

Abstract
p53 is one of the most studied tumor suppressors in the cancer research field. Of note, over 50% of human tumors carry loss of function mutations, and thus p53 has been considered to be a classical Knudson-type tumor suppressor. From the functional point of view, p53 is a nuclear transcription factor to transactivate a variety of its target genes implicated in the induction of cell cycle arrest, DNA repair, and apoptotic cell death. In response to cellular stresses such as DNA damage, p53 is activated and promotes cell cycle arrest followed by the replacement of DNA lesions and/or apoptotic cell death. Therefore, p53 is able to maintain the genomic integrity to prevent the accumulation of genetic alterations, and thus stands at a crossroad between cell survival and cell death. In this paper, we describe a variety of molecular mechanisms behind the regulation of p53.
1. Introduction
p53 has been initially identified in crude cell lysates prepared from cells transformed by simian virus 40 (SV40) [1–5]. Subsequent studies demonstrated that p53 forms a stable complex in SV40-transfromed cells with SV40 large T antigen which has an oncogenic potential, and p53 had an ability to promote tumor growth. In support with these results, p53 was detectable in a variety of tumor-derived cell lines [1, 3, 4]. Based on these observations, p53 came to be classified as an oncogene [6]. However, this classical point of view has been challenged by the findings showing that the initially discovered p53 is a mutant form of p53 [7]. In a sharp contrast to mutant forms of p53, subsequent studies revealed that wild-type p53 is capable to suppress the malignant growth of transformed cells as well as tumors, suggesting that p53 acts as a tumor suppressor [8–12]. Intriguingly, p53 gene locates on the short arm of human chromosome 17 (17p13), where loss of heterozygosity (LOH) was detectable in a wide variety of tumor tissues. It is worth noting that the remaining nondeleting p53 allele is mutated in some cases [13–18]. Extensive mutation searches demonstrated that over 50% of human tumors carry p53 mutations. Indeed, p53-deficient mice developed spontaneous tumors at a relatively young age [19].
Ninety-five percent of the mutations were detected within the central sequence-specific DNA-binding region of p53 [20–22]. These mutations disrupted the whole conformation of the sequence-specific DNA-binding domain of p53 and resulted in the loss of its sequence-specific DNA-binding ability [23]. Since p53 was a nuclear sequence-specific transcription factor which transactivated a set of its target genes involved in the induction of cell cycle arrest and/or apoptotic cell death, mutant forms of p53 lacked their critical function to maintain the genomic integrity. Furthermore, mutant forms of p53 has acquired a much longer half-life as compared with that of wild-type p53 [24, 25] and displayed a dominant-negative behavior toward wild-type p53 [26, 27]. This dominant-negative effect of mutant p53 on wild-type p53 might be mediated by the hetero-oligomerization through their oligomerization domains [7, 28–30]. In this connection, p53 mutation conferred the resistance of tumor cells to anticancer drugs by inhibiting p53-dependent proapoptotic pathway [31–33]. Thus, the development of the novel devices to remove or suppress the dominant-negative effect of mutant forms of p53 on wild-type p53 will serve as a basis for providing new therapeutic strategies to treat tumors bearing p53 mutations.
As described above, p53 had a strong proapoptotic activity. Under normal conditions, the expression level of this dangerous protein is kept at extremely low level. In response to multiple cellular stresses including DNA damage, oncogene activation, hypoxia, nucleotide imbalance, and oxidative damage, p53 was rapidly accumulated in cell nucleus through chemical modifications such as phosphorylation and acetylation and exerted its proapoptotic function to remove cells with seriously damaged DNA in which DNA damage was severe and repair was impossible [22, 34–36]. In this case, p53 transactivated proapoptotic target genes including BAX, PUMA, NOXA, and p53AIP1, and the collaboration of these gene products contributed to the disruption of mitochondrial membrane potential, which was a critical step in p53-dependent proapoptotic pathway [37]. On the other hand, p53 promoted G1 cell cycle arrest in the early stage of DNA damage response through the transactivation of p21WAF1, p53R2, and GADD45 implicated in the induction of cell cycle arrest and DNA repair [37]. After DNA repair had been completed, cells reentered into normal cell cycle. Upon DNA damage, cells underwent either cell cycle arrest or apoptotic cell death to allow DNA repair or suicide of cells, which was dependent on the degree and/or the nature of DNA damage.
For a long time, p53 has been considered to be a solitary gene product. Due to the improvement of cloning technologies, Kaghad et al. identified the first p53 homolog termed p73 [38]. Additionally, Yang et al. discovered the second p53 homolog termed p63 [39]. Cell-based studies demonstrated that p73 and p63 act as nuclear sequence-specific transcription factors which transactivate the overlapping set of p53-target genes and also have an ability to induce cell cycle arrest and/or apoptotic cell death in cancerous cells [40, 41]. Like p53, p73, and p63 were induced in response to a certain subset of DNA-damaging agents [42, 43]. Thus, p53 becomes a founding member of p53 tumor suppressor family composed of p53, p73 and p63.
Based on the above-mentioned brief background of p53, we focus primarily on a variety of regulatory mechanisms of p53 in the present paper.
2. General Feature of p53
p53 locates at a short arm of human chromosome 17p13 containing 11 exons spanning 20 kb. p53 acts as a nuclear sequence-specific transcription factor composed of NH2-terminal transactivation domain (TA, amino acid residues 1–45), central sequence-specific DNA-binding domain (DB, amino acid residues 102–292), and COOH-terminal oligomerization domain (OD, amino acid residues 319–359). In addition to these representative functional domains, p53 contains three nuclear localization signals (NLS, amino acid residues 305–322, 369–375, and 379–384) recognized by importin α/β complex [44], a Leu-rich nuclear export signal (NES, amino acid residues 339–352) recognized by CRM1 (chromosomal region maintenance 1) [44], and a Pro-rich domain (amino acid residues 63–97). Cytoplasmic retention of p53 was observed in certain breast cancer-derived cells expressing the truncated form of importin α, indicating that importin α plays an essential role in nuclear import of p53 [45]. Cytoplasmic p53 is nonfunctional. Pro-rich domain has been shown to be associated with proapoptotic activity of p53 [46, 47]. Deletion of this Pro-rich region resulted in a complete loss of proapoptotic activity of p53. Active form of nuclear p53, which functions as a tetramer, recognizes and binds to a consensus sequence motif made of tandem 10 bp elements (RRRCWWGYYY: R, G/A; W, A/T; Y, C/T) separated by 1–13 bp found within the promoter regions of p53-target genes. p53 exerts its proapoptotic function through the transactivation of its target genes [48, 49]. Genome-wide analysis revealed that there exist over 4,000 putative p53-responsive elements [22]. Although all of these canonical p53-responsive elements might not always be functional, identification and functional analysis of new p53-target genes provide novel insights into understanding the precise molecular mechanisms behind p53-dependent proapoptotic pathway.
Since the sequence-specific DNA-binding ability of p53 is tightly linked to its proapoptotic activity [48, 49], the genomic integrity of p53 gene encoding the sequence-specific DNA-binding domain (exons 5–8) is particularly important. Extensive mutation search revealed that over 50% of human tumors carry p53 mutations [20, 21]. Among these mutations, 95% of them occurred within the genomic region encoding the sequence-specific DNA-binding domain of p53. These mutations disrupted the proper conformation of the sequence-specific DNA-binding domain of p53, and thus mutant forms of p53 lacked the sequence-specific transactivation ability. In contrast to the short-lived wild-type p53, mutant forms of p53 had a longer half-life [24, 25]. Moreover, mutant forms of p53 exhibited an oncogenic potential [50] and displayed the dominant-negative behavior toward wild-type p53 [26, 27], suggesting that mutant forms of p53 attenuate p53-dependent proapoptotic pathway (Figure 1).
Figure 1.

Dominant-negative effect of mutant p53 on wild-type p53. In response to cellular stresses, wild-type p53 is activated and induces cell cycle arrest and/or apoptotic cell death. Mutant p53 binds to wild-type p53 and inhibits its tumor suppressor function, thereby promoting tumor formation.
Since the previous mutation search for p53 gene focused on the genomic region encoding the central core sequence-specific DNA-binding domain of p53, it is likely that there could exist the unidentified mutations outside the central core sequence-specific DNA-binding domain. Indeed, Lomax et al. found point mutations (L344P and R337C) within the COOH-terminal oligomerization domain [51, 52]. Similarly, DiGiammarino et al. reported the presence of a point mutation (R337H) within the COOH-terminal oligomerization domain [53]. In addition to these mutations, we have found p53ΔC lacking a part of the COOH-terminal oligomerization domain and nuclear localization signals in human neuroblastoma-derived cell lines [54]. According to our results, p53ΔC was largely expressed in cytoplasm and had a significantly lower proapoptotic ability as compared with wild-type p53. Therefore, p53 mutations detected outside central core DNA-binding domain caused loss of function of p53.
From the clinical point of view, mutational inactivation of p53 was a frequent and serious molecular event in most human tumors. Since mutant forms of p53 had a longer half-life with oncogenic potential, exhibited the dominant-negative effect on wild-type p53, and thus led to chemoresistance, it is required to develop novel anticancer therapeutic approaches to suppress mutant forms of p53 or rescue wild-type p53 activity from mutant p53. In this regard, a small compound termed PRIMA-1, which has an ability to reactivate mutant p53, might be one of the promising and efficient anticancer drugs [55].
3. p53-Target Gene Products
To date, numerous genes have been identified as p53-target genes [22, 34–36]. In this section, we would like to describe the functional significances of several representative p53-target gene products. Since the inductions of cell cycle arrest and apoptotic cell death are the major roles of p53, p53-target gene products are closely involved in these cellular processes.
El-Deiry et al. identified p53-target gene termed p21WAF1 by using a subtractive hybridization approach [56]. p21WAF1 gene promoter contained a p53-responsive element, and its gene product had an ability to suppress cell growth. Alternatively, Harper et al. discovered p21CIP1 as a Cdk2- (cyclin-dependent kinase 2-) binding partner by employing a yeast-based two-hybrid procedure [57]. p21CIP1 tightly bound to Cdk2 and inhibited its protein kinase activity to block the phosphorylation of pRB. Noda et al. identified p21SD1 by using an expression screening from senescent human diploid fibroblasts [58]. p21SD1 blocked DNA synthesis and maintained the senescent phenotype. Surprisingly, these gene products were identical. Now, we call it p21WAF1. In response to cellular stresses, p53 induces G1 cell cycle arrest through the upregulation of p21WAF1.
Tanaka et al. employed a differential display approach to isolate a novel p53-inducible gene termed p53R2 whose gene product was highly homologous to ribonucleotide reductase small subunit (R2) [59]. p53R2 was significantly induced in response to DNA damage in a p53-dependent manner. Intriguingly, p53R2 induced G2/M arrest and was directly involved in repair of damaged DNA.
Under normal condition, p53 was kept at an extremely low level. MDM2 (murine double minute 2), which has an intrinsic E3 ubiquitin protein ligase activity, controlled the expression level of p53 by targeting it to ubiquitin-/proteasome-dependent degradation [60–62]. Barak et al. described that MDM2 is a direct transcriptional target of p53 [63]. Thus, MDM2 participates in a negative autoregulatory feedback loop which controls p53 expression level (see below).
Among p53-target genes, those that encode mitochondrial proteins are particularly attractive, because p53-dependent apoptosis appears to proceed through mitochondrial dysfunction. Selvakumaran et al. reported that BAX (Bcl2-associated X protein) is an immediate early p53-responsive gene [64]. BAX contains two highly conserved Bcl2 homology 1 and 2 (BH1 and BH2) domains and displayed a dominant-negative effect over prosurvival Bcl2. Upon apoptotic stimuli, BAX, which resides on the mitochondrial outer membrane, dysregulated the mitochondrial outer membrane permeability and induced the release of cytochrome c from the mitochondrial intermembrane space to cytosol [65].
p53AIP1 (p53-regulated apoptosis-inducing protein 1), NOXA (Latin for damage), and PUMA (p53 upregulated modulator of apoptosis), which were included in BH-3 domain-containing mitochondrial Bcl2 family, were direct transcriptional target gene products of p53 [22, 34–36]. For p53AIP1, DNA damage-mediated induction of p53AIP1 was tightly associated with p53-dependent apoptotic cell death and phosphorylation of p53 at Ser-46. Matsuda et al. described that p53AIP1 promotes downregulation of the mitochondrial membrane potential through the direct interaction with Bcl2 and induces the release of cytochrome c [66].
NOXA has been rediscovered in a differential display approach [67]. Forced expression of NOXA induced cytochrome c release from mitochondria followed by subsequent caspase activation, and cells underwent apoptotic cell death. The intact BH 3 domain of NOXA was required for the induction of apoptotic cell death.
PUMA has been identified by using a microarray analysis [68, 69]. PUMA was induced in response to ADR (adriamycin), and p53-responsive element was found within intron 1 of PUMA gene. Indeed, PUMA was one of the transcriptional target genes of p53. PUMA was localized to mitochondria and induced apoptotic cell death. Like p53AIP1, PUMA interacted with Bcl2 and functioned to induce cytochrome c release, thereby activating caspase 9 and 3. Jeffers found that BAX is required for PUMA-mediated apoptotic cell death, placing BAX downstream of PUMA [70].
4. Stability Control of p53
Steady-state expression level of the endogenous p53 is maintained at extremely low level, keeping this dangerous protein in an inactive state. The expression level of p53 is dependent on a balance between protein production and degradation. It has been well-documented that p53 is rapidly induced at protein level in response to a variety of cellular stresses such as DNA damage [22, 34–36]. This accumulation is largely due to a significant increase in its protein stability. The proteolytic degradation of p53 was mediated by the physical interaction between p53 and oncogenic MDM2 [60–62]. MDM2, which is a RING-finger type E3 ubiquitin protein ligase, bound to NH2-terminal transactivation domain of p53, ubiquitylated COOH-terminal 6 Lys residues (Lys-370, Lys-372, Lys-373, Lys-381, Lys-382, and Lys-386), and thereby targeting p53 for proteasome-dependent degradation [71]. MDM2 masked NH2-terminal transactivation domain of p53 and thereby inhibiting its transcriptional activity [72]. A small compound termed Nutlin, which bound to p53-binding pocket of MDM2, inhibited the interaction between p53 and MDM2, and thereby stabilizing p53 followed by activation of p53-dependent proapoptotic pathway [73]. Thus, this small compound might provide a novel strategy for cancer therapy.
In general, polyubiquitin chains target proteins to proteasome and initiate the process of proteolytic degradation. Recently, Kulikov et al. found that MDM2 associated with several subunits of proteasome, suggesting that MDM2 might promote not only the ubiquitylation of p53 but also recruit the ubiquitylated forms of p53 into proteasome [74]. In addition to MDM2, RING-finger type E3 ubiquitin protein ligases Pirh2 (p53-induced RING H2 domain protein) [75] and COP1 (constitutive photomorphogenic 1) [76] also interacted with p53 and mediated the ubiquitin-/proteasome-dependent degradation of p53. As expected, all of them inhibited transcriptional as well as proapoptotic function of p53. Since MDM2, Pirh2, and COP1 were p53-induced target gene products, they participated in a negative autoregulatory feedback loop which controls p53. Alternatively, p53-interacting protein termed HAUSP (herpes virus-associated ubiquitin-specific protease) had an intrinsic enzymatic activity to deubiquitylate p53 and thereby increasing its stability [77].
5. Posttranslational Modification
The induction and activation of p53 in response to cellular stresses have been shown to be largely regulated at posttranslational level through multiple mechanisms. Upon cellular stresses, p53 is phosphorylated at Ser-15, Ser-20, and Ser-46 [22, 34–36]. NH2-terminal phosphorylation of p53 converted p53 from latent form to active and stable one. On the other hand, protein phosphatases PP-1 and PP2A had an ability to dephosphorylate p53 and negatively modulated its activity [78, 79]. Ser-15 was phosphorylated by ATM (ataxia-telangiectasia mutated) [80], ATR (ataxia-telangiectasia mutated and Rad3-related) [81], Chk1 (checkpoint kinase 1) [82], and DNA-PK (DNA-dependent protein kinase) [83]. Ser-20 was phosphorylated by Chk2 (checkpoint kinase 2) [84] and Plk3 (polo-like kinase 3) [85]. In addition, HIPK2 (homeodomain interacting protein kinase 2) and PKCdelta (protein kinase C delta) have been considered to be involved in phosphorylation of p53 at Ser-46 [86, 87]. As mentioned above, MDM2 bound to NH2-terminal transactivation domain of p53 to destabilize and also inactivate p53. Stress-induced NH2-terminal phosphorylation of p53 promoted the dissociation of MDM2 from MDM2/p53 complex and led to the stabilization and activation of p53 [22, 34–36]. Alternatively, COOH-terminal region of p53 was also phosphorylated. For example, CKII (Casein kinase II) phosphorylated p53 at Ser-392 [88], and PKC phosphorylated p53 at Ser-371, Ser-376, and Ser-378 [89]. These COOH-terminal phosphorylations enhanced the sequence-specific DNA-binding activity of p53 [22, 34–36]. It has been shown that COOH-terminal region of p53 acts as a negative regulator and might mask its sequence-specific DNA-binding domain in a latent conformation [90, 91]. It is likely that phosphorylation of COOH-terminal region of p53 might lead to a conformational shift that enables p53 to bind more efficiently to its target motif, and thereby enhancing its sequence-specific transactivation function. In contrast, Plk11 (polo-like kinase 1) inhibited transcriptional as well as proapoptotic function through physical interaction and phosphorylation [92]. Therefore, phosphorylation of p53 does not always act as an activation signal.
In addition to stress-induced phosphorylation of p53, p53 was subjected to the extensive acetylation mediated by p300 with intrinsic histone acetyltransferase activity [93]. p300 which acts as a ubiquitous transcriptional coactivator, bound to NH2-terminal region of p53 and promoted its acetylation of a cluster of COOH-terminal Lys residues (Lys-370, Lys-372, Lys-373, Lys-392, and Lys-381) [37]. PCAF (p300/CBP-associated factor), another histone acetyltransferase, had an ability to acetylate Lys-320 of p53 [94]. Intriguingly, theses COOH-terminal Lys residues were the sites for ubiquitin ligation. It is likely that p53 acetylation catalyzed by p300 reduces its ubiquitylation levels by competition between acetylation and ubiquitylation. Thus, p300-mediated acetylation of p53 increased the stability of p53 and enhanced the transcriptional as well as proapoptotic activity of p53 [95]. Consistent with this notion, SIRT1 (Silent mating type information regulation 2 homolog 1), which has an intrinsic deacetylase activity, interacted with p53 and attenuated p53-dependent cell cycle arrest as well as apoptotic cell death in response to DNA damage through deacetylation of Lys-382 [96].
Kawai et al. described that p300 has a dual role in the regulation of p53 stability [97]. According to their results, p300 acted as a positive regulator to increase p53 stability in the presence of lower level of MDM2, whereas p300 became a negative regulator for p53 to induce MDM2-dependent degradation in the presence of higher level of MDM2. Surprisingly, Grossman et al. found that, in addition to histone acetyltransferase activity, p300 has an E4 ubiquitin protein ligase activity which catalyzes polyubiquitylation of monoubiquitylated precursor p53 [98].
Sumoylation is the other type of posttranslational modification. SUMO (SUMO-1, SUMO-2, and SUMO-3) was a ubiquitin-related small protein which covalently binds to substrates through a mechanism similar to ubiquitylation. PIAS (protein inhibitor of activated STAT) family acted as a SUMO E3 ligase for p53 [99]. Unlike ubiquitylation, the modification by SUMO-1 did not target proteins for proteolytic degradation. Previous studies suggest that sumoylation targets p53 to the nucleoli, and sumoylated p53 tightly binds to chromatin structure. Sumoylation of p53 at Lys-386 resulted in a loss of a sequence-specific DNA-binding ability and thus inhibited its transcriptional activity, although sumoylated p53 bound to coactivator p300 [100].
Previously, Jackson and Tjian demonstrated that O-linked glycosylation enhances the sequence-specific transcriptional activity of Sp1 [101]. Close inspection of the amino acid residues of wild-type p53 revealed that there exists a putative O-linked glycosylation site within the COOH-terminal basic region. Shaw et al. described that O-linked glycosylation enhances sequence-specific transcriptional activity of p53, which might be due to the disruption of intramolecular interaction between the COOH-terminal inhibitory domain and sequence-specific DNA-binding domain [102].
6. Transcriptional Regulation
As described above, p53 is largely regulated at protein level through chemical modifications such as phosphorylation and acetylation. On the other hand, p53 expression is also regulated at transcriptional level in some cases. Previously, Reich and Levine found that p53 is transcriptionally regulated in response to mitogen stimulation and serum starvation [103]. Bruno et al. reported that p53 is transactivated in response to anticancer drug ADR [104]. Raman et al. described that homeobox protein HOXA5 acts as a transcriptional activator for p53 [105]. In addition, Noda et al. identified the cis-acting element termed PE21 at a nucleotide position from −79 to −60 (relative to the first transcriptional initiation site) within the p53 promoter region responsible for p53 gene basal as well as inducible expression in response to UV [106].
Recently, we have found that AMPK (AMP-activated protein kinase), which acts as an intracellular energy sensor by monitoring cellular energy levels, plays an important role in the regulation of apoptotic cell death in response to glucose deprivation [107]. According to our results, the activated form of AMPK was closely involved in the transcriptional activation of p53 under low glucose conditions. It has been shown that the activation of AMPK affects the gene expression, suggesting that AMPK itself and/or AMPK-containing cellular complex might have a transactivation potential [108]. Subsequent studies demonstrated that there exists a putative AMPK-responsive element within the genomic sequence between −531 and −239 relative to the transcriptional initiation site of p53. Within this region, we have found out canonical CREB- (cAMP-responsive element-binding protein-) binding site (5′-ATTACGGAA-3′). Finally, we have shown that AMPK collaborates with CREB through CREB-binding site to transactivate p53 in response to energetic stress [109]. Therefore, in addition to posttranslational modification, p53 is transcriptionally regulated in response to a certain subset of cellular stresses.
7. Subcellular Localization
Appropriate subcellular localization is critical for regulating function of p53. p53 acts as a sequence-specific transcription factor in cell nucleus. In addition to the mutational inactivation of p53, the abnormal cytoplasmic localization of p53 resulted in loss of function of p53. In contrast to the other human tumors, p53 was rarely mutated in human neuroblastoma [110]. Moll et al. found that wild-type p53 is detectable in cytoplasm of the majority of undifferentiated neuroblastoma, suggesting that the inability of nuclear access of p53 attenuates its tumor suppressor activity [111]. Subsequent study demonstrated that this abnormal cytoplasmic localization of wild-type p53 is due to the hyperactive nuclear export of p53 through the exposure of highly conserved COOH-terminal NES [112]. Intriguingly, Becker et al. described that the hyperubiquitylation of p53 contributes to its aberrant cytoplasmic retention in neuroblastoma in association with the impaired interaction between p53 and HAUSP which catalyzes the deubiquitylation of p53 [113]. In addition, MDM2 had an ability to significantly enhance nuclear export of p53 through its COOH-terminal NES [114].
By using an affinity purification strategy, Nikolaev et al. discovered a large cytoplasmic protein termed Parc (p53-associated, Parlin-like cytoplasmic protein), which associated with cytoplasmic p53 [115]. According to their results, NH2-terminal region of Parc interacted with the COOH-terminal region of p53. Parc had an intrinsic E3 ubiquitin ligase activity; however, Parc had an undetectable effect on the steady-state expression level of p53. Importantly, Parc was associated with the majority of cytoplasmic p53 and acted as a cytoplasmic anchor protein for p53. Indeed, depletion of Parc promoted nuclear localization of p53 and induced p53-dependent apoptotic cell death in neuroblastoma cells. Recently, it has been shown that the introduction of the COOH-terminal peptide of p53 containing Parc-interacting region disrupts the interaction between p53 and Parc in cytoplasm and results in the nuclear relocalization of p53 [116]. The treatment of this small peptide in cancerous cells increased the sensitivity of cancerous cells to anticancer drug and enhanced p53-dependent proapoptotic pathway.
It is worth noting that, in response to genotoxic stress, a certain fraction of p53 translocates to mitochondria, where p53 collaborates with BclXL and Bcl2 to induce permeabilization of the outer mitochondrial membrane, and thereby releasing cytochrome c [117]. Further study revealed that MDM2-mediated monoubiquitylation of p53 promotes the recruitment of p53 to mitochondria in which p53 undergoes a rapid deubiquitylation catalyzed by mitochondrial HAUSP, generating nonubiquitylated proapoptotic p53 [118]. Thus, targeting p53 to mitochondria, which resulted in the dysfunction of mitochondria, might be one of the transcription-independent proapoptotic pathways mediated by p53.
8. Variant Forms of p53
Yin et al. detected full-length wild-type p53 and another p53 with a relative molecular mass of 47 KDa termed p53/47 [119]. p53/47 was detectable by the 421 monoclonal antibody which recognized COOH-terminal portion of p53 and also detectable by the 1801 antibody which recognized an epitope between amino acid residues 46 and 55. On the other hand, the DO-1 (amino acid residues 20–25) and the DO-13 (amino acid residues 26–35) failed to recognize p53/47. Subsequent study demonstrated that p53/47 is generated from the NH2-terminal alternative initiation site (Met-40). Since p53/47 lacked an NH2-terminal MDM2-binding domain, it was not targeted for proteasome-dependent degradation by MDM2. Previous studies indicate that NH2-terminal transactivation domain of p53 is divided into two independent domains such as TA 1 (amino acid residues 1–40) and TA II (amino acid residues 43–63) [120, 121]. p53/47 lacked TA I domain but contained TA II domain. Of note, p53/47 failed to transactivate p21WAF1 but was able to induce the transcription of MDM2, GADD45, and BAX. These observations suggest that TA I and TA II domains contribute to enhance the specificity of p53-target promoter usage [119].
In addition to the alternative translation product of p53, Bourdon et al. found the presence of multiple variant forms of p53 arising from alternative promoter usage and alternative splicing events [122]. Based on their results, they identified the alternative promoter located within intron 4, and mRNA transcribed from this internal promoter generated NH2-terminally truncated form of p53 initiated at codon 133 (Δ133p53), which lacked NH2-terminal transactivation domain and Pro-rich domain. Further studies demonstrated that the alternative splicing of intron 9 results in the generation of p53β and p53γ, which deleted the COOH-terminal oligomerization domain. Thus, p53 is expressed as multiple variants including p53, p53β, p53γ, Δ133p53, Δ133p53β, Δ133p53γ, Δ40p53, Δ40p53β, and Δ40p53γ. Δ40p53 corresponds to p53/47 (Figure 2).
Figure 2.

Schematic diagram of p53 variants. TA: transactivation domain; DB: sequence-specific DNA-binding domain; OD: oligomerization domain.
Immunostaining experiments revealed that most of these p53 variants were localized largely in cell nucleus, whereas p53γ was detectable both in cell nucleus and cytoplasm. Additionally, Δ133p53γ was localized exclusively in cytoplasm. p53 variants had an ability to bind differentially to p53-responsive promoters and modulated p53-target gene expression. For example, p53β bound preferentially to BAX and p21WAF1 promoters rather than MDM2 promoter, whereas p53 bound preferentially to p21WAF1 and MDM2 promoters than to BAX promoter [122].
9. DNA Damage Response
During the early phase of DNA damage response, the activated forms of ATM (phospho-ATM at Ser-1981) phosphorylated histone variant H2AX at Ser-139 (γH2AX). This phosphorylation event took place at a large chromatin region surrounding DNA lesions and formed the nuclear foci, suggesting that γH2AX acts as a sensitive marker for the presence of DNA damage [123–125]. The disruption of H2AX resulted in an induction of genome instability and DNA double-strand break repair defects [126]. Then, NFBD1/MDC1 (nuclear factor with BRCT domain 1/mediator of DNA damage checkpoint protein 1), which had an antiapoptotic potential [127], interacted directly with γH2AX through its COOH-terminal BRCT domains and recruited MRN (MRE11, Rad50, and NBS1) complex onto the sites of DNA damage to facilitate the efficient DNA repair [128–130]. γH2AX then served as a platform for the recruitment of DNA checkpoint signaling factors as well as multifunctional MRN complex. Therefore, NFBD1-mediated local accumulation of DNA repair machinery such as MRN complex at the chromatin regions flanking the sites of DNA damage contributed to an increase in the fidelity of genomic integrity in response to DNA damage. Indeed, NFBD1-deficient mice exhibited chromosome instability, DNA repair defects, and radiation sensitivity [131]. In response to DNA damage, cell cycle checkpoint was activated to arrest cells at G1 phase, giving them time to repair damaged DNA. Recently, we have found that, during the early phase of DNA damage response, NFBD1 binds to NH2-terminal region of p53 to inhibit ATM-mediated p53 phosphorylation at Ser-15, and thereby blocking its proapoptotic activity [132]. During the late phase of DNA damage response, the expression level of NFBD1 is sharply downregulated, and then the activated forms of ATM phosphorylate free p53 at Ser-15 to enhance its proapoptotic activity [132].
Runt-related (RUNX) gene family is composed of three members including RUNX1, RUNX2, and RUNX3 [133]. Among them, RUNX3 has been considered to be a candidate tumor suppressor for human gastric cancer [134]; however, it remains unclear how RUNX3 exerts its tumor suppressor function. Recently, we have found that, in response to ADR treatment, RUNX3 is induced to accumulate in cell nucleus and binds to p53 [135]. Based on our results, RUNX3 acted as a coactivator for p53 to enhance its transcriptional and proapoptotic activities. Of note, knocking down of the endogenous RUNX3 significantly repressed ADR-mediated phosphorylation of p53 at Ser-15. Since RUNX3 had an ability to interact with activated forms of ATM, it is likely that RUNX3 recruits the activated forms of ATM to latent form of p53 and thereby assisting ATM-dependent phosphorylation of p53 at Ser-15 in response to ADR. Thus, RUNX3 is closely involved at least in part in the regulation of DNA damage-mediated activation of p53 (Figure 3).
Figure 3.

RUNX3 acts as a coactivator for p53 in response to DNA damage. In response to DNA damage, RUNX3 collaborates with activated form of ATM and induces phosphorylation of p53 at Ser-15. Phosphorylated form of p53 transactivates proapoptotic BAX and PUMA.
10. p53 Family
p53 tumor suppressor family is composed of three members including p53, p73 and p63. Like p53, p73, and p63 act as sequence-specific nuclear transcription factors and induce cell cycle arrest and/or apoptotic cell death in response to a certain subset of cellular stresses. p73 is expressed as multiple splicing variants with different COOH-terminal structures arising from the alternative splicing of the primary transcript such as p73α, p73β, p73γ, p73δ, p73ε, and p73ζ (37, 40, 135). Since all of them contain an intact NH2-terminal transactivation domain, they have an ability to transactivate the overlapping set of p53-target gene (TA variants). Intriguingly, p73 encodes the NH2-terminally truncated variants termed ΔNp73 [136]. Since ΔNp73 lacked the intact NH2-terminal transactivation domain, ΔNp73 was the transactivation defective and acted as the dominant-negative inhibitor toward TAp73 and wild-type p53 [136]. In this connection, ΔNp73 had an oncogenic potential [137]. Additionally, mutant forms of p53 inhibited the transcriptional and proapoptotic activities of TAp73 [138]. Of note, we and others demonstrated that TAp73 induces the transcription of its own inhibitor ΔNp73, creating a dominant-negative feedback loop which regulates the proapoptotic activities of both TAp73 and wild-type p53 [139–141]. Similarly, p63 encodes TAp63 and ΔNp63 [39]. Therefore, relative ratio of TA and ΔN variants might be an important determinant of the cell fate.
Since p73 and p63 induce apoptotic cell death in cancerous cells, extensive mutation searches were performed. In a sharp contrast to p53, p73 and p63 were rarely mutated in various human tumor tissues [142], suggesting that p73 and p63 might not be classical Knudson-type tumor suppressors. Initial genetic studies revealed that p73-deficient mice and also p63-deficient mice do not develop spontaneous tumors [143–145]. Instead, p73-deficient mice displayed severe developmental defects including hydrocephalus, hippocampal dysgenesis, and abnormal pheromone sensory pathways. In p63-deficient mice, differentiation of apical ectodermal ridge failed, and generation of skin was not observed. These results suggest that p73 as well as p63 plays an important role in normal development. Strikingly, it has been shown that p73 and p63 heterozygous mice develop malignant tumors at high frequency. Tumors derived from these mice exhibited loss of the remaining wild-type allele at high frequency. These observations strongly suggest that loss of p73 and/or p63 function causes tumor development [146].
p73 and p63 are regulated at transcription as well as posttranslation levels. It has been shown that there exist several E2F1-binding sites within p73 promoter region, and E2F1 actually acts as a transcriptional activator for p73 [147–149]. Recently, Logotheti et al. demonstrated that Sp1 binds to the external promoter of p73 gene and induces the expression of p73 [150]. On the other hand, Fontemaggi et al. found that ZEB (zinc finger/homeodomain repressor) acts as a transcriptional repressor for p73 [151]. Of note, we have described that TATA-binding protein- (TBP-) like protein (TLP) has an ability to directly transactivate p63 [43].
Like p53, the activity and stability of p73 and p63 is regulated by chemical modifications such as phosphorylation. c-Abl-dependent phosphorylation of p73 at Tyr-99 resulted in an increase in its stability [152–154]. PKCδ phosphorylated p73 at Ser-289 to enhance its transcriptional activity [155]. Chk1-mediated phosphorylation of p73 at Ser-47 resulted in an enhancement of its transcriptional activity [156]. Recently, we have found that, in response to cisplatin (CDDP), IKK-α (IκB kinase-α) is induced to be accumulated in cell nucleus and interacts with p73 to increase its stability, thereby enhancing its proapoptotic activity in a p53-independent manner [157, 158]. IKK-α had an ability to phosphorylate the NH2-terminal portion of p73 (Figure 4). On the other hand, CDK-mediated phosphorylation of p73 led to a significant inhibition of its transcriptional activity [159]. In addition, we have demonstrated that p73 is strongly inhibited by Plk1 through physical interaction and phosphorylation at Thr-27 [160]. Therefore, phosphorylation of p73 might not always act as an activation signal. Similarly, we have found that p63 is inhibited by Plk1-mediated phosphorylation at Ser-52 [161].
Figure 4.
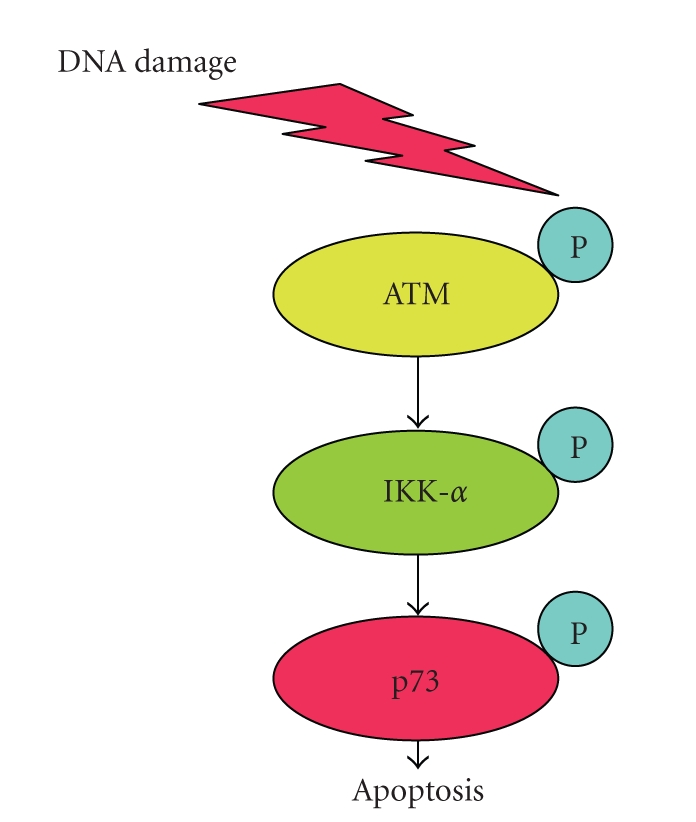
Upon DNA damage, activated form of ATM phosphorylates IKK-α and promotes nuclear accumulation of IKK-α. IKK-α enhances transactivation as well as proapoptotic function of p73 in a p53-independent manner.
Since p73 and p63 were rarely mutated in human tumors, elucidation of the precise regulatory mechanisms behind DNA damage-induced activation of p73 and p63 might provide a clue to develop a novel strategy for the treatment of malignant tumors bearing nonfunctional p53.
11. Future Perspective
p53 plays a pivotal role in the regulation of cell fate determination in response to a variety of cellular stresses. Dysfunction of p53 such as mutational inactivation permits the abnormal cell growth and finally results in the malignant tumor development. In addition, loss of function of p53 contributes to the significant decrease in the sensitivity of tumor cells to anticancer drugs. Therefore, the elucidation of the precise molecular mechanisms behind stress-induced activation of p53 might provide a clue(s) to find out the attractive therapeutic target(s) for cancer treatment. In contrast to p73 and p63, p53 is frequently mutated in human tumors. Mutant forms of p53 lack their proapoptotic function and display a dominant-negative behavior toward wild-type p53 family. Further efforts should be required to develop the novel strategies and/or the chemical compounds which could convert the abnormal conformations of mutant p53 to normal ones. In this connection, one of the remaining questions is that why p53 is frequently mutated in tumor tissues. Although it could be due to the serious defects in the DNA repair machinery in these tumors, further studies should be required to address this issue.
References
- 1.DeLeo AB, Jay G, Appella E. Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proceedings of the National Academy of Sciences of the United States of America. 1979;76(5):2420–2424. doi: 10.1073/pnas.76.5.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kress M, May E, Cassingena R, May P. Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. Journal of Virology. 1979;31(2):472–483. doi: 10.1128/jvi.31.2.472-483.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lane DP, Crawford LV. T antigen is bound to a host protein in SV40 transformed cells. Nature. 1979;278(5701):261–263. doi: 10.1038/278261a0. [DOI] [PubMed] [Google Scholar]
- 4.Linzer DIH, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40 transformed cells and uninfected embryonal carcinoma cells. Cell. 1979;17(1):43–52. doi: 10.1016/0092-8674(79)90293-9. [DOI] [PubMed] [Google Scholar]
- 5.Melero JA, Stitt DT, Mangel WF, Carroll RB. Identification of new polypeptide species (48–55K) immunoprecipitable by antiserum to purified large T antigen and present in SV40-infected and -transformed cells. Virology. 1979;93(2):466–480. doi: 10.1016/0042-6822(79)90250-2. [DOI] [PubMed] [Google Scholar]
- 6.Levine AJ, Momand J, Finlay CA. The p53 tumour suppressor gene. Nature. 1991;351(6326):453–456. doi: 10.1038/351453a0. [DOI] [PubMed] [Google Scholar]
- 7.Finlay CA, Hinds PW, Levine AJ. The p53 proto-oncogene can act as a suppressor of transformation. Cell. 1989;57(7):1083–1093. doi: 10.1016/0092-8674(89)90045-7. [DOI] [PubMed] [Google Scholar]
- 8.Eliyahu D, Michalovitz D, Eliyahu S, Pinhasi-Kimhi O, Oren M. Wild-type p53 can inhibit oncogene-mediated focus formation. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(22):8763–8767. doi: 10.1073/pnas.86.22.8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baker SJ, Markowitz S, Fearon ER, Willson JKV, Vogelstein B. Suppression of human colorectal carcinoma cell growth by wild-type p53. Science. 1990;249(4971):912–915. doi: 10.1126/science.2144057. [DOI] [PubMed] [Google Scholar]
- 10.Diller L, Kassel J, Nelson CE, et al. p53 functions as a cell cycle control protein in osteosarcomas. Molecular and Cellular Biology. 1990;10(11):5772–5781. doi: 10.1128/mcb.10.11.5772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michalovitz D, Halevy O, Oren M. Conditional inhibition of transformation and of cell proliferation by a temperature-sensitive mutant of p53. Cell. 1990;62(4):671–680. doi: 10.1016/0092-8674(90)90113-s. [DOI] [PubMed] [Google Scholar]
- 12.Martinez J, Georgoff I, Martinez J, Levine AJ. Cellular localization and cell cycle regulation by a temperature-sensitive p53 protein. Genes and Development. 1991;5(2):151–159. doi: 10.1101/gad.5.2.151. [DOI] [PubMed] [Google Scholar]
- 13.Baker SJ, Fearon ER, Nigro JM, et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science. 1989;244(4901):217–221. doi: 10.1126/science.2649981. [DOI] [PubMed] [Google Scholar]
- 14.Nigro JM, Baker SJ, Preisinger AC, et al. Mutations in the p53 gene occur in diverse human tumour types. Nature. 1989;342(6250):705–708. doi: 10.1038/342705a0. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi T, Nau MM, Chiba I, et al. p53: a frequent target for genetic abnormalities in lung cancer. Science. 1989;246(4929):491–494. doi: 10.1126/science.2554494. [DOI] [PubMed] [Google Scholar]
- 16.Kelman Z, Prokocimer M, Peller S, et al. Rearrangements in the p53 gene in Philadelphia chromosome positive chronic myelogenous leukemia. Blood. 1989;74(7):2318–2324. [PubMed] [Google Scholar]
- 17.Vogelstein B. Cancer. A deadly inheritance. Nature. 1990;348(6303):681–682. doi: 10.1038/348681a0. [DOI] [PubMed] [Google Scholar]
- 18.Iggo R, Gatter K, Bartek J, Lane D, Harris AL. Increased expression of mutant forms of p53 oncogene in primary lung cancer. Lancet. 1990;335(8691):675–679. doi: 10.1016/0140-6736(90)90801-b. [DOI] [PubMed] [Google Scholar]
- 19.Donehower LA, Harvey M, Slagle BL, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356(6366):215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 20.Hollstein M, Shomer B, Greenblatt M, et al. Somatic point mutations in the p53 gene of human tumors and cell lines: updated compilation. Nucleic Acids Research. 1996;24(1):141–146. doi: 10.1093/nar/24.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hollstein M, Hergenhahn M, Yang Q, Bartsch H, Wang Z-Q, Hainaut P. New approaches to understanding p53 gene tumor mutation spectra. Mutation Research. 1999;431(2):199–209. doi: 10.1016/s0027-5107(99)00162-1. [DOI] [PubMed] [Google Scholar]
- 22.Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nature Reviews Cancer. 2002;2(8):594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 23.Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994;265(5170):346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 24.Crawford LV, Pim DC, Lamb P. The cellular protein p53 in human tumours. Molecular Biology and Medicine. 1984;2(4):261–272. [PubMed] [Google Scholar]
- 25.Cattretti G, et al. p53 expression in breast cancer. International Journal of Cancer. 1986;41:178–183. doi: 10.1002/ijc.2910410204. [DOI] [PubMed] [Google Scholar]
- 26.Kern SE, Pietenpol JA, Thiagalingam S, Seymour A, Kinzler KW, Vogelstein B. Oncogenic forms of p53 inhibit p53-regulated gene expression. Science. 1992;256(5058):827–830. doi: 10.1126/science.1589764. [DOI] [PubMed] [Google Scholar]
- 27.Hachiya M, Chumakov A, Miller CW, Akashi M, Said J, Koeffler HP. Mutant p53 proteins behave in a dominant, negative fashion in vivo. Anticancer Research. 1994;14(5):1853–1859. [PubMed] [Google Scholar]
- 28.Rovinski B, Benchimol S. Immortalization of rat embryo fibroblasts by the cellular p53 oncogene. Oncogene. 1988;2(5):445–452. [PubMed] [Google Scholar]
- 29.Eliyahu D, Goldfinger N, Pinhasi-Kimhi O, et al. Fibrosarcoma cells express two transforming mutant p53 species. Oncogene. 1988;3(3):313–321. [PubMed] [Google Scholar]
- 30.Green MR. When the products of oncogenes and anti-oncogenes meet. Cell. 1989;56(1):1–3. doi: 10.1016/0092-8674(89)90975-6. [DOI] [PubMed] [Google Scholar]
- 31.Vogelstein B, Kinzler KW. p53 Function and dysfunction. Cell. 1992;70(4):523–526. doi: 10.1016/0092-8674(92)90421-8. [DOI] [PubMed] [Google Scholar]
- 32.Velculescu VE, El-Deiry WS. Biological and clinical importance of the p53 tumor suppressor gene. Clinical Chemistry. 1996;42(6):858–868. [PubMed] [Google Scholar]
- 33.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408(6810):307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 34.Sionov RV, Haupt Y. The cellular response to p53: the decision between life and death. Oncogene. 1999;18(45):6145–6157. doi: 10.1038/sj.onc.1203130. [DOI] [PubMed] [Google Scholar]
- 35.Prives C, Hall PA. The P53 pathway. Journal of Pathology. 1999;187(1):112–126. doi: 10.1002/(SICI)1096-9896(199901)187:1<112::AID-PATH250>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 36.El-Deiry WS. The role of p53 in chemosensitivity and radiosensitivity. Oncogene. 2003;22(47):7486–7495. doi: 10.1038/sj.onc.1206949. [DOI] [PubMed] [Google Scholar]
- 37.Lacroix M, Toillon R-A, Leclercq G. p53 and breast cancer, an update. Endocrine-Related Cancer. 2006;13(2):293–325. doi: 10.1677/erc.1.01172. [DOI] [PubMed] [Google Scholar]
- 38.Kaghad M, Bonnet H, Yang A, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90(4):809–819. doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- 39.Yang A, Kaghad M, Wang Y, et al. p63, a p53 homolog at 3q27–29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Molecular Cell. 1998;2(3):305–316. doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]
- 40.Melino G, De Laurenzi V, Vousden KH. p73: friend or foe in tumorigenesis. Nature Reviews Cancer. 2002;2(8):605–615. doi: 10.1038/nrc861. [DOI] [PubMed] [Google Scholar]
- 41.Ozaki T, Nakagawara A. p73, a sophisticated p53 family member in the cancer world. Cancer Science. 2005;96(11):729–737. doi: 10.1111/j.1349-7006.2005.00116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Irwin MS, Kondo K, Marin MC, Cheng LS, Hahn WC, Kaelin WG., Jr. Chemosensitivity linked to p73 function. Cancer Cell. 2003;3(4):403–410. doi: 10.1016/s1535-6108(03)00078-3. [DOI] [PubMed] [Google Scholar]
- 43.Suenaga Y, Ozaki T, Tanaka Y, et al. TATA-binding protein (TBP)-like protein is engaged in etoposide-induced apoptosis through transcriptional activation of human TAp63 gene. Journal of Biological Chemistry. 2009;284(51):35433–35440. doi: 10.1074/jbc.M109.050047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fabbro M, Henderson BR. Regulation of tumor suppressors by nuclear-cytoplasmic shuttling. Experimental Cell Research. 2003;282(2):59–69. doi: 10.1016/s0014-4827(02)00019-8. [DOI] [PubMed] [Google Scholar]
- 45.Kim I-S, Kim D-H, Han S-M, et al. Truncated form of importin α identified in breast cancer cell inhibits nuclear import of p53. Journal of Biological Chemistry. 2000;275(30):23139–23145. doi: 10.1074/jbc.M909256199. [DOI] [PubMed] [Google Scholar]
- 46.Walker KK, Levine AJ. Identification of a novel p53 functional domain that is necessary for efficient growth suppression. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(26):15335–15340. doi: 10.1073/pnas.93.26.15335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baptiste N, Friedlander P, Chen X, Prives C. The proline-rich domain of p53 is required for cooperation with anti-neoplastic agents to promote apoptosis of tumor cells. Oncogene. 2002;21(1):9–21. doi: 10.1038/sj.onc.1205015. [DOI] [PubMed] [Google Scholar]
- 48.El-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nature Genetics. 1992;1(1):45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- 49.Pietenpol JA, Tokino T, Thiagalingam S, El-Deiry WS, Kinzler KW, Vogelstein B. Sequence-specific transcriptional activation is essential for growth suppression by p53. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(6):1998–2002. doi: 10.1073/pnas.91.6.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen P-L, Chen Y, Bookstein R, Lee W-H. Genetic mechanisms of tumor suppression by the human p53 gene. Science. 1990;250(4987):1576–1580. doi: 10.1126/science.2274789. [DOI] [PubMed] [Google Scholar]
- 51.Lomax ME, Barnes DM, Gilchrist R, Picksley SM, Varley JM, Camplejohn RS. Two functional assays employed to detect an unusual mutation in the oligomerisation domain of p53 in a Li-Fraumeni like family. Oncogene. 1997;14(15):1869–1874. doi: 10.1038/sj.onc.1201133. [DOI] [PubMed] [Google Scholar]
- 52.Lomax ME, Barnes DM, Hupp TR, Picksley SM, Camplejohn RS. Characterization of p53 oligomerization domain mutations isolated from Li-Fraumeni and Li-Fraumeni like family members. Oncogene. 1998;17(5):643–649. doi: 10.1038/sj.onc.1201974. [DOI] [PubMed] [Google Scholar]
- 53.DiGiammarino EL, Lee AS, Cadwell C, et al. A novel mechanism of tumorigenesis involving pH-dependent destabilization of a mutant p53 tetramer. Nature Structural Biology. 2002;9(1):12–16. doi: 10.1038/nsb730. [DOI] [PubMed] [Google Scholar]
- 54.Nakamura Y, Ozaki T, Niizuma H, Ohira M, Kamijo T, Nakagawara A. Functional characterization of a new p53 mutant generated by homozygous deletion in a neuroblastoma cell line. Biochemical and Biophysical Research Communications. 2007;354(4):892–898. doi: 10.1016/j.bbrc.2007.01.057. [DOI] [PubMed] [Google Scholar]
- 55.Wiman KG. Pharmacological reactivation of mutant p53: from protein structure to the cancer patient. Oncogene. 2010;29(30):4245–4252. doi: 10.1038/onc.2010.188. [DOI] [PubMed] [Google Scholar]
- 56.El-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75(4):817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 57.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75(4):805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 58.Noda A, Ning Y, Venable SF, Pereira-Smith OM, Smith JR. Cloning of senescent cell-derived inhibitors of DNA synthesis using an expression screen. Experimental Cell Research. 1994;211(1):90–98. doi: 10.1006/excr.1994.1063. [DOI] [PubMed] [Google Scholar]
- 59.Tanaka H, Arakawa H, Yamaguchi T, et al. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature. 2000;404(6773):42–49. doi: 10.1038/35003506. [DOI] [PubMed] [Google Scholar]
- 60.Kubbutat MHG, Jones SN, Vousden KH. Regulation of p53 stability by MDM2. Nature. 1997;387(6630):299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 61.Haupt Y, Maya R, Kazaz A, Oren M. MDM2 promotes the rapid degradation of p53. Nature. 1997;387(6630):296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 62.Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Letters. 1997;420(1):25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- 63.Barak Y, Juven T, Haffner R, Oren M. MDM2 expression is induced by wild type p53 activity. EMBO Journal. 1993;12(2):461–468. doi: 10.1002/j.1460-2075.1993.tb05678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Selvakumaran M, Lin H-K, Miyashita T, et al. Immediate early up-regulation of bax expression by p53 but not TGFβ1: a paradigm for distinct apoptotic pathways. Oncogene. 1994;9(6):1791–1798. [PubMed] [Google Scholar]
- 65.Green DR. At the gates of death. Cancer Cell. 2006;9(5):328–330. doi: 10.1016/j.ccr.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 66.Matsuda K, Yoshida K, Taya Y, Nakamura K, Nakamura Y, Arakawa H. p53AIP1 regulates the mitochondrial apoptotic pathway. Cancer Research. 2002;62(10):2883–2889. [PubMed] [Google Scholar]
- 67.Oda E, Ohki R, Murasawa H, et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288(5468):1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- 68.Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Molecular Cell. 2001;7(3):673–682. doi: 10.1016/s1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- 69.Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Molecular Cell. 2001;7(3):683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- 70.Jeffers JR, Parganas E, Lee Y, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4(4):321–328. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- 71.Rodriguez MS, Desterro JMP, Lain S, Lane DP, Hay RT. Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Molecular and Cellular Biology. 2000;20(22):8458–8467. doi: 10.1128/mcb.20.22.8458-8467.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Prives C. Signaling to p53: breaking the MDM2-p53 circuit. Cell. 1998;95(1):5–8. doi: 10.1016/s0092-8674(00)81774-2. [DOI] [PubMed] [Google Scholar]
- 73.Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 74.Kulikov R, Letienne J, Kaur M, Grossman SR, Arts J, Blattner C. Mdm2 facilitates the association of p53 with the proteasome. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(22):10038–10043. doi: 10.1073/pnas.0911716107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Leng RP, Lin Y, Ma W, et al. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell. 2003;112(6):779–791. doi: 10.1016/s0092-8674(03)00193-4. [DOI] [PubMed] [Google Scholar]
- 76.Dornan D, Bheddah S, Newton K, et al. COP1, the negative regulator of p53, is overexpressed in breast and ovarian adenocarcinomas. Cancer Research. 2004;64(20):7226–7230. doi: 10.1158/0008-5472.CAN-04-2601. [DOI] [PubMed] [Google Scholar]
- 77.Li M, Chen D, Shiloh A, et al. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature. 2002;416(6881):648–653. doi: 10.1038/nature737. [DOI] [PubMed] [Google Scholar]
- 78.Haneda M, Kojima E, Nishikimi A, Hasegawa T, Nakashima I, Isobe K-I. Protein phosphatase 1, but not protein phosphatase 2A, dephosphorylates DNA-damaging stress-induced phospho-serine 15 of p53. FEBS Letters. 2004;567(2-3):171–174. doi: 10.1016/j.febslet.2004.04.066. [DOI] [PubMed] [Google Scholar]
- 79.Li H-H, Cai X, Shouse GP, Piluso LG, Liu X. A specific PP2A regulatory subunit, B56γ, mediates DNA damage-induced dephosphorylation of p53 at Thr55. EMBO Journal. 2007;26(2):402–411. doi: 10.1038/sj.emboj.7601519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Khanna KK, Keating KE, Kozlov S, et al. ATM associates with and phosphorylates p53: mapping the region of interaction. Nature Genetics. 1998;20(4):398–400. doi: 10.1038/3882. [DOI] [PubMed] [Google Scholar]
- 81.Tibbetts RS, Brumbaugh KM, Williams JM, et al. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes and Development. 1999;13(2):152–157. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Goudelock DM, Jiang K, Pereira E, Russell B, Sanchez Y. Regulatory interactions between the checkpoint kinase Chk1 and the proteins of the DNA-dependent protein kinase complex. Journal of Biological Chemistry. 2003;278(32):29940–29947. doi: 10.1074/jbc.M301765200. [DOI] [PubMed] [Google Scholar]
- 83.Shieh S-Y, Taya Y, Prives C. DNA damage-inducible phosphorylation of p53 at N-terminal sites including a novel site, Ser20, requires tetramerization. EMBO Journal. 1999;18(7):1815–1823. doi: 10.1093/emboj/18.7.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Craig A, Scott M, Burch L, Smith G, Ball K, Hupp T. Allosteric effects mediate CHK2 phosphorylation of the p53 transactivation domain. EMBO Reports. 2003;4(8):787–792. doi: 10.1038/sj.embor.embor901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xie S, Wang Q, Wu H, et al. Reactive oxygen species-induced phosphorylation of p53 on serine 20 is mediated in part by polo-like kinase-3. Journal of Biological Chemistry. 2001;276(39):36194–36199. doi: 10.1074/jbc.M104157200. [DOI] [PubMed] [Google Scholar]
- 86.D’Orazi G, Cecchinelli B, Bruno T, et al. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nature Cell Biology. 2002;4(1):11–20. doi: 10.1038/ncb714. [DOI] [PubMed] [Google Scholar]
- 87.Yoshida K, Liu H, Miki Y. Protein kinase C δ regulates Ser46 phosphorylation of p53 tumor suppressor in the apoptotic response to DNA damage. Journal of Biological Chemistry. 2006;281(9):5734–5740. doi: 10.1074/jbc.M512074200. [DOI] [PubMed] [Google Scholar]
- 88.Herrmann CPE, Kraiss S, Montenarh M. Association of casein kinase II with immunopurified p53. Oncogene. 1991;6(5):877–884. [PubMed] [Google Scholar]
- 89.Delphin C, Huang K-P, Scotto C, et al. The in vitro phosphorylation of p53 by calcium-dependent protein kinase C—characterization of a protein-kinase-C-binding site on p53. European Journal of Biochemistry. 1997;245(3):684–692. doi: 10.1111/j.1432-1033.1997.t01-1-00684.x. [DOI] [PubMed] [Google Scholar]
- 90.Hupp TR, Meek DW, Midgley CA, Lane DP. Regulation of the specific DNA binding function of p53. Cell. 1992;71(5):875–886. doi: 10.1016/0092-8674(92)90562-q. [DOI] [PubMed] [Google Scholar]
- 91.Friend S. p53: a glimpse at the puppet behind the shadow play. Science. 1994;265(5170):334–335. doi: 10.1126/science.8023155. [DOI] [PubMed] [Google Scholar]
- 92.Ando K, Ozaki T, Yamamoto H, et al. Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. Journal of Biological Chemistry. 2004;279(24):25549–25561. doi: 10.1074/jbc.M314182200. [DOI] [PubMed] [Google Scholar]
- 93.Shikama N, Lee C-W, France S, et al. A novel cofactor for p300 that regulates the p53 response. Molecular Cell. 1999;4(3):365–376. doi: 10.1016/s1097-2765(00)80338-x. [DOI] [PubMed] [Google Scholar]
- 94.Sakaguchi K, Herrera JE, Saito S, et al. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes and Development. 1998;12(18):2831–2841. doi: 10.1101/gad.12.18.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yuan Z-M, Huang Y, Ishiko T, et al. Role for p300 in stabilization of p53 in the response to DNA damage. Journal of Biological Chemistry. 1999;274(4):1883–1886. doi: 10.1074/jbc.274.4.1883. [DOI] [PubMed] [Google Scholar]
- 96.Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Molecular Biology of the Cell. 2005;16(10):4623–4635. doi: 10.1091/mbc.E05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kawai H, Nie L, Wiederschain D, Yuan Z-M. Dual role of p300 in the regulation of p53 stability. Journal of Biological Chemistry. 2001;276(49):45928–45932. doi: 10.1074/jbc.M107770200. [DOI] [PubMed] [Google Scholar]
- 98.Grossman SR, Deato ME, Brignone C, et al. Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science. 2003;300(5617):342–344. doi: 10.1126/science.1080386. [DOI] [PubMed] [Google Scholar]
- 99.Schmidt D, Müller S. Members of the PIAS family act as SUMO ligases for c-Jun and p53 and repress p53 activity. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(5):2872–2877. doi: 10.1073/pnas.052559499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wu S-Y, Chiang C-M. p53 sumoylation: mechanistic insights from reconstitution studies. Epigenetics. 2009;4(7):445–451. doi: 10.4161/epi.4.7.10030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jackson SP, Tjian R. O-glycosylation of eukaryotic transcription factors: implications for mechanisms of transcriptional regulation. Cell. 1988;55(1):125–133. doi: 10.1016/0092-8674(88)90015-3. [DOI] [PubMed] [Google Scholar]
- 102.Shaw P, Freeman J, Bovey R, Iggo R. Regulation of specific DNA binding by p53: evidence for a role for O-glycosylation and charged residues at the carboxy-terminus. Oncogene. 1996;12(4):921–930. [PubMed] [Google Scholar]
- 103.Reich NC, Levine AJ. Growth regulation of a cellular tumour antigen, p53, in nontransformed cells. Nature. 1984;308(5955):199–201. doi: 10.1038/308199a0. [DOI] [PubMed] [Google Scholar]
- 104.Bruno T, De Nicola F, Iezzi S, et al. Che-1 phosphorylation by ATM/ATR and Chk2 kinases activates p53 transcription and the G2/M checkpoint. Cancer Cell. 2006;10(6):473–486. doi: 10.1016/j.ccr.2006.10.012. [DOI] [PubMed] [Google Scholar]
- 105.Raman V, Tamori A, Vali M, Zeller K, Korz D, Sukumar S. HOXA5 regulates expression of the progesterone receptor. Journal of Biological Chemistry. 2000;275(34):26551–26555. doi: 10.1074/jbc.C000324200. [DOI] [PubMed] [Google Scholar]
- 106.Noda A, Toma-Aiba Y, Fujiwara Y. A unique, short sequence determines p53 gene basal and UV-inducible expression in normal human cells. Oncogene. 2000;19(1):21–31. doi: 10.1038/sj.onc.1203230. [DOI] [PubMed] [Google Scholar]
- 107.Okoshi R, Ozaki T, Yamamoto H, et al. Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. Journal of Biological Chemistry. 2008;283(7):3979–3987. doi: 10.1074/jbc.M705232200. [DOI] [PubMed] [Google Scholar]
- 108.Hardie DG. The AMP-activated protein kinase pathway—new players upstream and downstream. Journal of Cell Science. 2004;117(23):5479–5487. doi: 10.1242/jcs.01540. [DOI] [PubMed] [Google Scholar]
- 109.Okoshi R, Ando K, Suenaga Y, et al. Transcriptional regulation of tumor suppressor p53 by cAMP-responsive element-binding protein/AMP-activated protein kinase complex in response to glucose deprivation. Genes to Cells. 2009;14(12):1429–1440. doi: 10.1111/j.1365-2443.2009.01359.x. [DOI] [PubMed] [Google Scholar]
- 110.Vogan K, Bernstein M, Leclerc J-M, et al. Absence of p53 gene mutations in primary neuroblastomas. Cancer Research. 1993;53(21):5269–5273. [PubMed] [Google Scholar]
- 111.Moll UM, LaQuaglia M, Benard J, Riou G. Wild-type p53 protein undergoes cytoplasmic sequestration in undifferentiated neuroblastomas but not in differentiated tumors. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(10):4407–4411. doi: 10.1073/pnas.92.10.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Stommel JM, Marchenko ND, Jimenez GS, Moll UM, Hope TJ, Wahl GM. A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. EMBO Journal. 1999;18(6):1660–1672. doi: 10.1093/emboj/18.6.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Becker K, Marchenko ND, Maurice M, Moll UM. Hyperubiquitylation of wild-type p53 contributes to cytoplasmic sequestration in neuroblastoma. Cell Death and Differentiation. 2007;14(7):1350–1360. doi: 10.1038/sj.cdd.4402126. [DOI] [PubMed] [Google Scholar]
- 114.Carter S, Bischof O, Dejean A, Vousden KH. C-terminal modifications regulate MDM2 dissociation and nuclear export of p53. Nature Cell Biology. 2007;9(4):428–435. doi: 10.1038/ncb1562. [DOI] [PubMed] [Google Scholar]
- 115.Nikolaev AY, Li M, Puskas N, Qin J, Gu W. Parc: a cytoplasmic anchor for p53. Cell. 2003;112(1):29–40. doi: 10.1016/s0092-8674(02)01255-2. [DOI] [PubMed] [Google Scholar]
- 116.Vitali R, Cesi V, Tanno B, et al. Activation of p53-dependent responses in tumor cells treated with a PARC-interacting peptide. Biochemical and Biophysical Research Communications. 2008;368(2):350–356. doi: 10.1016/j.bbrc.2008.01.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mihara M, Erster S, Zaika A, et al. p53 has a direct apoptogenic role at the mitochondria. Molecular Cell. 2003;11(3):577–590. doi: 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 118.Marchenko ND, Wolff S, Erster S, Becker K, Moll UM. Monoubiquitylation promotes mitochondrial p53 translocation. EMBO Journal. 2007;26(4):923–934. doi: 10.1038/sj.emboj.7601560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yin Y, Luciani MG, Fåhraeus R. p53 stability and activity is regulated by MDM2-mediated induction of alternative p53 translation products. Nature Cell Biology. 2002;4(6):462–467. doi: 10.1038/ncb801. [DOI] [PubMed] [Google Scholar]
- 120.Zhu J, Zhou W, Jiang J, Chen X. Identification of a novel p53 functional domain that is necessary for mediating apoptosis. Journal of Biological Chemistry. 1998;273(21):13030–13036. doi: 10.1074/jbc.273.21.13030. [DOI] [PubMed] [Google Scholar]
- 121.Venot C, Maratrat M, Sierra V, Conseiller E, Debussche L. Definition of a p53 transactivation function-deficient mutant and characterization of two independent p53 transactivation subdomains. Oncogene. 1999;18(14):2405–2410. doi: 10.1038/sj.onc.1202539. [DOI] [PubMed] [Google Scholar]
- 122.Bourdon J-C, Fernandes K, Murray-Zmijewski F, et al. p53 isoforms can regulate p53 transcriptional activity. Genes and Development. 2005;19(18):2122–2137. doi: 10.1101/gad.1339905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. Journal of Biological Chemistry. 1998;273(10):5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 124.Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Current Biology. 2000;10(15):886–895. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- 125.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nature Reviews Cancer. 2003;3(3):155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 126.Celeste A, Petersen S, Romanienko PJ, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296(5569):922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ozaki T, Nagase T, Ichimiya S, et al. NFBD1/KIAA0170 is a novel nuclear transcriptional transactivator with BRCT domain. DNA and Cell Biology. 2000;19(8):475–485. doi: 10.1089/10445490050128403. [DOI] [PubMed] [Google Scholar]
- 128.Stewart GS, Wang B, Bigneli CR, Taylor AMR, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421(6926):961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- 129.Goldberg M, Stucki M, Falck J, et al. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature. 2003;421(6926):952–956. doi: 10.1038/nature01445. [DOI] [PubMed] [Google Scholar]
- 130.Lou Z, Minter-Dykhouse K, Wu X, Chen J. MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature. 2003;421(6926):957–961. doi: 10.1038/nature01447. [DOI] [PubMed] [Google Scholar]
- 131.Lou Z, Minter-Dykhouse K, Franco S, et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Molecular Cell. 2006;21(2):187–200. doi: 10.1016/j.molcel.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 132.Nakanishi M, Ozaki T, Yamamoto H, et al. NFBD1/MDC1 associates with p53 and regulates its function at the crossroad between cell survival and death in response to DNA damage. Journal of Biological Chemistry. 2007;282(31):22993–23004. doi: 10.1074/jbc.M611412200. [DOI] [PubMed] [Google Scholar]
- 133.Ito Y. RUNX genes in development and cancer: regulation of viral gene expression and the discovery of RUNX family genes. Advances in Cancer Research. 2008;99:33–76. doi: 10.1016/S0065-230X(07)99002-8. [DOI] [PubMed] [Google Scholar]
- 134.Li Q-L, Ito K, Sakakura C, et al. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell. 2002;109(1):113–124. doi: 10.1016/s0092-8674(02)00690-6. [DOI] [PubMed] [Google Scholar]
- 135.Yamada C, Ozaki T, Ando K, et al. RUNX3 modulates DNA damage-mediated phosphorylation of tumor suppressor p53 at Ser-15 and acts as a Co-activator for p53. Journal of Biological Chemistry. 2010;285(22):16693–16703. doi: 10.1074/jbc.M109.055525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pozniak CD, Radinovic S, Yang A, McKeon F, Kaplan DR, Miller FD. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science. 2000;289(5477):304–306. doi: 10.1126/science.289.5477.304. [DOI] [PubMed] [Google Scholar]
- 137.Stiewe T, Zimmermann S, Frilling A, Esche H, Pützer BM. Transactivation-deficient δTA-p73 acts as an oncogene. Cancer Research. 2002;62(13):3598–3602. [PubMed] [Google Scholar]
- 138.Di Como CJ, Gaiddon C, Prives C. p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Molecular and Cellular Biology. 1999;19(2):1438–1449. doi: 10.1128/mcb.19.2.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Grob TJ, Novak U, Maisse C, et al. Human ΔNp73 regulates a dominant negative feedback loop for TAp73 and p53. Cell Death and Differentiation. 2001;8(12):1213–1223. doi: 10.1038/sj.cdd.4400962. [DOI] [PubMed] [Google Scholar]
- 140.Nakagawa T, et al. Autoinhibitory regulation of p73 by Delta Np73 to modulate cell survival and death through a p73-specific target element within the Delta Np73 promoter. Molecular and Cellular Biology. 2002;22:2575–2585. doi: 10.1128/MCB.22.8.2575-2585.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zaika AI, Slade N, Erster SH, et al. δNp73, a dominant-negative inhibitor of wild-type p53 and TAp73, is up-regulated in human tumors. Journal of Experimental Medicine. 2002;196(6):765–780. doi: 10.1084/jem.20020179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ikawa S, Nakagawara A, Ikawa Y. p53 family genes: structural comparison, expression and mutation. Cell Death and Differentiation. 1999;6(12):1154–1161. doi: 10.1038/sj.cdd.4400631. [DOI] [PubMed] [Google Scholar]
- 143.Yang A, Walker N, Bronson R, et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature. 2000;404(6773):99–103. doi: 10.1038/35003607. [DOI] [PubMed] [Google Scholar]
- 144.Mills AA, Zheng B, Wang X-J, Vogel H, Roop DR, Bradley A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature. 1999;398(6729):708–713. doi: 10.1038/19531. [DOI] [PubMed] [Google Scholar]
- 145.Yang A, Schweitzer R, Sun D, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398(6729):714–718. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- 146.Flores ER, Sengupta S, Miller JB, et al. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell. 2005;7(4):363–373. doi: 10.1016/j.ccr.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 147.Stiewe T, Putzer BM. Role of the p53-homologue p73 in E2F1-induced apoptosis. Nature Genetics. 2000;26(4):464–469. doi: 10.1038/82617. [DOI] [PubMed] [Google Scholar]
- 148.Irwin M, Marin MC, Phillips AC, et al. Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature. 2000;407(6804):645–648. doi: 10.1038/35036614. [DOI] [PubMed] [Google Scholar]
- 149.Lissy NA, Davis PK, Irwin M, Kaelin WG, Dowdy SF. A common E2F-1 and p73 pathway mediates cell death induced by TCR activation. Nature. 2000;407(6804):642–645. doi: 10.1038/35036608. [DOI] [PubMed] [Google Scholar]
- 150.Logotheti S. Sp1 binds to the external promoter of the p73 gene and induces the expression of TAp73gamma in lung cancer. FEBS Journal. 2010;277:3014–3027. doi: 10.1111/j.1742-4658.2010.07710.x. [DOI] [PubMed] [Google Scholar]
- 151.Fontemaggi G, et al. The transcriptional repressor ZEB regulates p73 expression at the crossroad between proliferation and differentiation. Molecular and Cellular Biology. 2001;21:8461–8470. doi: 10.1128/MCB.21.24.8461-8470.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gong J, Costanzo A, Yang H-Q, et al. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature. 1999;399(6738):806–809. doi: 10.1038/21690. [DOI] [PubMed] [Google Scholar]
- 153.Agami R, Blandino G, Oren M, Shaul Y. Interaction of c-Abl and p73α and their collaboration to induce apoptosis. Nature. 1999;399(6738):809–812. doi: 10.1038/21697. [DOI] [PubMed] [Google Scholar]
- 154.Yuan Z-M, Shioya H, Ishiko T, et al. p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage. Nature. 1999;399(6738):814–817. doi: 10.1038/21704. [DOI] [PubMed] [Google Scholar]
- 155.Ren J, Datta R, Shioya H, et al. p73β is regulated by protein kinase Cδ catalytic fragment generated in the apoptotic response to DNA damage. Journal of Biological Chemistry. 2002;277(37):33758–33765. doi: 10.1074/jbc.M110667200. [DOI] [PubMed] [Google Scholar]
- 156.Gonzalez S, Prives C, Cordon-Cardo C. p73α regulation by Chk1 in response to DNA damage. Molecular and Cellular Biology. 2003;23(22):8161–8171. doi: 10.1128/MCB.23.22.8161-8171.2003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 157.Furuya K, Ozaki T, Hanamoto T, et al. Stabilization of p73 by nuclear IκB kinase-α mediates cisplatin-induced apoptosis. Journal of Biological Chemistry. 2007;282(25):18365–18378. doi: 10.1074/jbc.M610522200. [DOI] [PubMed] [Google Scholar]
- 158.Yoshida K, Ozaki T, Furuya K, et al. ATM-dependent nuclear accumulation of IKK-α plays an important role in the regulation of p73-mediated apoptosis in response to cisplatin. Oncogene. 2008;27(8):1183–1188. doi: 10.1038/sj.onc.1210722. [DOI] [PubMed] [Google Scholar]
- 159.Gaiddon C, Lokshin M, Gross I, et al. Cyclin-dependent kinases phosphorylate p73 at Threonine 86 in a cell cycle-dependent manner and negatively regulate p73. Journal of Biological Chemistry. 2003;278(30):27421–27431. doi: 10.1074/jbc.M300251200. [DOI] [PubMed] [Google Scholar]
- 160.Koida N, Ozaki T, Yamamoto H, et al. Inhibitory role of Plk1 in the regulation of p73-dependent apoptosis through physical interaction and phosphorylation. Journal of Biological Chemistry. 2008;283(13):8555–8563. doi: 10.1074/jbc.M710608200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Komatsu S, Takenobu H, Ozaki T, et al. Plk1 regulates liver tumor cell death by phosphorylation of TAp63. Oncogene. 2009;28(41):3631–3641. doi: 10.1038/onc.2009.216. [DOI] [PubMed] [Google Scholar]