

Abstract
Bioluminescence resonance energy transfer (BRET) is increasingly being used to monitor protein-protein interactions and cellular events in cells. However, the ability to monitor multiple events simultaneously is limited by the spectral properties of the existing BRET partners. Taking advantage of newly developed Renilla luciferases and blue-shifted fluorescent proteins (FPs), we explored the possibility of creating novel BRET configurations using a single luciferase substrate and distinct FPs. Three new (to our knowledge) BRET assays leading to distinct color bioluminescence emission were generated and validated. The spectral properties of two of the FPs used (enhanced blue (EB) FP2 and mAmetrine) and the selection of appropriate detection filters permitted the concomitant detection of two independent BRET signals, without cross-interference, in the same cells after addition of a unique substrate for Renilla luciferase-II, coelentrazine-400a. Using individual BRET-based biosensors to monitor the interaction between G-protein-coupled receptors and G-protein subunits or activation of different G-proteins along with the production of a second messenger, we established the proof of principle that two new BRET configurations can be multiplexed to simultaneously monitor two dependent or independent cellular events. The development of this new multiplexed BRET configuration opens the way for concomitant monitoring of various independent biological processes in living cells.
Introduction
Bioluminescence resonance energy transfer (BRET) is a natural phenomenon that occurs in a variety of coelenterates, including Aequorea, Obelia, Phialidium, and Renilla. It is based on the transfer of nonradiative energy originating from the luciferase-mediated oxidation of coelenterazine (the donor) to a fluorescent protein (FP) acting as the energy acceptor, which reemits part of the energy as photons (1). BRET occurs only when the donor and acceptor proteins are in close proximity (typically <100 Å) (2), and when the emission spectrum of the donor overlaps sufficiently with the excitation spectrum of the acceptor. The BRET phenomenon can be easily detected if the Stoke's shift between the excitation and emission spectra of the FP is sufficient for the light emission of the acceptor to be spectrally resolved from the donor's emission. Based on these properties, and the fact that the efficacy of the transfer varies with the 6th power of the distance between the donor and the acceptor, previous studies used BRET as an alternative to fluorescence resonance energy transfer (FRET) as a proximity-based assay to monitor macromolecular interactions and conformational rearrangements (3,4). More precisely, the luciferase from Renilla reniformis (Rluc) and different FP variants of Aequorea vitoria GFP were genetically fused to distinct proteins to monitor their possible interactions in living cells (5,6). In other cases, the energy donor and acceptor were fused to a single protein to monitor conformational changes that can be used as biosensors of specific cellular events (7,8). More recently, chemical fluorophores were used as energy acceptors and combined with Rluc-fused protein to monitor protein-lipid (9) and protein-RNA interactions (10) in living mammalian cells.
To date, three major BRET configurations using distinct Rluc substrates and FPs have been developed and used for protein-protein interaction and conformational rearrangement monitoring. BRET1 uses coelenterazine-h (coel-h) and a yellow FP (YFP) (5,6) or a green FP (GFP) from Renilla (RGFP) (11), BRET2 utilizes coelenterazine-400a (coel-400a) and a UV-excited green FP (uvGFP) (12,13), and BRET3 makes use of coel-h and the monomeric orange FP (mOrange) (14). Each of these BRET assays provides advantages that can be exploited for specific applications. BRET3, which results in the emission of red light, is less likely to be quenched by biological tissues (compared to yellow and green for BRET1 and BRET2, respectively) and thus facilitates in vivo detection. BRET2 has the best signal/noise ratio of the three configurations due to the larger Stoke's shift of the uvGFPs, and thus allows detection of smaller BRET changes. BRET1 results in the highest absolute signal, in part due to the very high quantum yield of YFP, permitting detection of BRET between proteins expressed at lower levels. However, none of the BRET partners used to date have allowed for simultaneous spectral resolution of signals, which is a prerequisite for true multiplexing within the same cells in a single well. The ability to multiplex BRET signals would allow investigators to detect distinct biological processes simultaneously in a unique cell population.
With the objective of developing BRET configurations that can be multiplexed, we searched for FPs that can serve as BRET acceptors for Rluc/coel-400a. The identification and characterization of new BRET partners would lead to more flexibility in the use of BRET under different experimental conditions, and could allow the use of an existing collection of proteins already in fusion with different FPs. For this purpose, we took advantage of the recently developed mutant form of Rluc (Rluc2) that increases its bioluminescence by a factor of 3–45 times, depending on the coelenterazine used as substrate (15). Such an increase in the amount of energy emitted by Rluc2 should theoretically allow monitoring of energy transfer to FPs, which cannot be readily detected using wild-type (WT) Rluc, due to their less than optimal excitation spectrum overlap with the donor emission spectrum. The FPs to be tested were selected on the basis that they can all be excited by the same energy donor (Rluc/coel-400a), and they have sufficiently different Stoke's shifts to allow spectral resolution of their emission spectra.
In this study we established the ability of three additional FPs (enhanced blue (EB) FP2, super cyan fluorescent protein (SCFP3A), and mAmetrine) to accept energy transfer from Rluc2/coel-400a, generating three new BRET configurations. The spectral properties of mAmetrine and EBFP2 allowed their multiplexing with Rluc2/coel-400a as an energy donor, thus permitting simultaneous monitoring of two independent biological phenomena in the same cells.
Materials and Methods
Plasmids
All fusion proteins in this study were subcloned in pcDNA3.1 vector (Invitrogen, Carlsbad, CA). Plasmids encoding all of the different G-protein subunits, prostanoid TPα receptor (TPαR), and dopamine D2 receptor (D2R) used in this study were purchased from the Missouri University of Science and Technology (www.cdna.org). The GFP10-Gγ2 construct was previously described (16). Constructs for EBFP2-Gγ2, SCFP3A-Gγ2, or mAmetrine-Gγ2 were derived from GFP10-Gγ2 by excising the GFP10 coding sequence with NheI-BamHI and replacing it with a polymerase chain reaction (PCR)-amplified coding sequence of EBFP2, SCFP3A, and mAmetrine, respectively. The mAmetrine-Gγ1 was constructed by replacing Gγ2 from mAmetrine-Gγ2 with Gγ1 using Acc65I-XbaI endonuclease. The EBFP2 (17) and mAmetrine1.1 (18) were obtained from AddGene (Cambridge, MA), and sCFP3 was generously provided by Dr. Gadella (19). The V2R-Rluc2 fusion was done using PCR fragments from V2R cDNAs and a mutant form of Rluc (15) in which C124 and M185 were replaced by alanine and valine residues, respectively. This mutant Rluc, named Rluc2, demonstrates an enhanced energy output from improved enzymatic properties (see Fig. S1 in the Supporting Material). The Gαq-121Rluc2 and Gαi1-91Rluc2 were constructing by PCR overlapping to insert the Rluc2 at position 121 and 91 of Gαq and Gαi1, respectively. The BRET-cAMP biosensors were derived from previously published GFP10-EPAC-Rluc2 fusion protein (20), in which GFP10 was replaced by the appropriate FPs using NheI-BamHI restriction enzymes (as described above for the GFP10-γ2) to generate EBFP2-EPAC-Rluc2, SCFP3A-EPAC-Rluc2, and mAmetrine-EPAC-Rluc2. Sequence integrity for the different clones was confirmed by DNA sequencing.
Cell culture and transfection
HEK293T cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin/streptomycin, and 2 mM L-glutamine (Wisent, Saint-Jean-Baptiste, Canada), and incubated at 37°C in 5% CO2. Two days before experiments were conducted, 40,000 cells were distributed in 96-well white culture plates (PerkinElmer, Waltham, MA) treated with poly-ornithine (Sigma-Aldrich, St. Louis, MO) and transfected with 250 ng of total DNA using polyethylenimine 25 kD linear (PEI; Polysciences, Warrington, PA) as transfecting agent (3:1 PEI/DNA ratio). Then 11–50 ng of each specified expression vector were diluted in 150 mM NaCl and the total quantity of DNA was completed at 250 ng with salmon sperm DNA (Invitrogen, Carlsbad, CA).
Bioluminescence and fluorescence spectral profiles
Cells transiently expressing the individual FP-EPAC-Rluc2 fusion protein were seeded at 100,000 cells/well in a 96-well white Optiplate (PerkinElmer). The fluorescence spectra were acquired every 2 nm at 430–600 nm after excitation at 400 nm using a FlexStationII microplate reader (Molecular Devices, Sunnyvale, CA) and expressed as a fraction of the maximal emission (normalized to one) for each FP. The bioluminescence spectra were acquired every 5 nm at 360–600 nm after 2 min of exposure to 5 μM coel-400a (Biotium, Hayward, CA) using the FlexStationII microplate reader. The bioluminescence was expressed as a fraction of the maximal emission of the energy donor (normalized to one).
BRET measurements
Transfected cells were washed twice with phosphate-buffered saline (PBS) directly in the 96-well culture plates and kept in 100 μL of PBS. BRET was monitored 10 min after the addition of 5 μM coel-400a in a Mithras LB 940 microplate reader (Berthold Technologies, Bad Wildbad, Germany) equipped with different donor/acceptor emission filter sets (filter set 1: donor 480 ± 20 nm/acceptor 530 ± 20 nm; filter set 2: donor 410 ± 70 nm/acceptor 515 ± 20 nm; filter set 3: donor 410 ± 70 nm/acceptor 480 ± 20 nm; and filter set 4: donor 410 ± 70 nm/acceptor 550 longpass). BRET signals were derived from the emission detected with the energy acceptor filter divided by the emission detected using the energy donor filter. Finally, the specific BRET signal was defined as the difference between the total BRET signals and the one obtained with Rluc2 alone. For the bioluminescence emission spectra experiments presented in Fig. 1, an mCherry-EPAC-Rluc2 construct, which is not competent for BRET, was used to define background values to be subtracted from those obtained from the spectrum of each FP. Identical background values were obtained using either the Rluc2 or the mCherry-EPAC-Rluc2 construct alone.
Figure 1.
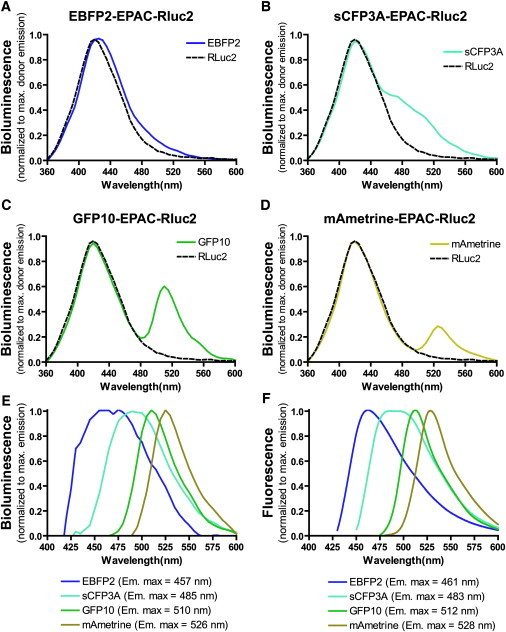
Bioluminescence and fluorescence emission spectra. Bioluminescence emission spectra of cells overexpressing EBFP2-EPAC-Rluc2 (A, blue line), SCFP3A-EPAC-Rluc2 (B, cyan line), GFP10-EPAC-Rluc2 (C, green line), and mAmetrine-EPAC-Rluc2 (D, dark yellow line), and negative control mCherry-EPAC-Rluc2 (A–D, black dotted line) were measured after the addition of coel-400a. (E) The bioluminescence of the mCherry-EPAC-Rluc2 control (which was not different from the emission from Rluc2 alone, since mCherry is not an acceptor for the Rluc2/coel-400a) was subtracted from the emission spectrum for each FP-EPAC-Rluc2 fusion proteins and normalized as a ratio of the maximal emission for each FP. (F) Fluorescence spectra obtained from cells overexpressing each of the FP-EPAC-Rluc2 fusion proteins was measured after direct excitation at 400 nm. The curves were generated using the LOWESS fitting equation from the Prism 4.0 software.
Multiplexing BRET assay
Transfected cells were washed and coel-400a was added as described for regular BRET measurement. To monitor two energy transfers, the luminescence signal was collected in the presence of three distinct filters, successively: 410 ± 70 nm (common donor; Rluc2/coel-400a), 480 ± 20 nm (EBFP2 acceptor), and 550 longpass (mAmetrine acceptor). The signal was collected for 1 s for each of the filters, for a total of 3 s per well of data acquisition. The signals from each BRET were derived from the emission detected with the appropriate energy acceptor filter divided by the emission detected using the energy donor filter (BRET400-BFP = 480 ± 20 nm over 410 ± 70 nm, and BRET400-mAmetrine = 550 longpass over 410 ± 70 nm). Finally, the specific BRET signal was defined as the difference between the total BRET signals and the one obtained in cells transfected with Rluc2 alone.
Statistical analysis
We assessed the statistical significance of the difference between conditions by performing two-way analyses of variance (ANOVAs) followed by Bonferroni post-tests using Prism 4.0 software (GraphPad Software, La Jolla, CA).
Results
Bioluminescence spectra
BRET2 is characterized by an energy transfer from the Rluc-mediated oxidation of coel-400a to the UV-shifted GFPs that can be excited at 400 nm and reemit light at 510 nm (e.g., GFP2 and GFP10) (4). The recently described FPs (EBFP2 (17), SCFP3A (19), and mAmetrine (18)), which have different emission wavelengths and Stoke's shifts but close excitation peaks in the dark-blue region of the spectrum, could therefore represent potential BRET partners for Rluc2/coel-400a to create novel BRET configurations emitting at different colors. However, the suboptimal spectrum overlap and lower quantum yield of these FPs compared to GFP2 and GFP10 make them less attractive. The development of Rluc2, which yields luminescence signals 50 times larger than those generated by the WT Rluc (Fig. S1) upon coel-400a oxidation may make it possible to detect significant BRET despite these apparent limitations. To assess possible energy transfer between Rluc2/coel-400a and these FPs, we measured bioluminescence spectra from cells expressing an unimolecular BRET-based cAMP-biosensor of each color: EBFP2-EPAC-Rluc2, SCFP3A-EPAC-Rluc2, mAmetrine-EPAC-Rluc2, and GFP10-EPAC-Rluc2. As shown in Fig. 1, A–D, the spectra are characteristic of energy transfer, since in addition to the peak corresponding to the emission of Rluc2, a second component corresponding to the expected emission of the respective FPs was observed. To determine whether the second component of the curves (which in some cases (e.g., EBFP2) is a relatively small shoulder) truly results from fluorophore reemission, we subtracted the spectrum for a BRET noncompetent mCherry-EPAC-Rluc2 construct (equivalent to Rluc2 alone) from the recorded spectrum for each FP. As shown in Fig. 1 E, the resulting spectra are essentially identical to those obtained after direct light excitation of each FP (Fig. 1 F), confirming that the second component of the curves originated from an energy transfer to an FP.
Since all constructs have essentially the same structure and sequence, except for a few mutations within FPs that lead to their specific spectral properties, the observed differences in emission intensity result from their distinct intrinsic ability to be excited and reemit transferred energy from the Rluc2/coel-400a. The differences are dictated by the extent of overlap between the Rluc2/coel-400a emission spectrum and the excitation spectrum of a given FP, as well as the quantum yield of the individual fluorophores. Based on the spectral overlap, the best partners for energy transfer from Rluc2/Coel400a are (in decreasing order) GFP10, mAmetrine, SCFP3A, and EBFP2. This is consistent with the amplitude of their emission peak (area under the curve): GFP10 (175.8) > SCFP3A (127.1) ≈ mAmetrine (121.0) > EBFP2 (40.8), which reflects both the donor emission/acceptor excitation overlap and the fluorophore quantum yield. The relatively small reemission observed for EBFP2 is to be expected, given its relatively low quantum yield and a left-shifted excitation maximum peak (386 nm) compared to the other FPs (21). The above data illustrate that energy transfer can be monitored between Rluc2/coel-400a and the four UV-shifted FPs tested, supporting the possible development of three new BRET assays with EBFP2, SCFP3A, and mAmetrine. To prevent confusion among the different Rluc-based BRET configurations, we propose a new nomenclature that first indicates the emission peak of the donor, and then the name of the prototypical FP (which usually reflects its emission color) used for the transfer. According to this nomenclature, the three new BRET assays would be called BRET400-BFP, BRET400-CFP, and BRET400-mAmetrine, whereas the existing BRET1, BRET2, and BRET3 would become BRET480-YFP, BRET400-GFP, and BRET480-Orange, respectively.
Multicolor BRET
Given the different spectral properties of the confirmed BRET pairs, we then explored whether some filter sets could optimally detect their signals. For this purpose, we tested four donor/acceptor filter sets (set 1: 480/530; set 2: 410/515; set 3: 410/480; and set 4: 410/550) for monitoring bimolecular BRET between the V2 vasopressin receptor fused to Rluc2 (V2R-Rluc2) and the γ2 subunit of an heterotrimeric G-protein fused to the different FPs (FP-Gγ2), upon stimulation of the receptor with increasing concentration of arginine-vasopressin (AVP). As shown in Fig. 2, A–D, both basal and AVP-promoted BRET increases were detected between FP-Gγ2 and V2R-Rluc2 for several FP and filter combinations. The basal BRET signal has been attributed to precoupling of the receptor with the G-protein, whereas the agonist-promoted increase reflects a functional engagement of the G-protein by the receptor (16,22,23). The BRET signals generated by specific FPs were differentially detected by distinct filter sets, but perfect correlations between basal and agonist-promoted BRET increases were observed independently of the filter set used (Fig. 2 E), indicating that the characteristics of the different filters affect only the intensity of the detected signal and not its dynamics. For identical acceptor/donor expression ratios, the agonist-promoted BRET increases (expressed as % of the basal BRET) were the same for all BRET pairs considered (Fig. S2), indicating that the different BRET configurations yield comparable responses. Also, the detected EC50-values for the AVP-stimulated increase in BRET between V2R-Rluc and each FP-Gγ2 were very similar for all FP and filter combinations used. The optimal filter sets for detecting BRET signals were different for each FP: filter set 1 was optimal for GFP10 and mAmetrine, whereas filter set 3 was best for SCFP3A and EBFP2, thus defining the optimal detection conditions for the four BRET assays. It is noteworthy that even for EBFP2, which yielded the weakest energy transfer (Fig. 1 A), the selection of a proper filter set allowed for the detection of a robust and highly reproducible signal.
Figure 2.
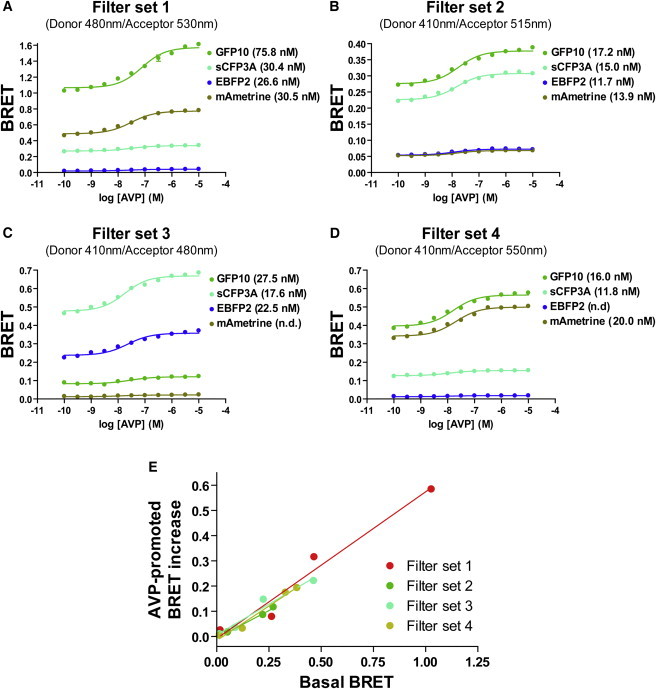
Multicolor BRET measurements. (A–D) BRET between V2R-Rluc2 and GFP10-Gγ2 (green), EBFP2-Gγ2 (blue), SCFP3A-Gγ2 (cyan), or mAmetrine-Gγ2 (dark yellow) were measured in cells coexpressing the indicated BRET partners and Gα12, in the presence or absence of increasing concentrations of AVP. Cells were stimulated for 20 min with the indicated concentration of AVP, and coel-400a was added 10 min before the readings were taken. BRET was measured using four different filter sets (see characteristics in the Materials and Methods section), as indicated in the panels. Results are expressed as the means ± SE of three independent experiments performed in triplicate. Curves were analyzed using a nonlinear regression sigmoid fit from the Prism 4.0 software. The EC50 (nM) is indicated for each FP within each filter set. (E) The maximal AVP-promoted BRET for each pair is plotted as a function of their basal BRET signals. The linear correlations for the four filter sets were obtained using a linear regression fit from the Prism 4.0 software and are depicted by the four lines in the graph.
Interestingly, some of the filter sets did not detect the BRET signal generated by some FPs (Fig. 2 and Fig. S2). For example, no BRET could be detected with the mAmetrine-Gγ2 in filter set 3 or with EBFP2-Gγ2 in filter set 4. Yet, good BRET signals were observed for mAmetrine-Gγ2 with filter set 4, set 1, and, to a lesser extent, set 2; and for EBFP2-Gγ2 with filter set 3 and, to a lesser extent, set 2 (Fig. 2). The absence of detected BRET between V2R-Rluc2 and Gγ2 with certain FPs and some filter sets did not result from a lack of interaction, but from the inadequacy of these filters to resolve the emissions resulting from energy transfer. The fact that BRET between Rluc2 and either mAmetrine or EBFP2 can be detected by distinct filter sets (sets 4 and 3, respectively) without contaminating the other signal raises the possibility that two independent transfers between a common donor and two acceptors can be monitored simultaneously within the same cells.
Multiplexing BRET
To experimentally determine whether simultaneous measurements of BRET between Rluc2 and both EBFP2 and mAmetrine can be used to differentially follow two events within the same cells, we combined two BRET-based biosensors: 1), a unimolecular cAMP-biosensor (mAmetrine-EPAC-Rluc2) that senses the cAMP level by monitoring the conformational change of EPAC upon cAMP binding; and 2), a bimolecular biosensor (EBFP2-Gγ2 and V2R-Rluc2) that detects the physical engagement of a G-protein subunit by V2R. Because Gα12 was previously shown to potentiate receptor-mediated activation of adenylyl cyclase (24), we performed the assays in the presence of coexpressed Gα12 to increase the size of the signal (Fig. S3). The two BRET events were monitored with three filters (filter set 5) composed of the common donor filter 410 ± 70 nm, and the optimal acceptor filters for mAmetrine (set 4: 550 longpass) and EBFP2 (set 3: 480 ± 20 nm) to follow the two energy transfers. In cells coexpressing V2R-Rluc2 and EBFP2-Gγ2, a significant basal increase and an AVP-promoted increase in BRET400-BFP were detected (Fig. 3 A), reflecting the engagement of the G-protein by the receptor, whereas no significant BRET400-mAmetrine was observed (Fig. 3 B). In cells coexpressing V2R-Rluc2 and mAmetrine-EPAC-Rluc2, a significant AVP- and forskolin-promoted decrease in BRET400-mAmetrine was detected (Fig. 3 B), indicating an increase in cAMP levels, whereas no BRET400-BFP was detected in these cells (Fig. 3 A). These results confirm the selectivity of each filter set for detecting only one of the two BRET events. In cells coexpressing V2R-Rluc2, EBFP2-Gγ2, and mAmetrine-EPAC-Rluc2, AVP promoted a significant increase in BRET400-BFP (Fig. 3 A), reflecting the engagement of EBFP2-Gγ2 by V2R-Rluc2, as well as a concomitant decrease in BRET400-mAmetrine (Fig. 3 B), resulting from an increase in cAMP levels. Forskolin-stimulated cAMP production also promoted a reduction in BRET400-mAmetrine (Fig. 3 B) but did not affect BRET400-BFP (Fig. 3 A), which is consistent with the ability of forskolin to stimulate adenylyl cyclase independently of the receptor/G-protein interaction. It is worth mentioning that the basal BRET400-BFP observed in cells coexpressing V2R-Rluc2, EBFP2-Gγ2, and mAmetrine-EPAC-Rluc2 was lower than in cells expressing only V2R-Rluc2 and EBFP2-Gγ2 (Fig. 3 A). This can easily be explained by the fact that BRET is a ratio of EBFP2-emission over Rluc2-emission, and the Rluc2 signal originated from both V2R-Rluc2 and mAmetrine-EPAC-Rluc2 in the former cells. It is therefore important when setting up such assay to ensure that the expression levels of each Rluc2-fused constructs are maintained in similar ranges. Interestingly, however, the AVP-promoted BRET400-BFP increase was very similar in the two conditions (73 ± 29% vs. 60 ± 11% for cells expressing both Rluc2 biosensors or only one, respectively), indicating that the presence of the two Rluc2-based biosensors did not influence their dynamic response. As shown in Fig. 3 C, in cells coexpressing V2R-Rluc2, EBFP2-Gγ2, and mAmetrine-EPAC-Rluc2, AVP promoted an increase in BRET400-BFP and a decrease in BRET400-mAmetrine with similar EC50-values.These results show for the first time, to our knowledge, that multiplexing of two separate biosensors expressed in the same cell population can be monitored after a single coelenterazine addition. It should be noted that although the coexpression of Gα12 increased the cAMP signal, its presence was not required for detection of the EPAC biosensor signal (Fig. S3).
Figure 3.
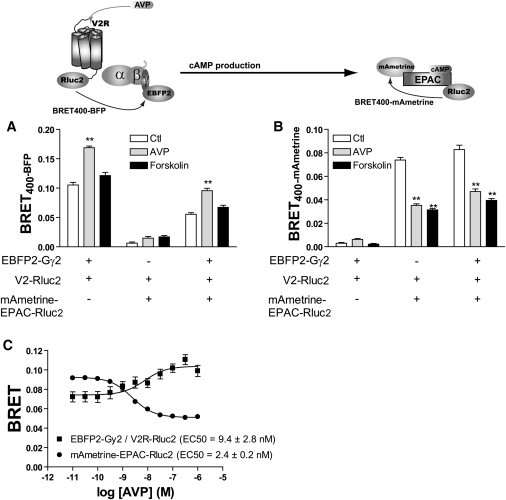
Multiplexing BRET. BRET400-BFP (A) and BRET400-mAmetrine (B) were measured in cells expressing the indicated combination of V2R-Rluc2, EBFP2-Gγ2, and mAmetrine-EPAC-Rluc2. BRET was measured in the absence of ligand (Ctl) or after 20 min stimulation with AVP (100 nM) or forskolin (100 μM). Coel-400a was added 10 min before readings were taken, using a single energy donor filter (410 ± 70 nm) and two different energy acceptor filters (480 ± 20 nm for BRET400-BFP and 550LP for BRET400-mAmetrine). Two-way ANOVAs followed by Bonferroni post-tests were used to assess the statistical significance of the differences (∗∗p < 0.001) using Prism 4.0 software. (C) BRET400-BFP between V2R-Rluc2 and EBFP2-Gγ2 (squares) and BRET400-mAmetrine for the cAMP biosensor mAmetrine-EPAC-Rluc2 (circles) were measured simultaneously in cells coexpressing these constructs, after 20 min stimulation with increasing concentration of AVP. The results are expressed as the means ± SE of three independent experiments performed in triplicate. The curves were generated using the nonlinear regression sigmoid fit from Prism 4.0 software.
To determine whether BRET400-mAmetrine and BRET400-BFP-based biosensors can be multiplexed to monitor the activation of other signaling pathways, we directly monitored the activation of Gαi by D2R by monitoring the BRET changes between Gαi1-91Rluc2 and mAmetrine-Gγ2 upon stimulation of the D2R with quimpirol, and monitoring cAMP accumulation with BRET480-BFP using EBFP2-EPAC-Rluc2 (Fig. 4 A). As expected, the stimulation with quimpirole promoted a decrease in BRET between Gαi1 and Gγ2, indicating activation of Gαi (16), as well as an increase in the EPAC intramolecular BRET, reflecting a decrease in cAMP resulting from the action of activated Gαi on the adenylyl cyclase. The effects of agonist stimulation were blocked by pretreatment with pertussis toxin (PTX), which inhibits Gαi activity. Direct activation of the adenylyl cyclase with forskolin had no effect on the BRET between Gαi1 and Gγ2, whereas it promoted a decrease in the intramolecular EPAC resulting from the increased cAMP. As expected, the latter effect was completely resistant to PTX treatment. We also assessed the activation of Gq by monitoring BRET400-mAmetrine between Gαq-121Rluc2 and mAmetrine-Gγ1 upon activation of the TPαR with the selective agonist U46619. As shown in Fig. 4 B, activation with the agonist promoted a decrease in BRET between Gαq and Gγ1, reflecting Gq activation, whereas it did not affect the EPAC intramolecular BRET400-BFP, consistent with the notion that activation of TPαR does not affect cAMP levels. In the same cells, direct stimulation of adenylyl cyclase by forskolin promoted a decrease in the EPAC BRET but did not affect the Gαq-Gγ1 BRET. Taken together, these results indicate that BRET400-mAmetrine and BRET400-BFP can be multiplexed to monitor many distinct signaling pathways, and the BRET signals obtained with the individual biosensors are independent of one another.
Figure 4.
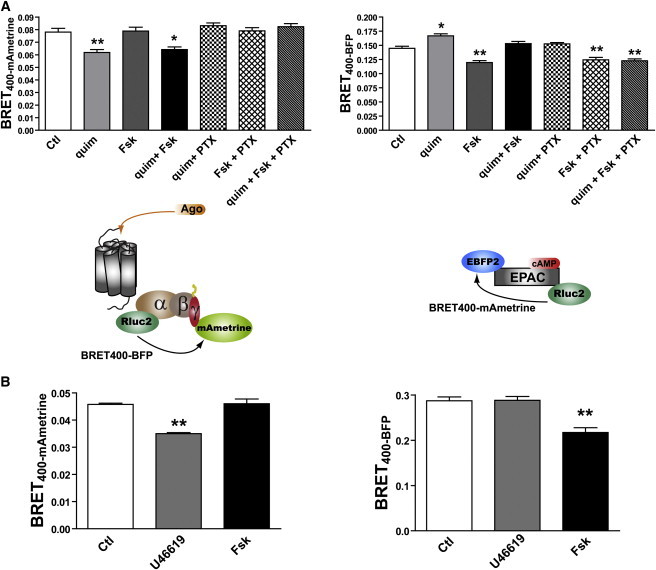
Multiplexing Gαi or Gαq activation with cAMP production. BRET from cells expressing (A) D2 dopamine receptor, Gαi1-91Rluc2, mAmetrine-Gγ2, and EBFP2-EPAC-Rluc2; or (B) TPαR, Gαq-121Rluc2, mAmetrine-Gγ1, and EBFP2-EPAC-Rluc2 was measured either in the absence of ligand or after 20 min stimulation with quimpirol (quim, 100 nM, A), U46619 (100 nM, B), and/or forskolin (Fsk, 100 μM, A and B), as indicated. For the negative control in A, PTX (100 ng/mL) was added 16 h before the experiment. Coel-400a was added 10 min before readings were taken, using a single energy donor filter (410 ± 70 nm) and two different energy acceptor filters (480 ± 20 nm for BRET400-BFP and 550LP for BRET400-mAmetrine). Two-way ANOVAs followed by Bonferroni post-tests were used to assess the statistical significance of the differences (∗p < 0.01, ∗∗p < 0.001) using Prism 4.0 software.
Discussion
In this study we report the development of what we believe are three new BRET configurations that complement the existing ones, and provide for the first time, to our knowledge, a toolbox for simultaneous multiplexing of spectrally resolved BRET assays within the same cells.
Multicolor BRET
To date, three configurations of genetically encoded BRET assays, with Rluc used as the energy donor, have been described. BRET1 and BRET3 use coel-h as the Rluc substrate for transferring energy to YFPs (eGFP, eYFP, citrine, venus, YPeT, and RGFP) and RFPs (mOrange, mTomato, and sdTomato), respectively. BRET2 uses coel-400a and Rluc as an energy donor, and uvGFPs (GFP10, GFP2, and Sapphire) as energy acceptors. In this study, three new BRET configurations are presented. They all use coel-400a as the Rluc substrate and acceptor FPs that can be excited at a similar wavelength (∼400 nm) but emit at different colors as a result of distinct Stoke's shifts.
Of the four BRET400 FPs used in our study, the original BRET400-GFP is still the one with the largest BRET signals due to its superior quantum yield and better overlap between the Rluc emission and FP excitation spectra. The lower transfer efficacy toward BFP, CFP, and mAmetrine as compared to GFP10 most likely explains why BRET signals obtained with these FPs have not been reported until now. Only the availability of Rluc mutant forms, such as Rluc8 and Rluc2 (15,25), that emit higher levels of bioluminescence upon oxidation of coel-400a, raised the possibility of using BFP, CFP, and mAmetrine as viable BRET400 acceptors. Indeed, in our hands, the amount of light emitted by Rluc2 was 50-fold higher than that emitted by the WT enzyme (Fig. S1), which led to detectable transfer to BFP, CFP, and mAmetrine despite a suboptimal overlap between Rluc2/coel-400a emission and FPs excitation spectra. Furthermore, because of the higher bioluminescence of Rluc2, one can achieve easily measurable BRET signals with coel-400a using much lower expression levels than previously needed for classical BRET2. For instance, when a standard curve correlating the luminescence signal to the density of V2R (assessed by radioligand binding) was generated (data not shown), the levels of V2R needed to obtain the BRET signals reported in Fig. 2 and Fig. S2 were estimated to be between 110 and 325 fmol/mg of protein. Such expression levels are comparable to those measured in kidney tissues and kidney cells (130 and 310 fmol/mg, respectively (26)).
The selection of optimal filter sets to quantify the BRET generated by specific fluorophores is also an important factor in obtaining the best signal/noise ratio. Larger BRET ratios are obtained by selecting filters that favor the detection of the FP over that of the Rluc emission. However, it is important to select a filter band-pass that will allow the detection of a sufficient amount of light to remain within the linear dynamic range of the detectors and avoid affecting the linearity of the signals. A good illustration of this phenomenon is provided by the observation that the filter sets that generate the largest signal for BRET400-GFP are Rluc em: 480 ± 20 nm and GFP em: 530 ± 20, and not Rluc em: 410 ± 35 nm and GFP em: 515 ± 15 nm (compare Fig. 2, A and B), which are commonly use for this type of BRET experiment (12,13). The filter sets used do not affect the dynamics of the response, since the increase in basal signal was found to be directly proportional to the ligand-promoted increase in BRET observed between V2-Rluc2 and GFP-Gγ2 (Fig. 2 E). However, the larger absolute signals obtained could allow the detection of small BRET signals that might be lost in the background noise when nonoptimal filter sets are used.
Although BRET400-GFP, which corresponds to traditional BRET2, remains the BRET system that generates the largest signal with Rluc2/coel-400a, access to additional BRET colors provides several advantages. First, the ability to perform BRET400-CFP provides an interesting alternative because it allows the direct use of the many proteins and protein libraries (generated to study proteins localization or protein interactions by FRET) that are already CFP-tagged. Moreover, some FPs, such as mAmetrine and EBFP2, have similar excitation profiles but different Stoke's shifts, resulting in spectrally resolvable emission spectra that allow multiplexing for the simultaneous detection of two distinct BRET events with a single Rluc substrate in living cells. It should also be noted that even though a single prototypical FP was used for each of the new color BRET configurations presented, a selection of FPs could be used for two of them: BRET400-BFP (EBFP, EBFP2, and mKalama1) and BRET400-CFP (ECFP, SCFP3A, CyPet, mCerulean, and mTFP1.0). In some cases, the specific biophysical properties of some FPs (e.g., pH sensitivity, propensity to dimerize, and quantum yield) could make them preferred choices.
Multiplexing BRET
A few studies have described genetically encoded dual FRET-based assays (27) that allow the detection of two simultaneous FRET events within the same cells. These assays provide the advantage of single-cell microscopy imaging (27). However, the quantification of two FRET events is complicated by the fact that only a limited numbers of FP pairs are available for efficient transfer. For most existing pairs, the excitation of the donor with a light source leads to direct excitation of the acceptor. Moreover, the emission spectra of the existing FP FRET acceptors are similar, which makes it difficult to achieve spectral resolution and leads to spectral bleed-through. This necessitates the use of mathematical correction factors, which are not always easy to translate to practice and require additional control measurements. BRET is more sensitive than FRET for measuring RET with the use of plate readers. This is due in part to the high sensitivity of the PMT detectors used in plate readers, which allows easy detection of BRET. Also, the autofluorescence originating from the light excitation of the donor reduces the signal/noise ratio for FRET (28). Thus, for different applications (i.e., imaging versus activity detection in plate readers), either technique may be preferred.
The results of this study demonstrate that two BRET events can be monitored simultaneously within the same cells, thus permitting the simultaneous detection of distinct biological outcomes. We have shown that BRET400-BFP and BRET400-mAmetrine can be multiplexed to detect the conformational rearrangement of a unimolecular cAMP biosensor simultaneously with the engagement of G-proteins by their cognate receptors. The activation of three distinct receptor/G-protein signaling pathways can be easily monitored with bimolecular sensors that monitor either the interaction between the receptor and Gγ or the receptor-mediated separation between Gα and Gγ. We observed BRET400-BFP and BRET400-mAmetrine, triggered by the activation of the receptor, within the same cells without cross-interference from the individual signals. This multiplexing method is distinct from and complementary to previously reported BRET approaches. For instance, because of its ability to monitor two events independently, the BRET400-BFP/BRET400-mAmetrine multiplexing approach is different from recently developed sequential RET (29) and bimolecular fluorescence complementation BRET assays (30,31) that were designed to monitor a single event involving three partners. BRET400-BFP/BRET400-mAmetrine multiplexing, which allows the monitoring of two BRET signals simultaneously in a single well, facilitates kinetic comparisons between the two events and reduces the number of manipulations needed, thus opening the door to high-throughput screening applications.
In this study, we used BRET400-BFP/BRET400-mAmetrine multiplexing to combine unimolecular biosensors (mAmetrine-EPAC-Rluc2 or BFP-EPAC-Rluc2) with bimolecular probes (V2R-Rluc2/EBFP2-Gγ2, Gαi1-91Rluc2/mAmetrine-Gγ2, or Gq-121Rluc2/mAmetrine-Gγ1) for the detection of protein-protein interactions. However, the assays would also allow one to combine two unimolecular biosensors or three probes to monitor the interaction of one partner in fusion with Rluc2 with two distinct partners in fusion with mAmetrine and EBFP2. In the latter case, the extent of BRET observed with each partner will be influenced by the relative expression levels of the partners. One can easily determine the optimal acceptor/donor ratio to use for signal detection by performing BRET titration experiments for each FP individually, where the Rluc2-fused construct is maintained constant and the level of the FP-fused construct is increased (Fig. S2, B and C). Because there is no spectral overlay between mAmetrine and EBFP2 when the appropriate filters are used, different ratios can easily be used for the two multiplexed pairs without signal contamination. Since the FPs used in the new BRET assays did not influence the interaction or the modulation of the interactions monitored, as shown by the similar amplitude of the BRET change detected between V2R and Gγ2 for the different FP combinations used (Fig. 2 E), we expect that the adaptation and optimization of known BRET interactions into BRET multiplexing should be straightforward.
In addition to introducing three new (to our knowledge) configurations of BRET, we have presented what we believe is a first proof of principle that spectrally resolved BRET assays can be multiplexed to monitor two independent cellular events. These assays will undoubtedly be valuable tools for monitoring protein-protein interactions as well as other events, such as second messenger production, for which RET biosensors can be designed. The robustness and easy implementation of these assays should ensure their wide utilization and facilitate their combination with other approaches, such as bimolecular fluorescence or luminescence complementation assays (30,32), to increase the number of interactions that can be simultaneously monitored within the same cell.
Acknowledgments
We thank Dr. Monique Lagacé for her critical reading of the manuscript.
This work was supported by grants from the Canadian Institute for Health Research, the Heart and Stroke Foundation of Canada, and the Kidney Foundation of Canada to M.B. B.B. received a studentship from the Fonds de la Recherche en Santé du Québec. M.B. holds the Canada Research Chair in Signal Transduction and Molecular Pharmacology.
Supporting Material
References
- 1.Morin J.G., Hastings J.W. Energy transfer in a bioluminescent system. J. Cell. Physiol. 1971;77:313–318. doi: 10.1002/jcp.1040770305. [DOI] [PubMed] [Google Scholar]
- 2.Dacres H., Wang J., Trowell S.C. Experimental determination of the Förster distance for two commonly used bioluminescent resonance energy transfer pairs. Anal. Chem. 2010;82:432–435. doi: 10.1021/ac9022956. [DOI] [PubMed] [Google Scholar]
- 3.Pfleger K.D., Eidne K.A. Illuminating insights into protein-protein interactions using bioluminescence resonance energy transfer (BRET) Nat. Methods. 2006;3:165–174. doi: 10.1038/nmeth841. [DOI] [PubMed] [Google Scholar]
- 4.Hamdan F.F., Percherancier Y., Bouvier M. MonitorinG-protein-protein interactions in living cells by bioluminescence resonance energy transfer (BRET) Curr. Protoc. Neurosci. 2006 doi: 10.1002/0471142301.ns0523s34. Chapter 5:Unit 5.23. [DOI] [PubMed] [Google Scholar]
- 5.Xu Y., Piston D.W., Johnson C.H. A bioluminescence resonance energy transfer (BRET) system: application to interacting circadian clock proteins. Proc. Natl. Acad. Sci. USA. 1999;96:151–156. doi: 10.1073/pnas.96.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Angers S., Salahpour A., Bouvier M. Detection of β2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET) Proc. Natl. Acad. Sci. USA. 2000;97:3684–3689. doi: 10.1073/pnas.060590697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang L.I., Collins J., Sternweis P.C. Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J. Biol. Chem. 2007;282:10576–10584. doi: 10.1074/jbc.M609695200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Charest P.G., Terrillon S., Bouvier M. Monitoring agonist-promoted conformational changes of β-arrestin in living cells by intramolecular BRET. EMBO Rep. 2005;6:334–340. doi: 10.1038/sj.embor.7400373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pontier S.M., Percherancier Y., Bouvier M. Cholesterol-dependent separation of the β2-adrenergic receptor from its partners determines signaling efficacy: insight into nanoscale organization of signal transduction. J. Biol. Chem. 2008;283:24659–24672. doi: 10.1074/jbc.M800778200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martel C., Dugré-Brisson S., Desgroseillers L. Multimerization of Staufen1 in live cells. RNA. 2010;16:585–597. doi: 10.1261/rna.1664210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Molinari P., Casella I., Costa T. Functional complementation of high-efficiency resonance energy transfer: a new tool for the study of protein binding interactions in living cells. Biochem. J. 2008;409:251–261. doi: 10.1042/BJ20070803. [DOI] [PubMed] [Google Scholar]
- 12.Bertrand L., Parent S., Ménard L. The BRET2/arrestin assay in stable recombinant cells: a platform to screen for compounds that interact with G-protein-coupled receptors (GPCRS) J. Recept. Signal Transduct. Res. 2002;22:533–541. doi: 10.1081/rrs-120014619. [DOI] [PubMed] [Google Scholar]
- 13.Mercier J.F., Salahpour A., Bouvier M. Quantitative assessment of β1- and β2-adrenergic receptor homo- and heterodimerization by bioluminescence resonance energy transfer. J. Biol. Chem. 2002;277:44925–44931. doi: 10.1074/jbc.M205767200. [DOI] [PubMed] [Google Scholar]
- 14.De A., Ray P., Gambhir S.S. BRET3: a red-shifted bioluminescence resonance energy transfer (BRET)-based integrated platform for imaginG-protein-protein interactions from single live cells and living animals. FASEB J. 2009;23:2702–2709. doi: 10.1096/fj.08-118919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loening A.M., Fenn T.D., Gambhir S.S. Consensus guided mutagenesis of Renilla luciferase yields enhanced stability and light output. Protein Eng. Des. Sel. 2006;19:391–400. doi: 10.1093/protein/gzl023. [DOI] [PubMed] [Google Scholar]
- 16.Galés C., Rebois R.V., Bouvier M. Real-time monitoring of receptor and G-protein interactions in living cells. Nat. Methods. 2005;2:177–184. doi: 10.1038/nmeth743. [DOI] [PubMed] [Google Scholar]
- 17.Ai H.W., Shaner N.C., Campbell R.E. Exploration of new chromophore structures leads to the identification of improved blue fluorescent proteins. Biochemistry. 2007;46:5904–5910. doi: 10.1021/bi700199g. [DOI] [PubMed] [Google Scholar]
- 18.Ai H.W., Hazelwood K.L., Campbell R.E. Fluorescent protein FRET pairs for ratiometric imaging of dual biosensors. Nat. Methods. 2008;5:401–403. doi: 10.1038/nmeth.1207. [DOI] [PubMed] [Google Scholar]
- 19.Kremers G.J., Goedhart J., Gadella T.W., Jr. Cyan and yellow super fluorescent proteins with improved brightness, protein folding, and FRET Förster radius. Biochemistry. 2006;45:6570–6580. doi: 10.1021/bi0516273. [DOI] [PubMed] [Google Scholar]
- 20.Leduc M., Breton B., Heveker N. Functional selectivity of natural and synthetic prostaglandin EP4 receptor ligands. J. Pharmacol. Exp. Ther. 2009;331:297–307. doi: 10.1124/jpet.109.156398. [DOI] [PubMed] [Google Scholar]
- 21.Day R.N., Davidson M.W. The fluorescent protein palette: tools for cellular imaging. Chem. Soc. Rev. 2009;38:2887–2921. doi: 10.1039/b901966a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bünemann M., Frank M., Lohse M.J. Gi protein activation in intact cells involves subunit rearrangement rather than dissociation. Proc. Natl. Acad. Sci. USA. 2003;100:16077–16082. doi: 10.1073/pnas.2536719100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galés C., Van Durm J.J., Bouvier M. Probing the activation-promoted structural rearrangements in preassembled receptor-G-protein complexes. Nat. Struct. Mol. Biol. 2006;13:778–786. doi: 10.1038/nsmb1134. [DOI] [PubMed] [Google Scholar]
- 24.Jiang L.I., Collins J., Sternweis P.C. Regulation of cAMP responses by the G12/13 pathway converges on adenylyl cyclase VII. J. Biol. Chem. 2008;283:23429–23439. doi: 10.1074/jbc.M803281200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kocan M., See H.B., Pfleger K.D. Demonstration of improvements to the bioluminescence resonance energy transfer (BRET) technology for the monitoring of G-protein-coupled receptors in live cells. J. Biomol. Screen. 2008;13:888–898. doi: 10.1177/1087057108324032. [DOI] [PubMed] [Google Scholar]
- 26.Hensen J., Haenelt M., Gross P. Lithium induced polyuria and renal vasopressin receptor density. Nephrol. Dial. Transplant. 1996;11:622–627. doi: 10.1093/oxfordjournals.ndt.a027350. [DOI] [PubMed] [Google Scholar]
- 27.Carlson H.J., Campbell R.E. Genetically encoded FRET-based biosensors for multiparameter fluorescence imaging. Curr. Opin. Biotechnol. 2009;20:19–27. doi: 10.1016/j.copbio.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 28.Boute N., Jockers R., Issad T. The use of resonance energy transfer in high-throughput screening: BRET versus FRET. Trends Pharmacol. Sci. 2002;23:351–354. doi: 10.1016/s0165-6147(02)02062-x. [DOI] [PubMed] [Google Scholar]
- 29.Carriba P., Navarro G., Franco R. Detection of heteromerization of more than two proteins by sequential BRET-FRET. Nat. Methods. 2008;5:727–733. doi: 10.1038/nmeth.1229. [DOI] [PubMed] [Google Scholar]
- 30.Héroux M., Hogue M., Bouvier M. Functional calcitonin gene-related peptide receptors are formed by the asymmetric assembly of a calcitonin receptor-like receptor homo-oligomer and a monomer of receptor activity-modifyinG-protein-1. J. Biol. Chem. 2007;282:31610–31620. doi: 10.1074/jbc.M701790200. [DOI] [PubMed] [Google Scholar]
- 31.Rebois R.V., Robitaille M., Hébert T.E. CombininG-protein complementation assays with resonance energy transfer to detect multipartner protein complexes in living cells. Methods. 2008;45:214–218. doi: 10.1016/j.ymeth.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 32.Stefan E., Aquin S., Michnick S.W. Quantification of dynamic protein complexes using Renilla luciferase fragment complementation applied to protein kinase A activities in vivo. Proc. Natl. Acad. Sci. USA. 2007;104:16916–16921. doi: 10.1073/pnas.0704257104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.