitutively active MK2EE (Fig. 4A, second panel from top). Instead, β-6bs mRNA appears to be deadenylated over time in a more distributive manner. This difference is particularly evident when deadenylated intermediates are trapped upon expression of the catalytically inactive mutant decapping enzyme Dcp2-E148Q (Fig. 4B). In this case, deadenylated mRNA fails to accumulate when TTP is phosphorylated by MK2EE (Fig. 4B, top panel) but not upon the expression of the kinase-dead kinase MK2KR (Fig. 4B, bottom panel, compare lanes 8 to 12 with lanes 1 to 5). These results suggest that phosphorylation of TTP by MK2 strongly impairs its ability to stimulate processive deadenylation of an associated mRNA.
Phosphorylation of TTP does not impair mRNA binding.
The deadenylation and decay of a target transcript by TTP are dependent upon the ability of TTP to bind the mRNA and to recruit decay enzymes (44) (Fig. 2B). Either or both of these steps are candidates for regulation by phosphorylation. The serines phosphorylated by MK2 flank the RNA-binding domain of TTP, thus raising the possibility that phosphorylation could impair its ability to bind the ARE. There are conflicting reports in the literature as to whether phosphorylation of TTP affects RNA binding as measured by the ability of TTP to bind to radiolabeled RNA in vitro (5, 8, 33, 45, 55, 57). We therefore wished to determine whether phosphorylation of TTP affects its ability to associate with mRNA in cells. To do this, Myc-tagged TTP expressed in the presence of the constitutively active kinase MK2EE or the inactive kinase MK2KR was tested for the ability to copurify with poly(A) mRNA isolated by immunopurification of FLAG-tagged cytoplasmic poly(A)-binding protein (PABPC1) from HEK293T cell extracts. As shown in Fig. 5A, no difference in the ability of TTP to associate with poly(A) mRNA was observed between the coexpression of TTP with MK2EE and its coexpression with MK2KR (compare lanes 2 and 5). As expected, no TTP was present in pelleted fractions in the absence of PABPC1 (lanes 1 and 4) or in the presence of RNase A (lanes 3 and 6). Myc-tagged hnRNP A1, which served as a positive RNA-binding control, copurifies with poly(A) mRNA in the absence of RNase, whereas a negative-control fragment of TTP (TTP1-100) lacking the RNA-binding domain does not. These results suggest that MK2-phosphorylated TTP retains its ability to bind to poly(A) mRNA.
FIG. 5.

Phosphorylation of TTP does not impair mRNA binding. (A) Coimmunoprecipitation assays performed with extracts derived from HEK293T cells coexpressing FLAG-tagged cytoplasmic poly(A)-binding protein (PABPC1) (lanes 2, 3, 5, and 6) together with Myc-tagged TTP, hnRNP A1, TTP1-100 (lanes 1 to 6), and MK2EE (lanes 1 to 3) or MK2KR (lane 4 to 6). Immunoprecipitations (IP) were performed in the absence (lanes 1, 2, 4, and 5) or presence (lanes 3 and 6) of RNase A. Lanes 1 and 4 are negative controls from cells expressing no FLAG-tagged protein. Myc-tagged hnRNP A1 served as a positive RNA binding control, whereas the N-terminal domain of TTP (TTP1-100) served as a negative control that does not bind RNA. Pellet and 5% of input fractions were loaded as indicated on the right and subjected to immunoblotting (Western blotting [WB]) with anti-myc or anti-FLAG antibodies as indicated on the left. (B) Coimmunoprecipitation assays as in panel A, except that cells were pretreated with the PP2A inhibitor okadaic acid (OA; lanes 1 to 3) or the vehicle (lanes 4 to 6) 2 h prior to lysate preparation. (C) Northern blot assays of RNA immunoprecipitated from cells coexpressing β-GAP and β-ARE reporter mRNAs together with FLAG-tagged wild-type (WT) TTP (lanes 1 and 2), or mutant versions of TTP with serines 52 and 178 mutated to alanines (AA, lanes 3 and 4) or bearing a point mutation that abolishes RNA binding (F126N, lanes 5 and 6). Prior to immunoprecipitation, cells were treated for 2 h with (+) or without (−) okadaic acid. TTP protein levels are shown in Western blot assays below each Northern blot assay. Upper panels, pellets; lower panels, 10% input. Lanes 7 and 8, immunoprecipitates from cells expressing no exogenous TTP protein.
Although MK2 phosphorylates TTP at serines 52 and 178, this phosphorylation is reversed in cells by the action of PP2A (57). It was therefore possible that even upon coexpression of MK2EE, a fraction of the TTP pool remains unphosphorylated at these sites and that the population of TTP that associated with poly(A) mRNA in Fig. 5A was not phosphorylated. However, as shown in Fig. 5B, TTP copurifies efficiently with poly(A) mRNA even when dephosphorylation of TTP is blocked by the treatment of cells with the PP2A inhibitor okadaic acid (compare lanes 2 and 5). As is evident from the shift in the mobility of TTP observed in Fig. 5B, okadaic acid treatment results in TTP hyperphosphorylation (compare lanes 1 to 3 with lanes 4 to 6). Under these conditions, a small fraction of phosphorylated TTP coimmunoprecipitates with PABPC1, even in the presence of RNase A (Fig. 5B, lane 3), suggesting that hyperphosphorylated TTP forms an RNA-independent interaction with PABPC1. However, the majority of the TTP copurifying with PABPC1 in this experiment is due to mRNA binding, as evidenced by the difference in TTP recovery in the presence or absence of RNase (Fig. 5B, compare lane 3 with lane 2).
The observations in Fig. 5A and B indicate that phosphorylated TTP retains its ability to associate with poly(A) mRNA. We next looked specifically at ARE mRNA. We tested the ability of FLAG-tagged TTP expressed in the presence or absence of okadaic acid to associate with a β-globin reporter mRNA containing the GM-CSF ARE in its 3′ UTR (β-ARE) in HEK293T cell extracts. As shown in the Northern blot assays in Fig. 5C, both hypophosphorylated and hyperphosphorylated TTP can efficiently coimmunoprecipitate the β-ARE reporter mRNA (compare lanes 1 and 2). Similarly, TTP mutated at the MK2 phosphorylation sites (TTP-AA) copurifies the β-ARE mRNA regardless of the presence of okadaic acid (lanes 3 and 4). RNA binding-deficient TTP (F126N, lanes 5 and 6) and empty beads (lanes 7 and 8) served as negative controls that only weakly pulled down the reporter mRNAs. Since okadaic acid treatment affects the stability of both TTP protein and β-ARE mRNA, thereby resulting in variable protein and mRNA input levels in the assays in Fig. 5C (see bottom panels), the association between TTP and β-ARE mRNA could not be quantitatively compared between different conditions in this experiment. Nevertheless, the results shown in Fig. 5, taken together with the observation that phosphorylated TTP is defective in the activation of deadenylation and decay even when tethered to an mRNA (Fig. 4), suggest that MK2-mediated phosphorylation inhibits TTP at a step downstream of mRNA binding.
Phosphorylation of TTP prevents deadenylase recruitment.
Another possible mechanism by which phosphorylation could impair the activation of deadenylation by TTP is by preventing it from recruiting deadenylases. We therefore tested the effect of TTP phosphorylation on deadenylase association by using coimmunoprecipitation assays. In the absence of okadaic acid, wild-type TTP associates with the deadenylases hCcr4b (Fig. 6A, lane 2) and hPan2 (Fig. 6A, lane 6) in RNase-treated cell extracts. However, when TTP is hyperphosphorylated upon treatment with okadaic acid, the association with both deadenylases is strongly diminished (lanes 1 and 7). Thus, hyperphosphorylated TTP is impaired in deadenylase association.
FIG. 6.

Phosphorylation of TTP prevents deadenylase recruitment. (A) Anti-FLAG coimmunoprecipitation assays performed with extracts from HEK293T cells coexpressing FLAG-tagged deadenylase hPan2 or hCcr4b together with Myc-tagged TTP (WT) or the TTP-AA mutant protein as indicated. Cells were treated with the phosphatase inhibitor okadaic acid (OA) (lanes 1, 3, 7, and 10) or the vehicle (lanes 2, 4, 5, 6, 8, and 9). Immunoprecipitates (IP) from cells with no FLAG-tagged protein served as a negative control (lanes 5 and 8). (B) Western blot (WB) assays showing coimmunoprecipitation of myc-tagged TTP (WT or AA mutant) and FLAG-tagged 14-3-3ɛ (lanes 1 to 4). Cells were treated with (lanes 2, 4, 6, and 8) or without (lanes 1, 3, 5, and 7) okadaic acid. Myc-tagged hnRNP A1 (lanes 1 to 8) and immunoprecipitation from cells with no FLAG-tagged protein (lanes 5 to 8) served as negative controls. (C) Western blot assays of coimmunoprecipitation of transiently expressed FLAG-tagged 14-3-3ɛ and Myc-tagged TTP-WT, TTP-AA, or TTP-EE from HEK293T extracts. Pellet fractions (P) and 5% of the total (T) fractions are indicated above the panels. (D) Northern blot assays showing decay rates of β-6bs in the presence of tethered TTP-WT, TTP-AA, or TTP-EE with serines 52 and 178 mutated to negatively charged glutamate residues in an attempt to mimic serine phosphorylation. t1/2s are indicated on the right.
To test the contribution of the serines targeted for phosphorylation by MK2, we tested the ability of the MK2 phosphorylation-resistant TTP-AA mutant protein to associate with deadenylases. Although the TTP-AA mutant protein cannot be phosphorylated by MK2, it does retain a number of other phosphorylation sites present in wild-type TTP (7). Consistent with this, okadaic acid treatment results in the accumulation of hyperphosphorylated TTP-AA (Fig. 6A, lanes 3 and 10; input samples). Similar to wild-type TTP, TTP-AA associates with hCcr4b and hPan2 in the absence of okadaic acid (Fig. 6A, lanes 4 and 9). However, in contrast to wild-type TTP, TTP-AA remains associated with hCcr4b even in the presence of okadaic acid (Fig. 6A, compare lanes 3 and 1). Likewise, enhanced association with hPan2 is observed for TTP-AA compared to wild-type TTP in the presence of okadaic acid (compare lanes 10 and 7), although this association is clearly impaired compared to that in the absence of okadaic acid (compare lane 10 with lane 9). These results demonstrate that hyperphosphorylated TTP is impaired in the ability to recruit deadenylase enzymes and that the serines targeted by MK2 contribute to this impairment, particularly in the case of the deadenylase hCcr4b. The partially reduced affinity of TTP-AA for deadenylases in the presence of okadaic acid indicates that phosphorylations other than those mediated by MK2 may also contribute to the impaired association of phosphorylated TTP with deadenylases. We did not observe clear defects in deadenylase association upon coexpression of TTP with MK2EE (S.L.C., unpublished observation), suggesting either that TTP is not fully phosphorylated by MK2 under those conditions or that additional phosphorylations are important for preventing this association.
Several isoforms of the phospho-serine/threonine binding protein 14-3-3 have been shown to bind TTP in a manner dependent upon the phosphorylation of serine 52 or 178 (14, 55). Although 14-3-3 binding to these phosphorylated residues has been shown to affect the localization and stability of phospho-TTP (35, 57), it is unknown whether 14-3-3 association plays a direct inhibitory role in the decay of TTP target transcripts. To test whether the observed loss of deadenylase recruitment correlates with 14-3-3 binding, we tested the effect of phosphorylation on the association of TTP with the 14-3-3ɛ isoform, which we have previously observed in complex with TTP by mass spectrometry (S.L.C. and J.L., unpublished observations). As expected, TTP-AA associates much more poorly with 14-3-3ɛ than does wild-type TTP (Fig. 6B, compare lanes 3 and 4 to lanes 1 and 2) (55). This inversely correlates with the ability of these proteins to associate with deadenylases (compare Fig. 6A and B). The correlation between 14-3-3 recruitment and inhibition of TTP function is further supported by the observation that mutant TTP protein containing negatively charged glutamates in place of the serines at positions 52 and 178 (TTP-EE) fails to associate with 14-3-3ɛ (Fig. 6C, compare lane 6 to lane 2) and fails to act as a phosphomimetic TTP protein, as it is fully active in the deadenylation and decay of a tethered mRNA (Fig. 6D, bottom panel). Since multiple 14-3-3 paralogs exist in human cells (27), it was not possible to test by depletion of 14-3-3 proteins whether 14-3-3 plays a direct role in inhibiting deadenylase recruitment by phosphorylated TTP. Taken together, our observations demonstrate that the phosphorylation of TTP impairs its ability to recruit deadenylases and activate processive deadenylation of target mRNAs and that this correlates with TTP-14-3-3 association.
DISCUSSION
The control of mRNA turnover plays a critical role in the regulation of gene expression. Cell signaling pathways can influence mRNA decay rates, yet the mechanism by which cell signaling information is communicated to mRNA decay enzymes is poorly understood. In this study, we show that in the case of the ARE mRNA decay pathway, the AUBP TTP activates rapid deadenylation of ARE mRNAs by the recruitment of cytoplasmic deadenylases. Upon activation of the p38 MAPK signaling pathway, the phosphorylation of TTP by the downstream kinase MK2 results in impaired deadenylase recruitment and subsequent stabilization of ARE mRNAs (Fig. 7). Thus, recruitment of deadenylases is a regulated step in ARE mRNA decay.
FIG. 7.

Phosphorylation of TTP impairs target mRNA decay by preventing deadenylase recruitment. TTP activates deadenylation by binding mRNA and recruiting cytoplasmic deadenylases (shown as biting circles). Phosphorylation of TTP by MK2 inhibits deadenylation downstream of mRNA binding by preventing deadenylase recruitment in a manner inversely correlated with binding of the adaptor protein 14-3-3.
TTP activates deadenylation of ARE mRNAs by the recruitment of cytoplasmic deadenylase complexes.
TTP has been previously implicated in the deadenylation of ARE mRNAs (9, 37, 39, 49), but it has remained unknown how TTP stimulates this process. Three major deadenylases have been described in human cells. These include the Ccr4-Caf1-Not complex, the Pan2-Pan3 complex, and PARN (19, 61, 70). Here we show several lines of evidence that the ability of TTP to activate deadenylation depends on its recruitment of cytoplasmic Ccr4-Caf1-Not and Pan2-Pan3 deadenylase complexes to ARE mRNA. First, tethered TTP triggers rapid deadenylation of target RNAs in cells (Fig. 1 and 4). Second, TTP copurifies with deadenylase activity from human cells, and the ability of the TTP-deadenylase complexes to activate deadenylation in vitro is dependent on the presence of an ARE in the RNA substrate and the ability of TTP to bind RNA (Fig. 2). This deadenylase activity is not a property of the TTP protein itself, as recombinant TTP produced in E. coli shows no deadenylation in vitro (C.S., unpublished). Third, exogenously expressed components of both the Ccr4-Caf1-Not and Pan2-Pan3 cytoplasmic deadenylase complexes associate with TTP in an RNA-independent manner (Fig. 3) (44).
Although TTP associates with deadenylase components of both the Ccr4-Caf1-Not and Pan2-Pan3 complexes, the deadenylation activity stimulated by TTP both in cells (Fig. 1 and 4) and in vitro (Fig. 2) is most consistent with that of Ccr4-Caf1-Not complexes (70) in that the activity appears to be processive without deadenylation intermediates (Fig. 1, 2, and 4) and is inhibited by PABPC1 in vitro (Fig. 3B). In contrast, the Pan2-Pan3 complex is known to show distributive activity (70) and to be stimulated by PABPC1 in vitro (61). However, we cannot rule out the possibility that the poly(A) tail of the substrate RNA used in our in vitro assays is too short to observe a stimulating effect due to PABPC1.
A previous report presented evidence that TTP can stimulate deadenylation by PARN in cell extracts, although no interaction between PARN and TTP was observed (39). While our coimmunoprecipitation assays suggest that TTP exists in a complex with components of both the Ccr4-Caf1-Not complex and the Pan2-Pan3 complex, no interaction with PARN was observed (Fig. 3), confirming this previous observation. Consistent with the inability of TTP to copurify with PARN, the deadenylating activity associated with TTP is, in contrast to activity observed in vitro for PARN (19), not dependent upon a 5′ cap (Fig. 3C). An important question for future studies is whether TTP stimulates deadenylation in cells not only through the recruitment of deadenylases but also by remodeling the target mRNP. Such an activity would be consistent with the ability of a deadenylase such as PARN to activate the deadenylation of TTP-associated mRNA when provided in trans.
The recruitment of Ccr4-Caf1-Not deadenylase complexes by RNA-binding proteins is emerging as a general theme in the repression of gene expression. Similar to TTP, yeast Puf proteins and Drosophila Smaug have also been observed to interact with Ccr4 and Caf1 deadenylases and to activate the deadenylation of target mRNAs (30, 31, 51). Other RNA-binding proteins appear to stimulate deadenylation by creating a physical link between PABP and deadenylases. For example, both the antiproliferative protein TOB (24) and the RISC component GW182 (25, 71) appear to stimulate deadenylation by making contacts with both PABP and Caf1 proteins. Thus, the recruitment of deadenylases may be a key step in the activation of mRNA decay in eukaryotic cells.
Phosphorylation of TTP by MK2 inhibits deadenylase recruitment.
How does the phosphorylation of TTP inhibit ARE mRNA decay? While it has been shown previously that phosphorylation of TTP by MK2 is associated with impaired deadenylation and decay of ARE mRNA (8, 55), the mechanism behind this was unknown. Previous observations from other labs using in vitro RNA gel shift experiments have suggested that phosphorylation of TTP negatively affects its ability to bind ARE mRNA in vitro (5, 8, 33), whereas other studies using similar assays have suggested otherwise (45, 55, 57). Our observations are consistent with the latter. First, we did not observe a decrease in the ability of MK2-phosphorylated TTP to bind poly(A) mRNA as measured by its ability to associate with FLAG-tagged PABPC1 in an RNA-dependent manner, nor did we observe a loss of the ability of phosphorylated TTP to bind ARE-containing mRNA (Fig. 5). Second, phosphorylation of TTP by MK2 impairs its ability to activate the deadenylation and decay of an mRNA substrate to which it is tethered (Fig. 4), suggesting that phosphorylation of TTP affects a step downstream of mRNA binding. However, we cannot rule out the possibilities that phosphorylation of TTP may affect the kinetics of mRNA binding in cells and that this may influence decay rates.
Rather than a defect in substrate binding, our observations suggest that the phosphorylation of TTP causes reduced association with Ccr4-Caf1-Not and Pan2-Pan3 deadenylase complexes, as shown by the loss of interaction between TTP with Ccr4b and Pan2 upon okadaic acid treatment (Fig. 6A). This defect is, at least in part, due to the phosphorylation of MK2 target residues S52 and S178 of TTP, as evidenced by the partial restoration of TTP-deadenylase interactions during okadaic acid treatment when TTP is mutated in these residues (TTP-AA) (Fig. 6A). Taken together, these observations suggest that upon activation of the p38 MAPK pathway, the phosphorylation of TTP by MK2 leaves TTP associated with target mRNAs but unable to recruit deadenylase complexes to initiate mRNA decay (Fig. 7). If phosphorylated TTP remains associated with its target transcripts, it may be poised to rapidly reactivate mRNA decay when dephosphorylated upon the attenuation of p38 MAPK signaling. Additionally, phosphorylated TTP may play an active role in protecting the mRNA from decay by preventing other AUBPs from binding the ARE. Future studies should reveal the mechanism by which TTP is dephosphorylated and reactivated on these target transcripts to prevent constitutive upregulation of gene expression after a transient signaling event.
How does phosphorylation of TTP prevent deadenylase recruitment? This could be due to either a decreased affinity of TTP for deadenylase complexes or the creation of a binding site(s) for factors that prevent TTP-deadenylase complex formation. The introduction of negative charges as a result of phosphorylation does not appear sufficient, in and of itself, to inhibit TTP function, as mutation of serines 52 and 178 of TTP to glutamates (TTP-EE, Fig. 6C and D) does not mimic the inhibitory effect of serine phosphorylation. Phosphorylation of TTP by MK2 at serine 52 or 178 induces binding by the adaptor protein 14-3-3 (14, 35, 55) (Fig. 6B). It is known that the association of TTP with 14-3-3 protects TTP from dephosphorylation by PP2A (57). However, because mutations that destroy 14-3-3 binding sites in TTP also prevent its phosphorylation, it is unclear whether 14-3-3 binding is required to inhibit TTP function. Our observations are consistent with the idea that 14-3-3 binding to TTP directly inhibits deadenylase recruitment. First, there is an inverse correlation between the associations of TTP with 14-3-3 and deadenylases (Fig. 6A and B). Second, the TTP-EE mutant, which fails to functionally mimic phospho-TTP (Fig. 6D), also fails to associate with 14-3-3 in coimmunoprecipitation assays (Fig. 6C). While these observations are consistent with the idea that the association of 14-3-3 impairs deadenylase recruitment by TTP, further experiments are needed to determine whether 14-3-3 binding directly competes for deadenylase recruitment.
Do other phosphorylation sites of TTP have similar regulatory effects? TTP has the potential to be phosphorylated at a number of residues by various kinases (6, 7, 13), and inhibition of PP2A results in dramatic hyperphosphorylation of TTP (Fig. 6) (57). Interestingly, hyperphosphorylation of the TTP-AA mutant protein, which cannot be phosphorylated by MK2, partially impairs deadenylase association (Fig. 6A), suggesting that there may be other inhibitory phosphorylation events that regulate TTP function. At least one of these events has been shown to result in the recruitment of 14-3-3 (13) (Fig. 6B). Future studies should reveal whether signaling pathways other than the p38 MAPK pathway use a similar mechanism to inhibit TTP's ability to stimulate the deadenylation and decay of target transcripts. Alternatively, different signaling pathways may affect TTP in different ways, whether by repression or activation of TTP. Other activators of ARE mRNA decay are inhibited by phosphorylation events, including the TTP paralogs BRF-1 and BRF-2, as well as the unrelated AUBPs AUF1 and KSRP (1, 3, 20, 29, 46, 50, 66). As with TTP, phosphorylation of these AUBPs leads to 14-3-3 recruitment, although the mechanistic consequences of this recruitment vary. For example, phosphorylation of KSRP by PI3K-AKT or p38 signaling leads to 14-3-3 association and the inhibition of KSRP's ability to activate mRNA decay by preventing exosome recruitment (20, 29) and mRNA binding (3), respectively. The ability of these phospho-AUBPs to associate with cytoplasmic deadenylases was not tested. An important goal for future studies is to understand in greater detail the mechanisms by which phosphorylation and 14-3-3 recruitment regulate these activators of mRNA decay and to determine whether the nature of the recruited 14-3-3 isoform and/or the identity of the phosphorylated residue to which it is recruited has differential consequences for their function. These studies should also reveal whether the regulated recruitment of deadenylases is a general principle in the regulation of gene expression at the mRNA turnover level.
ADDENDUM IN PROOF
Following acceptance of this paper, Marchese et al. (J. Biol. Chem. 285:27590-27600, 2010) reported in vitro studies supporting the conclusion that phosphorylation of TTP inhibits deadenylase recruitment.
Acknowledgments
This research was supported by grants R01 GM066811 from the National Institutes of Health and RSG-GMC 111896 from the American Cancer Society to J.L. S.L.C. was supported by a postdoctoral fellowship (PF-06-156-01-GMC) from the American Cancer Society.
Footnotes
Published ahead of print on 15 November 2010.
REFERENCES
- 1.Blum, J. L., A. M. Samarel, and R. Mestril. 2005. Phosphorylation and binding of AUF1 to the 3′-untranslated region of cardiomyocyte SERCA2a mRNA. Am. J. Physiol. Heart Circ. Physiol. 289:H2543-H2550. [DOI] [PubMed] [Google Scholar]
- 2.Boeck, R., S. Tarun, Jr., M. Rieger, J. A. Deardorff, S. Muller-Auer, and A. B. Sachs. 1996. The yeast Pan2 protein is required for poly(A)-binding protein-stimulated poly(A)-nuclease activity. J. Biol. Chem. 271:432-438. [DOI] [PubMed] [Google Scholar]
- 3.Briata, P., S. V. Forcales, M. Ponassi, G. Corte, C. Y. Chen, M. Karin, P. L. Puri, and R. Gherzi. 2005. p38-dependent phosphorylation of the mRNA decay-promoting factor KSRP controls the stability of select myogenic transcripts. Mol. Cell 20:891-903. [DOI] [PubMed] [Google Scholar]
- 4.Cao, D., and R. Parker. 2001. Computational modeling of eukaryotic mRNA turnover. RNA 7:1192-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao, H. 2004. Expression, purification, and biochemical characterization of the antiinflammatory tristetraprolin: a zinc-dependent mRNA binding protein affected by posttranslational modifications. Biochemistry 43:13724-13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cao, H., L. J. Deterding, and P. J. Blackshear. 2007. Phosphorylation site analysis of the anti-inflammatory and mRNA-destabilizing protein tristetraprolin. Expert Rev. Proteomics 4:711-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao, H., L. J. Deterding, J. D. Venable, E. A. Kennington, J. R. Yates III, K. B. Tomer, and P. J. Blackshear. 2006. Identification of the anti-inflammatory protein tristetraprolin as a hyperphosphorylated protein by mass spectrometry and site-directed mutagenesis. Biochem. J. 394:285-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carballo, E., H. Cao, W. S. Lai, E. A. Kennington, D. Campbell, and P. J. Blackshear. 2001. Decreased sensitivity of tristetraprolin-deficient cells to p38 inhibitors suggests the involvement of tristetraprolin in the p38 signaling pathway. J. Biol. Chem. 276:42580-42587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carballo, E., W. S. Lai, and P. J. Blackshear. 2000. Evidence that tristetraprolin is a physiological regulator of granulocyte-macrophage colony-stimulating factor messenger RNA deadenylation and stability. Blood 95:1891-1899. [PubMed] [Google Scholar]
- 10.Carballo, E., W. S. Lai, and P. J. Blackshear. 1998. Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science 281:1001-1005. [DOI] [PubMed] [Google Scholar]
- 11.Chen, C. Y., and A. B. Shyu. 1995. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem. Sci. 20:465-470. [DOI] [PubMed] [Google Scholar]
- 12.Chen, C. Y., Y. Yamashita, T. C. Chang, A. Yamashita, W. Zhu, Z. Zhong, and A. B. Shyu. 2007. Versatile applications of transcriptional pulsing to study mRNA turnover in mammalian cells. RNA 13:1775-1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiba, S., M. Tokuhara, E. H. Morita, and S. Abe. 2009. TTP at Ser245 phosphorylation by AKT is required for binding to 14-3-3. J. Biochem. 145:403-409. [DOI] [PubMed] [Google Scholar]
- 14.Chrestensen, C. A., M. J. Schroeder, J. Shabanowitz, D. F. Hunt, J. W. Pelo, M. T. Worthington, and T. W. Sturgill. 2004. MAPKAP kinase 2 phosphorylates tristetraprolin on in vivo sites including Ser178, a site required for 14-3-3 binding. J. Biol. Chem. 279:10176-10184. [DOI] [PubMed] [Google Scholar]
- 15.Clark, A., J. Dean, C. Tudor, and J. Saklatvala. 2009. Post-transcriptional gene regulation by MAP kinases via AU-rich elements. Front. Biosci. 14:847-871. [DOI] [PubMed] [Google Scholar]
- 16.Clement, S. L., and J. Lykke-Andersen. 2008. A tethering approach to study proteins that activate mRNA turnover in human cells. Methods Mol. Biol. 419:121-133. [DOI] [PubMed] [Google Scholar]
- 17.Datta, S., R. Biswas, M. Novotny, P. G. Pavicic, Jr., T. Herjan, P. Mandal, and T. A. Hamilton. 2008. Tristetraprolin regulates CXCL1 (KC) mRNA stability. J. Immunol. 180:2545-2552. [DOI] [PubMed] [Google Scholar]
- 18.Dean, J. L., S. J. Sarsfield, E. Tsounakou, and J. Saklatvala. 2003. p38 Mitogen-activated protein kinase stabilizes mRNAs that contain cyclooxygenase-2 and tumor necrosis factor AU-rich elements by inhibiting deadenylation. J. Biol. Chem. 278:39470-39476. [DOI] [PubMed] [Google Scholar]
- 19.Dehlin, E., M. Wormington, C. G. Korner, and E. Wahle. 2000. Cap-dependent deadenylation of mRNA. EMBO J. 19:1079-1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Díaz-Moreno, I., D. Hollingworth, T. A. Frenkiel, G. Kelly, S. Martin, S. Howell, M. Garcia-Mayoral, R. Gherzi, P. Briata, and A. Ramos. 2009. Phosphorylation-mediated unfolding of a KH domain regulates KSRP localization via 14-3-3 binding. Nat. Struct. Mol. Biol. 16:238-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emmons, J., W. H. Townley-Tilson, K. M. Deleault, S. J. Skinner, R. H. Gross, M. L. Whitfield, and S. A. Brooks. 2008. Identification of TTP mRNA targets in human dendritic cells reveals TTP as a critical regulator of dendritic cell maturation. RNA 14:888-902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engel, K., H. Schultz, F. Martin, A. Kotlyarov, K. Plath, M. Hahn, U. Heinemann, and M. Gaestel. 1995. Constitutive activation of mitogen-activated protein kinase-activated protein kinase 2 by mutation of phosphorylation sites and an A-helix motif. J. Biol. Chem. 270:27213-27221. [DOI] [PubMed] [Google Scholar]
- 23.Eulalio, A., E. Huntzinger, T. Nishihara, J. Rehwinkel, M. Fauser, and E. Izaurralde. 2009. Deadenylation is a widespread effect of miRNA regulation. RNA 15:21-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ezzeddine, N., T. C. Chang, W. Zhu, A. Yamashita, C. Y. Chen, Z. Zhong, Y. Yamashita, D. Zheng, and A. B. Shyu. 2007. Human TOB, an antiproliferative transcription factor, is a poly(A)-binding protein-dependent positive regulator of cytoplasmic mRNA deadenylation. Mol. Cell. Biol. 27:7791-7801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fabian, M. R., G. Mathonnet, T. Sundermeier, H. Mathys, J. T. Zipprich, Y. V. Svitkin, F. Rivas, M. Jinek, J. Wohlschlegel, J. A. Doudna, C. Y. Chen, A. B. Shyu, J. R. Yates III, G. J. Hannon, W. Filipowicz, T. F. Duchaine, and N. Sonenberg. 2009. Mammalian miRNA RISC recruits CAF1 and PABP to affect PABP-dependent deadenylation. Mol. Cell 35:868-880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farooq, F., S. Balabanian, X. Liu, M. Holcik, and A. MacKenzie. 2009. p38 mitogen-activated protein kinase stabilizes SMN mRNA through RNA binding protein HuR. Hum. Mol. Genet. 18:4035-4045. [DOI] [PubMed] [Google Scholar]
- 27.Gardino, A. K., S. J. Smerdon, and M. B. Yaffe. 2006. Structural determinants of 14-3-3 binding specificities and regulation of subcellular localization of 14-3-3-ligand complexes: a comparison of the X-ray crystal structures of all human 14-3-3 isoforms. Semin. Cancer Biol. 16:173-182. [DOI] [PubMed] [Google Scholar]
- 28.Garneau, N. L., J. Wilusz, and C. J. Wilusz. 2007. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 8:113-126. [DOI] [PubMed] [Google Scholar]
- 29.Gherzi, R., M. Trabucchi, M. Ponassi, T. Ruggiero, G. Corte, C. Moroni, C. Y. Chen, K. S. Khabar, J. S. Andersen, and P. Briata. 2006. The RNA-binding protein KSRP promotes decay of beta-catenin mRNA and is inactivated by PI3K-AKT signaling. PLoS Biol. 5:e5. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Goldstrohm, A. C., B. A. Hook, D. J. Seay, and M. Wickens. 2006. PUF proteins bind Pop2p to regulate messenger RNAs. Nat. Struct. Mol. Biol. 13:533-539. [DOI] [PubMed] [Google Scholar]
- 31.Goldstrohm, A. C., D. J. Seay, B. A. Hook, and M. Wickens. 2007. PUF protein-mediated deadenylation is catalyzed by Ccr4p. J. Biol. Chem. 282:109-114. [DOI] [PubMed] [Google Scholar]
- 32.Hau, H. H., R. J. Walsh, R. L. Ogilvie, D. A. Williams, C. S. Reilly, and P. R. Bohjanen. 2007. Tristetraprolin recruits functional mRNA decay complexes to ARE sequences. J. Cell. Biochem. 100:1477-1492. [DOI] [PubMed] [Google Scholar]
- 33.Hitti, E., T. Iakovleva, M. Brook, S. Deppenmeier, A. D. Gruber, D. Radzioch, A. R. Clark, P. J. Blackshear, A. Kotlyarov, and M. Gaestel. 2006. Mitogen-activated protein kinase-activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraprolin expression, stability, and binding to adenine/uridine-rich element. Mol. Cell. Biol. 26:2399-2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ishmael, F. T., X. Fang, M. R. Galdiero, U. Atasoy, W. F. Rigby, M. Gorospe, C. Cheadle, and C. Stellato. 2008. Role of the RNA-binding protein tristetraprolin in glucocorticoid-mediated gene regulation. J. Immunol. 180:8342-8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson, B. A., J. R. Stehn, M. B. Yaffe, and T. K. Blackwell. 2002. Cytoplasmic localization of tristetraprolin involves 14-3-3-dependent and -independent mechanisms. J. Biol. Chem. 277:18029-18036. [DOI] [PubMed] [Google Scholar]
- 36.Khabar, K. S. 2005. The AU-rich transcriptome: more than interferons and cytokines, and its role in disease. J. Interferon Cytokine Res. 25:1-10. [DOI] [PubMed] [Google Scholar]
- 37.Lai, W. S., E. Carballo, J. R. Strum, E. A. Kennington, R. S. Phillips, and P. J. Blackshear. 1999. Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol. Cell. Biol. 19:4311-4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lai, W. S., E. A. Kennington, and P. J. Blackshear. 2002. Interactions of CCCH zinc finger proteins with mRNA: non-binding tristetraprolin mutants exert an inhibitory effect on degradation of AU-rich element-containing mRNAs. J. Biol. Chem. 277:9606-9613. [DOI] [PubMed] [Google Scholar]
- 39.Lai, W. S., E. A. Kennington, and P. J. Blackshear. 2003. Tristetraprolin and its family members can promote the cell-free deadenylation of AU-rich element-containing mRNAs by poly(A) ribonuclease. Mol. Cell. Biol. 23:3798-3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Linker, K., A. Pautz, M. Fechir, T. Hubrich, J. Greeve, and H. Kleinert. 2005. Involvement of KSRP in the post-transcriptional regulation of human iNOS expression-complex interplay of KSRP with TTP and HuR. Nucleic Acids Res. 33:4813-4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lykke-Andersen, J. 2002. Identification of a human decapping complex associated with hUpf proteins in nonsense-mediated decay. Mol. Cell. Biol. 22:8114-8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lykke-Andersen, J., M. D. Shu, and J. A. Steitz. 2001. Communication of the position of exon-exon junctions to the mRNA surveillance machinery by the protein RNPS1. Science 293:1836-1839. [DOI] [PubMed] [Google Scholar]
- 43.Lykke-Andersen, J., M. D. Shu, and J. A. Steitz. 2000. Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell 103:1121-1131. [DOI] [PubMed] [Google Scholar]
- 44.Lykke-Andersen, J., and E. Wagner. 2005. Recruitment and activation of mRNA decay enzymes by two ARE-mediated decay activation domains in the proteins TTP and BRF-1. Genes Dev. 19:351-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mahtani, K. R., M. Brook, J. L. Dean, G. Sully, J. Saklatvala, and A. R. Clark. 2001. Mitogen-activated protein kinase p38 controls the expression and posttranslational modification of tristetraprolin, a regulator of tumor necrosis factor alpha mRNA stability. Mol. Cell. Biol. 21:6461-6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maitra, S., C. F. Chou, C. A. Luber, K. Y. Lee, M. Mann, and C. Y. Chen. 2008. The AU-rich element mRNA decay-promoting activity of BRF1 is regulated by mitogen-activated protein kinase-activated protein kinase 2. RNA 14:950-959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Milligan, J. F., D. R. Groebe, G. W. Witherell, and O. C. Uhlenbeck. 1987. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 15:8783-8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ogilvie, R. L., M. Abelson, H. H. Hau, I. Vlasova, P. J. Blackshear, and P. R. Bohjanen. 2005. Tristetraprolin down-regulates IL-2 gene expression through AU-rich element-mediated mRNA decay. J. Immunol. 174:953-961. [DOI] [PubMed] [Google Scholar]
- 49.Rowlett, R. M., C. A. Chrestensen, M. J. Schroeder, M. G. Harp, J. W. Pelo, J. Shabanowitz, R. DeRose, D. F. Hunt, T. W. Sturgill, and M. T. Worthington. 2008. Inhibition of tristetraprolin deadenylation by poly(A) binding protein. Am. J. Physiol. Gastrointest. Liver Physiol. 295:G421-G430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmidlin, M., M. Lu, S. A. Leuenberger, G. Stoecklin, M. Mallaun, B. Gross, R. Gherzi, D. Hess, B. A. Hemmings, and C. Moroni. 2004. The ARE-dependent mRNA-destabilizing activity of BRF1 is regulated by protein kinase B. EMBO J. 23:4760-4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Semotok, J. L., R. L. Cooperstock, B. D. Pinder, H. K. Vari, H. D. Lipshitz, and C. A. Smibert. 2005. Smaug recruits the CCR4/POP2/NOT deadenylase complex to trigger maternal transcript localization in the early Drosophila embryo. Curr. Biol. 15:284-294. [DOI] [PubMed] [Google Scholar]
- 52.Shyu, A. B., J. G. Belasco, and M. E. Greenberg. 1991. Two distinct destabilizing elements in the c-fos message trigger deadenylation as a first step in rapid mRNA decay. Genes Dev. 5:221-231. [DOI] [PubMed] [Google Scholar]
- 53.Skloot, R. 2010. The immortal life of Henrietta Lacks. Crown Publishers, New York, NY.
- 54.Stoecklin, G., B. Gross, X. F. Ming, and C. Moroni. 2003. A novel mechanism of tumor suppression by destabilizing AU-rich growth factor mRNA. Oncogene 22:3554-3561. [DOI] [PubMed] [Google Scholar]
- 55.Stoecklin, G., T. Stubbs, N. Kedersha, S. Wax, W. F. Rigby, T. K. Blackwell, and P. Anderson. 2004. MK2-induced tristetraprolin:14-3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J. 23:1313-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stoecklin, G., S. A. Tenenbaum, T. Mayo, S. V. Chittur, A. D. George, T. E. Baroni, P. J. Blackshear, and P. Anderson. 2008. Genome-wide analysis identifies interleukin-10 mRNA as target of tristetraprolin. J. Biol. Chem. 283:11689-11699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun, L., G. Stoecklin, S. Van Way, V. Hinkovska-Galcheva, R. F. Guo, P. Anderson, and T. P. Shanley. 2007. Tristetraprolin (TTP)-14-3-3 complex formation protects TTP from dephosphorylation by protein phosphatase 2a and stabilizes tumor necrosis factor-alpha mRNA. J. Biol. Chem. 282:3766-3777. [DOI] [PubMed] [Google Scholar]
- 58.Tchen, C. R., M. Brook, J. Saklatvala, and A. R. Clark. 2004. The stability of tristetraprolin mRNA is regulated by mitogen-activated protein kinase p38 and by tristetraprolin itself. J. Biol. Chem. 279:32393-32400. [DOI] [PubMed] [Google Scholar]
- 59.Tucker, M., M. A. Valencia-Sanchez, R. R. Staples, J. Chen, C. L. Denis, and R. Parker. 2001. The transcription factor associated Ccr4 and Caf1 proteins are components of the major cytoplasmic mRNA deadenylase in Saccharomyces cerevisiae. Cell 104:377-386. [DOI] [PubMed] [Google Scholar]
- 60.Tudor, C., F. P. Marchese, E. Hitti, A. Aubareda, L. Rawlinson, M. Gaestel, P. J. Blackshear, A. R. Clark, J. Saklatvala, and J. L. Dean. 2009. The p38 MAPK pathway inhibits tristetraprolin-directed decay of interleukin-10 and pro-inflammatory mediator mRNAs in murine macrophages. FEBS Lett. 583:1933-1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Uchida, N., S. Hoshino, and T. Katada. 2004. Identification of a human cytoplasmic poly(A) nuclease complex stimulated by poly(A)-binding protein. J. Biol. Chem. 279:1383-1391. [DOI] [PubMed] [Google Scholar]
- 62.Wagner, E., S. L. Clement, and J. Lykke-Andersen. 2007. An unconventional human Ccr4-Caf1 deadenylase complex in nuclear Cajal bodies. Mol. Cell. Biol. 27:1686-1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wakiyama, M., K. Takimoto, O. Ohara, and S. Yokoyama. 2007. Let-7 microRNA-mediated mRNA deadenylation and translational repression in a mammalian cell-free system. Genes Dev. 21:1857-1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang, B., H. Yang, Y. C. Liu, T. Jelinek, L. Zhang, E. Ruoslahti, and H. Fu. 1999. Isolation of high-affinity peptide antagonists of 14-3-3 proteins by phage display. Biochemistry 38:12499-12504. [DOI] [PubMed] [Google Scholar]
- 65.Wang, Z., and M. Kiledjian. 2001. Functional link between the mammalian exosome and mRNA decapping. Cell 107:751-762. [DOI] [PubMed] [Google Scholar]
- 66.Wilson, G. M., J. Lu, K. Sutphen, Y. Sun, Y. Huynh, and G. Brewer. 2003. Regulation of A + U-rich element-directed mRNA turnover involving reversible phosphorylation of AUF1. J. Biol. Chem. 278:33029-33038. [DOI] [PubMed] [Google Scholar]
- 67.Winzen, R., G. Gowrishankar, F. Bollig, N. Redich, K. Resch, and H. Holtmann. 2004. Distinct domains of AU-rich elements exert different functions in mRNA destabilization and stabilization by p38 mitogen-activated protein kinase or HuR. Mol. Cell. Biol. 24:4835-4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Winzen, R., B. K. Thakur, O. Dittrich-Breiholz, M. Shah, N. Redich, S. Dhamija, M. Kracht, and H. Holtmann. 2007. Functional analysis of KSRP interaction with the AU-rich element of interleukin-8 and identification of inflammatory mRNA targets. Mol. Cell. Biol. 27:8388-8400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu, N., P. Loflin, C. Y. Chen, and A. B. Shyu. 1998. A broader role for AU-rich element-mediated mRNA turnover revealed by a new transcriptional pulse strategy. Nucleic Acids Res. 26:558-565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yamashita, A., T. C. Chang, Y. Yamashita, W. Zhu, Z. Zhong, C. Y. Chen, and A. B. Shyu. 2005. Concerted action of poly(A) nucleases and decapping enzyme in mammalian mRNA turnover. Nat. Struct. Mol. Biol. 12:1054-1063. [DOI] [PubMed] [Google Scholar]
- 71.Zekri, L., E. Huntzinger, S. Heimstadt, and E. Izaurralde. 2009. The silencing domain of GW182 interacts with PABPC1 to promote translational repression and degradation of microRNA targets and is required for target release. Mol. Cell. Biol. 29:6220-6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang, T., V. Kruys, G. Huez, and C. Gueydan. 2002. AU-rich element-mediated translational control: complexity and multiple activities of trans-activating factors. Biochem. Soc. Trans. 30:952-958. [DOI] [PubMed] [Google Scholar]