

Abstract
Object
Complement activation has been suggested to play a role in the development of secondary injuries following traumatic brain injury (TBI). The present study was initiated in order to analyze complement activation in relation to the primary brain injury and to secondary insults, frequently occurring following TBI.
Methods
Twenty patients suffering from severe TBI (Glasgow coma score ≤8) were included in the study. The “membrane attack complex,” C5b9, which is the cytolytic end product of the complement system was analyzed in cerebrospinal fluid (CSF). The degree of brain tissue damage was assessed using the release of S100B and neuron-specific enolase (NSE) to the CSF and blood. The blood–brain barrier was assessed using the CSF/serum quotient of albumin (Q A).
Results
Following impact, initial peaks (0–48 h) of C5b9, S100B, and NSE with a concomitant loss of integrity of the blood–brain barrier were observed. Secondary insults at the intensive care unit were monitored. Severe secondary insults were paralleled by a more pronounced complement activation (C5b9 in CSF) as well as increased levels of S100B (measured in CSF), but not with NSE.
Conclusion
This human study indicates that complement activation in the brain is triggered not only by the impact of trauma per se but also by the amount of secondary insults that frequently occur at the scene of accident as well as during treatment in the neurointensive care unit. Complement activation and in particular the end product C5b9 may in turn contribute to additional secondary brain injuries by its membrane destructive properties.
Keywords: Traumatic brain injury, Complement, C5b9, S-100B, NSE, Human, Secondary insults
Introduction
Traumatic brain injury (TBI) accounts for the majority of trauma deaths in Europe today, with an incidence of 235/100,000 and case mortality rate of approximately 11% [39]. Many of the survivors suffer from various severe sequels. The effects of TBI are extensive, not only for the victim, but also for family and friends, and furthermore, the society suffers huge costs for rehabilitation, care, and productivity loss.
The primary brain injury occurs as a result of the energy transmitted to the brain tissue at impact. Following impact, different secondary insults [25] most often occur, such as hypoxemia, hypercarbia, seizures, etc. These secondary insults lead to a disturbed balance between cerebral blood flow (CBF) and cerebral metabolism [23] with a subsequent risk to develop ischemia and thus impair outcome [6]. At autopsy, ischemic injuries have been shown to occur in 80%–90% of victims that succumb following TBI [14]. The outcome is further influenced by other biochemical processes initiated by the trauma. These processes include the release of excitatory amino acids [30] with a subsequent calcium intoxication of the neurons [11], formation of reactive oxygen species [12], activation of neuroinflammation [18, 28] including the complement system [4, 5, 37], and trauma-induced thrombocyte dysfunction [29]. Furthermore, genetic factors influence the pathophysiologic events following TBI. Apolipoprotein E (ApoE) modulates the risk to develop Alzheimer’s disease but also inflict on the prognosis following TBI [45]. Taken together, the prognosis for patients suffering from TBI is due not only to the primary injury but also to the extent of secondary insults and the biochemical processes that the patients suffer from as well as the phenotype of the individual. To counteract these effects of secondary insults, prehospital trauma teams have been organized, trauma-units launched, and neurointensive care units (NICUs) established resulting in improved outcome [33, 38, 44].
The use of markers for tissue damage in the central nervous system, such as S100B and neuron-specific enolase (NSE), has been proposed as potentially useful in order to quantify the extent of the TBI early in the process [16].
The present study aims to analyze complement activation, the tissue damage markers S100B and NSE, the integrity of the blood–brain barrier (BBB), and clinical parameters obtained at the scene of accident (SOA) and at the NICU.
Clinical material and methods
This project was approved by the local ethical committee at the Karolinska University Hospital in Stockholm (No: 01-297 and 03-540). Informed consent was obtained from next of kin. Twenty patients (18 males and 2 female; age, 22–72 years; median age, 59.5 years) suffering from severe TBI (Glasgow coma score [GCS], ≤8) admitted to the neurosurgical intensive care unit at Karolinska University Hospital in Solna (KS) were included (Table 1). Ten of the patients arrived from the SOA, while 10 were admitted from a primary hospital.
Table 1.
Patients
| Patients | SOA | Status | CT | CSF | Serum | BBB | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | Age (y) | Sex | Respiratory | Circulation | Seizures | Etiology | Energy | GCSad | GCSb | Pup | CTM | S100B | NSE | C5b9 | S100B | NSE | Q A | GOS |
| 1 | 70 | M | − | − | − | Fall | Low | 4 | 9 | 0 | 5 | 54 | 27 | 14 | 0.77 | 14 | 6 | 4 |
| 2 | 54 | F | − | − | − | RTA | Low | 8 | 8 | 0 | 5 | 31 | 10 | 8 | 0.9 | 16 | 1 | 4 |
| 3 | 22 | M | − | + | − | Fall >3 m | High | 7 | 7 | 0 | 5 | 23 | 17 | 21 | 1.7 | 27 | 7 | 4 |
| 4 | 67 | M | NK | NK | NK | Fall | Low | 7 | 8 | 0 | 5 | 153 | 58 | 124 | 0.39 | 13 | 72 | 3 |
| 5 | 59 | M | + | − | (+) | Fall | Low | 7 | 9 | 0 | 5 | 50 | 52 | 110 | 0.84 | 13 | 59 | 4 |
| 6 | 31 | M | − | − | − | Assault | Low | 8 | 8 | 0 | 2 | 61 | 154 | 54 | 0.97 | 22 | 16 | 4 |
| 7 | 60 | M | − | − | − | RTA | Low | 3 | 3 | 0 | 5 | 214 | 55 | 38 | 0.93 | 15 | 10 | 1 |
| 8 | 68 | M | + | − | + | Fall >3 m | High | 7 | 7 | 2 | 2 | 186 | 282 | 186 | 1.3 | 15 | 92 | 3 |
| 9 | 24 | M | + | − | + | RTA | High | 3 | 4 | 2 | 4 | 1,058 | 620 | 44 | 2.1 | 26 | 21 | 1 |
| 10 | 72 | F | − | NK | − | Fall | Low | 8 | 14 | 0 | 5 | 574 | 196 | 380 | 1.4 | 17 | 75 | 4 |
| 11 | 60 | M | NK | − | NK | Fall | Low | 5 | 9 | 1 | 5 | 50 | 21 | 80 | 0.17 | 17 | 7 | 3 |
| 12 | 66 | M | − | NK | − | Fall | Low | 7 | 10 | 0 | 5 | 226 | 52 | 29 | 0.3 | 9,5 | 36 | 3 |
| 13 | 65 | M | + | − | (+) | Fall | Low | 3 | 10 | 1 | 5 | 94 | 28 | 152 | 0.37 | 9,9 | 33 | 1 |
| 14 | 30 | M | − | − | + | Fall | Low | 7 | 8 | 0 | 5 | 207 | 56 | 113 | 0.35 | 10 | 13 | 5 |
| 15 | 50 | M | − | − | − | RTA | High | 8 | 8 | 0 | 2 | 121 | 39 | 25 | 0.5 | 9,9 | 7 | 3 |
| 16 | 64 | M | NK | NK | + | Fall | Low | 6 | 6 | 0 | 5 | 64 | 14 | 67 | 0.48 | 9,4 | 14 | 1 |
| 17 | 65 | M | − | + | − | Assault | Low | 3 | 7 | 0 | 5 | 24 | 13 | 9 | 0.6 | 13 | 5 | 3 |
| 18 | 41 | M | + | NK | − | RTA | High | 8 | 8 | 0 | 6 | 566 | 1,273 | NK | 0.4 | 12 | 35 | 3 |
| 19 | 48 | M | − | − | + | Fall | Low | 7 | 7 | 1 | 5 | 150 | 328 | 161 | 1.7 | 17 | 48 | 3 |
| 20 | 40 | M | − | − | (+) | Fall >3 m | High | 3 | 10 | 0 | 5 | 198 | 138 | 44 | 0.51 | 13 | 19 | 4 |
SOA scene of accident; Verified +, suspected (+), or absent (−) respiratory, circulatory insufficiency or ongoing seizure; NK not known; RTA road traffic accident; High high impact; Low low impact; GCSad Glasgow coma score at admission; GCSb best GCS registered within first 24 h; Pup Pupil reaction to light; CT M CT performed before operation scored ad modum Marshall; CSF cerebrospinal fluid; BBB blood–brainbarrier; Q A csf-albumin/s-albumin ratio; CSF/Serum/BBB, C5b9, S100B, NSE, and Q A , highest measured values (0–48 h posttrauma) defined as initial peak values; GOS Glasgow outcome score 3–12 months after admission trauma; F female; M male. 0 = normal; 1 = unilateral nonreactive; 2 = bilateral nonreactive
Patients presenting bilateral wide nonreacting pupils and GCS 3 judged by the neurosurgeon on call to be beyond salvage, or patients unconscious mainly due to drug or alcohol intoxication, as well as patients presenting terminal illness or for some other reason judged not to be possible for follow up were excluded from the study.
Fifteen of the patients were subjected to evacuation of mass lesions and/or hemicraniectomy. All 20 patients had an intraventricular catheter inserted for continuous intracranial pressure (ICP) monitoring. Zero-point for ICP, systolic arterial blood pressure (SAP), and mean arterial blood pressure (MAP) was the temple. All patients were treated in 30° sitting position. Following surgery, the patients were treated at the NICU. All patients except three were suffering from isolated TBI. Patient 6 suffered from an additional pneumothorax, patient 8 suffered from costal fractures and a lung contusion, and patient 9 suffered from a C3 fracture.
Occurrences of secondary insults at the SOA were obtained from the prehospital trauma team charts. Respiratory insufficiency was defined as SpO2 below 85% or notes in the chart indicating “insufficient breathing,” “cyanosis,” etc. Insufficient circulation was defined as a SAP <90 mm Hg. Glasgow coma score (GCS) and pupil reaction were documented at admission (GCSad), and the best GCS within 24 h following admission (GCSb) was registered (Table 1).
During ventilator treatment, all patients were sedated using midazolam and/or propofol together with infusion or intermittent administration of Morfine. Intractable intracranial hypertension was treated using intermittent cerebrospinal drainage, moderate hyperventilation, and barbiturate coma.
All patients performed at least one initial computed tomography (CT) and a subsequent follow-up within the first 24 h posttrauma. CT scans were classified ad modum Marshall [22] (Table 1).
All patients were monitored online using a computerized surveillance system, intensive care unit (ICU) pilot (CMA Microdialysis, Solna, Sweden). The following values exceeding a duration of 5 min were defined as secondary insults at the NICU; ICP >20 mm Hg, cerebral perfusion pressure (CPP) <60 mm Hg, MAP <70 mm Hg or MAP >110 mm Hg, SAP <90 mm Hg or SAP >160 mm Hg, saturation (SaO2) <90%, pulse <50 beats/min or >120 beats/min, and temperature >38°C. The insults were graded as mild, moderate, or severe (Table 2).
Table 2.
Secondary insults at the NICU
| Mild | Moderate | Severe | ||
|---|---|---|---|---|
| Intracranial hypertension | ICP (mm Hg) | 20–30 | 30–40 | >40 |
| Poor cerebral perfusion | CPP (mm Hg) | 50–60 | 40–50 | <40 |
| Hypotension | MAP (mm Hg) | 55–70 | 40–55 | <40 |
| Hypotension | SAP (mm Hg) | 70–90 | 50–70 | <50 |
| Hypertension | MAP (mm Hg) | 110–130 | 130–150 | >150 |
| Hypertension | SAP (mm Hg) | 160–190 | 190–220 | >220 |
| Hypoxia | SpO2 (%) | 85–90 | 80–85 | <80 |
| Bradycardia | bpm | 40–50 | 30–40 | <30 |
| Tachycardia | bpm | 120–135 | 135–150 | >150 |
| Pyrexia | °C | 38–39 | 39–40 | >40 |
ICP intracranial pressure; CPP cerebral perfusion pressure; MAP mean arterial blood pressure; SAP systolic arterial blood pressure; SpO 2 oxygen saturation; bpm beats per minute
Except for standard laboratory test procedures, additional samples of CSF and blood were obtained at admission and every morning for analysis of S-100B, NSE, and albumin. The soluble form of the complement systems cytolytic terminal membrane attack complex, C5b9, was analyzed in daily samples of CSF, as a marker for CNS complement activation. S100B and NSE were analyzed using commercially available chemiluminometric immunoassays (Liaison Sangtec 100 and Liaison Sangtec NSE; DiaSorin, Salugia, Italy). The complement compound C5b9 was analyzed using enzyme immunoassay (Quidel A009, Quidel CA. USA).
The levels in CSF of soluble C5b9 has been evaluated in healthy controls and found to be ranging between not detectable levels up to 12 μg/mL [37]. From seven orthopedic patients anesthetized using spinal anesthesia, we change a range from csf-S100B between 0.86 and 2.4 μg/L (mean ± SD, 1.29 ± 0.55) and csf-NSE 5.2 and 14.0 μg/L (mean ± SD, 8.10 ± 3.2). Each patient was followed as long as ventilator treatment and ICP monitoring proceeded.
Traumatic disruption of the BBB leads to a leakage of plasma proteins from blood to the cerebral parenchyma [42]. BBB integrity was measured as the ratio of CSF–albumin (mg/L)/serum–albumin (g/L) (Q A) [41]. A ratio below 7 was regarded as normal [20, 41].
Outcome was assessed using Glasgow outcome scale (GOS) at discharge, and following 3 months to 1 year after trauma [31, 40] (Table 1). Disability Rating Scale [32], Functional Independent Score [7], and Mini-Mental State Examination [9] were used 3 months to 12 months posttrauma.
The data were collected online to a computerized patient surveillance system, ICU pilot (CMA Microdialysis, Stockholm, Sweden) and analyzed statistically using the Mann–Whitney U-test or regression analysis (Statistica for Windows 8.0; StatSoft, Tulsa, OK). For Mann–Whitney U-test, p < 0.05 was used as significance. For regression analysis, p < 0.05 and R 2 > 0.20 were used as significance.
Results
Primary brain injury
Following impact, initial peaks were found in CSF, defined as the highest obtained value 0–48 h postinjury, of C5b9 (csf-C5b9), S100B (csf-S100B), NSE (csf-NSE), and Q A (Fig. 1a–d; Table 1) as well as the corresponding serum levels of S100B (s-S100B) and NSE (s-NSE) were noted. The highest initial peak values within 48 h after trauma were csf-S100B 23–1,058 μg/L (median, 136 μg/L), csf-NSE 10–1,273 μg/L (median, 54 μg/L), and csf-C5b9 8–380 ng/L (median, 54 ng/L) (Table 1). The highest initial peak value within 48 h of s-S100B ranged between 0.17 and 2.10 μg/L (median, 0.83 μg/L). The corresponding level for s-NSE was 9.40–27.00 μg/L (mean, 14.94; median, 13.50 μg/L).
Fig. 1.
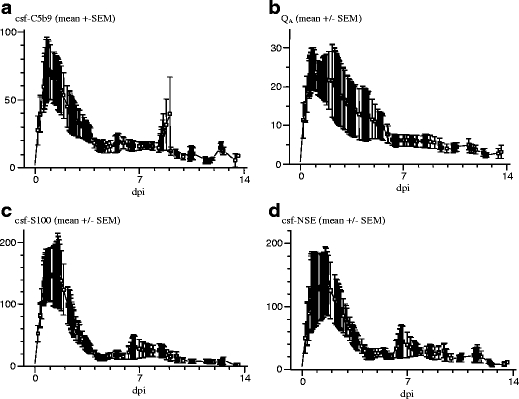
Temporal pattern of the CSF levels for C5b9 and of the tissue damage markers S100b and NSE as well as the temporal pattern for BBB integrity (Q A; [normal value for Q A <7]). a Mean values (±SEM) of C5b9. b Blood–brain barrier, Q A. c csf-S100B. d csf-NSE from patients treated at the NICU due to TBI. Note the initial peak in C5b9, initial loss of BBB integrity (Q A), and initial peaks in csf-S100B and csf-NSE and their subsequent development during the ICU stay. Y-bar: ng/L for C5b9, μg/L for csf-S100B and csf-NSE. dpi days postinjury
No correlations were found between the tissue damage markers or csf-C5b9 versus energy at impact, occurrence of additional extracranial injuries, GCS at admission or best GCS within the first 24 h, surgical procedures, or outcome.
There was a significant positive correlation between csf-NSE and csf-S100B (R 2 = 0.46, p < 0.005; Fig. 2a) as well as between s-NSE and s-S100B (R 2 = 0.63, p < 0.001; Fig. 2b). There was a significant positive correlation also between s-S100B and csf-S100B (R 2 = 0.20, p < 0.05; Fig. 2c), which was not found between s-NSE and csf-NSE (R 2 = 0.02, p = 0.55; not shown). Serum levels of NSE, but not csf-NSE, inversely correlated to age (R 2 = 0.32, p = 0.01; not shown).
Fig. 2.
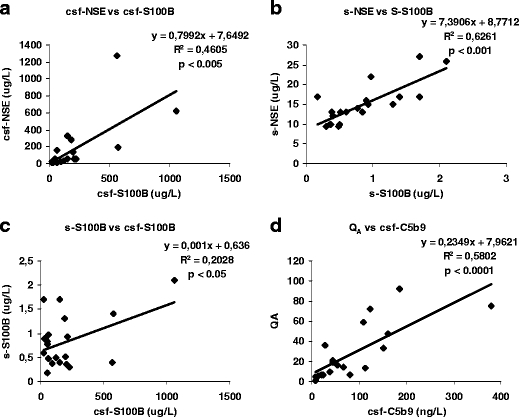
a In CSF, there is a significant correlation between the peak levels of csf-NSE and csf-S100B within 48 h after trauma (R 2 = 0.4605, p < 0.005). b In serum, there is a significant correlation between the peak levels of s-NSE and s-S100B within 48 h after trauma (R 2 = 0.6261, p < 0.001). c The peak levels of S100B in CSF and serum correlate (R 2 = 0.2028, p < 0.05). d Loss of BBB integrity (Q A) and the levels in CSF of the terminal complement complex C5b9 correlate significantly (R 2 = 0.5802, p < 0.0001).
Blood–brain barrier
The initial peak values for S100B and NSE were analyzed versus Q A. There were no correlations between either csf-S100B or s-S100B and disturbed BBB integrity (Q A) as well as between csf-NSE or s-NSE and Q A. To analyze the influence of disturbed BBB integrity on the serum levels of the tissue damage markers, each csf-S100B was multiplied with the corresponding Q A. There was no correlation between csf-S100B × Q A and s-S100B (R 2 = 0.16, p = 0.08; not shown), nor between csf-NSE × Q A and s-NSE (R 2 = 0.003, p = 0.82; not shown). There was no correlation between the initial peak values for csf-C5b9 and csf-S100B or between csf-C5b9 and csf-NSE, but there was a correlation between csf-C5b9 and Q A (R 2 = 0.58, p < 0.0001; Fig. 2d).
Secondary insults at SOA
Five patients presented insufficient respiration on the SOA and two patients presented unstable circulation with a SAP ≤90 mm Hg. Eight patients presented epileptic seizures or “suspect” seizures (Table 1). None of the patients were subjected to cardiorespiratory resuscitation at the SOA, during transport, at the primary hospital, the trauma unit, or at the NICU. All together, secondary insults at the SOA were verified in 11 of 20 patients. Increased levels of C5b9 were found in patients presenting at least one secondary insults at the scene of trauma (p = 0.05; Fig. 3a). These patients also presented a significant disturbed BBB integrity (p < 0.05; Fig. 3b). The levels of csf-S100B and csf-NSE were also increased in patients presenting secondary insults at the SOA, but were not statistically significant.
Fig. 3.
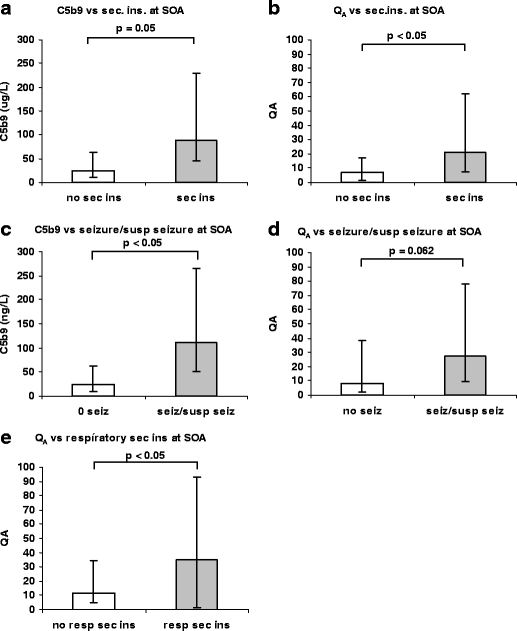
a There was an increase in the initial peak values for C5b9 in the 11 patients where at least one secondary insult was verified at the SOA in comparison with the five patients without secondary insults (Mann–Whitney U-test, p < 0.05). The remaining four patients with uncertain history of early secondary insults are not included in this analysis. b A statistically significant increased loss of integrity of the BBB, assessed by the initial peak values for Q A, was found when comparing the group of 11 patients with at least one secondary insults at the SOA, compared with the 5 patients without secondary insults (Mann–Whitney U-test, p < 0.05). c The complement complex C5b9 in CSF was significantly higher among patients presenting seizures or “suspected” seizures (n = 8) compared with patients with no seizures (n = 10) at the SOA (Mann–Whitney U-test, p < 0.05). d The patients suffering from seizures or suspected seizures at the SOA also presented a trend for impaired BBB integrity compared with patients without seizures (Mann–Whitney U-test, p = 0.062). e Patients presenting respiratory insults (n = 5) at the SOA presented more disturbed BBB integrity (Mann–Whitney U-test, p < 0.05) than patients with no respiratory secondary insults at SOA (n = 12). Sec ins secondary insults; SOA scene of accident. Bar: first to third quartile
When specifying type of secondary insult into “respiratory insult,” “circulatory insult,” or “seizure/suspect seizure,” we found that “seizures/suspected seizures” at SOA correlated to csf-C5b9 (p < 0.05; Fig. 3c) and a trend for disturbed BBB integrity Q A (p = 0.062; Fig. 3d). “Respiratory insults” at the SOA correlated to disturbed BBB integrity, Q A (p < 0.05; Fig. 3e).
Pupil reaction
Fifteen patients presented normal pupil reactions to light stimulation at admission. The remaining 5 patients presented unilateral or bilateral nonreacting pupils. There was no significant correlation between S100B, NSE, or Q A between the groups. However, a significant increase in csf-C5b9 was found in patients presenting nonresponsive pupils (p < 0.05; Fig. 4).
Fig. 4.
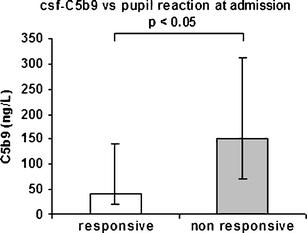
Five of 20 patients presented unilateral or bilateral nonreactive pupils at admission. Patients presenting unilateral or bilateral nonreactive pupils showed statistically significant higher peak values of C5b9 in CSF in comparison with the patients presenting normal pupil reaction (Mann–Whitney U-test, p < 0.05). SOA scene of accident. Bar: first to third quartile
CT classification ad modum Marshall
CT classification ad modum Marshall (CTM; Table 1) was performed on the first CT scan. When several CT scans preceded surgery, the CT obtained prior to operation was used for classification. The majority of patients (14 of 20) presented CTM grade 5 with hematomas that was surgically evacuated. One patient presented CTM grade 6. Four patients (patients 6, 8, 9, and 15) presented diffuse injuries (3 patients CTM grade 2 and one patient CTM grade 4).
By comparing the group of patients with diffuse injuries (CTM grades 1–4) versus patients with focal injuries (CTM grades 5–6), there were somewhat higher values for S100B and NSE in CSF in the group presenting diffuse injuries, but not statistically significant.
Secondary insults at NICU
The total monitored time possible to analyze was 2,520 h. The levels of the tissue damage markers and the complement protein C5b9 were analyzed in relation to the occurrences of secondary insults during the NICU stay registered by the ICU pilot system. To become registered as a secondary insult, the value had to be outside normal range for at least 5 consecutive minutes. The magnitude of these insults was graded into mild, moderate, and severe (Table 2).
The monitoring data were divided into 24-h periods. All 24-h periods where CSF was not possible to obtain, due to dysfunctional ventricular drains or slit ventricles, were excluded as well as all 24-h periods with insufficient monitoring data concerning the occurrence of secondary insults. A total number of twenty-one 24-hour periods were omitted. Furthermore, patient no 11 (Table 1) was excluded from this part of the study, as he was not monitored for more than the first 24 h. Patient 20 (Table 1) was excluded because of loss of data concerning vital parameters due to a computer error. The total number of 24-h periods possible to evaluate was 104. The number of individuals that completed each 24-h period is shown in Fig. 5a. The temporal distribution of registered moderate and severe secondary insults is shown in Fig. 5b. No vital parameters have been registered to the computer system while patients have been on operation or transported to the radiological department for CT or magnetic resonance imaging scans.
Fig. 5.
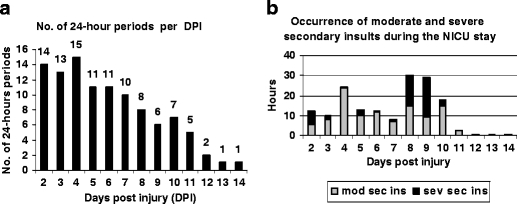
a Number of completed 24-h periods per DPI during the NICU period, from which the data of secondary insults and levels of S100B, NSE, and C5b9 are derived from. The number of completed 24-h periods decreases with the duration of the period at the ICU. b The occurrence of moderate and severe secondary insults over time during the NICU stay. DPI days postinjury; mod-sev sec ins moderate and/or severe secondary insults; sev sec ins severe secondary insults
As CPP is dependent on ICP and MAP, CPP was excluded as secondary insult and analyzed separately. All one hundred four 24-h intervals were analyzed as independent parameters using regression analysis.
The most prominent secondary insult was mild intracranial hypertension (ICP, 20–30 mm Hg) with a total time of 30 027 min, followed by mild pyrexia (temperature, 37–38°C) 25,501 min and mild poor CPP (50–60 mm Hg) for 12,147 min.
Correlations were found between csf-S100B and csf-NSE (R 2 = 0.3823, p < 0.001; Fig. 6a) and between s-S100B and s-NSE (R 2 = 0.4635, p < 0.001; Fig. 6b). The relation between serum and CSF levels for S100B (R 2 = 0.0269, p = 0.0998; not shown) did not correlate, but for NSE, it did but with a poor R 2-value (R 2 = 0.1377, p < 0.001; not shown).
Fig. 6.
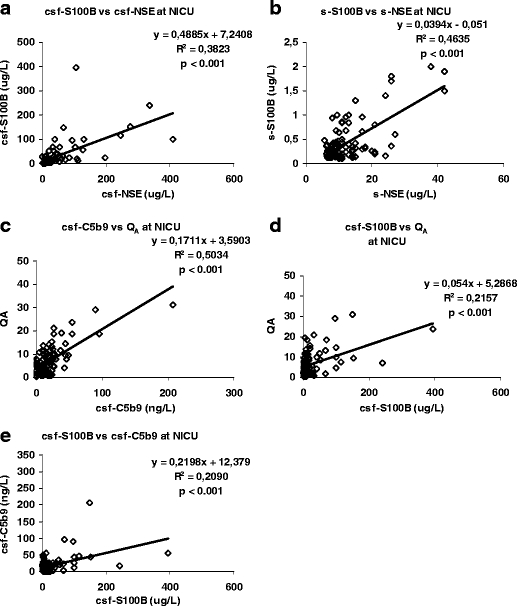
a There is a correlation between the two tissue damage markers S100B and NSE in CSF during the NICU stay (R 2 = 0.3823, p < 0.001). b A correlation between S100B and NSE is also found in serum during the NICU stay (R 2 = 0.4635, p < 0.001). c The complement complex C5b9 correlates to loss of BBB integrity during the NICU stay (R 2 = 0.5034, p < 0.001). d The loss of BBB integrity also correlates to the levels of S100B in CSF (R 2 = 0.2157, p < 0.001). e The levels of the tissue damage marker S100B correlate to the complement complex C5b9 in CSF during the NICU stay (R 2 = 0.2090, p < 0.001).
A correlation between csf-C5b9 and disturbed BBB integrity, Q A, was found (R 2 = 0.5034, p < 0.001; Fig. 6c). Furthermore, similar correlations were found between csf-S100B and Q A (R 2 = 0.2157, p < 0.001; Fig. 6d), csf-S100B and csf-C5b9 (R 2 = 0.2090, p < 0.001; Fig. 6e), but not between csf-NSE and csf-C5b9.
The 104 periods of 24 h were divided into “group A”: No moderate or severe secondary insults, “group B”: <1 h of moderate and/or severe secondary insults, “group C”: 1–2 h of moderate and/or severe secondary insults, “group D”: 2–3 h of moderate and/or severe secondary insults, and “group E”: >3 h of moderate and/or severe secondary insults. The data were analyzed using Mann–Whitney U-test. There were no differences in tissue damage markers between “group A” and “group B” or “group C.” Between “group A” and “group D,” a statistically increased level of C5b9 was found in “group D” (p < 0.05; Fig. 7a). Between “group A” and “group E,” statistically significant increased levels for C5b9 (p < 0.01; Fig. 7a), csf-S100B (p < 0.01; Fig. 7a), and Q A (p < 0.05; Fig. 7b) were found.
Fig. 7.
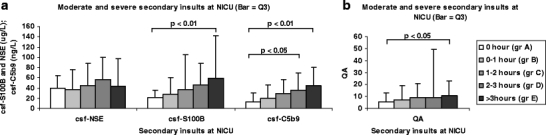
a There are increasing levels of S100B and C5b9 in CSF with increasing incidences of moderate and/or severe secondary insults during the NICU stay. Statistical significance was achieved for csf-S100B when moderate and/or severe secondary insults occurred more than 3-h /24-h period (p < 0.05). The complement complex C5b9 was statistically increased already when moderate and/or severe secondary insults occurred 2–3 h (p < 0.05) and was even stronger when secondary insults occurred more than 3 h (p < 0.01). b The integrity of BBB was significantly impaired when moderate and/or severe secondary insults occurred more than 3 h at the NICU stay (p < 0.05). Bar: third quartile
No correlation between secondary insults and NSE in CSF was observed. CPP as a solely secondary insult did not correspond to tissue damage markers or C5b9.
Outcome
No correlation was found between outcome (GOS, Disability Rating Scale, or Functional Independent Score) and early (0–48 h) or late peak levels (>24 h) of C5b9, S100B, NSE, or Q A. No correlation was found between secondary insults at the NICU and outcome. Of the four patients that died following TBI, one presented a huge subdural hematoma, two presented large contusions, and the fourth patient suffered from a severe diffuse TBI (CTM grade 4) as a result of a high-energy impact. This patient presented the highest value of S100B obtained in serum (2.1 μg/L) as well as in CSF (1,058 μg/L).
Discussion
The present study demonstrates an increase of csf-C5b9 in CSF following TBI, paralleled by an increase of the tissue damage markers S100B and NSE. The finding of increased csf-C5b9 from patients suffering from TBI is an indirect evidence for activation of the complement system in line with previous studies [4, 37].
C5b9 has been shown to accumulate at the surface of neurons located in the border zone in the immediate vicinity to contusion injuries [4]. Complement activation has the ability to play a significant role in the development of secondary brain injuries mediated by the cytolytic effects of C5b9 [26].
The activation of complement has been regarded to be initiated by impact, but in the present study, the occurrence of secondary insults at the SOA, as well as at the NICU stay, seems to intensify the complement activation and, furthermore, leads to a more pronounced loss of BBB integrity.
A weakness in assessing BBB integrity using Q A is that patients suffering from TBI sometimes exhibit intraventricular hemorrhage. Intraventricular hemorrhage implies albumin content in the CSF, not originating from a disintegrated BBB itself but from injured vessels, a fact that might disturb the efficacy of Q A to assess BBB integrity.
S100B is a protein, which in the CNS is present in astrocytes, and the increase in CSF following TBI may reflect a release when these cells degenerate in the primary lesion area. However, an increased synthesis in hypertrophic astrocytes in the glial scar that gradually develops in the border zone may contribute to the increased levels of this protein as well. In peripheral tissues, S100B is present also in other cells, e.g., Schwann cells in peripheral nerves [35], fat cells, muscle, and marrow in mediastinal blood [2]. Elevated S100B concentrations in serum immediately after multitrauma have been shown to be difficult to interpret because of extracerebral contributions [1]. However, S100B in CSF should mainly and probably exclusively represent the CNS tissue and more specifically the astrocytes.
NSE is a “neuron specific enzyme,” albeit found in extracerebral sources such as the erytrocytes. NSE has been shown to increase significantly with hemolysis [13]. The increase in CSF found in this study was interpreted as a result from leaking neurons due to membrane destructions caused by either primary (mechanical) or secondary (neuroinflammatory) mechanisms.
The serum levels of S100B and NSE did not correlate to the BBB integrity, neither at SOA, nor during the NICU period. The levels of S100B in CSF and serum correlate, but the corresponding serum levels of S100B do not correlate to the product of csf-S100B × Q A. Thus, BBB integrity seems to be of a minor importance when assessing the extent of the primary brain injury using serum levels of S100B and NSE.
One would expect that a more profound traumatic impact would release more S100B and NSE, but when the patients in this study were grouped into high- versus low-energy trauma at impact, no differences in initial peaks of S100B or NSE were detected between the groups. Still, to analyze the mechanism of impact only from information gained by the charts from the prehospital trauma teams is an uncertain method. All patients except three were suffering from isolated TBI. By excluding these patients and recalculate, there were still no correlations between Q A and serum levels of S100B or NSE, indicating that extracerebral sources of the tissue damage markers were not a significant explanation for the mismatch with impaired BBB integrity.
Furthermore, the majority of the patients in the present study suffered from focal lesions, why conclusions concerning strictly diffuse brain injuries should be drawn with caution.
In the present study, diffuse injuries on CT at admission represented by CTM grades 2–4 showed no significant difference in levels of tissue damage markers compared with patients presenting more focal lesions (CTM grades 5–6). In a recent study, serum levels of NSE was found to correlate significantly with the injury severity score and CT findings [46]. The serum levels of NSE as well as S100B also have been shown to significantly correlate with the volume of contusions [17]. Those findings could not be verified in our study.
There are no valid methods available to quantify the degree of tissue damage following diffuse, focal, or combined TBI. However, a combination of clinical examination, magnetic resonance imaging, and an increased knowledge of various tissue markers like S100B, NSE, and C5b9 and their dynamics following TBI might be useful in the future in order to estimate the long-term prognosis for the neurotraumatized patient.
Secondary insults (Table 2) have been argued to cause ischemic lesions in the contusion border zones or globally in the brain, eventually contributing to the development of secondary brain injuries, and thus worsen the outcome [19]. In this study, we found more pronounced complement activation (csf-C5b9) as well as increased levels of csf-S100B in patients where more secondary insults were detected during the ICU period. These findings suggest that complement activation is further triggered by such secondary insults, which in turn may induce secondary neuronal injuries by the effect of C5b9. One would expect NSE to mimic the S100B pattern; however, the statistical data in this study failed to confirm this, perhaps due to a too low number of observations.
The correlation between initial peaks of csf-C5b9 and Q A indicates that neuroinflammatory mechanisms are involved in the loss of integrity of the BBB that occurs following TBI. These findings are congruent with a previous study [37]. Cytokines have been shown to have a potential impact on BBB integrity by causing an increasing permeability in cultured endothelial cells [8, 24]. Tumor necrosis factor (TNF) α contributed to loss of BBB integrity following TBI [27] and has been shown to have an influence on the complement system [36]. Stimulation by TNF-α of rat astrocytes resulted in elevation of C3 mRNA [34] and treatment of adult human astrocytes in vitro with TNF-α increased mRNA and protein of complement C3 [3]. A recent animal study has shown that microglial activation and increased synthesis of the complement component C1q preceded loss of BBB integrity in rats [21].
Based on such findings, one may argue that anti-inflammatory therapy should have effect on brain edema related to the loss of BBB integrity, which, however, is a question that remains obscure and need to be further addressed. Even though steroids are known to reduce the inflammatory response after TBI [43], a randomized placebo-controlled trial of intravenous corticosteroid in adults with head injury (the CRASH study) showed an increased mortality in humans treated with steroids [10].
Patients suffering from long-term neuropsychological impairment have been shown to present significantly higher serum concentrations of NSE as well as a more prolonged release of NSE than patients presenting no neuropsychological impairment [15]. In the present study, no correlation between tissue damage markers, BBB integrity, and outcome was found.
Conclusion
The complement system does not only become activated by the trauma per se but also appears to be further triggered by secondary insults at the SOA as well as at the NICU stay. The study underscores the importance of meticulous monitoring of TBI patients in the ICU in order to rapidly recognize, identify, and treat secondary insults aiming at minimizing secondary brain damages. The relation between secondary insults and neuroinflammation has to be further explored aiming to find directed treatment strategies. The serum levels of the tissue damage marker S100B do not seem to be overwhelmingly influenced by the loss of BBB integrity, indicating that S100B may reflect the true cellular damage following TBI.
Acknowledgements
The study was approved by the local ethical committee (01-297; 03-540). The staff at NICU, especially Pia Svensson, R.N., and Birgitta Ohlgren, R.N., are gratefully acknowledged for their everlasting enthusiasm and help concerning the present project. Mrs. Rumjana Dijlai-Merzoug is greatly acknowledged for excellent laboratory technical assistance. Magnus Backheden and Margaretha Krook are acknowledged for their help and guidance concerning the statistical analysis. This study was funded by the Stockholm county and the Swedish Medical Research Council.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Conflicts of interest
None.
References
- 1.Anderson RE, Hansson LO, Nilsson O, Dijlai-Merzoug R, Settergren G. High serum S100B levels for trauma patients without head injuries. Neurosurgery. 2001;48:1255–1258. doi: 10.1097/00006123-200106000-00012. [DOI] [PubMed] [Google Scholar]
- 2.Anderson RE, Hansson LO, Nilsson O, Liska J, Settergren G, Vaage J. Increase in serum S100A1-B and S100BB during cardiac surgery arises from extracerebral sources. Ann Thorac Surg. 2001;71:1512–1517. doi: 10.1016/S0003-4975(01)02399-2. [DOI] [PubMed] [Google Scholar]
- 3.Barnum SR, Jones JL, Benveniste EN. Interleukin-1 and tumor necrosis factor-mediated regulation of C3 gene expression in human astroglioma cells. Glia. 1993;7:225–236. doi: 10.1002/glia.440070306. [DOI] [PubMed] [Google Scholar]
- 4.Bellander BM, Singhrao SK, Ohlsson M, Mattsson P, Svensson M. Complement activation in the human brain after traumatic head injury. J Neurotrauma. 2001;18:1295–1311. doi: 10.1089/08977150152725605. [DOI] [PubMed] [Google Scholar]
- 5.Bellander BM, von Holst H, Fredman P, Svensson M. Activation of the complement cascade and increase of clusterin in the brain following a cortical contusion in the adult rat. J Neurosurg. 1996;85:468–475. doi: 10.3171/jns.1996.85.3.0468. [DOI] [PubMed] [Google Scholar]
- 6.Chesnut RM, Marshall LF, Klauber MR, Blunt BA, Baldwin N, Eisenberg HM, Jane JA, Marmarou A, Foulkes MA. The role of secondary brain injury in determining outcome from severe head injury. J Trauma. 1993;34:216–222. doi: 10.1097/00005373-199302000-00006. [DOI] [PubMed] [Google Scholar]
- 7.Corrigan JD, Smith-Knapp K, Granger CV. Validity of the functional independence measure for persons with traumatic brain injury. Arch Phys Med Rehabil. 1997;78:828–834. doi: 10.1016/S0003-9993(97)90195-7. [DOI] [PubMed] [Google Scholar]
- 8.de Vries HE, Blom-Roosemalen MC, van Oosten M, de Boer AG, van Berkel TJ, Breimer DD, Kuiper J. The influence of cytokines on the integrity of the blood–brain barrier in vitro. J Neuroimmunol. 1996;64:37–43. doi: 10.1016/0165-5728(95)00148-4. [DOI] [PubMed] [Google Scholar]
- 9.Dick JP, Guiloff RJ, Stewart A, Blackstock J, Bielawska C, Paul EA, Marsden CD. Mini-mental state examination in neurological patients. J Neurol Neurosurg Psychiatry. 1984;47:496–499. doi: 10.1136/jnnp.47.5.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edwards P, Arango M, Balica L, Cottingham R, El-Sayed H, Farrell B, Fernandes J, Gogichaisvili T, Golden N, Hartzenberg B, Husain M, Ulloa MI, Jerbi Z, Khamis H, Komolafe E, Laloë V, Lomas G, Ludwig S, Mazairac G, Muñoz Sanchéz Mde L, Nasi L, Olldashi F, Plunkett P, Roberts I, Sandercock P, Shakur H, Soler C, Stocker R, Svoboda P, Trenkler S, Venkataramana NK, Wasserberg J, Yates D, Yutthakasemsunt S, CRASH Trial Collaborators Final results of MRC CRASH, a randomised placebo-controlled trial of intravenous corticosteroid in adults with head injury-outcomes at 6 months. Lancet. 2005;365:1957–1959. doi: 10.1016/S0140-6736(05)66552-X. [DOI] [PubMed] [Google Scholar]
- 11.Faden AI, Demediuk P, Panter SS, Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- 12.Freeman BA, Crapo JD. Biology of disease: free radicals and tissue injury. Lab Invest. 1982;47:412–426. [PubMed] [Google Scholar]
- 13.Gao F, Harris DN, Sapsed-Byrne S, Sharp S. Neurone-specific enolase and Sangtec 100 assays during cardiac surgery: part III—dose haemolysis affect their accuracy? Perfusion. 1997;12:171–177. doi: 10.1177/026765919701200305. [DOI] [PubMed] [Google Scholar]
- 14.Graham DI, Adams JH, Gennarelli TA. Pathology of brain damage in head injury. In: Cooper PR, editor. Head injury. Baltimore: Williams & Wilkins; 1993. pp. 91–113. [Google Scholar]
- 15.Herrmann M, Curio N, Jost S, Grubich C, Ebert AD, Fork ML, Synowitz H. Release of biochemical markers of damage to neuronal and glial brain tissue is associated with short and long term neuropsychological outcome after traumatic brain injury. J Neurol Neurosurg Psychiatry. 2001;70:95–100. doi: 10.1136/jnnp.70.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herrmann M, Curio N, Jost S, Wunderlich MT, Synowitz H, Wallesch CW. Protein S-100B and neuron specific enolase as early neurobiochemical markers of the severity of traumatic brain injury. Restor Neurol Neurosci. 1999;14:109–114. [PubMed] [Google Scholar]
- 17.Herrmann M, Jost S, Kutz S, Ebert AD, Kratz T, Wunderlich MT, Synowitz H. Temporal profile of release of neurobiochemical markers of brain damage after traumatic brain injury is associated with intracranial pathology as demonstrated in cranial computerized tomography. J Neurotrauma. 2000;17:113–122. doi: 10.1089/neu.2000.17.113. [DOI] [PubMed] [Google Scholar]
- 18.Holmin S, Mathiesen T, Sheteye J, Biberfeld P. Intracerebral inflammatory response to experimental brain contusion. Acta Neurochir. 1995;132:110–119. doi: 10.1007/BF01404857. [DOI] [PubMed] [Google Scholar]
- 19.Jones PA, Andrews PJD, Midgley S, Anderson SI, Piper IR, Tocher JL, Housley AM, Corrie JA, Slattery J, Dearden NM, Miller JD. Measuring the burden of secondary insults in head-injured patients during intensive care. J Neurosurg Anesthesiol. 1994;6:4–14. [PubMed] [Google Scholar]
- 20.Link H, Tibbling G. Principles of albumin and IgG analyses in neurological disorders. II. Relation of the concentration of the proteins in serum and cerebrospinal fluid. Scand J Clin Lab Invest. 1977;37:391–396. doi: 10.1080/00365517709091497. [DOI] [PubMed] [Google Scholar]
- 21.Lynch NJ, Willis CL, Nolan CC, Roscher S, Fowler MJ, Weihe E, Ray DE, Schwaeble WJ. Microglial activation and increased synthesis of complement component C1q precedes blood–brain barrier dysfunction in rats. Mol Immunol. 2004;40:709–716. doi: 10.1016/j.molimm.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 22.Marshall LF, Marshall SB, Klauber MR, Van Berkum CM, Eisenberg H, Jane JA, Luerssen TG, Marmarou A, Foulkes MA. The diagnosis of head injury requires a classification based on computed axial tomography. J Neurotrauma. 1992;9:S287–S292. [PubMed] [Google Scholar]
- 23.Martin NA, Patwardhan RV, Alexander MJ, Africk CZ, Lee JH, Shalmon E, Hovda DA, Becker DP. Characterization of cerebral hemodynamic phases following severe head trauma: hypoperfusion, hyperemia, and vasospasm. J Neurosurg. 1997;87:9–19. doi: 10.3171/jns.1997.87.1.0009. [DOI] [PubMed] [Google Scholar]
- 24.Maruo N, Morita I, Shirao M, Murota S. IL-6 increases endothelial permeability in vitro. Endocrinology. 1992;131:710–714. doi: 10.1210/en.131.2.710. [DOI] [PubMed] [Google Scholar]
- 25.Miller JD, Piper IR, Jones PA. Pathophysiology of head injury. In: Narayan RK, Wilberger JE Jr, Povlishock JT, editors. Neurotrauma. New York: McGraw-Hill; 1995. pp. 61–69. [Google Scholar]
- 26.Morgan BP. Regulation of the complement membrane attack pathway. Crit Rev Immunol. 1999;19:173–198. [PubMed] [Google Scholar]
- 27.Morganti-Kossman MC, Lenzlinger PM, Hans V, Stahel P, Csuka E, Ammann E, Stocker R, Trentz O, Kossmann T. Production of cytokines following brain injury: beneficial and deleterious for the damaged tissue. Mol Psychiatry. 1997;2:133–136. doi: 10.1038/sj.mp.4000227. [DOI] [PubMed] [Google Scholar]
- 28.Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T. Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr Opin Crit Care. 2002;8:101–105. doi: 10.1097/00075198-200204000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Nekludov M, Bellander BM, Blombäck M, Wallen HN. Platelet dysfunction in patients with severe traumatic brain injury. J Neurotrauma. 2007;11:1699–1706. doi: 10.1089/neu.2007.0322. [DOI] [PubMed] [Google Scholar]
- 30.Olney JW. Inciting excitotoxic cytocide among central neurons. Adv Exp Med Biol. 1986;203:631–645. doi: 10.1007/978-1-4684-7971-3_48. [DOI] [PubMed] [Google Scholar]
- 31.Pettigrew LE, Wilson JT, Teasdale GM. Reliability of ratings on the Glasgow Outcome Scales from in-person and telephone structured interviews. J Head Trauma Rehabil. 2003;18:252–258. doi: 10.1097/00001199-200305000-00003. [DOI] [PubMed] [Google Scholar]
- 32.Rappaport M, Hall KM, Hopkins K, Belleza T, Cope DN. Disability Rating Scale for severe head trauma: coma to community. Arch Phys Med Rehabil. 1982;63:118–123. [PubMed] [Google Scholar]
- 33.Rudehill A, Bellander BM, Weitzberg E, Bredbacka S, Backheden M, Gordon E. Outcome of traumatic brain injuries in 1, 508 patients: impact of prehospital care. J Neurotrauma. 2002;19:855–868. doi: 10.1089/08977150260190447. [DOI] [PubMed] [Google Scholar]
- 34.Rus HG, Kim LM, Niculescu FI, Shin ML. Induction of C3 expression in astrocytes is regulated by cytokines and Newcastle disease virus. J Immunol. 1992;148:928–933. [PubMed] [Google Scholar]
- 35.Sandelin M, Zabihi S, Liu L, Wicher G, Kozlova EN. Metastasis-associated S100A4 (Mts1) protein is expressed in subpopulations of sensory and autonomic neurons and in Schwann cells of the adult rat. J Comp Neurol. 2004;473:233–243. doi: 10.1002/cne.20115. [DOI] [PubMed] [Google Scholar]
- 36.Stahel PF, Kariya K, Shohami E, Barnum SR, Eugster H, Trentz O, Kossmann T, Morganti-Kossmann MC. Intracerebral complement C5a receptor (CD88) expression is regulated by TNF and lymphotoxin-alpha following closed head injury in mice. J Neuroimmunol. 2000;109:164–172. doi: 10.1016/S0165-5728(00)00304-0. [DOI] [PubMed] [Google Scholar]
- 37.Stahel PF, Morganti-Kossmann C, Perez D, Redaelli C, Gloor B, Trentz O, Kossmann T. Intrathecal levels of complement-derived soluble membrane attack complex (sC5b-9) correlate with blood–brain barrier dysfunction in patients with traumatic brain injury. J Neurotrauma. 2001;18:773–781. doi: 10.1089/089771501316919139. [DOI] [PubMed] [Google Scholar]
- 38.Suarez JI, Zaidat OO, Suri MF, Feen ES, Lynch G, Hickman J, Georgiadis A, Selman WR. Length of stay and mortality in neurocritically ill patients: impact of a specialized neurocritical care team. Crit Care Med. 2004;32:2311–2317. doi: 10.1097/01.ccm.0000146132.29042.4c. [DOI] [PubMed] [Google Scholar]
- 39.Tagliaferri F, Compagnone C, Korsic M, Servadei F, Kraus J. A systematic review of brain injury in Europe. Acta Neurochir (Wien) 2006;148:255–268. doi: 10.1007/s00701-005-0651-y. [DOI] [PubMed] [Google Scholar]
- 40.Teasdale GM, Pettigrew LE, Wilson JT, Murray G, Jennett B. Analyzing outcome of treatment of severe head injury: a review and update on advancing the use of the Glasgow Outcome Scale. J Neurotrauma. 1998;15:587–597. doi: 10.1089/neu.1998.15.587. [DOI] [PubMed] [Google Scholar]
- 41.Tibbling G, Link H, Öhman S. Principles of albumin and IgG analyses in neurological disorders. I. Establishment of reference values. Scand J Clin Lab Invest. 1977;37:385–390. doi: 10.1080/00365517709091496. [DOI] [PubMed] [Google Scholar]
- 42.Todd NV, Graham DI. Blood–brain barrier damage in traumatic brain contusions. Acta Neurochir Suppl. 1990;51:296–299. doi: 10.1007/978-3-7091-9115-6_100. [DOI] [PubMed] [Google Scholar]
- 43.VanLandingham JW, Cekic M, Cutler S, Hoffman SW, Stein DG. Neurosteroids reduce inflammation after TBI through CD55 induction. Neurosci Lett. 2007;25:94–98. doi: 10.1016/j.neulet.2007.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Varelas PN, Conti MM, Spanaki MV, Potts E, Bradford D, Sunstrom C, Fedder W, Hacein Bey L, Jaradeh S, Gennarelli TA. The impact of a neurointensivist-led team on a semiclosed neurosciences intensive care unit. Crit Care Med. 2004;32:2191–2198. doi: 10.1097/01.ccm.0000146131.03578.21. [DOI] [PubMed] [Google Scholar]
- 45.Waters RJ, Nicoll JA. Genetic influences on outcome following acute neurological insults. Curr Opin Crit Care. 2005;11:105–110. doi: 10.1097/01.ccx.0000155354.78617.91. [DOI] [PubMed] [Google Scholar]
- 46.Vos PE, Lamers KJ, Hendriks JC, van Haaren M, Beems T, Zimmerman C, van Geel W, de Reus H, Biert J, Verbeek MM. Glial and neuronal proteins in serum predict outcome after severe traumatic brain injury. Neurology. 2004;62:1303–1310. doi: 10.1212/01.wnl.0000120550.00643.dc. [DOI] [PubMed] [Google Scholar]