

Abstract
We have previously characterized a 21-kDa protein encoded by UL138 (pUL138) as a viral factor inherent to low-passage strains of human cytomegalovirus (HCMV) that is required for latent infection in vitro. pUL138 is encoded on 3.6-, 2.7-, and 1.4-kb 3′ coterminal transcripts that are produced during productive and latent infections. pUL138 is encoded at the 3′ end of each transcript and is preceded by an extensive 5′ sequence (∼0.5 to 2.5 kb) containing several putative open reading frames (ORFs). We determined that three putative ORFs upstream of UL138 (UL133, UL135, and UL136) encode proteins. The UL138 transcripts are polycistronic, such that each transcript expresses pUL138 in addition to the most-5′ ORF. The upstream coding sequences (CDS) present a significant challenge for the translation of pUL138 in mammalian cells. We hypothesized that sequences 5′ of UL138 mediate translation initiation of pUL138 by alternative strategies. Accordingly, a 663-nucloetide (nt) sequence overlapping the UL136 CDS supported expression of a downstream cistron in a bicistronic reporter system. We did not detect cryptic promoter activity or RNA splicing events that could account for downstream cistron expression. These data are consistent with the sequence element functioning as an internal ribosome entry site (IRES). Interestingly, pUL138 expression from the 3.6- and 2.7-kb transcripts was induced by serum stress, which concomitantly inhibited normal cap-dependent translation. Our work suggests that an alternative and stress-inducible strategy of translation initiation ensures expression of pUL138 under a variety of cellular contexts. The UL138 polycistronic transcripts serve to coordinate the expression of multiple proteins, including a viral determinant of HCMV latency.
Mechanisms of viral coexistence with hosts are poorly understood. Human cytomegalovirus (HCMV), like all herpesviruses, persists in infected individuals indefinitely through a lifelong, latent infection. HCMV persists in 60 to 99% of the population worldwide and is associated with increased risk of atherosclerosis and age-related immune senescence (46, 68, 71). A primary HCMV infection during pregnancy is associated with a high incidence of congenital birth defects, whereas reactivation of latent HCMV in individuals with compromised T-cell immunity is associated with increased morbidity and mortality (5, 6, 9, 46). Understanding viral persistence and latency is essential to understanding both the overt and nonovert pathologies of HCMV and to developing antivirals that target the latent infection.
The establishment of latency, while poorly understood, involves coordinated interactions between multiple viral (3, 22, 34, 52) and cellular determinants (14, 57, 60). HCMV latency has been best studied in hematopoietic cells of the myeloid lineage (20-22, 42, 58). We have recently identified sequences in the ULb′ region of the HCMV genome, unique to clinical strains of HCMV, that are required for an in vitro latent infection in CD34+ hematopoietic progenitor cells (22). Specifically, disruption of the UL138 coding sequences (CDS) in the ULb′ region results in a virus that fails to establish latency and instead replicates productively. UL138 encodes a 21-kDa type-1 transmembrane protein that localizes to the Golgi apparatus (52). The mechanism by which this protein promotes latent infection is not yet known. While pUL138 is required for HCMV latency, it is not sufficient, and other viral determinants likely contribute to the establishment of latency (52). pUL138 is encoded at the 3′ end of multiple, long transcripts, with at least three putative open reading frames (ORFs) 5′ of its CDS (UL133, UL135, and UL136) for which the coding potential is unknown. Given the extensive length (∼0.5 to 2.5 kb) of the sequence and multiple start codons 5′ of UL138, we hypothesize that pUL138 requires a noncanonical alternative mechanism of translation initiation.
Eukaryotic cells have several alternative strategies for positioning the 40S ribosomal subunit at the appropriate 5′ AUG codon and initiating protein translation beyond the canonical 5′ m7G cap-dependent ribosome binding and scanning model (26, 51). Alternative mechanisms of translation initiation, commonly used by viruses, include (i) short upstream ORFs (uORFs) (37, 38, 74), (ii) internal ribosomal entry sites (IRESs) (33, 49, 61, 62), and (iii) ribosome shunting (59). The expression of a uORF can positively or negatively regulate the translation of a downstream ORF alone or in combination with other alternative mechanisms of translation initiation (72). Translation from IRES elements of RNA viruses occurs independently of the 5′ cap. However, some cellular transcripts containing IRESs are synthesized with 5′ caps and can use cap-binding translation initiation factors to stabilize RNA secondary structure and recruit ribosomes (16, 17, 51, 62). IRES elements have complex secondary structures, and IRES-mediated translation is often dependent on cell type or context-specific IRES trans-acting factors (ITAFs) (16, 27, 54). Finally, ribosomal shunting is a cap-dependent mechanism in which the 40S ribosomal subunit binds the 5′ cap, scans the mRNA, and then translocates from an upstream shunt donor site past large spans of mRNA, often containing regions of complex secondary structure and uORFs, to a downstream shunt acceptor site, where translation ensues (25). Interestingly, alternative mechanisms of translation initiation are most active during mitosis, cell stress, or viral infection, when canonical cap-dependent mechanisms of translation are inhibited (2, 66, 75).
Our present study analyzes the transcripts and translation mechanisms involved for expression of the pUL138 HCMV latency determinant. We have examined the protein-coding potential of the three UL138 transcripts in the context of virus infection and in cells expressing individual transcripts encoding pUL138. We determined that these transcripts encode three novel proteins, pUL133, pUL135, and pUL136, in addition to pUL138. The existence of these proteins suggests that the transcripts are functionally polycistronic and presents a significant challenge for the translation of UL138 in mammalian cells. Indeed, each transcript supported the expression of pUL138 in addition to the most-5′ cistron. We hypothesized that alternative strategies of translation initiation were required for the translation of UL138 from these transcripts. We identified a 663-nucleotide (nt) sequence, overlapping the 5′ end of UL136, that promoted translation of a downstream cistron in a bicistronic reporter system. Our data are consistent with this element functioning as an IRES. Interestingly, translation of the UL138 cistron from the largest two transcripts is induced under serum starvation stress, which inhibits normal cap-dependent translation. In contrast, expression of the UL138 cistron from the smallest 1.4-kb transcript was inhibited by serum stress. These results suggest that HCMV uses multiple mechanisms of translation initiation to ensure the expression of pUL138 under a variety of cellular contexts. Given that these transcripts give rise to multiple proteins, this region may represent a latency locus that functions to coordinate expression of the proteins required for latency.
MATERIALS AND METHODS
Cells.
Human embryonic lung fibroblasts (MRC5) and human embryonic kidney 293 (HEK-293) (ATCC, Manassas, VA) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES, 1 mM sodium pyruvate, 2 mM l-glutamine, 0.1 mM nonessential amino acids, 100 U/ml penicillin, and 100 μg/ml streptomycin. For experiments requiring serum starvation, MRC5 cells were cultured in normal growth medium without serum for 24 h prior to nucleofection and through the duration of the experiment. Mononuclear cells were isolated from human cord blood or bone marrow from donors at University Medical Center at the University of Arizona by using Ficoll/Paque Plus (Amersham Pharmacia Biosciences) density gradient centrifugation with a protocol approved by the Institutional Review Board. Magnetic cell separation (Miltenyi Biotec) was used to enrich for CD34+ hematopoietic progenitor cells. CD34+ cells were maintained in Iscove's modified Dulbecco's medium (IMDM) supplemented with 10% BIT serum substitute (StemCell Technologies), 2 mM l-glutamine, 20 ng/ml human low-density lipoprotein (Sigma), and 50 μM 2-mercaptoethanol.
Viruses.
A fusion-inducing factor X (FIX) virus strain bacterial artificial chromosome (BAC) that expresses the green fluorescent protein (GFP) was used as the parental strain (FIXwt) for all recombinations described. Viruses were propagated by electroporating infectious BAC DNA into MRC5 cells and purified by density gradient centrifugation as described previously (52). Virus titers were determined by 50% tissue culture infective dose (TCID50) assays on MRC5 cells.
Recombinant viruses (FIX-UL133myc, FIX-UL135myc, FIX-UL136myc, FIX-LUC-ΔUL138) were constructed in Escherichia coli by linear recombination in a two-step positive/negative selection method that leaves no trace of the engineering process (52, 73, 76). Briefly, in the first step, PCR products containing galK flanked by homologous viral sequences target the galK insertion into the viral BAC by homologous recombination. GalK+ recombinants were selected on minimal medium plates containing galactose as a sole carbon source. In the second step, the galK insertion was replaced with PCR products containing the myc epitope tag flanked by viral sequences homologous to the desired insertion site. Similarly, the FIX-LUC-ΔUL138 recombinant virus was generated by replacing galK with the PCR product encoding firefly luciferase (FL) amplified from the pGL3-Basic plasmid (Promega). Recombinants were selected on M63 minimal plates containing 0.2% 2-deoxygalactose and 15 μg/ml chloramphenicol and further screened by BAC digestion, PCR, and sequencing. The generation of the FIX-UL138myc recombinant virus was described previously (52). All primers used for virus recombination are listed in Table 1.
TABLE 1.
Primers used for recombinant virus generation
| Primer name | Sequence (5′ to 3′) |
|---|---|
| FIX-LUC-ΔUL138-Fwd | TGTACAAAAGAGAGAGACTGGGACGTAGATCCGGACAGAGGACGGTCACCA |
| FIX-LUC-ΔUL138-Rev | GTCAAAACGACATTACCGCGATCCGCTCCCCTCTTTTTTCTTTTTCTCATTTACACGGCGATCTTTCCGC |
| FIX-UL133myc-GalK-Fwd | CCGTGGCCGCGGCTCTACAACAACAGCAGCAGCAGCTACAGACCGGAACGCCTGTTGACAATTAATCATCGGCA |
| FIX-UL133myc-GalK-Rev | GGAGATGTGGGCCAAGTCGGAAAATTCCTTATCACACCGGGGGCGGGTTATCAGCACTGTCCTGCTCCTT |
| FIX-UL133myc-Myc-Fwd | GTGGCCGCGGCTCTACAACAACAGCAGCAGCAGCATCAGACCGGAACGGAACAAAAACTCATCTCAGAAGAGGATCTG |
| FIX-UL133myc-Myc-Rev | AGATGTGGGCCAAGTCGGAAAATTCCTTATCACACCGGGGGCGGGTTACAGATCCTCTTCTGAGATGAGTTTTTGTTC |
| FIX-UL135myc-GalK-Fwd | GGAAAAGGCGGTGCAGAGCGTCATGAAGGACGCCGAGTCAATGCAGATGACCCCTGTTGACAATTAATCATCGGC |
| FIX-UL135myc-GalK-Rev | GAGGGAAGGCGTGTGCTGCTATACAACTGTACAACGGACGCGCTCGCTGTTTCGGTCTCAGCACTGTCCTGCTCCTT |
| FIX-UL135myc-Myc-Fwd | GGAAAAGGCGGTGCAGAGCGTCATGAAGGACGCCGAGTCAATGCAGATGACCGAACAAAAACTCATCTCAGAAGAGGATCTG |
| FIX-UL135myc-Myc-Rev | GGCGTGTGCTGCTATACAACTGTACAACGGACGCGCTCGCTGTTTCGGTCCAGATCCTCTTCTGAGATGAGTTTTTGTTC |
| FIX-UL136myc-GalK-Fwd | CGACCGTGCAAGTAAGTGGCCCGCGGGAGAACGCCGTATCTCCCGCTACGCCTGTTGACAATTAATCATCGGCA |
| FIX-UL136myc-GalK-Rev | TCTCTCTTTTGTACAGCACTCGCGCGGGAACGGCCCCCTCAACCCTCTTATCAGCACTGTCCTGCTCCTT |
| FIX-UL136myc-Myc-Fwd | ACCGTGCAAGTAAGTGGCCCGCGGGAGAACGCCGTATCTCCCGCTACGGAACAAAAACTCATCTCAGAAGAGGATCTG |
| FIX-UL136myc-Myc-Rev | TCTCTTTTGTACAGCACTCGCGCGGGAACGGCCCCCTCAACCCTCTTACAGATCCTCTTTGA |
Plasmids.
All primers used for plasmid cloning are listed in Table 2. The bicistronic, dual luciferase reporter construct (pCDNA3-rLuc-polIRES-fLuc) containing the poliovirus IRES (pv-IRES) was kindly provided by N. Sonenberg (McGill University, Canada) (55). The poliovirus IRES was excised using KpnI and BamHI restriction sites, and portions of UL138 cDNA upstream of UL138 or the Kaposi sarcoma-associated herpesvirus (KSHV) ORF72 IRES (nucleotide position 123206 to 122973) (23) were cloned into the plasmid using primers flanked with KpnI and BamHI restriction sites and the FIXwt BAC DNA or KSHV genomic DNA as the template. The KSHV genome DNA was derived from the KSHV-positive primary effusion lymphoma (PEL) cell line BCBL-1, a kind gift from P. Schaffer (University of Arizona). Sequences 5′ of UL138 or the KSHV-IRES were cloned into the promoterless pGL3-Basic plasmid (Promega) using primers with KpnI and HindIII restriction sites. Generation of pGL3-major immediate-early promoter (pGL3-MIEP1400) was described previously (52). cDNAs (pFL) encoding each of the UL138 transcripts in which FL had been substituted for UL138 were cloned into pEF1/myc-His-B (Invitrogen) using XbaI and PmeI restriction sites and FIX-LUC-ΔUL138 as the template.
TABLE 2.
Primers used for cloning expression vectors
| Construct name | Orientationd | Sequence (5′ to 3′)a |
|---|---|---|
| ICS-1 | FWD | ggtaccTGACTCGGCAGCCACTGTA |
| REV | ggatccTTTGTTCTTCAGTTTACCCTTCGGTTT | |
| ICS-2 | FWD | ggtaccGGGAATTTTTCAAAGATTCCATAATC |
| REV | ggatccAAGTCGGAAAATTCCTTATCACA | |
| ICS-3 | FWD | ggtaccAAACCGAAGGGTAAACTGAAGAACAAA |
| REV | ggatccATGATCTTCTCTCCTGCTTGGAAT | |
| ICS-4 | FWD | ggtaccGGACTGGCTTCCCTGGT |
| REV | ggatccCTTTTCCAAGAGTTCCTCGATGT | |
| ICS-5 | FWD | ggtaccTCGTCGAGCTCTTTGTCGTCCT |
| REV | ggatccAAAACGGCACAGATAACGTGAAA | |
| ICS-6 | FWD | ggtaccATGGTCGGACATCGAGGAACT |
| REV | ggatccTAATGACAGCCGCAAAATAGATCG | |
| ICS-7 | FWD | ggtaccGTACAGTTGTATAGCAGCACACG |
| REV | ggatccTACGAGTCGCGGATGATGTTA | |
| ICS-8 | FWD | ggtaccTTCACGTTATCTGTGCCGTTTTGC |
| REV | ggatccACCGTCCTCTGTCCGGATCTAC | |
| ICS-9 | FWD | ggtaccGGTGTACGTCGCTCTACATAGGAG |
| REV | ggatccTCTCGTCTCGTCCACCATTT | |
| ICS-10 | FWD | ggtaccTCCTCGTCGAGCTCTTTGTC |
| REV | ggatccTACGAGTCGCGGATGATGTTA | |
| ICS-11 | FWD | ggtaccTAAGAACCTGAGCACGCCGC |
| REV | ggatccTCTCGTCTCGTCCACCATTT | |
| ICS-12 | FWD | ggtaccGGGCTGGAGAATCTCTCGAA |
| REV | ggatccTCTCGTCTCGTCCACCATTT | |
| ICS-13 | FWD | ggtaccGGACTTGGACGTTGGAAATAAAT |
| REV | ggatccTCTCTCTTTTGTACAGCACTCGC | |
| ICS-14 | FWD | ggtaccGTCATTATCCTGAGAAGTGCCG |
| REV | ggatccTCTCTCTTTTGTACAGCACTCGC | |
| ICS7-RC | FWD | ggtaccTACGAGTCGCGGATGATGTTA |
| REV | ggatccGTACAGTTGTATAGCAGCACACG | |
| ICS-7A | FWD | ggtaccGTACAGTTGTATAGCAGCACACG |
| REV | ggatccTAATGACAGCCGCAAAATAGATCG | |
| ICS-7B | FWD | ggtaccGTACAGTTGTATAGCAGCACACG |
| REV | ggatccATTTCCACCGTGTCGAAGC | |
| ICS-7C | FWD | ggtaccTGAGTCGCATTCACCGGTTCT |
| REV | ggatccTACGAGTCGCGGATGATGTTA | |
| ICS-7D | FWD | ggtaccATCTATTTTGCGGCTGTCATTATCC |
| REV | ggatccTACGAGTCGCGGATGATGTTA | |
| KSHV-IRES | FWD | ggtaccTTGGACAGACTCCTACTTATAAAGC |
| REV | ggatccTTGTATATGTGAAGGCACCGAT | |
| KSHV-IRES-RC | FWD | ggtaccTTGTATATGTGAAGGCACCGATGT |
| REV | ggatccTTGGACAGACTCCTACTTATAAAGCAGG | |
| KSHV-IRES-pGL3 | FWD | ggtaccTTGGACAGACTCCTACTTATAAAGC |
| REV | aagcttTTGTATATGTGAAGGCACCGATGTG | |
| ICS-4-pGL3 | FWD | ggtaccGGACTGGCTTCCCTGGT |
| REV | aagcttGCCTTTTCCAAGAGTTCCTCGATGT | |
| ICS-7-pGL3 | FWD | ggtaccGTACAGTTGTATAGCAGCACACG |
| REV | aagcttTACGAGTCGCGGATGATGTTA | |
| ICS-7-RC-pGL3 | FWD | ggtaccTACGAGTCGCGGATGATGTTA |
| REV | aagcttTACGAGTCGCGGATGATGTTA | |
| FL | FWD | gtttaaacTTACACGGCGATCTTTCCGC |
| REV | tctagaATGGAAGACGCCAAAAACATAAAGAAAGGC | |
| FL-RC | FWD | tctagaTTGGCCTTTATGAGGATCTCTCTGATTTTTCTTG |
| REV | gtttaaacATGGAAGACGCCAAAAACATAAAGAAAGGC | |
| FIX-3.6 | FWD | gatatcGCAGTAGCGATGGGTTGCGACG |
| FWD | tctagaGCAGTAGCGATGGGTTGCGACG | |
| FIX-2.7 | FWD | gatatcCGGAGCTTGAGATTCCAAGCAGG |
| FWD | tctagaCGGAGCTTGAGATTCCAAGCAGG | |
| FIX-1.4 | FWD | gatatcGAGACATGCTCCACGATCTATTTT |
| FWD | tctagaGAGACATGCTCCACGATCTATTTT | |
| FIX-T-3′b | REV | gtttaaacTTACTGACTCATGTGAAAAGTGTGCTTTTTATTAACAGAGCAGAGGG |
| REV | gcgatcgcCATGTGAAAAGTGTGCTTTTTATTAACAGAGCAGAG | |
| UL133 | FWD | ATGGGTTGCGACGTGCACG |
| UL134 | FWD | ATGGCCAGGACCAGGG |
| UL135 | FWD | ATGTCCGTACACCGGCC |
| UL136 | FWD | ATGTCAGTCAAGGGCGTGGAG |
| UL137 | FWD | ATGGCCACGATCAGCACGAG |
| ORF-Mycc | REV | gatatcGCTCACAGATCCTCTTCTGAGATGAGTT |
Enzyme sites are in lowercase.
Used for all three cDNAs.
Used for all ORFs.
FWD, forward; REV, reverse.
The pCIG lentivirus expression vector (32) obtained from L. Lybarger (University of Arizona) was further modified to (i) provide a more versatile multiple-cloning site (MCS)-IRES-GFP cassette resulting in pCIG2-IRES-EGFP (where EGFP is enhanced GFP) and (ii) to remove all posttranscriptional regulatory elements, including the IRES GFP and woodchuck posttranscriptional regulatory element (WPRE), resulting in pCIG2-IGW. Briefly, pCIG2-IRES-EGFP was generated with an expanded MCS by annealing and elongating two primers with partial homology (bold) with EcoRI and BamHI restriction enzyme sites (underlined) at each end (forward, 5′-GGGG GAATTC GG TGA TCA GGG TAT ACG GCT CGA GGG GAT ATC GGG TTT AAA CGG GGC GC-3′; reverse, 5′-GGGG GGATCC CCT TAA TTA ACC TTC GAA CCG CGA TCG CCC GGC GCG CCC CGT TTA AAC C-3′). Next, the encephalomyocarditis virus IRES (EMCV-IRES) was PCR amplified from pIRESpuro (Clontech) using primers containing BamHI and BglII enzyme sites (underlined) (forward, 5′-GGGG GGATCC GCC CCT CTC CCT CCC CCC CCC C-3′; reverse, 5′-GGGG AGATCT GGT TGT GGC AAG CTT ATC ATC G-3′). EGFP was amplified from pCMS-EGFP (Clontech) using primers containing BglII and SalI enzyme sites (underlined) while maintaining the contextual Kozak sequence (bold) immediately preceding the ATG (forward, 5′-GGGG AGATCT CGC CAC CAT GGT GAG CAA GGG CGA GGA GC-3′; reverse, 5′-GGGG GTCGAC CTG CCC CAG CTG GTT CTT TCC GCC-3′). The MCS, ECMV-IRES and EGFP PCR amplicons were digested, ligated, and inserted into the EcoRI and SalI sites of pCIG lacking the parental IRES and GFP to form pCIG2-IRES-EGFP. The modified MCS contains the following enzyme sites in 5′-to-3′ order: XbaI, NheI, EcoRI, BclI, BstZ17I, XhoI, EcoRV, PmeI, AscI, AsiSI, BstBI, PacI, and BamHI. The pCIG2-IRES-EGFP vector was used to clone individual myc-tagged ORFs amplified from pEF1/myc-His-B expression plasmids containing each myc-tagged ORF using the EcoRV restriction site. This vector contains an IRES sequence downstream of the multiple cloning site that allows for the coexpression of enhanced GFP as a marker for infection. For vectors containing UL138 cDNAs, the pCIG2-IRES-EGFP expression vector was further modified to pCIG-IGW. Briefly, the IRES, EGFP, and WPRE posttranscriptional regulatory element were excised using PacI and KpnI followed by religation. UL138 cDNAs were cloned into pCIG-IGW using XbaI and PmeI or EcoRV and AsiSI restriction sites with FIXwt BAC as the template.
Transient transfection and luciferase reporter assays.
For transient expression in HEK-293 cells, molar equivalents (1 × 1010 to 7 × 1010 copies) of the indicated constructs were transfected into confluent HEK-293 cells using Lipofectamine 2000 (Invitrogen) and Opti-MEM I (Invitrogen) according to the manufacturer's recommendations. At 4 h posttransfection, the medium was replaced with normal growth medium, and protein expression was assayed at 48 h posttransfection. Luciferase activity was measured using either the Dual-Luciferase reporter assay system (Promega) or the luciferase assay system (Promega) as described previously (52). Nucleofection of subconfluent MRC5 cells was performed with molar equivalents (3 × 1011 copies) of constructs using the basic Nucleofector kit for primary mammalian fibroblasts (Lonza) program U-023 according to the manufacturer's recommendations. Protein expression was analyzed at 6 h postnucleofection. For RNA experiments, molar equivalents (3 × 1011 copies) of in vitro-transcribed RNA (iv-RNA) were transfected into HEK-293 cells using Lipofectamine 2000 (Invitrogen) and Opti-MEM I (Invitrogen), and FL activity was assayed 6 h posttransfection.
Immunoblotting.
Protein lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to a 0.45-μm polyvinylidene difluoride (Immobilon-FL [Millipore]) membrane, and imaged as described previously (52). Primary antibody conditions for rabbit anti-myc, mouse anti-myc, mouse anti-α-tubulin, and mouse anti-HCMV IE1 and IE2 proteins (generous gift from T. Shenk; clone 3H4) were described previously (52). Custom rabbit polyclonal, primary antibodies (Open Biosystems) were generated for pUL133, pUL135, pUL136, and UL138. Each antibody was affinity purified using the immunizing peptide and used at the following concentrations: anti-UL133 (2 μg/ml), anti-UL135 (2 μg/ml), and anti-UL136 (5 μg/ml).
Northern blotting.
Total RNA was isolated from HEK-293 cells transfected with the bicistronic luciferase constructs and prepared as described previously (52). A pEF1/myc-His-B (Invitrogen) plasmid containing the reverse complement of firefly luciferase was linearized with PmeI and used to generate an [α-32P]CTP-radiolabeled antisense riboprobe using T7 in the SP6/T7 Riboprobe combination system (Promega) according to the manufacturer's instructions. Probe hybridization and visualization were in accordance with previous methods (52).
RACE.
RNA expressed during HCMV (FIXwt) infection was analyzed by RNA ligase-mediated rapid amplification of cDNA ends (RLM-RACE) using the FirstChoice RLM-RACE kit (Ambion). Briefly, primary human CD34+ cells or MRC5 fibroblasts were infected at a multiplicity of infection (MOI) of 5. At 18 h postinfection (hpi), total RNA was isolated and DNase was treated using the RNA II kit (Macherey-Nagel), processed as directed by the FirstChoice kit recommendations, and reverse transcribed with 90 U of Moloney murine leukemia virus reverse transcriptase (MMLV-RT) (Ambion) and 20 U of SuperScript III (Invitrogen) and 10% dimethyl sulfoxide (DMSO). The reverse transcription proceeded for 45 min at 45°C and then for 20 min at 55°C. The RNA template was degraded by the addition of 10 U RNase H for 30 min at 37°C. As a negative control (NC), RNA was processed as above; however, the tobacco acid pyrophosphatase enzyme (TAP-null) was omitted during the RNA preparation and ligation. The cDNA generated from capped mRNA species was amplified using the 5′ FirstChoice outer primer and gene-specific primers UL138 gsp2 (5′-TAGCTGCACTGGGAAGACACTT-3′), UL135 gsp (5′-ACCGCCTTTTCCAAGAGTT-3′), and UL133 gsp (5′-ATGCTGCTGCTGCTGTTGTTGTA-3′) using Phusion polymerase (New England Biolabs), 500 nM primers, 200 nM deoxynucleoside triphosphates (DNTPs), 3% DMSO, and 1× Phusion GC buffer. Reactions were denatured at 98°C for 1 min and then at 35 cycles of 98°C for 12 s, 65°C for 30 s, and 72°C for 40 s, followed by a final extension time of 5 min at 72°C. The primary PCR amplicon was then subjected to 35 cycles of nested PCR using the 5′ FirstChoice inner primer, which contains a BamHI restriction site, and primers UL138 REV-411nt (5′-GTATCTAGAGTCGTGCCAATGGTAAGCTAGA-3′), UL135 REV-62nt (5′-GTATCTAGAGCAGGCACCACGCTTATCAGAA-3′), and UL133 REV-187nt (5′-GTATCTAGACACGAAGGTTTGTTCTTCAGTTTA-3′) (with the XbaI insertion site indicated by underlining) using the same thermocycling parameters that were used during the primary PCR. Resulting products were digested with BamHI-HF and XbaI (New England Biolabs), gel purified, cloned, and sequenced. For the 3′ RACE, total RNA was reverse transcribed as described above and primed by a poly(A) adapter primer supplied with the FirstChoice RLM-RACE kit (Ambion). cDNA was then amplified with the 3′ outer primer and a primer for UL138 FWD (5′-ATGGACGATCTGCCGCTGAACGT-3′) using Phusion polymerase with GC buffer, 500 nM primers, 200 nM DNTPs, and 3% DMSO. Cycling parameters were 98°C for 30 s and then 35 cycles of 98°C for 10 s, 65°C for 30 s, and 72°C for 30 s, followed by a final extension at 72°C for 5 min. The primary PCR amplicon was then subjected to 35 cycles of nested PCR using the 3′ FirstChoice inner primer and UL138 gsp1 (5′-TCTAGCTTACCATTGGCACGAC-3′), with the same cycling parameters. The resulting product was digested with BamHI-HF (New England Biolabs), gel purified, cloned, and sequenced.
In vitro transcription and translation.
pEF1/myc-His-B expression plasmids containing FIX-LUC-Δ138 cDNAs were linearized using PmeI, phenol-chloroform extracted, and transcribed using the mMessage mMachine T7 kit (Ambion) and subsequently poly(A) tailed using the poly(A) tailing kit (Ambion) according to the manufacturer's instructions. Retic lysate IVT (Ambion) was used for translation of in vitro-transcribed RNA using a 1:1 mix of the 20× translation mix lacking Met and Leu (potassium acetate concentration, 80 mM) according to the manufacturer's instructions.
RNA structure prediction.
RNA secondary structure prediction was generated using the RNA Mfold server, version 3.2 (77) (http://mfold.bioinfo.rpi.edu/cgi-bin/rna-form1.cgi). Default parameters were used, except for the following: structure draw mode was set at “untangle with loop fix,” exterior loop type was set to “flat,” and structure annotation was set to “p-num.” Free energy of stable stem loops was calculated by submitting the sequence for each stem loop individually to the Mfold server.
qRT-PCR.
RNA was isolated and DNase treated using the NucleoSpin RNA II kit (Machery-Nagel) according to the manufacturer's instructions. cDNA was generated from 1 μg input RNA or in vitro-transcribed and poly(A)-tailed RNA using an anchored oligo[dT]18 primer and the transcriptor first-strand cDNA synthesis kit (Roche) according to the manufacturer's instructions. Quantitative reverse transcription PCR (qRT-PCR) was performed with the LightCycler 480 Probes Master (Roche) according to the manufacturer's instructions along with Universal Probe Library (Roche) probes and primers specific for UL133 (forward, 5′-TTTCTGGACCTGGCATCG-3′; reverse, 5′-TACGCGCAGAGGAAAATCAT-3′; probe number 114, 5′-CTGTGGTC-3′), UL135 (forward, 5′-GCGGTGTACGTCGCTCTAC-3′; reverse, 5′-GGAAACTCGGGTTTATCTATCG-3′; probe number 55, 5′-GGCAGACCATCCTCTCCTATG-3′), UL136 (forward, 5′-GCGGCTGTCATTATCCTGAG-3′; reverse, 5′-TAGTACATGGCCCCGTTCA-3′; probe number 152, 5′-TCGCCGTC-3′), UL138 (forward, 5′-GGTTCATCGTCTTCGTCGTC-3′; reverse, 5′-CACGGGTTTCAACAGATCG-3′; probe number 126, 5′-TCTCTGGT-3′), and firefly luciferase (FL) (forward, 5′-TGAGTACTTCGAAATGTCCGTTC-3′; reverse, 5′-GTATTCAGCCCATATCGTTTCAT-3′; probe number 29, 5′-GAAGACGG-3′) and the prevalidated UPL human beta-actin gene assay (Roche). Linear fragments for the generation of UL133, UL135, UL136, UL138, and firefly luciferase standard curves were synthesized using the following primer sets, using the FIX BAC or pGL3-Basic plasmid (Promega) as a template: UL133 (forward, 5′-ATGGGTTGCGACGTGCACG-3′; reverse, 5′-TTACGTTCCGGTCTGATGCTGC-3′), UL135 (forward, 5′-ATGTCCGTACACCGGCC-3′; reverse, 5′-TCAGGTCATCTGCATTGACTCG-3′), UL136 (forward, 5′-AGAGAGAAAGAGAGTATGTCAGTC-3′; reverse, 5′-TTACGTAGCGGGAGATACGGC-3′), UL138 (forward, 5′-ATGGACGATCGTCCGCTGAA-3′; reverse, 5′-TCACGTGTATTCTTGATGATAA-3′), firefly luciferase (forward, 5′-ATGGAAGACGCCAAAAACATAAAGAAAG-3′; reverse, 5′-TTGGCCTTTATGAGGATCTCTCTGATTT-3′). qRT-PCR conditions were as follows: 95°C for 10 min and then 60 cycles of 95°C for 10 s and 60°C for 30 s. Reactions were run on a LightCycler 480 (Roche), and data analysis was performed using the LightCycler 480 software (version 1.5) using the second derivative max method.
A fold change of 5′ and 3′ probe amplification for each transcript was used to quantify transcript ends compared to cDNA ends derived from in vitro-transcribed RNA (iv-RNA) using the following equation (53):
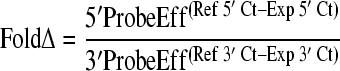 |
where Eff is efficiency, Ref is reference sample (iv-RNA), Exp is the experimental sample, and Ct is the crossing point. The efficiency of each probe set was determined by standard curve amplification: UL133 (1.646), UL135 (1.979), UL136 (1.921), UL138 (1.953), and FL (1.897). In our analysis, we considered a 2-fold change to be within the confidence interval for full-length transcripts.
RESULTS
pUL138 is encoded on multiple transcripts in fibroblasts and hematopoietic progenitor cells.
We have previously identified two transcripts of 3.6 and 2.7 kb encoding pUL138 by using Northern blotting and rapid amplification of cDNA ends (RACE) (52). Further studies using alternative RACE methods to amplify the cDNA ends generated from polyadenylated RNA isolated from FIX-infected fibroblasts and CD34+ hematopoietic progenitor cells (HPCs) identified a third transcript corresponding to 1.4 kb (Fig. 1). The 1.4-kb transcript 5′ start maps to nucleotide position 188206, 292 nucleotides downstream of the predicted start of UL136. All three transcripts share a 3′ end at nucleotide position 186833, which corresponds to a polyadenylation site just downstream of the putative UL138b ORF (previously ORF11). It is not clear why the 1.4-kb transcript was not identified in our original studies, as all RACE methods should favor the amplification of the smaller transcript. One possibility is that the 1.4-kb transcript is present in lower abundance during infection or that the RNA ligase-mediated rapid amplification of cDNA ends (RLM-RACE) technology used in this study provides enhanced sensitivity. We also failed to detect this transcript by Northern blotting. The presence of this band on Northern blots may have been obscured, as it runs at the same molecular mass as 18S rRNA. Alignment of the UL138 cDNAs with the HCMV genome suggests that splicing events are not involved in generation of the UL138 transcripts (data not shown). All three 5′ RACE products were detected in fibroblasts and CD34+ HPCs, indicating that these transcripts are expressed in multiple contexts of infection.
FIG. 1.

Schematic representation of UL138 transcripts present in CD34+ HPCs and MRC5 fibroblasts. RLM-RACE using gene-specific primers (white arrows) was used to map the precise 5′ and 3′ ends of the polyadenylated transcripts encoding UL138. Template RNAs were purified from infected HPCs or fibroblasts at 24 hpi and enriched for polyadenylated transcripts. These analyses identified a 1.4-kb transcript in addition to the 3.6- and 2.7-kb transcripts described previously (52). The 5′-end sequence of each transcript start is shown below the transcript. Black vertical arrows indicate the transcript start site as determined by RACE, and underlined sequences indicate the experimentally proposed translation start sites for the most-5′ ORF of each transcript. In the case of the 2.7-kb transcript, the mapped 5′ end of the transcript does not include the annotated translation start for UL135 (47), which is marked by an asterisk. US, unique short region; UL, unique long region.
Putative ORFs upstream of UL138 encode proteins during HCMV infection.
Our transcript-mapping studies demonstrate extensive sequence (∼0.5 to 2.5 kb) upstream of UL138 with the potential to encode multiple ORFs. These putative ORFs were originally annotated as start-to-stop codons encoding at least 80 amino acids (7, 13, 47). The presence of putative ORFs upstream of UL138 raises the possibility that these transcripts encode more than one protein or that they contain a substantial 5′ untranslated region (UTR). To examine the coding potential of each putative ORF, we cloned each individual ORF into the pCIG2-IRES-EGFP expression vector with an in-frame C-terminal myc epitope tag. HEK-293 cells were transiently transfected with each construct, and expression of the UL133, UL134, UL135, UL136, UL137, and UL138 ORFs was analyzed by immunoblotting using a mouse monoclonal antibody to the myc epitope. Only UL133, UL135, and UL136 exhibited evidence of protein coding capacity (Fig. 2A). Excluding the myc epitope tag, UL133, UL135, UL136, and UL138 coded for proteins of approximately 28, 36, 27, and 21 kDa, respectively. The molecular masses of pUL133, pUL135, and pUL136 myc-tagged proteins are approximately 10 kDa greater than their predicted molecular mass based on sequence annotation, suggesting that these proteins are posttranslationally modified. While we detected multiple bands for some proteins, the significance of these bands is not known. In the case of pUL138, the additional lower-molecular-mass protein bands appear to be artifacts of transient overexpression of the myc-tagged construct and are not observed in infection with the wild-type virus (52) or a recombinant virus expressing a myc-tagged version of pUL138 (Fig. 2B). Protein products from ORFs UL134 and UL137 were not detected in this assay, suggesting that these ORFs may not encode stable proteins in a transient expression assay. It should be noted that both UL134 and UL137 are transcribed from the opposite DNA strand relative to the other putative ORFs analyzed.
FIG. 2.

UL133, UL135, and UL136 encode proteins. (A) Protein lysates derived from transfected HEK-293 cells expressing UL133, UL134, UL135, UL136, UL137, and UL138 myc-tagged ORFs or the empty vector were analyzed by immunoblotting with the rabbit anti-myc (71D10) and mouse anti-α-tubulin (DM1A) antibodies. (B) Protein lysates from fibroblasts infected with FIX-UL133myc, FIX-UL135myc, FIX-UL136myc, and FIX-UL138myc (MOI, 0.5) recombinant viruses were analyzed at 48 hpi by immunoblotting with mouse anti-myc (9E10) and mouse anti-IE1/2 (3H4) antibodies. α-Tubulin serves as a loading control.
To determine if UL133, UL135, and UL136 encode proteins during HCMV infection, we constructed recombinant viruses with in-frame myc epitope tags inserted at the 5′ end of UL133, UL135, or UL136 of the FIX strain BAC. Infection of MRC5 fibroblasts with the recombinant viruses, termed FIX-UL133myc, FIX-UL135myc, and FIX-UL136myc, resulted in protein production from each ORF, as detected by immunoblotting at 48 hpi with a myc monoclonal antibody (Fig. 2B). Each virus produced a single protein product, except for FIX-UL136myc, which produced two major protein species during infection. The larger-molecular-mass pUL136 band corresponds to the protein species produced following transfection of the pUL136 expression plasmid encoded by the full-length UL136 ORF (Fig. 2A). The smaller-molecular-mass species may represent a truncated form of pUL136. HCMV IE proteins, IE1 and IE2, show similar levels of infection between the recombinant viruses such that the observed levels of pUL133, pUL135, pUL136, and pUL138 reflect their relative levels of expression during infection. These results are the first to demonstrate the coding potential of the ULb′ ORFs UL133, UL135, and UL136.
UL138 is expressed from polycistronic mRNAs.
The existence of bona fide ORFs upstream of UL138 suggests that the UL138 transcripts are polycistronic. To explore this possibility, cDNAs generated for each transcript were cloned into pCIG-IGW, and molar equivalents of each were transfected in HEK-293 cells. Protein lysates were harvested 48 h posttransfection and analyzed by immunoblotting with rabbit polyclonal antisera generated against peptides specific to pUL133, pUL135, pUL136, or pUL138 coding sequences (Fig. 3A). Lysates from cells transfected with an expression construct encoding the myc-tagged version of each individual ORF (as described for the experiments shown in Fig. 2) were used as positive controls.
FIG. 3.

UL138 transcripts are polycistronic. (A) HEK-293 cells were transfected with cDNAs representing the 3.6-, 2.7- and 1.4-kb UL138 transcripts or constructs encoding individual myc-tagged ORFs and analyzed by immunoblotting using polyclonal rabbit antisera specific to pUL133, pUL135, pUL136, and pUL138 or a monoclonal antibody specific to tubulin as a loading control. (B) The intensities of the pUL138 band were quantitated with a Li-Cor Odyssey imaging system and are represented graphically. (C) RNA isolated from lysates described in panel A was analyzed by qRT-PCR. Inverse crossing points (1/Ct) were determined using the Roche LightCycler 480 software with the second derivative max method (graphed). Fold change compares amplifications of the 5′ and 3′ ends of experimental cDNAs isolated from transfected cells to cDNA derived from full-length iv-RNA. Transcripts with fold changes ≤2 compared to iv-RNA were considered to be full length.
The 3.6-kb cDNA expressed a 36-kDa protein corresponding to pUL133 in addition to low-intensity bands corresponding to pUL138. The 2.7-kb cDNA resulted in reactive bands corresponding to pUL135 and pUL138. The expression of pUL135 from the 2.7-kb cDNA was surprising, as the 5′ end of the transcript does not include the predicted start codon for pUL135 (Fig. 1). The expression of this protein from the 2.7-kb cDNA suggests that the translation start for pUL135 may correspond with an in-frame AUG 28 nt downstream of the 5′ end of the 2.7-kb transcript (Fig. 1). Several bands are observed for UL135 expressed during infection (Fig. 2B) or transfection (Fig. 3), indicative of possible alternative starts or posttranslational modifications. The 1.4-kb cDNA expressed two proteins, one corresponding to pUL138 and the other corresponding to a smaller-molecular-mass pUL136. This result was unexpected, since the 5′ start of the 1.4-kb transcript maps downstream of the predicted AUG of UL136 (Fig. 1). This band most likely corresponds to the smaller-molecular-mass species of pUL136 observed during virus infection (Fig. 2B). Full-length pUL136 was not expressed from either the 3.6- or the 2.7-kb cDNAs during transfection, although it was detected during infection (Fig. 2B). These results suggest that expression of full-length pUL136 from the polycistronic transcripts may be specific to the context of viral infection. Importantly, pUL138 was expressed from each of the three cDNAs, with the greatest expression resulting from the transfection of the 1.4-kb cDNA (Fig. 3B).
Our results suggest that UL138 transcripts are polycistronic. To rule out the possibility that internal promoter activity or splicing could result in monocistronic transcripts encoding pUL138, we analyzed the 5′ and 3′ ends of the RNAs by qRT-PCR following transfection of the cDNA constructs. Equivalent amplifications of 5′ and 3′ ends argue against the presence of smaller distinct RNAs that may arise from internal promoter activity or splicing events, since it is unlikely that distinct RNAs would be present at equivalent levels. These results were evaluated against comparable probe amplification of cDNA generated from full-length, in vitro-transcribed RNA (iv-RNA), thus eliminating the possibility of mammalian promoter or splicing activity. Our results indicate equivalent amplifications (within a 2-fold confidence level) of the 5′ and 3′ transcript ends derived from the transfected cells relative to iv-RNAs (Fig. 3C). We interpret this result to indicate that the transcripts supporting pUL138 expression are full length and that monocistronic UL138 transcripts cannot be responsible for pUL138 protein production, as shown in Fig. 3A. In addition, transcript numbers from each cDNA transfection were comparable, indicating that levels of pUL138 expression cannot be explained by differences in transfection efficiency.
A sequence overlapping the 5′ end of the UL136 ORF promotes translation of a downstream cistron.
The expression of multiple proteins from a single transcript necessitates alternative mechanisms of translation initiation. We hypothesized that pUL138 translation is mediated by upstream sequences that facilitate translation initiation. Given the extensive sequence (∼0.5 to 2.5 kb) 5′ of UL138, we favored the possibility that an internal ribosome entry site (IRES) was responsible for pUL138 expression. To explore this possibility, we cloned sequences 5′ of UL138 into a bicistronic reporter construct in which the 5′ cistron encodes Renilla luciferase (RL) and the 3′ cistron encodes firefly luciferase (FL) (Fig. 4A). The cloned sequences were designated IRES candidate sequences (ICS). A construct containing the poliovirus IRES (PV-IRES) or the Kaposi's sarcoma-associated herpesvirus IRES (KSHV-IRES) served as a positive control. A construct lacking an ICS or IRES was used as a negative control (empty). The RL and FL activities of each construct were measured 48 h posttransfection of HEK-293 cells, and IRES activity was expressed as a ratio of FL to RL. Negligible FL expression was measured in the empty control, whereas the PV- or KSHV-IRES resulted in substantial expression of the downstream FL cistron. Of the ICS analyzed, only ICS-7 FL expression was comparable to that of the PV-IRES, and to a greater extent than that of the KSHV-IRES (Fig. 4B). ICS-7 is a 663-nt sequence that starts 65 nt upstream of the start for UL136. ICS-5, -6, and -8 overlapped with ICS-7; however, these sequences supported moderate to low expression of FL. These results suggest that a sequence element upstream of UL138 has the potential to mediate translation of pUL138.
FIG. 4.

A 663-nt sequence overlapping UL136 supports expression of a downstream cistron. (A) Schematic of the bicistronic luciferase construct used to screen sequences 5′ of UL138, designated IRES candidate sequences (ICS). Known viral IRESs (v-IRES) and a construct with an empty intercistronic region (Empty) were used as positive and negative controls, respectively. RL and FL activity was quantified 48 h following transfection of equal molar quantities of each construct into HEK-293 cells. The activity of each intercistronic sequence is expressed as a ratio of FL to RL activity. (B) Analysis of ICS 5′ of UL138 relative to poliovirus IRES (PV), KSHV-IRES (KSHV), or empty controls. (C) Analysis of sequences proximal to and including ICS-7 to determine sequences required for maximal activity. (D) Analysis of portions of ICS-7 to determine the minimal sequence required for ICS-7 activity. The reverse complement of KSHV-IRES (KSHV-RC) and ICS-7 were also analyzed. The activity of each construct relative to ICS-7 is expressed to the right of the graphs in panels C and D.
To further define the sequences required for maximal IRES activity, we cloned fragments extending from the 5′ and 3′ ends of ICS-7 (Fig. 4C). Interestingly, none of these sequences showed FL activity beyond that seen with ICS-7. ICS-10 and ICS-12 exhibited 52.7% and 47.2% of the activity of ICS-7, respectively. However, ICS-9, ICS-11, ICS-13, and ICS-14 exhibited less than 10% of the activity of ICS-7. These data indicate that an additional sequence around ICS-7 is not required for activity and that the context of the sequence is likely critical for proper folding of the RNA sequence into a functional element.
Next, we sought to determine the minimal ICS-7 sequence required for IRES function. Shorter fragments of ICS-7 as well as the reverse complement of ICS-7 (ICS-7RC) were cloned into the bicistronic dual luciferase reporter construct and analyzed for FL expression (Fig. 4D). None of the sequences supported comparable FL expression to ICS-7. Importantly, the reverse complement of ICS-7 (ICS-7RC) exhibited diminished activity relative to ICS-7. The ICS-7RC exhibited only 26% of the activity of ICS-7, whereas the reverse complement of the KSHV-IRES (KSHV-RC) exhibited 52% of the activity of the KSHV-IRES (Fig. 4D). Our results indicate that ICS-7 is the optimal sequence element required for expression of the downstream cistron. While our results do not exclude other mechanisms of alternative translation initiation, they are consistent with this element functioning as an IRES.
Transcripts originating from the dual luciferase vector are full length, and ICS-7 does not have inherent promoter activity.
Possible alternatives to our interpretation that downstream cistron expression results from internal ribosome entry include the presence of monocistronic UL138 transcripts resulting from (i) alternative splicing or RNA fragmentation or (ii) cryptic promoter activity. To rule out these possibilities, we examined the length of transcripts generated in HEK-293 cells transfected with the bicistronic luciferase vector by Northern blotting. A radiolabeled anti-sense RNA probe specific to FL was hybridized to total RNA isolated from HEK-293 cells transfected with the empty bicistronic luciferase construct or the constructs containing PV-IRES, ICS-7, or ICS-7RC. RNA from untransfected cells was used as a negative control (NC). We detected a predominant reactive transcript corresponding to the predicted size of each full-length bicistronic luciferase transcript (Fig. 5A). Smaller transcripts unique to the ICS-7 construct that could independently result in FL activity were not detected. These results were confirmed by using qRT-PCR to demonstrate equivalent levels of RL and FL transcripts for each of the bicistronic constructs compared to cDNA derived from in vitro-transcribed RNA (data not shown), suggesting that monocistronic transcripts are not responsible for expression of the FL cistron.
FIG. 5.

Downstream cistron expression is not due to the presence of monocistronic transcripts specific to ICS-7. (A) Total RNA isolated from HEK-293 cells untransfected (NC) or transfected with the indicated bicistronic constructs were analyzed by Northern blotting using a radiolabeled antisense probe specific to FL (top). The expected transcript sizes were 2.7 kb (Empty), 3.4 kb (PV-IRES), 3.4 kb (ICS-7), and 3.4 kb (ICS-7RC). 18S and 28S rRNAs were stained with methylene blue and served as a loading control (bottom). (B) iv-RNAs generated from linearized bicistronic constructs were translated in vitro using RRL. IRES activity is expressed as the ratio of FL to RL. (C) A promoterless expression plasmid (pGL3-Basic), depicted in the schematic, was used to assay for promoter activity of ICS compared to that of the HCMV major immediate-early promoter (MIEP1400), the KSHV-IRES, or an empty vector. FL activity was measured, and expression relative to the MIEP1400 is indicated below the graph.
Previously, in vitro transcription and translation assays have been used to rule out the possibility of promoter activity in testing the existence of an IRES. We analyzed the ability of full-length bicistronic luciferase iv-RNA containing ICS-7 as a template for expression of the downstream FL cistron. The empty vector or two ICS sequences that were either determined to be negative (ICS-9) or positive (ICS-10) for IRES activity during our initial IRES screen were used as controls. Transcripts were translated in vitro using rabbit reticulocyte lysates (RRL). Similar to the IRES activity measured by DNA transfection (Fig. 4), ICS-7 and ICS-10 supported expression of the downstream cistron, whereas only modest levels of FL were supported by ICS-9 (Fig. 5B). These data suggest that cryptic promoter activity or splicing cannot explain FL expression, as shown in Fig. 4.
We also tested ICS-7 directly for promoter activity by cloning it upstream of FL in a promoterless plasmid relative to the HCMV major immediate-early promoter (MIEP1400). The empty plasmid and a plasmid containing the KSHV-IRES were used as negative controls. In addition to ICS-7, we included ICS-4 and ICS-7RC in our analysis, since these sequences did not stimulate expression of the downstream cistron, as shown in Fig. 4. FL activity was measured 48 h after transfection of HEK-293 cells. Relative to the MIEP1400, ICS-7 did not promote significant FL activity (Fig. 5C). Instead, ICS-7 FL activity was similar to those of the KSHV-IRES and HCMV sequences that did not exhibit IRES activity in bicistronic assays. The FL activity from each of these constructs was 0.02 to 0.05% of MIEP1400 activity. It is therefore unlikely that ICS-7 facilitates gene expression through promoter function.
ICS-7 Mfold prediction suggests extensive RNA secondary structure.
IRES sequences have extensive RNA secondary structures that allow for the recruitment of the translation machinery. We analyzed the RNA secondary structure of ICS-7 using Mfold, a computer-based RNA modeling program (Fig. 6) (77). We used the p-num function to identify highly favorable structures. The shaded stem loops represent predicted highly stable structures (−17 Kcal/mol) with p-num scores that correspond to 95.8 to 99.8% probability that these bases will pair only with each other. The likelihood of these structures forming was considerably higher than that of other structures within the same molecule, independent of whether only ICS-7 or the entire 3.6- or 2.7-kb transcripts were folded (data not shown). With the exception of a single base pair substitution at the 5′ end of ICS within structure ii (Fig. 6), the ICS-7 sequence is highly conserved among all HCMV strains for which a sequence is known (data not shown). These results support the idea that the hairpins are structurally conserved in the mRNA molecule and may be involved in IRES function (78). Our primary sequence analysis also revealed a region containing a 15-base polypyrimidine tract (bold line) with a polypyrimidine tract binding protein (PTB) consensus sequence (bold) (43, 50). The polypyrimidine tract is conserved with 100% identity in all HCMV strain sequences available through NCBI. Polypyrimidine tracts have been found in many IRES elements and may facilitate PTB binding to stabilize the mRNA and then recruitment of ribosomes (36). This region is relatively unstable (−7.6 Kcal/mol) compared to the conserved hairpins. The presence of highly stable, conserved hairpins, combined with a polypyrimidine tract, supports our hypothesis that this region functions as an IRES.
FIG. 6.

Predicted RNA secondary structure of the HCMV IRES. Secondary structure predictions were created using Mfold. The Mfold p-num option was used to identify highly favorable structures. The free energies of the three highly predictable (shaded) hairpins are shown. Each of the featured hairpins (i to iii) is enlarged. Hairpin i contains a polypyrimidine tract (bold line) with a conserved PTB binding consensus sequence (bold) (43, 50). Hairpin iii contains a predicted pseudoknot between nucleotide bases at the base of the hairpin (bold) with those in the loop of the hairpin. Start codons for full-length and truncated versions of pUL136 are indicated with * and ⋄, respectively.
ICS-7 is stress-inducible.
Alternative mechanisms of translation initiation are often active during cellular stress when canonical translation is repressed. This allows for a select subset of mRNAs to be expressed during stress. Therefore, we investigated the ability of ICS-7 to maintain translation in the presence of stress in a cell type that is permissive for HCMV infection. Bicistronic constructs containing either no IRES (empty), the PV-IRES, or ICS-7 were nucleofected into MRC5 fibroblasts that were fed under normal growth conditions or serum stressed for 18 h prior to nucleofection, and luciferase levels were quantitated 6 h later. ICS-7 IRES activity was increased approximately 2-fold under serum-stressed conditions relative to fed conditions, whereas the PV-IRES was unaffected by serum stress (Fig. 7). These results suggest that ICS-7 may act as an inducible IRES in a context-specific manner.
FIG. 7.

ICS-7 is stress inducible. Lysates derived from MRC5 fibroblasts grown under either fed or serum-stressed conditions were nucleofected with the bicistronic constructs described in Fig. 4 and analyzed for downstream FL cistron expression. IRES activity is expressed as a ratio of FL to RL activity.
UL138 cistron expression from the 3.6- and 2.7-kb transcripts, but not the 1.4-kb transcript, is stress inducible.
Next, we analyzed the impact of serum stress on UL138 cistron expression from the 3.6-, 2.7-, or 1.4-kb cDNAs in which we substituted UL138 with FL as a reporter gene (Fig. 8A). MRC5 fibroblasts were cultured under fed or serum-stressed conditions for 18 h and then nucleofected with each pFL-cDNA construct. Lysates were analyzed for FL expression 6 h later, and FL expression under starved conditions was normalized to FL expression under fed conditions. Consistent with the ability of ICS-7 to induce expression of a downstream cistron (Fig. 7), we demonstrated enhanced FL expression from the 3.6-kb (pFL-3.6) and 2.7-kb (pFL-2.7) cDNAs. FL expression was induced 2.14- and 9.43-fold from the 3.6- and 2.7-kb cDNAs, respectively, whereas FL expression from the pFL-1.4 cDNA decreased ∼2-fold in serum-stressed cells (Fig. 8B). Similar to FL expression from the pFL 1.4-kb cDNA, FL expression was decreased (∼2-fold) from a monocistronic construct encoding only FL under serum-starved conditions, consistent with the inhibition of cap-dependent translation. From these data, we conclude that FL is expressed by a canonical mechanism from the pFL-1.4 transcript, whereas FL is expressed by alternative IRES-like mechanisms from the pFL-3.6 and pFL-2.7 cDNAs. Taken together, these results suggest that pUL138 expression is ensured under a variety of cellular contexts and that UL138 cistron expression from the pFL-3.6 and pFL-2.7 cDNAs is inducible under stress.
FIG. 8.

IRES-mediated expression from the UL138 cistron is enhanced during stress. (A) Schematic for the generation of UL138 cDNAs with FL substituted as a marker for UL138 expression. cDNA constructs for each transcript were cloned into pEF1/myc-His-B (pFL-3.6, pFL-2.7, pFL-1.4). The location of the IRES is marked. (B) MRC5 cells were cultured for 18 h in fed or stressed conditions and nucleofected with molar equivalents of pFL-cDNA constructs and analyzed for FL expression 6 h postnucleofection. FL expression during stressed conditions was normalized to expression during fed conditions. (C) qRT-PCR analysis of RNA isolated from lysates described in panel B. Inverse crossing points (1/Ct) were determined using the second derivative max method (graphed). Fold change compares amplification of the 5′ and 3′ ends of experimental cDNAs isolated from transfected cells to that of cDNA derived from full-length iv-RNA. Transcripts with fold changes ≤2 compared to iv-RNA were considered to be full length. Similarly, the 5′ and 3′ ends of transcripts derived from fed and stressed cells were compared for each cDNA construct. Values ≤2 indicate that equal numbers of 5′ and 3′ transcript ends were present under fed and stressed conditions. (D) Linearized pFL-3.6, pFL-2.7, and pFL-1.4 cDNAs or a vector encoding FL alone (pFL) were transcribed and translated in vitro. The resulting FL expression was quantified. (E) Linearized pFL-2.7 and pFL-1.4 or empty vector were transcribed in vitro, and RNA was transfected into HEK-293 cells. Protein lysates were analyzed 6 h later for FL expression.
In parallel, we isolated RNA from cells nucleofected under serum-stressed or fed conditions and used qRT-PCR to confirm that the increase in FL expression measured from the pFL-3.6 and pFL-2.7 cDNAs in Fig. 8B was not due to (i) increased transcript levels or (ii) shorter, monocistronic transcripts specific to serum stress. We quantitated the 5′ and 3′ ends of transcripts generated after nucleofection by using qRT-PCR, as we did for the experiment shown in Fig. 3C. Our results demonstrate equivalent levels of 5′ and 3′ ends under fed or stressed conditions for each pFL-cDNA (e.g., stressed/fed) (Fig. 8C), suggesting that increased FL expression from pFL-3.6 and pFL-2.7 is not due to an increase in FL transcripts in the stressed condition. Additionally, we measured a ≤2-fold change in amplification of 5′ and 3′ transcript ends relative to cDNA derived from iv-RNA for all samples (see Materials and Methods), with the exception of pFL-3.6 (stressed) and pFL-1.4 (fed) (Fig. 8C). These data suggest that most transcripts are full length and independent of cellular context. The two points outside the 2-fold confidence interval indicate that there are a greater number of 5′ ends relative to 3′ ends and therefore cannot account for the increased FL expression. Importantly, equivalent 5′ and 3′ transcript ends in the pFL-2.7 fed and stressed samples indicate that the 9-fold induction of FL expression (Fig. 8B) is not due to changes in transcription.
To confirm that our cDNA nucleofection results are reflective of bona fide IRES activity, we analyzed full-length RNAs transcribed in vitro for their ability to support expression from the downstream UL138 cistron. RNA was transcribed in vitro using linearized pFL-cDNA (Fig. 8A) as a template, and then equal molar equivalents were either translated in RRL (Fig. 8D) or transfected into HEK-293 cells (Fig. 8E). Levels of FL expression from the pFL-2.7 and pFL-1.4 cDNAs in the RRL in vitro translation system were comparable (Fig. 8D). In contrast, FL expression from the pFL-3.6 cDNA was minimal, consistent with previous experiments. Similarly, when the pFL-2.7 and pFL-1.4 iv-RNAs were transfected into cells, similar levels of FL expression were measured (Fig. 8E). Taken together, these results suggest that the pFL-2.7 and pFL-1.4 cDNAs function efficiently as templates for translation. These data further support our interpretation that translation of the UL138 cistron from the pFL-3.6 or pFL-2.7 cDNAs requires alternative mechanisms of translation initiation and cannot be explained by the presence of monocistronic RNAs encoding the UL138 cistron.
DISCUSSION
The coding potential of the ULb′ region of the HCMV genome unique to clinical virus isolates has been explored for only a small number of ORFs, as this region is nonessential for lytic virus replication in culture. Genes in the ULb′ region likely represent viral adaptations important for latency and persistence in the host. Accordingly, pUL138 is required for the latent infection in CD34+ HPCs infected in vitro (22, 52). pUL138 is encoded at the 3′ end of three 3′ coterminal transcripts (Fig. 1). We have determined that these transcripts are polycistronic and encode three proteins in addition to pUL138 (Fig. 2 and 3). The presence of multiple coding regions upstream of UL138 presents a significant challenge for the translation of pUL138, which likely requires alternative mechanisms of translation initiation for expression. We identified a 663-nt sequence (ICS-7) within the 5′ end of UL136 that facilitates the translation of a downstream cistron in a bicistronic reporter assay (Fig. 4). This sequence does not function to produce a monocistronic UL138 transcript through promoter activity or splicing (Fig. 5). Interestingly, ICS-7-mediated translation was stress inducible (Fig. 7 and 8B). Therefore, expression of the UL138 cistron was induced from the 3.6- and 2.7-kb transcripts during stress, whereas expression from the 1.4-kb transcript was inhibited (Fig. 8B). These results suggest that the UL138 transcripts coordinate the expression of multiple proteins (pUL133, pUL135, pUL136) in addition to pUL138 and that the expression of pUL138 is ensured under multiple cellular contexts. The coordinated expression of these proteins likely has important implications for the function of pUL138 in regulating HCMV latent infection.
IRES-mediated translation was first described for poliovirus and encephalomyocarditis virus (EMCV) RNAs (33, 49) and has since been described for a number of other viral and cellular transcripts (16, 25, 44, 65). Classifying an RNA sequence as an IRES must satisfy several experimental criteria to rule out other possible reasons for expression of a downstream cistron. We have carefully evaluated transcripts generated from our bicistronic luciferase constructs and UL138 cDNAs to rule out the existence of monocistronic RNAs resulting from (i) a cryptic promoter or (ii) cryptic splicing events or fragmentation that could result in protein translation even in the absence of a 5′ cap. ICS-7 did not support FL expression in a promoterless vector relative to a bona fide promoter (Fig. 5C). Consistent with this data, and also providing evidence against cryptic splicing or RNA fragmentation, we were not able to detect smaller monocistronic transcripts originating from ICS-7 by Northern blotting (Fig. 5A) or qRT-PCR transcript analysis from cDNA transfections (Fig. 3C and Fig. 8C). Further, iv-RNAs containing ICS-7 supported expression of the downstream cistron in in vitro translation assays (Fig. 5B and 8D) or when transfected into cells (Fig. 8E). Taken together, these results support the conclusion that ICS-7 functions as an IRES to mediate protein expression through internal initiation of translation. However, in transfected cells compromised for canonical translation by cleavage of the 4G translation initiation factor (eIF4G), expression from ICS-7 was reduced (data not shown). The dependence on eIF4G suggests that ICS-7 more closely resembles a cellular IRES rather than the original 4G-indepenent IRES elements described for RNA viruses, such as poliovirus.
An intriguing finding of our studies is the stress-inducible nature of the UL138 IRES. Alternative mechanisms of translation are typically active during cellular stress (15, 19, 24, 29), mitosis (10, 56, 63), and apoptosis (28, 29, 40), when canonical translation initiation is inhibited (19). Our studies demonstrate that ICS-7 mediates FL expression from the bicistronic luciferase construct (Fig. 4) and UL138 cistron expression from the 3.6- and 2.7-kb transcripts via an alternative mechanism of translation initiation that is enhanced under stress conditions (Fig. 8B). Translation from other canonical transcripts, including the 1.4-kb transcript, was inhibited within the same cellular context (Fig. 8B). This result is interesting given the fact that the 1.4-kb transcript encoded a truncated form of pUL136 in addition to pUL138, indicating that this transcript is also polycistronic. Our data suggest translation of pUL138 from the 1.4-kb transcript. This may require translation reinitiation or readthrough. Of note, pUL138 is the only protein on the UL138 transcripts that is in an optimal context for translation with a Kozak consensus sequence at the AUG. The fact that the UL138 cistron appears to be optimally expressed from different transcripts depending on the cellular context implies that the multiple polycistronic transcripts represent a viral strategy to ensure the expression of pUL138.
Activation of a cellular IRES element is facilitated through interactions with a class of RNA binding proteins known as IRES trans-acting factors (ITAFs) (27, 40, 43, 66). ITAFs provide an additional level of regulation for IRES-mediated expression through specific ITAF relocalization (39, 66) or cell type specificity (12, 45, 54). In support of this hypothesis, some mRNAs may require a “nuclear experience” to acquire association with particular nuclear ITAFs required for IRES activity (64). The requirement of the nuclear experience may explain why some cytoplasmic RNA virus IRES elements exhibit greater activity relative to cellular IRES or DNA virus IRES elements following RNA transfection into cells (16, 67). The context specificity of ICS-7 activity is highlighted by the induction of downstream cistron expression from both the bicistronic luciferase construct and the pFL cDNAs under serum stress (Fig. 7 and 8B). These results are consistent with the hypothesis that the HCMV IRES requires a nuclear experience and association with a stress-associated ITAF. Polypyrimidine tract binding protein (PTB) is an attractive ITAF that may regulate ICS-7 function, as a PTB binding consensus sequence exists within ICS-7 (Fig. 6). Further, recent work demonstrates that PTB localization is temporally regulated during HCMV infection (18) and inhibits HCMV major immediate-early gene expression by regulating alternative splicing of MIEP IE products (11).
Interestingly, polycistronic transcripts generated from latency- associated regions of herpesviruses are not uncommon and may represent a conserved strategy for the regulation of protein expression during latency (4, 8, 23, 31, 41, 69, 70). For example, Kaposi's sarcoma-associated herpesvirus (KSHV) encodes a cluster of gene products, LANA (ORF73), v-cyclin (ORF72), and v-flip (ORF71), that are important for latency (1, 48). Protein expression of v-flip is mediated by an IRES element that overlaps the upstream v-cyclin ORF (4, 23, 41). Similarly, the HCMV IRES we describe overlaps the UL136 ORF upstream of UL138. The overlap between these ORFs and IRES sequences is not surprising given the limited intergenic space in the herpesviral genomes and the need to maximize viral genome coding capacity (47). The Epstein-Barr virus (EBV) nuclear antigen-1 (EBNA1), the only nuclear protein expressed during all types of EBV latency, is also expressed via an IRES which is present in the U leader exon inherent to all EBNA1 transcripts regardless of the promoter utilized (31). Further, the translation of immediate-early proteins RLORF9 (70) and pp14 (69), proteins expressed during Marek's disease virus infection, and the immune evasion protein, MK3, expressed during murine gammaherpesvirus 68 infection (8), is mediated by IRES elements. Collectively, these findings demonstrate an attractive mechanism by which herpesviruses tightly coordinate the expression of multiple proteins required for complex host-virus interactions that impact the outcome of infection.
Our data indicate that pUL136 may be expressed in two forms, full length and truncated, depending on the translation template and the context of expression. While both forms of pUL136 are expressed during infection (Fig. 2B), only the truncated form can be detected during transfection of the cDNAs containing UL136 (Fig. 2A and 3A). This finding suggests that expression of full-length pUL136 may be dependent on other viral factors or infection-induced cellular factors. Strikingly, full-length UL136 is not a 5′ cistron on any of the transcripts characterized. The mechanisms allowing for expression of full-length pUL136 are unknown. Our experiments did not detect an additional upstream IRES (Fig. 4) that could allow full-length pUL136 expression, although a more exhaustive analysis of overlapping fragments 5′ of UL136 will be required to fully explore this possibility. It is tempting to hypothesize a role for the truncated pUL136 in regulating the expression or activity of full-length pUL136. Consistent with this speculation, lymphoid enhancer factor-1 (LEF-1) is translated in two forms, a full-length protein translated via IRES-mediated cap-independent mechanisms and a truncated form translated from a shorter transcript by canonical cap-dependent mechanisms (35). The truncated form of LEF-1 inhibits the growth-promoting activity of full-length LEF-1 (30). Ongoing work is focused on understanding the role of each UL138 transcript and the different forms of pUL136 in regulating the outcome of HCMV infection.
Our work demonstrates the first requirement of an alternative mechanism of translation initiation for expression of an HCMV protein important to latency. Further, our work demonstrates the novel coding potential of three additional ULb′ proteins, pUL133, pUL135, and pUL136. The existence of multiple polycistronic transcripts and the possibility of multiple mechanisms to ensure the expression of pUL138 have important implications for latency. The UL138 transcripts likely serve to coordinate the expression of UL133 to UL138. The relative abundance of pUL133, pUL135, pUL136, and pUL138 expressed during infection may vary depending on the relative abundance of individual UL138 transcripts and the context of infection. We postulate that unique subsets of these proteins, or complexes of these proteins, may be formed in myeloid lineage cells to promote the latent infection. Further, the function of these proteins in infected cells may rely on interaction with cell type-specific factors. The profile of expression and the interactions between these ULb′ proteins and with other viral or cellular proteins during HCMV latency are the focus of ongoing research.
Acknowledgments
We gratefully thank Nahum Sonenberg for pCDNA3-rLuc-polIRES-fLuc, Priscilla Schaffer and R. Bringhurst for the KSHV-positive primary effusion lymphoma (PEL) cell line BCBL-1, Tom Shenk for the HCMV IE1 and IE2 antibody, and Erica Roberts for technical assistance in constructing myc-tagged recombinant viruses. We acknowledge E. Katsanis and E. Hadley for their invaluable assistance in acquiring human hematopoietic cells. We further acknowledge M. Umashankar for critical reading of the manuscript.
The research described was supported by Public Health Service grants CA111343 and A1079059 to F.G. and AI026765 to B.L.S. from the National Institute of Allergy and Infectious Diseases (NIAID).
The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the NIAID or the National Institutes of Health.
F.G. is a Pew Scholar in Biomedical Sciences, supported by The Pew Charitable Trusts. K.D.F. is a predoctoral trainee of Public Health Service training grant AI007319.
L.G. and L.C. designed and executed experiments, analyzed the data, and wrote the paper. F.G. designed experiments, analyzed the data, and wrote the paper. K.D.F. and B.L.S. designed experiments, provided reagents, and provided essential conversation and feedback.
Footnotes
Published ahead of print on 30 June 2010.
REFERENCES
- 1.Allen, R. D. III, S. Dickerson, and S. H. Speck. 2006. Identification of spliced gammaherpesvirus 68 LANA and v-cyclin transcripts and analysis of their expression in vivo during latent infection. J. Virol. 80:2055-2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baird, S. D., M. Turcotte, R. G. Korneluk, and M. Holcik. 2006. Searching for IRES. RNA 12:1755-1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bego, M., J. Maciejewski, S. Khaiboullina, G. Pari, and S. St. Jeor. 2005. Characterization of an antisense transcript spanning the UL81-82 locus of human cytomegalovirus. J. Virol. 79:11022-11034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bieleski, L., and S. J. Talbot. 2001. Kaposi's sarcoma-associated herpesvirus vCyclin open reading frame contains an internal ribosome entry site. J. Virol. 75:1864-1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boeckh, M., W. G. Nichols, G. Papanicolaou, R. Rubin, J. R. Wingard, and J. Zaia. 2003. Cytomegalovirus in hematopoietic stem cell transplant recipients: current status, known challenges, and future strategies. Biol. Blood Marrow Transplant. 9:543-558. [DOI] [PubMed] [Google Scholar]
- 6.Bowen, E. F., P. D. Griffiths, C. C. Davey, V. C. Emery, and M. A. Johnson. 1996. Lessons from the natural history of cytomegalovirus. AIDS 10(Suppl. 1):S37-S41. [PubMed] [Google Scholar]
- 7.Cha, T. A., E. Tom, G. W. Kemble, G. M. Duke, E. S. Mocarski, and R. R. Spaete. 1996. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J. Virol. 70:78-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coleman, H. M., I. Brierley, and P. G. Stevenson. 2003. An internal ribosome entry site directs translation of the murine gammaherpesvirus 68 MK3 open reading frame. J. Virol. 77:13093-13105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coll, O., G. Benoist, Y. Ville, L. E. Weisman, F. Botet, M. M. Anceschi, A. Greenough, R. S. Gibbs, and X. Carbonell-Estrany. 2009. Guidelines on CMV congenital infection. J. Perinat. Med. 37:433-445. [DOI] [PubMed] [Google Scholar]
- 10.Cornelis, S., Y. Bruynooghe, G. Denecker, S. Van Huffel, S. Tinton, and R. Beyaert. 2000. Identification and characterization of a novel cell cycle-regulated internal ribosome entry site. Mol. Cell 5:597-605. [DOI] [PubMed] [Google Scholar]
- 11.Cosme, R. S., Y. Yamamura, and Q. Tang. 2009. Roles of polypyrimidine tract binding proteins in major immediate-early gene expression and viral replication of human cytomegalovirus. J. Virol. 83:2839-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Creancier, L., D. Morello, P. Mercier, and A. C. Prats. 2000. Fibroblast growth factor 2 internal ribosome entry site (IRES) activity ex vivo and in transgenic mice reveals a stringent tissue-specific regulation. J. Cell Biol. 150:275-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dolan, A., C. Cunningham, R. D. Hector, A. F. Hassan-Walker, L. Lee, C. Addison, D. J. Dargan, D. J. McGeoch, D. Gatherer, V. C. Emery, P. D. Griffiths, C. Sinzger, B. P. McSharry, G. W. Wilkinson, and A. J. Davison. 2004. Genetic content of wild-type human cytomegalovirus. J. Gen. Virol. 85:1301-1312. [DOI] [PubMed] [Google Scholar]
- 14.Dosa, R., K. Burian, and E. Gonczol. 2005. Human cytomegalovirus latency is associated with the state of differentiation of the host cells: an in vitro model in teratocarcinoma cells. Acta Microbiol. Immunol. Hung. 52:397-406. [DOI] [PubMed] [Google Scholar]
- 15.Fernandez, J., I. Yaman, R. Mishra, W. C. Merrick, M. D. Snider, W. H. Lamers, and M. Hatzoglou. 2001. Internal ribosome entry site-mediated translation of a mammalian mRNA is regulated by amino acid availability. J. Biol. Chem. 276:12285-12291. [DOI] [PubMed] [Google Scholar]
- 16.Fitzgerald, K. D., and B. L. Semler. 2009. Bridging IRES elements in mRNAs to the eukaryotic translation apparatus. Biochim. Biophys. Acta 1789:518-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fraser, C. S., and J. A. Doudna. 2007. Structural and mechanistic insights into hepatitis C viral translation initiation. Nat. Rev. Microbiol. 5:29-38. [DOI] [PubMed] [Google Scholar]
- 18.Gaddy, C. E., D. S. Wong, A. Markowitz-Shulman, and A. M. Colberg-Poley. 2010. Regulation of the subcellular distribution of key cellular RNA processing factors during permissive human cytomegalovirus infection. J. Gen. Virol. 91:1547-1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilbert, W. V., K. Zhou, T. K. Butler, and J. A. Doudna. 2007. Cap-independent translation is required for starvation-induced differentiation in yeast. Science 317:1224-1227. [DOI] [PubMed] [Google Scholar]
- 20.Goodrum, F., C. T. Jordan, S. S. Terhune, K. High, and T. Shenk. 2004. Differential outcomes of human cytomegalovirus infection in primitive hematopoietic cell subpopulations. Blood 104:687-695. [DOI] [PubMed] [Google Scholar]
- 21.Goodrum, F. D., C. T. Jordan, K. High, and T. Shenk. 2002. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: a model for latency. Proc. Natl. Acad. Sci. U. S. A. 99:16255-16260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goodrum, F. D., M. Reeves, J. Sinclair, K. High, and T. Shenk. 2007. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 110:937-945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grundhoff, A., and D. Ganem. 2001. Mechanisms governing expression of the v-FLIP gene of Kaposi's sarcoma-associated herpesvirus. J. Virol. 75:1857-1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harding, H. P., I. Novoa, Y. Zhang, H. Zeng, R. Wek, M. Schapira, and D. Ron. 2000. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6:1099-1108. [DOI] [PubMed] [Google Scholar]
- 25.Hellen, C. U., and P. Sarnow. 2001. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev. 15:1593-1612. [DOI] [PubMed] [Google Scholar]
- 26.Hershey, J., and W. Merrick. 2000. Pathway and mechanism of initiation of protein synthesis. In N. Sonenberg, J. W. B. Hershey, and M. Mathews (ed.), Translational control of gene expression. Cold Spring Harbor Laboratory Press, New York, NY.
- 27.Holcik, M., B. W. Gordon, and R. G. Korneluk. 2003. The internal ribosome entry site-mediated translation of antiapoptotic protein XIAP is modulated by the heterogeneous nuclear ribonucleoproteins C1 and C2. Mol. Cell Biol. 23:280-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holcik, M., N. Sonenberg, and R. G. Korneluk. 2000. Internal ribosome initiation of translation and the control of cell death. Trends Genet. 16:469-473. [DOI] [PubMed] [Google Scholar]
- 29.Holcik, M., C. Yeh, R. G. Korneluk, and T. Chow. 2000. Translational upregulation of X-linked inhibitor of apoptosis (XIAP) increases resistance to radiation induced cell death. Oncogene 19:4174-4177. [DOI] [PubMed] [Google Scholar]
- 30.Hovanes, K., T. W. Li, J. E. Munguia, T. Truong, T. Milovanovic, J. Lawrence Marsh, R. F. Holcombe, and M. L. Waterman. 2001. Beta-catenin-sensitive isoforms of lymphoid enhancer factor-1 are selectively expressed in colon cancer. Nat. Genet. 28:53-57. [DOI] [PubMed] [Google Scholar]
- 31.Isaksson, A., M. Berggren, and A. Ricksten. 2003. Epstein-Barr virus U leader exon contains an internal ribosome entry site. Oncogene 22:572-581. [DOI] [PubMed] [Google Scholar]
- 32.Jabbour, M., E. M. Campbell, H. Fares, and L. Lybarger. 2009. Discrete domains of MARCH1 mediate its localization, functional interactions, and posttranscriptional control of expression. J. Immunol. 183:6500-6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jang, S. K., H. G. Krausslich, M. J. Nicklin, G. M. Duke, A. C. Palmenberg, and E. Wimmer. 1988. A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J. Virol. 62:2636-2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jenkins, C., A. Abendroth, and B. Slobedman. 2004. A novel viral transcript with homology to human interleukin-10 is expressed during latent human cytomegalovirus infection. J. Virol. 78:1440-1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jimenez, J., G. M. Jang, B. L. Semler, and M. L. Waterman. 2005. An internal ribosome entry site mediates translation of lymphoid enhancer factor-1. RNA 11:1385-1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kafasla, P., N. Morgner, T. A. Poyry, S. Curry, C. V. Robinson, and R. J. Jackson. 2009. Polypyrimidine tract binding protein stabilizes the encephalomyocarditis virus IRES structure via binding multiple sites in a unique orientation. Mol. Cell 34:556-568. [DOI] [PubMed] [Google Scholar]
- 37.Kochetov, A. V. 2008. Alternative translation start sites and hidden coding potential of eukaryotic mRNAs. Bioessays 30:683-691. [DOI] [PubMed] [Google Scholar]
- 38.Kochetov, A. V., S. Ahmad, V. Ivanisenko, O. A. Volkova, N. A. Kolchanov, and A. Sarai. 2008. uORFs, reinitiation and alternative translation start sites in human mRNAs. FEBS Lett. 582:1293-1297. [DOI] [PubMed] [Google Scholar]
- 39.Lewis, S. M., and M. Holcik. 2008. For IRES trans-acting factors, it is all about location. Oncogene 27:1033-1035. [DOI] [PubMed] [Google Scholar]
- 40.Lewis, S. M., and M. Holcik. 2005. IRES in distress: translational regulation of the inhibitor of apoptosis proteins XIAP and HIAP2 during cell stress. Cell Death Differ. 12:547-553. [DOI] [PubMed] [Google Scholar]
- 41.Low, W., M. Harries, H. Ye, M. Q. Du, C. Boshoff, and M. Collins. 2001. Internal ribosome entry site regulates translation of Kaposi's sarcoma-associated herpesvirus FLICE inhibitory protein. J. Virol. 75:2938-2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maciejewski, J. P., E. E. Bruening, R. E. Donahue, E. S. Mocarski, N. S. Young, and S. C. St. Jeor. 1992. Infection of hematopoietic progenitor cells by human cytomegalovirus. Blood 80:170-178. [PubMed] [Google Scholar]
- 43.Majumder, M., I. Yaman, F. Gaccioli, V. V. Zeenko, C. Wang, M. G. Caprara, R. C. Venema, A. A. Komar, M. D. Snider, and M. Hatzoglou. 2009. The hnRNA-binding proteins hnRNP L and PTB are required for efficient translation of the Cat-1 arginine/lysine transporter mRNA during amino acid starvation. Mol. Cell. Biol. 29:2899-2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martinez-Salas, E., R. Ramos, E. Lafuente, and S. Lopez de Quinto. 2001. Functional interactions in internal translation initiation directed by viral and cellular IRES elements. J. Gen. Virol. 82:973-984. [DOI] [PubMed] [Google Scholar]
- 45.Merrill, M. K., E. Y. Dobrikova, and M. Gromeier. 2006. Cell-type-specific repression of internal ribosome entry site activity by double-stranded RNA-binding protein 76. J. Virol. 80:3147-3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mocarski, E. S., T. Shenk, and R. F. Pass. 2007. Cytomegaloviruses. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 5th ed., vol. 1. Lippincott Williams & Wilkins, Philadelphia, PA.
- 47.Murphy, E., D. Yu, J. Grimwood, J. Schmutz, M. Dickson, M. A. Jarvis, G. Hahn, J. A. Nelson, R. M. Myers, and T. E. Shenk. 2003. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc. Natl. Acad. Sci. U. S. A. 100:14976-14981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pearce, M., S. Matsumura, and A. C. Wilson. 2005. Transcripts encoding K12, v-FLIP, v-cyclin, and the microRNA cluster of Kaposi's sarcoma-associated herpesvirus originate from a common promoter. J. Virol. 79:14457-14464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pelletier, J., and N. Sonenberg. 1988. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature 334:320-325. [DOI] [PubMed] [Google Scholar]
- 50.Perez, I., C. H. Lin, J. G. McAfee, and J. G. Patton. 1997. Mutation of PTB binding sites causes misregulation of alternative 3′ splice site selection in vivo. RNA 3:764-778. [PMC free article] [PubMed] [Google Scholar]
- 51.Pestova, T. V., V. G. Kolupaeva, I. B. Lomakin, E. V. Pilipenko, I. N. Shatsky, V. I. Agol, and C. U. Hellen. 2001. Molecular mechanisms of translation initiation in eukaryotes. Proc. Natl. Acad. Sci. U. S. A. 98:7029-7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petrucelli, A., M. Rak, L. Grainger, and F. Goodrum. 2009. Characterization of a novel Golgi apparatus-localized latency determinant encoded by human cytomegalovirus. J. Virol. 83:5615-5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pfaffl, M. W. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pilipenko, E. V., T. V. Pestova, V. G. Kolupaeva, E. V. Khitrina, A. N. Poperechnaya, V. I. Agol, and C. U. Hellen. 2000. A cell cycle-dependent protein serves as a template-specific translation initiation factor. Genes Dev. 14:2028-2045. [PMC free article] [PubMed] [Google Scholar]
- 55.Poulin, F., A. C. Gingras, H. Olsen, S. Chevalier, and N. Sonenberg. 1998. 4E-BP3, a new member of the eukaryotic initiation factor 4E-binding protein family. J. Biol. Chem. 273:14002-14007. [DOI] [PubMed] [Google Scholar]
- 56.Pyronnet, S., L. Pradayrol, and N. Sonenberg. 2000. A cell cycle-dependent internal ribosome entry site. Mol. Cell 5:607-616. [DOI] [PubMed] [Google Scholar]
- 57.Reeves, M. B., P. J. Lehner, J. G. Sissons, and J. H. Sinclair. 2005. An in vitro model for the regulation of human cytomegalovirus latency and reactivation in dendritic cells by chromatin remodelling. J. Gen. Virol. 86:2949-2954. [DOI] [PubMed] [Google Scholar]
- 58.Reeves, M. B., P. A. MacAry, P. J. Lehner, J. G. Sissons, and J. H. Sinclair. 2005. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc. Natl. Acad. Sci. U. S. A. 102:4140-4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ryabova, L. A., M. M. Pooggin, and T. Hohn. 2006. Translation reinitiation and leaky scanning in plant viruses. Virus Res. 119:52-62. [DOI] [PubMed] [Google Scholar]
- 60.Saffert, R. T., and R. F. Kalejta. 2007. Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J. Virol. 81:9109-9120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sarnow, P. 2003. Viral internal ribosome entry site elements: novel ribosome-RNA complexes and roles in viral pathogenesis. J. Virol. 77:2801-2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sarnow, P., R. C. Cevallos, and E. Jan. 2005. Takeover of host ribosomes by divergent IRES elements. Biochem. Soc. Trans. 33:1479-1482. [DOI] [PubMed] [Google Scholar]
- 63.Schepens, B., S. A. Tinton, Y. Bruynooghe, E. Parthoens, M. Haegman, R. Beyaert, and S. Cornelis. 2007. A role for hnRNP C1/C2 and Unr in internal initiation of translation during mitosis. EMBO J. 26:158-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Semler, B. L., and M. L. Waterman. 2008. IRES-mediated pathways to polysomes: nuclear versus cytoplasmic routes. Trends Microbiol. 16:1-5. [DOI] [PubMed] [Google Scholar]
- 65.Sonenberg, N., and A. G. Hinnebusch. 2007. New modes of translational control in development, behavior, and disease. Mol. Cell 28:721-729. [DOI] [PubMed] [Google Scholar]
- 66.Spriggs, K. A., M. Stoneley, M. Bushell, and A. E. Willis. 2008. Re-programming of translation following cell stress allows IRES-mediated translation to predominate. Biol. Cell 100:27-38. [DOI] [PubMed] [Google Scholar]
- 67.Stoneley, M., T. Subkhankulova, J. P. Le Quesne, M. J. Coldwell, C. L. Jopling, G. J. Belsham, and A. E. Willis. 2000. Analysis of the c-myc IRES; a potential role for cell-type specific trans-acting factors and the nuclear compartment. Nucleic Acids Res. 28:687-694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Streblow, D. N., S. L. Orloff, and J. A. Nelson. 2001. Do pathogens accelerate atherosclerosis? J. Nutr. 131:2798S-2804S. [DOI] [PubMed] [Google Scholar]
- 69.Tahiri-Alaoui, A., D. Matsuda, H. Xu, P. Panagiotis, L. Burman, L. S. Lambeth, L. Petherbridge, W. James, V. Mauro, and V. Nair. 2009. The 5′ leader of the mRNA encoding the Marek's disease virus serotype 1 pp14 protein contains an intronic internal ribosome entry site with allosteric properties. J. Virol. 83:12769-12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tahiri-Alaoui, A., L. P. Smith, S. Baigent, L. Kgosana, L. J. Petherbridge, L. S. Lambeth, W. James, and V. Nair. 2009. Identification of an intercistronic internal ribosome entry site in a Marek's disease virus immediate-early gene. J. Virol. 83:5846-5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vasto, S., G. Colonna-Romano, A. Larbi, A. Wikby, C. Caruso, and G. Pawelec. 2007. Role of persistent CMV infection in configuring T cell immunity in the elderly. Immun. Ageing 4:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang, X. Q., and J. A. Rothnagel. 2004. 5′-untranslated regions with multiple upstream AUG codons can support low-level translation via leaky scanning and reinitiation. Nucleic Acids Res. 32:1382-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Warming, S., N. Costantino, D. L. Court, N. A. Jenkins, and N. G. Copeland. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 33:e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yaman, I., J. Fernandez, H. Liu, M. Caprara, A. A. Komar, A. E. Koromilas, L. Zhou, M. D. Snider, D. Scheuner, R. J. Kaufman, and M. Hatzoglou. 2003. The zipper model of translational control: a small upstream ORF is the switch that controls structural remodeling of an mRNA leader. Cell 113:519-531. [DOI] [PubMed] [Google Scholar]
- 75.Yamasaki, S., and P. Anderson. 2008. Reprogramming mRNA translation during stress. Curr. Opin. Cell Biol. 20:222-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yu, D., H. M. Ellis, E. C. Lee, N. A. Jenkins, N. G. Copeland, and D. L. Court. 2000. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 97:5978-5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zuker, M. 2003. Mfold Web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31:3406-3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zuker, M., and A. B. Jacobson. 1995. “Well-determined” regions in RNA secondary structure prediction: analysis of small subunit ribosomal RNA. Nucleic Acids Res. 23:2791-2798. [DOI] [PMC free article] [PubMed] [Google Scholar]