

Abstract
Cyclooxygenase-2 (COX-2; Ptgs2) acts as a tumor promoter in rodent models for colorectal cancer, but its precise role in carcinogenesis remains unclear. We evaluated the contribution of host-derived COX-1 and COX-2 in tumor growth using both genetic and pharmacological approaches. Lewis lung carcinoma (LLC) cells grow rapidly as solid tumors when implanted in C57BL/6 mice. We found that tumor growth was markedly attenuated in COX-2–/–, but not COX-1–/– or wild-type mice. Treatment of wild-type C57BL/6 mice bearing LLC tumors with a selective COX-2 inhibitor also reduced tumor growth. A decrease in vascular density was observed in tumors grown in COX-2–/– mice when compared with those in wild-type mice. Because COX-2 is expressed in stromal fibroblasts of human and rodent colorectal carcinomas, we evaluated COX-2–/– mouse fibroblasts and found a 94% reduction in their ability to produce the proangiogenic factor, VEGF. Additionally, treatment of wild-type mouse fibroblasts with a selective COX-2 inhibitor reduced VEGF production by 92%.
Introduction
Numerous studies of rodent cancer models and in humans show that nonsteroidal anti-inflammatory drugs (NSAIDs) have antineoplastic properties. One known effect of NSAIDs is their ability to inhibit the key enzymes required for prostaglandin production (prostaglandin endoperoxide synthase or cyclooxygenase). Two isoforms of cyclooxygenase have been characterized, COX-1 and COX-2. Most of the preclinical evidence for the antineoplastic effects of NSAIDs is based on studies using animals that have either a genetic or a chemically induced predisposition to develop colon cancer (1). These type of experimental approaches lack the power to determine whether the antineoplastic effects of NSAIDs are specifically due to inhibition of cyclooxygenase, the result of other effects, or both. Oshima et al. (2) evaluated COX-2–/– mice to determine whether COX-2 contributed to adenoma formation. They found that when mice with a genetic predisposition for polyp formation were mated with COX-2–/– mice, tumor burden in the offspring was significantly reduced when compared with that of control mice, strongly implicating a role for COX-2 in tumor promotion. Others have shown that treatment with COX-2 inhibitors leads to a marked reduction in the growth of a variety of neoplasms including colon (3), head and neck (4), skin (5), and bladder (6). Recent clinical studies have indicated that the presence of COX-2 in human lung and colon cancers is associated with a negative prognosis (7, 8). Therefore, COX-2 may play a wider role in carcinogenesis than was originally thought.
The data regarding the sublocalization of COX-2 in solid tumors are conflicting. COX-2 is expressed in some colon carcinoma cell lines (9–11), and its expression can be induced in cultured rat intestinal epithelial cells by treatment with mitogens and tumor promoters (12). Elevated COX-2 expression has been reported in the epithelial component of adenomas in the multiple intestinal neoplasia (Min) mouse (13), azoxymethane-treated rat cancers (14), replication error repair–positive human carcinomas (15), and sporadic human colorectal cancers (16, 17). We and others have recently found that COX-2 expression is also elevated in the subepithelial component in adenomas from the Min mouse (18, 19), in carcinogen-induced colon cancers in mice (18), and in colorectal carcinomas from IL-10–/– mice (20). Some of the conflicting data regarding localization of COX-2 may be due to nonspecific binding of the polyclonal antibodies that were used for COX-2 immunostaining or a change in COX-2 expression patterns as adenomas progress to carcinomas. Also, it seems clear from one recent report that COX-2 expression is found in both the epithelial and stromal components of sporadic human colorectal cancers (19). Oshima et al., localized COX-2 reporter expression in polyps from APCΔ716 mice by crossing them with mice in which the LacZ gene was placed under the control of the COX-2 promoter (2). They found LacZ expression in areas surrounding the adenoma, but no significant expression in the epithelial component of the adenomas. We have recently carried out in situ hybridization experiments to localize COX-2 expression in sporadic colon cancers from 50 patients and in colon carcinomas from IL-10–/– mice. We found high levels of COX-2 mRNA expressed in stromal fibroblasts of both human and rodent colorectal cancers (20). It is clear from these results that COX-2 is expressed in both epithelial and tumor stromal cells. Therefore, it is possible that COX-2 in stromal fibroblasts could act to promote tumor growth by producing bioactive prostaglandins that have paracrine effects on nearby carcinoma cells. Recent reports have indicated that prostaglandins can regulate VEGF expression (21, 22) and that COX-2 inhibitors can directly affect angiogenesis (23). Others have shown that expression of VEGF by host fibroblasts plays an important role in angiogenesis (24). Therefore, COX-2 could be acting as a “landscaping tumor promoter” according to the model proposed by Kinzler and Vogelstein (25). This model supports the notion that COX-2 expression in the stromal component of a solid tumor could influence its growth or expression of proangiogenic factors. The mechanism(s) for inhibition of tumor formation and growth by NSAIDs may involve inhibition of COX-2 located in stromal tumor cells.
Methods
Reagents.
Celecoxib (SC-58635) and SC-58125 were a kind gift from K. Siebert and P. Isakson of G.D. Searle and Co. (St. Louis, Missouri, USA).
Cell culture.
Lewis lung carcinoma (LLC) cells were purchased from the American Type Tissue Collection (Manassas, Virginia, USA) and maintained according to standard cell culture techniques.
COX-1 and COX-2 knockout animals.
The disruption of Ptgs2 (also referred to as COX-2) was performed by introducing a PGK-neo cassette in place of a 1.8-kb EcoRV genomic fragment housing exon 1 and surrounding sequences (26). PCR of tail DNA and blood urea nitrogen (BUN) tests (26) determined the genotypes. The disruption of Ptgs1 (also called COX-1) was performed by replacing 1 kb of intron 10, together with the splice junction and first 44 bp of exon 11, with the neomycin resistance gene (27). PCR analysis of tail DNA determined the genotypes.
Western blotting.
Immunoblot analysis of cell protein lysates were performed as described previously (28). Briefly, tumor samples were lysed for 30 minutes in RIPA buffer (1 × PBS, 1% Nonidet P40, 0.5% sodium deoxycholate, 0.1% SDS). The clarified cell lysates (50 μg) were denatured and fractionated by 10% SDS-PAGE. The proteins were transferred to PVDF membranes after electrophoresis. The filters were blocked for 3 hours in BLOTTO (0.15 M NaCl, 10 mM Tris-HCl [pH 7.4], 0.1% Tween-20, and 5% nonfat dry milk) and then incubated overnight with either COX-1 (1:500) or COX-2 (1:500) specific antibodies (COX-1 catalog no. 1753, COX-2 catalog no. 1746; Santa Cruz Biotechnology Inc., Santa Cruz, California, USA). The membranes were then washed in BLOTTO before a 1-hour incubation with donkey anti-goat horseradish peroxidase–conjugated secondary antibody. The membranes were washed in TBST (0.15 M NaCl, 10 mM Tris-HCl pH 7.4, 0.1% Tween-20) and developed by the ECL plus chemiluminescence system (Amersham Life Sciences Inc., Arlington Heights, Illinois, USA) and exposed to Hyperfilm (Amersham Life Sciences).
Isograft model of tumor biology.
LLC cells were grown on plastic culture dishes according to standard cell culture techniques (9), gently washed in sterile PBS, detached by reverse pipetting with PBS, and then pelleted by brief centrifugation at 300 g. The supernatant was aspirated, and cells were resuspended in PBS and counted using a hemocytometer. A final concentration of 2 × 107 cells/mL was made, and 100 μL of cell suspension was injected subcutaneously using a tuberculin syringe and a 27-gauge needle. In celecoxib treatment experiments, mice were pretreated with celecoxib for 3 days before tumor implantation. Celecoxib was mixed in 4% Harlan Teklad chow (Harlan Teklad, Madison, Wisconsin, USA) at 1,500 mg/kg. The size of the tumor was determined by direct measurement of tumor dimensions. The volume was calculated according to the equation (V=[L × W2] × 0.5), where V = volume, L = length, and W = width (29).
In situ hybridization.
Sense or antisense [35S]-labeled cRNA probes were generated using appropriate polymerases from cDNAs to VEGF, COX-1, or COX-2 for in situ hybridization. The VEGF probe was derived from a VEGF cDNA fragment from nucleotides 270 to 849 of human VEGF. This fragment cross-hybridizes with murine VEGF (30). Mouse COX-1 and COX-2 probes were generated from murine cDNAs encompassing Asn82-Gln369 of COX-1 and Met1-Gln270 of COX-2 (31). The probes had specific activities of approximately 2 × 109 disintegrations per minute/μg. Sections hybridized with sense probes served as negative controls. After hybridization and washing, the slides were incubated with Rnase-A (20 μg/mL), and Rnase-A–resistant hybrids were detected by autoradiography using Kodak NTB-2 liquid emulsion (Eastman Kodak Co., Rochester, New York, USA).
Preparation of skin fibroblasts and VEGF determination.
Skin tissue was chopped into small pieces and then placed into 60-mm culture dishes and covered with DMEM/20% FCS containing 0.01% collagenase. After 24 hours of culture, the tissue was washed and transferred into 10-cm dishes containing DMEM/20% FCS. After 10 days, spindle-shaped fibroblasts were collected and seeded at a 1:3 dilution. When these cells became 80% confluent, they were collected and counted, and 50,000 cells were seeded into 35-mm culture dishes and incubated for 24 hours in DMEM/20% FCS. The media were then changed, and serum-free DMEM was added to the cells and they were incubated for an additional 12 hours. The conditioned media were collected, and the concentration of VEGF was determined using a mouse VEGF immunoassay (catalog no. MMV00; R&D Systems Inc., Minneapolis, Minnesota, USA).
Quantification of vascular density.
The intratumoral blood vessels were quantitated using immunohistochemistry with a factor VIII rabbit anti-human polyclonal antibody (A0082; DAKO Corp., Carpinteria, California, USA). For each tumor, five random images were captured at ×200. Only areas of viable tumor tissue were imaged; necrotic regions and overlying subdermal regions were excluded. Zeiss Image (Thornwood, New York, USA) morphometric analysis software was used to automatically count stained vessels, thus removing any possible observation bias. All tumors were imaged and analyzed using identical instrument settings over a 3-hour period. Vessels that were visibly interconnected were scored only once. The final vascular density score for the tumor represents an average of all scored fields.
Results and Discussion
We were interested in carefully examining the role of host-derived COX-1 and COX-2 in tumor growth. We grafted LLC cells into either wild-type, COX-1–/–, or COX-2–/– C57BL/6 mice. Using this model system, we directly tested the hypothesis that COX-1 or COX-2 produced by the host could affect tumor growth. We chose to study the LLC cells because they are capable of growing in the C57BL/6 mice without being rejected by the host, as they have a compatible genetic background; however, carcinoma cells derived from other species or mouse strains do not grow in these mice.
We found that the cyclooxygenase status of the host mouse does not influence tumor engraftment rates. All mice exhibited similar tumor growth rates during the first 7 days after implantation (Figure 1a). By day 15, tumors grafted in the COX-2 null mice were significantly smaller than those engrafted in wild-type or COX-2+/– mice. In addition, tumors in COX-2–/– mice continued to grow more slowly than did control tumors for the remainder of the experiment. Interestingly, tumor growth in COX-2–/– mice increased slightly between days 20 and 25, indicating that the absence of COX-2 in the host did not permanently halt carcinoma growth. No difference in the growth of LLC tumors in COX-1–/– mice was observed when compared with control mice (Figure 1b). This suggests that COX-1 expression in the host is not essential for tumor growth under these conditions. Even though the lack of COX-1 is known to affect platelet function, there was no inhibition of tumor growth in the COX-1–/– mice. Collectively, these data strongly implicate a role for host-derived COX-2 in promoting tumor growth in this model system.
Figure 1.

Host-derived COX-2 is important for LLC tumor growth. (a) A total of 2 × 106 LLC cells were implanted into COX-2+/+ (gray bars), COX-2+/– (white bars), or COX-2–/– (black bars) C57BL/6 mice; (b) a total of 2 × 106 LLC cells were implanted into COX-1+/+ (gray bars) or COX-1–/– (black bars) C57BL/6 mice. Tumor volumes were calculated from tumor measurements scored at the indicated day. Results are presented as the mean tumor volume ± SEM.
We next evaluated whether treatment with celecoxib, a selective COX-2 inhibitor, could inhibit tumor growth. Celecoxib treatment was initiated 3 days before tumor implantation to mimic more closely the experiments conducted with the COX-2–/– animals. This treatment regimen would inhibit COX-2 activity in both the host and tumor tissues. Peak serum levels of celecoxib in treated mice were 889 ± 272 ng/mL (2.3 μM). Celecoxib treatment inhibited tumor growth; however, the results were not as striking as those obtained in COX-2–/– mice (Figure 2). This variation may be due to differences between total lack of COX-2 versus intermittently inhibiting COX-2 activity by treatment with dietary celecoxib.
Figure 2.

Celecoxib inhibits LLC tumor growth. A total of 2 × 106 LLC cells were implanted into C57BL/6 mice. Mice were fed either control chow (gray bars) or 1,500 mg/kg celecoxib-containing chow (black bars). Tumor volumes were calculated from tumor measurements taken at the indicated day and are represented as the average of each group ± SEM.
To evaluate further LLC isografts, we examined intratumoral gene expression patterns. Cultured LLC cells express both COX-1 and COX-2. Immunoblotting of tumor lysates obtained from COX-2+/+, COX-2+/–, or COX-2–/– mice did not reveal any differences in expression of either COX-1 or COX-2 (Figure 3). To determine whether differential localization of COX-1 or COX-2 expression occurs within the tumors, in situ hybridization for COX-1 and COX-2 was performed on tumor sections. The COX-1 hybridization pattern was diffuse, with uniform accumulation throughout the tumor (data not shown). COX-2 mRNA hybridization, in contrast, was focal with punctate accumulation in carcinoma and stromal cells that surrounded areas of necrotic tissue (Figure 4). Immunolocalization of COX-2 protein showed a similar expression pattern (data not shown). Interestingly, the expression pattern for COX-2 and VEGF was similar, with high levels of expression in the stromal compartment, and a significant reduction of VEGF expression in tumors grown in the COX-2–/– mice (Figure 4). We also examined the expression pattern of COX-2 in human colorectal adenocarcinomas from 50 patients, using in situ hybridization, and found high levels of COX-2 expressed in stromal fibroblasts of these tumors (data not shown).
Figure 3.

COX-1 and COX-2 are expressed within LLC tumors. Tumor lysates from LLC tumors grown in COX-2+/+ (wild-type), COX-2+/– (heterozygote), and COX-2–/– (null) mice were produced and 50 μg of lysate was fractionated on a 10% SDS-PAGE gel. (a) COX-2 and (b) COX-1 specific antisera were used to blot the membranes. Het, heterozygote.
Figure 4.

COX-2 and VEGF are expressed in LLC isografts. COX-2 (upper panels, a and b, wild-type host), VEGF (middle panels, c and d, wild-type host), and VEGF (lower panels, e and f, COX-2–/– host) riboprobes were hybridized to sections from tumors grown in C57BL/6 mice with the designated genotype (×400). Control hybridizations with sense cRNA riboprobes were performed to validate the specificity of Rnase-A–resistant hybrids. WT, wild-type.
Tumor growth requires the maintenance and expansion of a vascular network (32). It has been demonstrated using in vitro assays that COX-2 can influence angiogenesis (23, 33, 34) and that treatment with selective COX-2 inhibitors blocks angiogenesis (23, 34, 35). Therefore, we questioned whether vascularization of tumors grown in COX-2–/– mice was affected by the lack of COX-2 expression in tumor host cells. We evaluated vascular density by selectively staining endothelial cells, as this is a measure of tumor-associated angiogenesis. We found that vascular density was 30% lower in tumors grafted in COX-2–/– mice when compared with tumors from wild-type mice (Figure 5). From these results we conclude that COX-2 contributes to tumor vascularization. To explore the mechanism underlying this effect, we isolated fibroblasts from COX-1–/–, COX-2–/–, and wild-type mice and evaluated their ability to produce proangiogenic factors. We found that COX-2–/– fibroblasts have a 94% reduction in production of VEGF when compared with wild-type and COX-1–/– fibroblasts (Figure 6). Additionally, when wild-type fibroblasts were treated with a selective COX-2 inhibitor, we observed a 92% reduction in VEGF production (Figure 6). Both of these results demonstrate a link between COX-2 and regulation of VEGF expression. This is not so surprising in light of recent data indicating that the products of the COX-2 pathway (prostaglandins) can stimulate VEGF production (21, 22). Of interest, we found no difference in the levels of IGF in the fibroblasts isolated from COX-1–/–, COX-2–/–, or wild-type mice (data not shown). However, some groups have questioned the role of VEGF in maintaining the malignant phenotype of melanoma cells (36).
Figure 5.
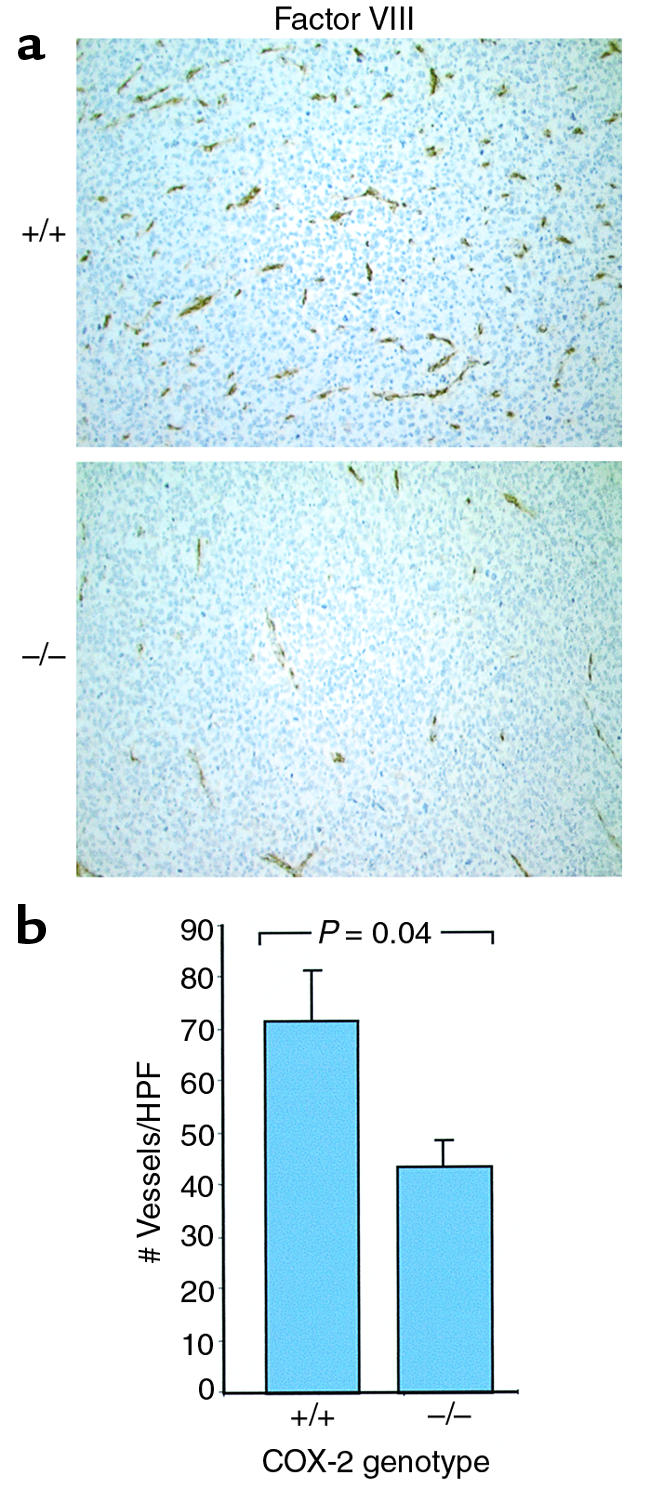
Decreased vascular density in LLC tumors when grown in COX-2–/– mice. (a) Representative photomicrographs of factor VIII–stained tumor sections from LLC tumors grown in wild-type (+/+) or Ptgs2–/– (–/–) mice (×200). (b) Factor VIII–positive vessel counts obtained by morphometric analysis of LLC tumors. Values represent the average number of vessels at ×200 ± the SD (Student’s t test; P = 0.04). HPF, high power field.
Figure 6.

VEGF production in fibroblasts is regulated by COX-2. Production of VEGF by wild-type, COX-1–/–, and COX-2–/– fibroblasts was determined. Each treatment condition is listed below its respective bar graph. WT denotes wild-type mouse fibroblasts. We observed a 93% reduction in VEGF production in COX-2–/– fibroblasts when compared with wild-type fibroblasts. Additionally, treatment of wild-type fibroblasts with a selective COX-2 inhibitor (10 μM SC-58125) led to a ∼90% reduction in VEGF levels.
In summary, we have demonstrated that efficient tumor growth requires the presence of COX-2 in the tumor host. Our data also suggest that host-derived COX-2 may regulate intratumoral vascular density, which, in turn, may modulate tumor growth. We speculate that one mechanism for the antineoplastic effects of NSAIDs is via their inhibition of COX-2 located in the stromal compartment of the tumor, which leads to inhibition of the production of proangiogenic factors by tumor and stromal cells.
Acknowledgments
This work was supported in part by United States Public Health Services grants (DK 47297, P01CA77839, and P30 CA68485, to R.N. DuBois). R.N. DuBois is a recipient of a Veterans Affairs Research Merit Grant, and S.K. Dey is a recipient of an NICHD MERIT Award (HD 12304). We thank the T.J. Martell Foundation for their generous support. Thanks are due to Y. Zhu and N. Brown for genotyping of mice, and X. Zhao for in situ hybridization. We also thank Kay Washington for examining the histology of the tumors and Wade Krause for assistance with in vivo studies.
References
- 1.Williams CS, Mann M, DuBois RN. The role of cyclooxygenases in inflammation, cancer and development. Oncogene. 1999;18:7908–7916. doi: 10.1038/sj.onc.1203286. [DOI] [PubMed] [Google Scholar]
- 2.Oshima M, et al. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2) Cell. 1996;87:803–809. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- 3.Kawamori T, Rao CV, Seibert K, Reddy BS. Chemopreventive activity of celecoxib, a specific cyclooxygenase-2 inhibitor, against colon carcinogenesis. Cancer Res. 1998;58:409–412. [PubMed] [Google Scholar]
- 4.Nishimura G, Yanoma S, Mizuno H, Kawakami K, Tsukuda M. A selective cyclooxygenase-2 inhibitor suppresses tumor growth in nude mouse xenografted with human head and neck squamous carcinoma cells. Jpn J Cancer Res. 1999;90:1152–1162. doi: 10.1111/j.1349-7006.1999.tb00690.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pentland AP, Schoggins JW, Scott GA, Khan KN, Han R. Reduction of UV-induced skin tumors in hairless mice by selective COX-2 inhibition. Carcinogenesis. 1999;20:1939–1944. doi: 10.1093/carcin/20.10.1939. [DOI] [PubMed] [Google Scholar]
- 6.Okajima E, et al. Chemopreventive effects of nimesulide, a selective cyclooxygenase-2 inhibitor, on the development of rat urinary bladder carcinomas initiated by N-butyl-N-(4-hydroxybutyl) nitrosamine. Cancer Res. 1998;58:3028–3031. [PubMed] [Google Scholar]
- 7.Achiwa H, et al. Prognostic significance of elevated cyclooxygenase 2 expression in primary, resected lung adenocarcinomas. Clin Cancer Res. 1999;5:1001–1005. [PubMed] [Google Scholar]
- 8.Sheehan KM, et al. The relationship between cyclooxygenase-2 expression and colorectal cancer. JAMA. 1999;282:1254–1257. doi: 10.1001/jama.282.13.1254. [DOI] [PubMed] [Google Scholar]
- 9.Sheng H, et al. Inhibition of human colon cancer cell growth by selective inhibition of cyclooxygenase-2. J Clin Invest. 1997;99:2254–2259. doi: 10.1172/JCI119400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanif R, et al. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem Pharmacol. 1996;52:237–245. doi: 10.1016/0006-2952(96)00181-5. [DOI] [PubMed] [Google Scholar]
- 11.Coffey RJ, et al. EGF receptor activation induces nuclear targeting of COX-2, basolateral release of prostaglandins and mitogenesis in polarizing colon cancer cells. Proc Natl Acad Sci USA. 1997;94:657–662. doi: 10.1073/pnas.94.2.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DuBois RN, Awad J, Morrow J, Roberts LJ, Bishop PR. Regulation of eicosanoid production and mitogenesis in rat intestinal epithelial cells by transforming growth factor-α and phorbol ester. J Clin Invest. 1994;93:493–498. doi: 10.1172/JCI116998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams CS, et al. Elevated cyclooxygenase-2 levels in Min mouse adenomas. Gastroenterology. 1996;111:1134–1140. doi: 10.1016/s0016-5085(96)70083-5. [DOI] [PubMed] [Google Scholar]
- 14.Shao J, et al. Coordinate regulation of cyclooxygenase-2 and TGF-β1 in hereditary nonpolyposis colorectal cancer and azoxymethane- induced rat colonic tumors. Carcinogenesis. 1999;20:185–191. doi: 10.1093/carcin/20.2.185. [DOI] [PubMed] [Google Scholar]
- 15.Karnes WE, et al. Reduced COX-2 protein in colorectal cancer with defective mismatch repair. Cancer Res. 1998;58:5473–5477. [PubMed] [Google Scholar]
- 16.Kutchera W, et al. Prostaglandin H synthase-2 is expressed abnormally in human colon cancer: evidence for a transcriptional effect. Proc Natl Acad Sci USA. 1996;93:4816–4820. doi: 10.1073/pnas.93.10.4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sano H, et al. Expression of cyclooxygenase-1 and -2 in human colorectal cancer. Cancer Res. 1995;55:3785–3789. [PubMed] [Google Scholar]
- 18.Shattuck-Brandt RL, Lamps LW, Heppner-Goss KJ, DuBois RN, Matrisian LM. Differential expression of matrilysin and cyclooxygenase-2 in intestinal and colorectal neoplasms. Mol Carcinog. 1999;24:177–187. [PubMed] [Google Scholar]
- 19.Chapple KS, et al. Localization of cyclooxygenase-2 in human sporadic colorectal adenoma. Am J Pathol. 2000;156:545–553. doi: 10.1016/S0002-9440(10)64759-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shattuck-Brandt RL, et al. Cyclooxygenase-2 expression is increased in the subepithelial myofibroblasts of colon and cecal carcinomas from IL-10 (–/–) mice. Gastroenterology. 2000;118:337–345. doi: 10.1016/s0016-5085(00)70216-2. [DOI] [PubMed] [Google Scholar]
- 21.Cheng T, Cao W, Wen R, Steinberg RH, LaVail MM. Prostaglandin E2 induces vascular endothelial growth factor and basic fibroblast growth factor mRNA expression in cultured rat Muller cells. Invest Ophthalmol Vis Sci. 1998;39:581–591. [PubMed] [Google Scholar]
- 22.Hoper MM, et al. Prostaglandins induce vascular endothelial growth factor in a human monocytic cell line and rat lungs via cAMP. Am J Respir Cell Mol Biol. 1997;17:748–756. doi: 10.1165/ajrcmb.17.6.2888. [DOI] [PubMed] [Google Scholar]
- 23.Jones MK, et al. Inhibition of angiogenesis by nonsteroidal anti-inflammatory drugs: insight into mechanisms and implications for cancer growth and ulcer healing. Nat Med. 1999;5:1418–1423. doi: 10.1038/70995. [DOI] [PubMed] [Google Scholar]
- 24.Fukumura D, et al. Tumor induction of VEGF promoter activity in stromal cells. Cell. 1998;94:715–725. doi: 10.1016/s0092-8674(00)81731-6. [DOI] [PubMed] [Google Scholar]
- 25.Kinzler KW, Vogelstein B. Landscaping the cancer terrain. Science. 1998;280:1036–1037. doi: 10.1126/science.280.5366.1036. [DOI] [PubMed] [Google Scholar]
- 26.Dinchuk JE, et al. Renal abnormalities and an altered inflammatory response in mice lacking cyclooxygenase II. Nature. 1995;378:406–409. doi: 10.1038/378406a0. [DOI] [PubMed] [Google Scholar]
- 27.Langenbach R, et al. Prostaglandin synthase-1 gene disruption in mice reduces arachidonic acid induced inflammation and indomethacin induced gastric ulceration. Cell. 1995;83:483–492. doi: 10.1016/0092-8674(95)90126-4. [DOI] [PubMed] [Google Scholar]
- 28.DuBois RN, Shao J, Sheng H, Tsujii M, Beauchamp RD. G1 delay in intestinal epithelial cells overexpressing prostaglandin endoperoxide synthase-2. Cancer Res. 1996;56:733–737. [PubMed] [Google Scholar]
- 29.Wang J, et al. Demonstration that mutation of the type II transforming growth factor beta receptor inactivates its tumor suppressor activity in replication error-positive colon carcinoma cells. J Biol Chem. 1995;270:22044–22049. doi: 10.1074/jbc.270.37.22044. [DOI] [PubMed] [Google Scholar]
- 30.Chakraborty I, Das SK, Dey SK. Differential expression of vascular endothelial growth factor and its receptor mRNAs in the mouse uterus around the time of implantation. J Endocrinol. 1995;147:339–352. doi: 10.1677/joe.0.1470339. [DOI] [PubMed] [Google Scholar]
- 31.Chakraborty I, Das SK, Wang J, Dey SK. Developmental expression of the cyclo-oxygenase-1 and cyclo-oxygenase-2 genes in the peri-implantation mouse uterus and their differential regulation by the blastocyst and ovarian steroids. J Mol Endocrinol. 1996;16:107–122. doi: 10.1677/jme.0.0160107. [DOI] [PubMed] [Google Scholar]
- 32.Holash J, et al. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science. 1999;284:1994–1998. doi: 10.1126/science.284.5422.1994. [DOI] [PubMed] [Google Scholar]
- 33.Tsujii M, et al. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell. 1998;93:705–716. doi: 10.1016/s0092-8674(00)81433-6. [DOI] [PubMed] [Google Scholar]
- 34.Daniel TO, Liu H, Morrow JD, Crews BC, Marnett LJ. Thromboxane A2 is a mediator of cyclooxygenase-2-dependent endothelial migration and angiogenesis. Cancer Res. 1999;59:4574–4577. [PubMed] [Google Scholar]
- 35.Yamada M, Kawai M, Kawai Y, Mashima Y. The effect of selective cyclooxygenase-2 inhibitor on corneal angiogenesis in the rat. Curr Eye Res. 1999;19:300–304. doi: 10.1076/ceyr.19.4.300.5301. [DOI] [PubMed] [Google Scholar]
- 36.Chin L, et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468–472. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]